
Targeting metabolic vulnerabilities to overcome resistance to therapy in acute myeloid leukemia
 0
0 ,
, Abstract
Malignant hematopoietic cells gain metabolic plasticity, reorganize anabolic mechanisms to improve anabolic output and prevent oxidative damage, and bypass cell cycle checkpoints, eventually outcompeting normal hematopoietic cells. Current therapeutic strategies of acute myeloid leukemia (AML) are based on prognostic stratification that includes mutation profile as the closest surrogate to disease biology. Clinical efficacy of targeted therapies, e.g., agents targeting mutant FMS-like tyrosine kinase 3 (FLT3) and isocitrate dehydrogenase 1 or 2, are mostly limited to the presence of relevant mutations. Recent studies have not only demonstrated that specific mutations in AML create metabolic vulnerabilities but also highlighted the efficacy of targeting metabolic vulnerabilities in combination with inhibitors of these mutations. Therefore, delineating the functional relationships between genetic stratification, metabolic dependencies, and response to specific inhibitors of these vulnerabilities is crucial for identifying more effective therapeutic regimens, understanding resistance mechanisms, and identifying early response markers, ultimately improving the likelihood of cure. In addition, metabolic changes occurring in the tumor microenvironment have also been reported as therapeutic targets. The metabolic profiles of leukemia stem cells (LSCs) differ, and relapsed/refractory LSCs switch to alternative metabolic pathways, fueling oxidative phosphorylation (OXPHOS), rendering them therapeutically resistant. In this review, we discuss the role of cancer metabolic pathways that contribute to the metabolic plasticity of AML and confer resistance to standard therapy; we also highlight the latest promising developments in the field in translating these important findings to the clinic and discuss the tumor microenvironment that supports metabolic plasticity and interplay with AML cells.
Keywords
INTRODUCTION
Acute myeloid leukemia (AML) is a cancer derived from the myeloid lineage of blood cells. It is characterized by overproduction of leukemic blasts and maturation arrest. With approximately 120,000 cases a year globally, AML is the most common type of acute leukemia in adults[1]. Although conventional chemotherapy is known to eliminate bulk tumor cells, its utility is limited in AML, as older patients lack tolerance to chemotherapy and the drug may lack efficacy at eliminating leukemia stem cells (LSCs). Residual clones that survive chemotherapy lead to disease relapse[2]. Moreover, AML exhibits extensive genetic heterogeneity, as it often harbors a complex mixture of heterogenous subclones that possess genetic aberrations in diverse driver genes, with distinct clonal evolutionary patterns existing in single individuals. Cytogenetic abnormalities and acquired somatic mutations are used as surrogate markers for risk assessment and some mutations can be specifically targeted. However, the utility of therapies that target mutations viz. FMS-like tyrosine kinase 3 (FLT3) and isocitrate dehydrogenase enzyme 2 (IDH2) are limited to the context of the particular mutation.
Cancer cells are known to reprogram their metabolism to support their survival and proliferation; this was recently recognized as a hallmark of cancer[3]. AML cells exhibit unique metabolic dependencies compared with those of their normal counterparts, the noncancerous blasts. Hematopoietic stem cells (HSCs) use mitochondrial oxidative phosphorylation (OXPHOS) during differentiation to meet their higher energy requirements; however, they use anaerobic glycolysis for their routine energy needs and generally avoid aerobic mitochondrial OXPHOS[4]. Of note, mitochondrial OXPHOS generates reactive oxygen species (ROS), which are deleterious for HSCs. In fact, ROS functions as a signaling mediator in the crosstalk between metabolism and stem cell fate decisions by inhibiting the repopulating capacity of HSCs[5]. ROS-high-HSCs are characterized by low self-renewal capacity and high myeloid differentiation capacity compared to ROS-low HSCs.
Upon cell division, HSCs have fate choices when they undergo cell division; they either undergo self-renewal, wherein they produce new HSCs, or differentiate, wherein they produce cells that mature into committed cells. On the basis of the status of the daughter cells, HSC division can occur in one of the following ways: (1) asymmetric division, which maintains the HSC pool, in which one daughter cell remains as stem cell while the other differentiates; (2) symmetric commitment, in which both daughter cells differentiate (stem cell exhaustion); or (3) symmetric division, in which two daughter stem cells are produced, which helps in HSC expansion. Interestingly, mitochondria distribution during stem cell division appears to be crucial to determining the fate of HSCs. Of note, mitochondrial division is asymmetrical during stem cell division and daughter cells that receive fewer old mitochondria maintain stem cell traits[6]. Notably, through a ROS-mediated physiological process, changes in mitochondrial dynamics regulate stem cell fate decisions, triggering a dual program to suppress self-renewal and promote differentiation[6,7]. Evasion from mitochondrial OXPHOS and reliance on glycolysis help prevent HSC pool exhaustion. In contrast, LSCs rely on mitochondrial OXPHOS to generate high-energy compounds while maintaining ROS levels at non-toxic levels by employing a wide repertoire mechanism for ROS mitigation[8]. Therefore, it may be possible to target leukemic cells differently based on their metabolic needs for therapeutic purposes.
In this review, we discuss the role of cancer metabolic pathways in AML development that contribute to the metabolic plasticity of the disease and confer resistance to standard therapy. We also highlight the latest developments in the field including clinical trials for translating metabolic inhibitors to clinic [Figure 1] and discuss the role of tumor microenvironment and extracellular vesicles in supporting metabolic plasticity and their interplay with AML cells.
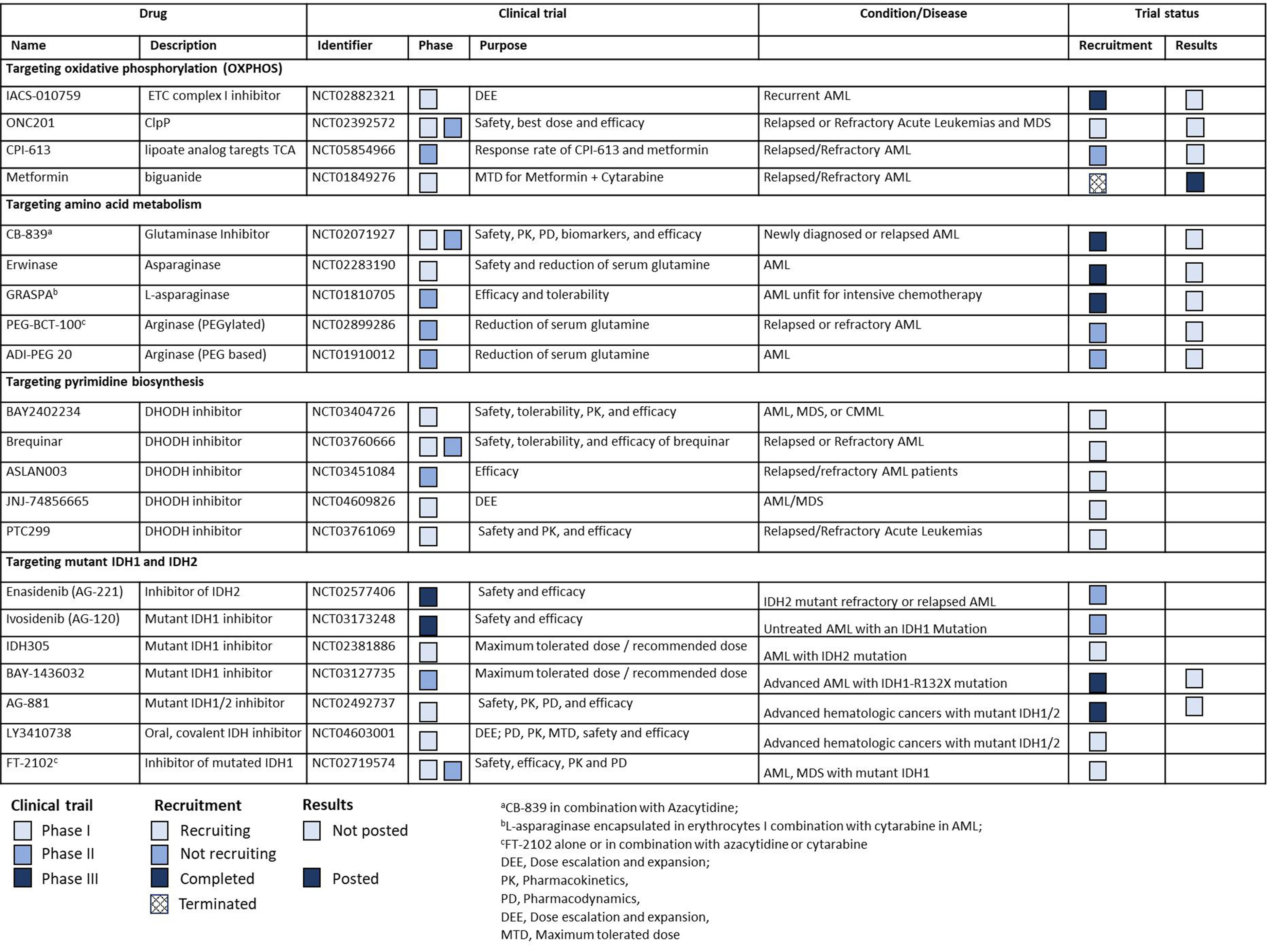
Figure 1. List of clinical trials for metabolic inhibitors in AML. AML: Acute myeloid leukemia; ClpP: caseinolytic protease proteolytic subunit; CMML: chronic myelomonocytic leukemia; DHODH: dihydroorotate dehydrogenase; ETC: electron transport chain; IDH: isocitrate dehydrogenase enzyme; MDS: myelodysplastic syndromes; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid cycle.
TARGETING MITOCHONDRIAL OXPHOS
AML cells are more sensitive to mitochondria-targeted drugs, likely because despite having increased respiratory activity, their mitochondria have lower coupling efficiency and spare reserve capacity due to proton leak[9]. In different cancers, including AML, depletion of mtDNA results in OXPHOS deficiency. OXPHOS-deficient cancer cells fail to form tumors unless mtDNA is restored from stromal cells via the horizontal transfer of whole mitochondria[10-13], suggesting that functional OXPHOS is essential for cancer development, although it is not clear which component of OXPHOS contributes the most to tumor formation. For instance, aspartate supplementation enables cells to proliferate under pharmacological inhibition of electron transport chain (ETC), suggesting that although adenosine triphosphate (ATP) production is the primary function of OXPHOS, proliferating cancer cells obtain oxidizing power and aspartate for pyrimidine biosynthesis through aerobic respiration[14,15], thus, in addition to ATP or ROS levels, there may be other mechanisms underlying the antiproliferative effects of ETC inhibition. Several compounds block complex I (i.e., NADH: ubiquinone oxidoreductase), with high affinity viz.
The OXPHOS inhibitor IACS-010759 is currently being evaluated in phase 1 clinical trials in relapsed/refractory AML [Figure 1][16]. BAY87-2243, an inhibitor of hypoxia-inducible factor-1 (HIF-1) that inhibits mitochondrial complex I activity, causes grade III nausea/vomiting [NCT01297530], while ME-344 showed no clinical efficacy in patients with solid cancers[18,20].
Oxidative phosphorylation integrity is maintained by a protease, caseinolytic protease proteolytic subunit (ClpP) in AML cells[21]. The altered function of this protease contributes to misfolding or degradation of respiratory chain subunits and their accumulation, causing respiratory chain dysfunction. The imipridones ONC201 and ONC212 inhibited the growth and viability of leukemia cells by hyperactivating mitochondrial ClpP and thus selectively proteolyzing certain mitochondrial matrix proteins[21]. A significant reduction in the leukemic burden and improved survival was observed in mice that were administered ONC201 orally[21]. In a clinical trial, a single oral dose of ONC201 resulted in a decrease in circulating blasts and a subsequent increase in platelet counts in patients with relapsed/refractory AML[21].
Mubritinib (TAK-165) is a direct and ubiquinone-dependent ETC complex I inhibitor which induces ROS accumulation. Mubritinib also decreases ATP/ADP and NAD/NADH concentration ratios and inhibits the activities of PDH, tricarboxylic acid cycle (TCA), and OXPHOS, which results in the death of AML cells treated with mubritinib[22]. In vivo, mubritinib delayed AML development without affecting normal hematopoiesis in mice[22]. An isoquinoline alkaloid, berberine, targets mitochondrial ETC complex I, leading to reduced mitochondrial activity and enhanced antileukemic effects in vitro and in vivo when combined with IDH1 mutant inhibitor AG-120[23].
A rationally designed lipoic acid analog, CPI-613, is currently being investigated in clinical trials for patients with pancreatic cancer and acute myeloid leukemia[24]. Mechanistically, CPI-613 inhibits the TCA cycle by displacing lipoic acid, a cofactor for the TCA cycle enzymes pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. This displacement, in turn, prevents the enzymes’ activity and halts the TCA cycle[25].
Metformin is being evaluated in combination with chemotherapy in different cancers; it sensitizes AML cells to Ara-C by reducing the mitochondrial transfer from bone marrow stromal cells (BMSCs) and OXPHOS in the recipient cells[26]. Metformin was found to synergistically sensitize AML cells to Ara-C by inhibiting the mammalian target of rapamycin (mTOR)C1/P70S6K pathway[27]. By promoting autophagy and apoptosis mediated by mTOR, metformin sensitizes FLT3-ITD-positive acute myeloid leukemia to sorafenib[28]. Additionally, metformin and phenformin are also potentiators of the cytotoxicity of Bcl2 antagonist - ABT-737 against leukemia[29]. By inhibiting protein production, metformin downregulates antiapoptotic Mcl-1 and Bcl-xl expression, resulting in a synergistic anti-tumor effect with ABT-199 in AML[30]. It also exhibited anticancer activity with the MCL1 inhibitor S63845 via redox remodeling[31]. IM156 is another oral biguanide OXPHOS inhibitor of mitochondrial ETC Protein Complex 1, which activates AMPK and is well tolerated at doses active in preclinical models with modest clinical efficacy [NCT03272256] in solid cancer patients[32]. Though the biguanides showed promising efficacy in preclinical studies, they are still under development and warrant further investigation in clinical trials for AML to establish their clinical utility.
Role of mitochondrial OXPHOS in chemoresistance
Cytarabine remains the backbone of AML chemotherapy. Interestingly, pre-existing and persisting cytarabine-resistant cells do not necessarily reside in a quiescent or LSC compartment but rather have higher OXPHOS levels, with polarized mitochondria and an increased mitochondrial mass[33]. The ability of cytarabine to induce mitochondrial ROS is a major determinant of cytarabine sensitivity in primary AML cells. Resistance to cytarabine is often mediated by SIRT3, a NAD+-dependent protein deacetylase that deacetylates mitochondrial anti‐oxidant enzymes, including IDH2 and superoxide dismutase 2 (SOD2)[34]. SIRT3‐mediated deacetylation increases the enzymatic activity of its antioxidant targets, thereby restricting cytarabine-induced mitochondrial ROS production and resulting in reduced apoptosis and increased chemoresistance[34]. In addition, SIRT3 drives OXPHOS and increases the levels of both dihydronicotinamide adenine dinucleotide phosphate (NADPH) and reduced glutathione (GSH) while decreasing the concentrations of their respective oxidized forms. SIRT3 activation conferred resistance to chemotherapy in-vivo and its inhibition showed a synergistic effect with cytarabine[34]. SIRT3 SUMOylation was higher in Ara-C sensitive primary cells, but lower in resistant primary cells[35]. De-SUMOylation of SIRT3 is mediated by SUMO-specific peptidase 1 (SENP1), which enhances its deacetylase activity by inhibiting its protein degradation. When SIRT3 is de-SUMOylated, the expression of HES1, a negative regulator of FAO, is decreased and FAO is upregulated; either FAO inhibitors or overexpression of HES1 can attenuate this effect[35]. The use of Momordin-Ic a, SENP1 inhibitor or HES1 overexpression in vitro and in vivo showed synergy with cytarabine to eradicate AML cells[35]. Although these studies demonstrate a role for SIRT3 in chemoresistance in AML, the number of patients included in these studies is limited, and therefore validation is required in larger cohorts.
Notably, following cytarabine therapy, residual cells showed increased FAO, increased CD36 expression, (a fatty acid translocase) and higher expression of an OXPHOS gene signature that is predictive of clinical response to treatment in PDX models and AML patients[33]. Inhibition of mitochondrial protein biosynthesis, electron transfer, or FAO resulted in reduced OXPHOS and strikingly increased efficacy of cytarabine in AML[33]. Another mechanism of cytarabine resistance is mediated by upregulation of CD39, an ectonucleotidase that is localized on the cell surface and hydrolyses extracellular ATP and ADP to produce adenosine. Increased activity of CD39 promotes mitochondrial activity and biogenesis by activating the cAMP-mediated adaptive mitochondrial stress response, which promotes resistance to cytarabine. The activation of cAMP-PKA signaling driven by CD39 through P2RY13 purinergic receptor causes metabolic reprogramming and promotes the survival of chemo-resistant AML cells[36]. Thus, CD39-P2RY13-cAMP-OxPHOS axis plays a key role in cytarabine resistance; inhibiting CD39 ecto-ATPase activity enhanced cytarabine’s cytotoxicity in AML by blocking the mitochondrial reprogramming caused by the drug[36].
TARGETING IDH1/2M
The isocitrate dehydrogenases enzymes, IDH1 and IDH2, catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) in the cell cytoplasm and mitochondria, respectively, and reduce NADP+; this, in turn, contributes to the generation of NADPH. α-KG is essential for the optimal functioning of multiple metabolic and epigenetic processes, while NADPH functions as a reducing agent and plays a crucial role in maintaining the cellular redox state. IDH1/2 mutations have been found in 1 of every 5 AML patients with a higher preponderance among patients with normal karyotypes[37,38]. IDH1/2 mutations detected in AML are predominantly heterozygous point mutations that not only render the protein incapable of carrying out decarboxylation of isocitrate, but confer it the ability to produce
In recent years, ivosidenib (AG120), an IDH1 inhibitor, has been used in the treatment of relapsed and refractory AML[45] while many other inhibitors including Olutasidenib (FT-2102), IDH305, Vorasidenib (AG881), BAY1436032, LY3410738 and DS-1001 are currently being evaluated in Phase I/II clinical trials in patients with IDH1/2 mutant AML. The IDH2 inhibitor Enasidenib (AG-221) and Ivosidenib are approved for relapsed/refractory AML in the presence of relevant mutations and ivosidenib was recently approved, in combination with hypomethylating agents, as first-line therapy for IDH1 mutant AML [Figure 1]. The IDH1 mutant AML cells carrying IDH1R132H, IDH1R132C, IDH1R132G, IDH1R132L and IDH1R132S mutations undergo myeloid differentiation when treated with BAY1436032, a novel pan mutant IDH1 inhibitor which specifically inhibits R-2HG production and colony growth in these cells. Aside from that, the compound affects DNA methylation and attenuates hypermethylation of histones. In two independent xenograft mouse models derived from AML patients with IDH1 mutation, treatment with BAY1436032 increased leukemic blast clearance, myeloid differentiation, and stem cell depletion while prolonging survival[46]. Combining azacitidine and BAY1436032 increased survival and depleted LSCs by inhibiting MAPK/ERK and RB/E2F signaling[47]. In a study conducted by Shih et al., AG-221 treatment reduced aberrant hypermethylation in several genes involved in hematopoietic differentiation, suggesting an epigenetically driven differentiation effect. As a single agent, AG221 or 5-azacitidine in vivo stimulated the production of mature myeloid cells from mutant leukemic progenitor blasts. Importantly, in
The emergence of second-site IDH2 mutations in trans, at glutamine 316 (Q316E) and isoleucine 319 (I319M), which are located at the interface of the enasidenib binding site in the IDH2 dimer, were found to confer therapeutic resistance to enasidenib. Although the expression of these mutant proteins alone is not sufficient for the synthesis of 2HG, the presence of R140Q mutation in trans produces 2HG, which is resistant to inhibition by enasidenib[49]. Mutations in the IDH dimer-interface in cis were found to confer resistance to ivosidenib in AML[49]. In a high-content shRNA screen of isogenic leukemia cells expressing wild-type and mutant IDH1, Chan et al. (2015) found that survival of IDH1/2 mutant cells was highly dependent upon BCL-2 and BCL-W expression[50]. Venetoclax-based therapy was effective in 6 out of 7 patients with IDH1 mutations who had previously been treated with ivosidenib; however, a lower response rate was reported for patients with FLT3-ITD mutations, indicating that venetoclax is an effective salvage therapy in patients who previously received IDH1/2 inhibitors[51]. IDH305 is a selective oral IDH1 inhibitor that specifically targets R132* IDH1 mutation. There is a phase I clinical trial currently being conducted on this drug for the treatment of advanced malignancies, including relapsed/refractory AML and myelodysplastic syndromes (MDS) [NCT02381886][52].
Vulnerabilities in IDH mutated AML
Mutations in IDH1/2 have been reported to confer sensitivity to several chemotherapeutic agents in most cancer types, including colorectal cancer[53], glioma[54,55], cholangiocarcinoma[53,56], and AML[50,57]. Conversely, inhibition of mutant IDH1/2 may counteract the cytotoxic effect of chemotherapeutic drugs[58]. In IDH1/2 mutants, D2HG mediates the downregulation of ATM, a DNA damage response gene. Therefore, IDH1/2 mutant AML cells are sensitive to DNA damage-causing agents, such as daunorubicin, and the PARP inhibitors olaparib and talazoparib, while IDH1/2 mutant inhibitors had a protective effect against these treatments[58]. Intriguingly, IDH1/2 mutation sensitized primary AML cells to ABT199 by inhibiting cytochrome c oxidase (i.e., Complex IV of ETC) by 2HG[50]. The cytochrome c oxidase complex contains two heme moieties (a and a3) and two copper atoms (CuA and CuB)[59]. A binding of (R)-2-HG at or near the binuclear center of heme a3 and CuB inhibits cytochrome c oxidase activity, thereby blocking oxygen reduction at that site[50]. Suppression of cytochrome c oxidase activity results in oxygen deprivation, which triggers activation of BAX/BAK and leads to outer membrane permeabilization and apoptosis. When BAX/BAK are bound to BCL-2, permeabilization is prevented, but ABT-199 disrupts this binding so that IDH1/2 mutant cells die, while wild-type IDH1/2 cells are relatively unaffected[33]. IDH1/2 mutant AML exhibited a superior response (36%) to BCL-2 inhibition compared to IDH1/2 wildtype (9%)[60]. Patients with IDH-mutant AML show robust responses to venetoclax + azacytidine[61]. This is a great example of a driver mutation that creates a unique metabolic vulnerability[62].
Primary resistance to IDH inhibitors is primarily associated with leukemia stemness, while acquired resistance is often conferred by mutations in the genes belonging to the RUNX1/CEBPA or RAS-receptor tyrosine kinase (TK) pathways or genes such as BCOR, and TET2[63]. These mutations often affect transcription factors that are involved in hematopoietic and myeloid differentiation, particularly RUNX1 and CEBPA. An association was found between co-mutation(s) of RUNX1 at baseline and a lower CR rate with IDH inhibitor[63]. RUNX1 mutations were also the most frequently acquired mutations in relapsed disease. In addition, co-occurrence of CEBPA mutation was associated with a lack of response to IDH inhibitor, and the mutation was also reported to be acquired at relapse[63]. Because of their roles as differentiation factors, RUNX1 and CEBPA mutations may interfere with differentiation signals induced by IDH inhibitors, resulting in clinical resistance[63]. Almost 30% of relapse cases were found to have acquired mutations in either NRAS or KRAS during relapse[63]. Interestingly, Tateishi et al. found that IDH1 mutation may cause addiction to NAD+ in solid cancers[64]. The mutant form of IDH1 lowered NAD+ levels through downregulation of the enzyme nicotinate phosphoribosyltransferase (NAPRT1) of the NAD+ salvage pathway and thus sensitized cells to NAD+ depletion when combined with inhibition of nicotinamide phosphoribosyltransferase (NAMPT). Depletion of NAD+ activated AMPK, triggering pro-cell death autophagy, resulting in cell death[64].
TARGETING AMINO ACID METABOLISM
Role of glutamine and glutaminolysis: as cellular building blocks and in ROS mitigation homeostasis
Besides serving as substrates for protein biosynthesis, amino acids participate in the biosynthesis of purine and pyrimidine nucleotides and provide precursors for energy generation. For instance, acetyl-CoA can be generated by the ketogenic amino acids leucine and lysine. Similarly, the glycogenic amino acids alanine and glycine generate pyruvate and TCA cycle intermediates. Glutamine addiction has been reported in some primary AML patient samples and cell lines[65-67], and can drive the TCA cycle by providing glutamate; through glutaminolysis, glutamate is then converted to α-ketoglutarate. In addition, it can serve as a substrate in the synthesis of GSH, a ROS mitigator[66,68] [Figure 2]. Gregory et al. demonstrated that inhibition of glutaminase, an enzyme responsible for converting glutamine to glutamate, could perturb GSH balance and adversely affect the redox state in AML[69]. AML development in NSG mice was dramatically inhibited by knocking down the glutaminase gene GLS1, leading to the failure of conversion of glutamine to glutamate (glutaminolysis); this, in turn, induced apoptosis, and exhibited a synergistic effect in sensitizing leukemic cells to venetoclax[66].
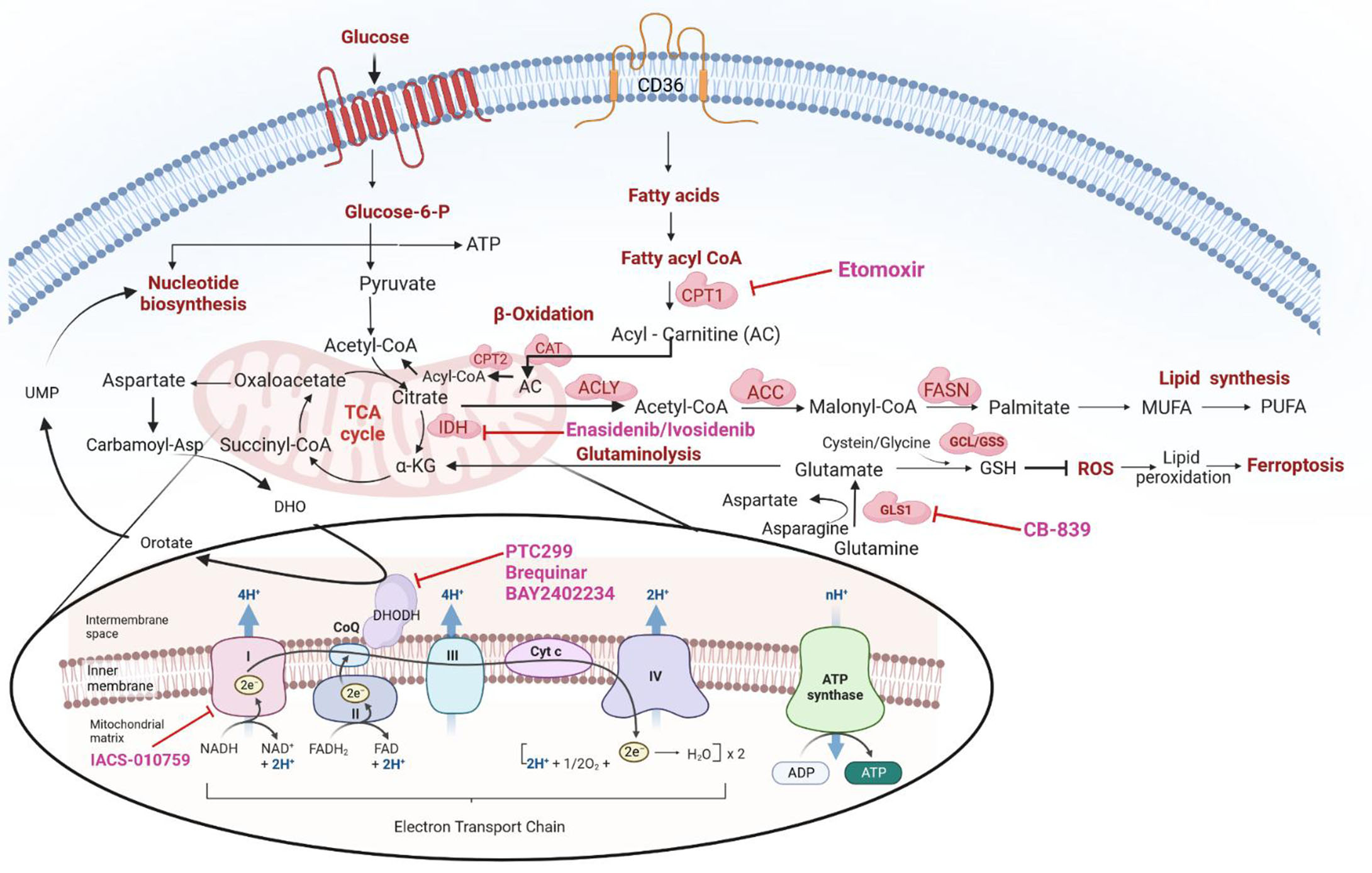
Figure 2. Overview of cellular metabolic pathways and their crosstalk -including: (1) glycolysis; (2) TCA cycle; (3) OXPHOS;
Drivers of glutaminolysis
The cellular influx and efflux of glutamine in exchange for leucine influences the intracellular levels of glutamine, which is critical for mTORC1 activity, and translation and control of autophagy to coordinate cell survival and proliferation[70]. Solute-carrier family 1 member 5 (SLC1A5) is responsible for glutamine transport into the cells. Glutamine transport is essentially linked to leucine uptake. The bidirectional transporter, SLC7A5/3A2 is responsible for the uptake of leucine in exchange of glutamine[71]. Knockdown of SLC1A5 in a murine model of AML resulted in increased apoptosis and reduced tumor formation[72].
Translation of cellular mRNAs is tightly regulated by mTOR. The mTOR pathway regulates the phosphorylation of 4E-BP1 (at serine 65), which is required for the initiation of translation[73]. Asparaginases are routinely used in the treatment of acute lymphoblastic leukemia ; they catalyze the conversion of asparagine and glutamine to aspartate and glutamate, respectively, and help to reduce the concentrations of asparagine and glutamine in the blood[74]. Glutamine depletion induced by L-asparaginases inhibits 4EBP1 phosphorylation at residue S65 and decreases protein synthesis in AML cell lines[72]. When 4EBP1 is dephosphorylated, it binds to eIF4E, resulting in inactivation of translational initiation complexes and a reduction in cap-dependent protein synthesis. In Complex karyotype AML, co-treatment with venetoclax and pegcrisantaspase, an asparaginase, significantly diminished cellular protein synthesis including that of MCL-1, an important antiapoptotic protein in AML, by promoting the interaction between eIF4E and 4EBP1 on the cap-binding complex and interfering with active cap-mRNA translation downstream of mTOR signaling[75].
Glutaminolysis is a therapeutically targetable vulnerability in FLT3-ITD and other tyrosine kinase (TK) activating leukemias. A synthetic lethality screen using CRISPR/Cas9, metabolomics analyses, and gene expression analysis reveals that FLT3-ITD cells develop glutamine metabolism dependence following FLT3 TK inhibition. FLT3 TK inhibition hinders glucose uptake and use, suppressing the enhanced central carbon metabolism that is found in FLT3-ITD cells and reversing the glycolytic phenotype; as a result, FLT3-ITD cells develop a metabolic dependency on glutamine metabolism. Despite a decrease in glucose uptake, TCA cycle activity and respiratory function are less affected by FLT3 TK inhibition; instead, they are supported by the continuous uptake of glutamine. FLT3 TK inhibition with concomitant suppression of glutamine metabolism using either GLS chemical inhibition or gene silencing, increased cell death in
Agents targeting glutaminolysis
Glutaminolysis was recently identified as a therapeutic target for hematological and solid malignancies, and potent inhibitors are now being evaluated in clinical trials [NCT02071862 and NCT02071927]. Inhibition of glutaminolysis with CB-839 blocks glutamine utilization, resulting in decreased levels of glutamate, aspartate and several TCA cycle intermediates. As described in a previous study[5], inhibition of glutaminolysis with BPTES in mutant IDH1/2 AML may be beneficial, as it arrested cell proliferation, decreased 2-HG levels and enhanced differentiation[67]. Interestingly, in AML harboring FLT3-ITD mutations, glutaminolysis inhibition alone confers only minor antiproliferative effects. Glutaminolysis inhibition is more effective when combined with FLT3-ITD tyrosine kinase inhibition[76], which perturbs glycolysis and glucose utilization, with less effect on glutamine metabolism. Therefore, even though glycolysis and glucose utilization are impaired following FLT3 inhibition, glutamine can drive the TCA cycle and participate in GSH synthesis[76]. Notably, in AML cells treated with quizartinib, glutamine starvation resulted in the depletion of GSH and elevation of intracellular ROS. In addition to glutamine starvation, pharmacological inhibition using CB839 or glutaminolysis gene silencing enhanced the efficacy of FLT3-ITD tyrosine kinase inhibition by reducing the availability of intracellular glutamine (due to TCA cycle inhibition), and GSH generation, thus impacting redox metabolism[76].
LSCs exhibit increased uptake and catabolism of amino acids. Venetoclax + azacytidine is highly effective in AML patients because of its action at inhibiting amino acid metabolism in LSCs[79]. Jones et al. demonstrated that depletion of amino acids, especially cysteine, was strongly associated with response to venetoclax + azacytidine regimen compared to ROS induction. LSCs in patients with de novo AML rely on amino acid metabolism to drive OXPHOS. In LSCs, the depletion of cysteine causes a reduction in GSH levels, which, in turn, leads to decreased glutathionylation of succinate dehydrogenase A (SDHA), a component of ETC complex II. This decreased glutathionylation of SDHA results in OXPHOS inhibition and ATP depletion, ultimately leading to LSC death[80]. BCL-2 inhibition is believed to promote the generation of mitochondrial permeability transition pores, resulting in leakage of mitochondrial membrane and reduced amino acid metabolism[81]. The combination of venetoclax and azacytidine has shown good efficacy in older AML patients, who are generally considered unfit for traditional chemotherapy due to high toxicity and poor outcomes with chemotherapeutic agents. In fact, in a Phase III study, this regimen resulted in higher complete remission compared to the placebo control (36.7% vs. 17.9%; P < 0.001), the composite complete remission was reported in 66.4% AML patients receiving ven + aza vs. 28.3% in placebo control; P < 0.001[61]. The study reported that almost half of all patients receiving azacitidine plus venetoclax had a first response (complete remission or complete remission with incomplete hematologic recovery) before starting cycle 2, and their remissions lasted an average of 17.5 months. Of note, the remission rate after venetoclax + azacytidine in patients with de novo AML is independent of the genetic background, suggesting that venetoclax targets a unique metabolic program of LSCs[61].
Targeting arginine
Primary AML cells are auxotrophic for glutamine, cysteine, and arginine[80,82]. As they lack argininosuccinate synthetase-1 (ASS1), a key enzyme required for the generation of arginine, these cells rely on arginine import[83]. The depletion of extracellular arginine via the use of pegylated arginine deiminase, which converts plasma arginine to citrulline, induced responses in AML through the activation of caspases[83]. Depleting extracellular arginine shows synergy with cytarabine and enhances its cytotoxicity[82]. Arginine and cysteine metabolism are also controlled by the stroma in CLL, which may promote the resistance and growth of leukemic cells[84]; this can be targeted by arginine depletion.
Targeting branched-chain amino acids
The survival of HSCs and LSCs is influenced by branched-chain amino acids (BCAAs), such as leucine, isoleucine, and valine. For instance, valine is essential for survival and maintenance of HSCs[85,86]. BCAAs promote cancer growth in de novo AML[87]. LSCs exhibit elevated levels of branched-chain amino acid transaminase 1 (BCAT1)[88], a cytoplasmic aminotransferase that generates glutamate by transferring an amino group from a BCAA to α-KG[89], which can in turn fuel OXPHOS.
TARGETING PYRIMIDINE BIOSYNTHESIS
Proliferating cells require an abundance of nucleotides. They heavily depend on de novo nucleotide biosynthesis to meet their nucleotide requirements. As expected, the expression of enzymes involved in nucleotide biosynthesis is elevated in cancer. Uridine monophosphate (UMP) is generated in de novo nucleotide biosynthesis from glutamine. Similarly, dihydroorotate dehydrogenase (DHODH) is a mitochondrial enzyme that converts dihydroorotate (DHO) into orotate through oxidation, in de novo pyrimidine biosynthesis and donates the electrons to ubiquinone to generate ubiquinol, which is then re-oxidized by ETC complex III[90]. In experimental models of AML, inhibition of DHODH enhances myeloid differentiation[90]. In addition, a decrease in leukemia-initiating cells, as well as increased survival in vivo, have also been observed following the inhibition of DHODH[91]. DHODH inhibition promotes myeloid differentiation by depleting uridine, a precursor of uridine diphosphate-N-acetylglucosamine
Notably, several clinical trials have reported that brequinar, a DHODH inhibitor, has limited efficacy, likely related to a schedule that did not allow sustained exposure[91,95]. ETC complex III helps in maintaining the oxidation of ubiquinol, which is essential to supporting DHODH activity[96]. Interventions targeting respiratory Complex III would increase the generation of ROS and block the activity of DHODH and pyrimidine biosynthesis[97,98], and could thus be an effective two-pronged strategy involving the exploitation of cell death promoted by ROS[99] and differentiation induced by DHODH inhibition. Alternative oxidase (AOX) can restore DHODH activity and reactivate CoQ redox-cycling following DHODH inhibition[100].
Aspartate biosynthesis is a crucial step in pyrimidine biosynthesis, which requires ETC complex I activity[15]. AML’s dependency on ETC complex I has been well-documented. As a result, there has been an increasing interest in targeting ETC complex I and induction chemotherapy together.
TARGETING NICOTINAMIDE METABOLISM
LSCs from relapsed/refractory AML patients are resistant to venetoclax + azacytidine, in contrast to those from de novo AML patients, suggesting that they have alternative resistance mechanisms. Jones et al. (2020) determined whether LSCs from relapsed/refractory AML patients exhibited altered metabolome profiles compared to those from de novo AML; they identified 18 metabolites that were upregulated, including amino acids and nicotinamide[101]. Interestingly, relapsed LSCs exhibited an enhanced rate of NAD+ synthesis driven by the salvage pathway, with no difference in the protein levels of NAMPT, the rate-limiting enzyme responsible for the generation of NAD+ from nicotinamide, suggesting an increase in nicotinamide uptake in relapsed/refractory LSCs[101]. The elevation of nicotinamide metabolism in relapsed LSCs, activates amino acid metabolism and FAO, which in turn drives OXPHOS. An increase in OXPHOS would help LSCs to confer resistance to venetoclax + azacytidine[101].
Unlike de novo LSCs, relapsed/refractory LSCs exhibit extensive metabolic plasticity, which enables them to adapt alternative mechanisms for activating OXPHOS, a key mechanism for developing resistance to chemotherapy[101]. Given that therapeutic options are limited for relapsed/refractory AML, the inhibition of NAMPT, the rate-limiting enzyme for NAD+ synthesis, may be an effective approach[101]. Notably, relapsed/refractory LSCs were selectively eradicated by the genetic and pharmacological inhibition of NAMPT, while normal hematopoietic stem/progenitor cells were spared. Of note, in secondary transplantation of relapsed/refractory AML LSCs, in-vivo treatment with APO866, a small molecule inhibitor of NAMPT, compromised the engraftment[101].
TARGETING FATTY ACID METABOLISM
FA catabolism has been implicated in regulating HSC fate (i.e., maintenance vs. exhaustion) by providing the required energy to allow the proper execution of asymmetric division, favoring the maintenance and function of the HSC compartment. Loss of the mitochondrial FAO activator peroxisome proliferator-activated receptor δ (PPAR-δ) perturbs HSC maintenance, whereas treatment with PPAR-δ agonists restores HSC maintenance by promoting asymmetric HSC division[102]. PPAR-δ activates FAO in mitochondria. LSCs from de novo AML patients drive OXPHOS exclusively using amino acid metabolism. In contrast, LSCs from relapsed/refractory AML patients tend to compensate significantly through increased FA metabolism[79]. Despite the importance of FAs for mitochondrial metabolism in LSCs, aberrant lipid metabolism may also promote cancer cell proliferation by increasing its energy availability, structural lipid-based building blocks, and signaling molecules[103]. Vriens et al. demonstrated that cancer cells exhibit extensive plasticity in FA metabolism and can facilitate membrane biosynthesis during proliferation through the FA desaturation pathway[103]. Further investigation is needed to determine whether such metabolic plasticity exists in leukemic cells and if it could be targeted.
Mcl1 is an antiapoptotic protein that is known to interact with proapoptotic proteins in mitochondria. Mcl1 upregulation is common in venetoclax-resistant AML[104]. Interestingly, MCl1 in cancer cells regulates lipid metabolism through a functional gain. This is achieved by binding its α- helix of the BH3 domain to very long-chain acyl-CoA dehydrogenase (VLCAD), a key enzyme of the mitochondrial FAO pathway, through its hydrophobic surface[105]. Thus, MCL1 not only suppresses apoptosis in cancer cells, but also regulates
Fatty acid metabolism and resistance to therapy
Cellular levels of ceramide are key determinants of response to several chemotherapeutic drugs. Modulation of the ceramide pathway has been implicated in apoptosis induced by daunorubicin in AML cells[108]. Ceramidases convert ceramide to sphingosine. Kao et al. found that daunorubicin and cytarabine treatment resulted in the increased expression and activity of enzymes involved in the ceramide pathway, leading to a decrease in ceramide levels and an increase in ceramide 1- and sphingosine 1-phosphate levels. These perturbations induced remodeling of mitochondria in cancer cells and promoted chemoresistance[109]. For instance, FLT3-ITD signaling activation suppresses pro-cell death lipid ceramide generation in AML cells[110]. Intriguingly, LCL-461, a mitochondria-targeted ceramide analog, effectively induced lethal mitophagy in human AML blasts and crenolanib (FLT3-ITD inhibitor)-resistant AML xenografts, suggesting that activation of mitochondrial ceramide synthesis overcomes resistance to FLT3 inhibition[110].
A previous study reported that de novo cholesterol synthesis through the mevalonate pathway plays a key role in AML[111]. Recently, using genome-scale metabolic modeling, Karakitsou et al. demonstrated that AraC-resistant cells could be targeted by suppressing cholesterol biosynthesis through the inhibition of squalene synthase[112].
Acetyl-CoA is converted to malonyl-CoA by Ac-CoA carboxylases (ACCs). FA synthase (FASN) catalyzes the condensation of malonyl-CoA and Ac-CoA to generate palmitate as the first product of FA synthesis[113]. FASN is significantly elevated in AML blasts compared to healthy granulocytes or CD34+ hematopoietic progenitors at the RNA level. Inhibition of FASN expression on one side accelerated differentiation of APL cells, on the other hand, sensitized ATRA resistant -non-APL AML cells to ATRA, by promoting translocation of transcription factor EB (TFEB) to nucleus and lysosomal biogenesis[114].
Cellular stress directly regulates FA metabolism by affecting the enzymatic activity of Acetyl-CoA carboxylases (ACC), namely ACC1 and ACC2. AMPK inhibits ACC under energy stress and thus activates fat synthesis and catabolism. Nutrient abundance, on the other hand, downregulates the activity of AMPK, and ACC1 and ACC2 are no longer repressed[115]. This suggests that ACCs act as a regulatory node in FA metabolism by sensing nutrient availability and balancing the switch between anabolic and catabolic processes. Of note, ACC1 suppressed growth-promoting activity and promoted the generation of ROS in primary BM cultures. In addition, ACC1 has been shown to promote myeloid differentiation and delay the development of AML in mice[116]. Hydroxylation of ACC2 by prolyl-hydroxylase 3 (PHD3) increases the activity of ACC2, which, in turn, prevents FA utilization[117,118]. PHD3 levels are decreased in AML[119], thus generating a vulnerability due to dependence on FAs, which can make FAO inhibition an attractive strategy to control AML.
Venetoclax + azacytidine-resistant LSCs exhibit increased levels of several metabolites that are responsible for the transport of FAs into mitochondria at baseline, including acetyl-carnitine, iso-butylryl carnitine, and hexanoyl-carnitine, compared to sensitive LSCs[120]. Venetoclax + azacydine treatment results in a reduction in amino acid uptake by LSCs. Enhanced FA transport facilitates FAO and allows LSCs to compensate for amino acid uptake loss caused by venetoclax + azacytidine treatment, resulting in resistance. However, this resistance can be overcome by the knockdown of very long-chain acyl-CoA dehydrogenase (ACADVL), an enzyme involved in mitochondrial FA beta-oxidation[120], suggesting that FA beta-oxidation plays an important role in conferring resistance to venetoclax + azacytidine treatment in LSCs.
FAs are conjugated to carnitine by CPT1 to facilitate mitochondrial translocation, and both the PPARs and the coactivator-1 of PPARs modulate CPT1A expression[121]. Pharmacological inhibition of CPT1 by etomoxir depleted refractory LSC function[122], and eliminated venetoclax+ azacytidine-resistant LSCs[85]. In addition, CPT1A inhibition by etomoxir induced cell death in AML cells and increased cells’ sensitivity to cytarabine[123]. CD36+ LSCs are characterized by an enhanced FAO rate, and disruption of mitochondrial metabolism by targeting CD36-FAO-OXPHOS drives leukemia cells to low OXPHOS and enhances AraC sensitivity[19]. In addition, topoisomerase inhibitor (mitoxantrone)-resistant AML cells showed activation of lipid metabolic pathways. Inhibition of FAO, using etomoxir in these cells, reduced colony formation ability, suggesting that FA metabolism and dependency on OXPHOS are key vulnerabilities, conferring chemoresistance in AML[124].
TUMOR MICROENVIRONMENT-MEDIATED METABOLIC PLASTICITY
Tumor microenvironment-mediated OXPHOS regulation
BMSCs, which are composed of endothelial cells, osteoclasts, osteoblasts, adipocytes, and fibroblasts, form the BM microenvironment. BMSCs aid the survival of AML cells and mediate resistance to therapy. Cell-cell communications in the BM microenvironment play a key role in aiding AML cell survival, disease progression, and therapeutic response through the transfer of biomolecules or cellular organelles, such as mitochondria[125]. Of note, BMSCs supply mitochondria to AML cells in vitro, thus providing them with additional energy[126]. AML cells were observed to enhance their mitochondrial mass up to 14% by mitochondrial uptake from BMSCs; this transfer increased in response to chemotherapy and contributed to resistance by reducing mitochondrial depolarization[126]. AML cells exhibited up to a 1.5-fold increase in mitochondrial ATP production following the uptake of mitochondria from BMSCs, were less prone to depolarization of mitochondria following chemotherapy, and showed improved survival[126]. Of note, transfer of mitochondria to AML cells from BMSCs in the presence of chemotherapeutic agents occurs predominantly through endocytosis and requires cell-cell contacts[12]. The NADPH oxidase, NOX2 generates superoxide in AML cells, which promotes mitochondrial transfer to AML blasts from BMSCs through AML-derived tunneling nanotubes. Interestingly, suppression of NOX2 reduced the transfer of mitochondria to AML blasts and resulted in enhanced cell death and improved survival of AML-bearing mice[126]. AML cells show high OXPHOS levels when cocultured with BMSCs. Hou et al. showed that the elevation of OXPHOS levels in AML cells in coculture was dependent on the activation of mitochondrial serine-phosphorylated STAT3 (pS-STAT3)[127]. AML cells in coculture induced the secretion of interleukin-6 from BMSCs, which in turn promoted the activation of total and mitochondrial STAT3 in AML cells, resulting in increased proliferation and chemoresistance[127].
AML cells are known to harbor abnormally high levels of ROS. Mesenchymal stem cells, a component of BMSCs, support the survival of AML cells by influencing several key metabolic pathways and eliminating ROS. BMSCs expressing the intermediate filament protein nestin provide HSC niche function[128]. Forte
Tumor microenvironment mediated regulation of amino acid and pyrimidine metabolism
Induction chemotherapy causes transient stress in BM, which results in the elevation of glutamine, glutamate, and aspartate with a concomitant increase in the expression of several glutamine transporters, such as Slc1a5, Slc38a1, Slc7a5, and Slc7a6, but no change in the enzymes that metabolize glutamine or glutamate, in residual AML cells[130,131]. This suggests that glutamine metabolism in AML cells could be regulated by the tumor microenvironment[130,131]. Interestingly, van Gastel et al. showed that glutamine metabolism differed between AML and stromal BM cells, especially LepR+ mesenchymal stromal cells[131]. AML cells took up glutamine to maintain GSH but not to promote the TCA cycle. In contrast, in LepR+ mesenchymal stromal cells, glutamine entered the TCA cycle to produce aspartate[131]. Aspartate is transported to and supports pyrimidine biosynthesis in persistent MLL-AF9 AML cells, thus preventing the metabolic collapse triggered by chemotherapy[130,131]. These findings suggest that the tumor microenvironment plays a major role in chemoresistance, independent of cell-intrinsic signaling/apoptosis alterations. However, it is unclear whether aspartate-rich locations in the BM provide a protective niche, and if specific AML subclones compete for these spots. Further niche-specific studies are warranted to delineate whether mutational subclones compete for specific hotspots.
Tumor microenvironment mediated regulation of fatty acid metabolism
Accumulating evidence supports the existence of metabolic symbiosis and bidirectional dialogues between leukemic cells and the BM adipose tissue niche, which provides diverse sources of lipids, such as FAs, cholesterol, and eicosanoids, to resident leukemic cells. Leukemic cells meet their energy needs by using FAs to fuel β-oxidation, which eventually supports anabolism[33,132]. When FAO was inhibited pharmacologically with etomoxir or ranolazine, AML cells were sensitized to apoptosis induction by ABT-737, and proliferation was inhibited in the setting of coculture with BMSCs[123]. Interestingly, a recent report described two forms of LSCs, distinguished by differences in their expression of CD36 and in their metabolism and cell cycle status. LSCs expressing CD36 (CD36+ LSCs) are often localized to gonadal adipose tissue (GAT) which is rich in fatty acids, indicating a tropism rich for microenvironments rich in FAs[132]. Specifically, these GAT-resident LSCs are more resistant to conventional chemotherapy because they exist in a quiescent cell cycle state. In the context of these findings, leukemic cells co-opt adipose tissue to create a microenvironment to support leukemia growth and provide a drug-resistant niche[132]. Further, leukemic burden in GAT for the MLL-AF9 model was considerably lower than that in BM, indicating that localization to GAT may be dependent on the specific oncogenes that drive the malignant transformation process[132]. An interesting question for future investigations would be to understand the role of FA metabolism in conferring drug resistance to LSCs in a niche-specific BM microenvironment.
ROLE OF EXTRACELLULAR VESICLES IN CONFERRING RESISTANCE BY MEDIATING COMMUNICATION BETWEEN LEUKEMIC CELLS AND MICROENVIRONMENT TO ALTER METABOLISM
AML cells alter the BM by releasing exosomes, which promote the proliferation and survival of leukemic cells and suppress normal hematopoiesis. In a study by Javidi-Sharifi et al., the expression of FGF2 and its receptor, FGFR1, were both increased in several stromal cell lines and primary AML stroma. This heightened FGF2/FGFR1 signaling augmented greater secretion of exosomes[133]. Huang et al. (2021), studied the impact of inhibition of small extracellular vesicle (SEV) secretion from various sources on the progression of MLL-AF9 induced AML, as well as normal hematopoiesis[134]. A significant delay in AML progression occurred when SEV secretion from endothelial cells was inhibited, but not that from perivascular cells, megakaryocytes, or spleen stromal cells[134]. SEVs derived from endothelial cells contained high levels of Angiopoietin-like protein 2 (ANGPTL2) protein, which binds to the leukocyte immunoglobulin-like receptor B2 (LILRB2 receptor) and accelerates leukemia progression[134]. Moreover, Vacuolar protein sorting-associated protein 33B (VPS33B) governed the release of ANGPTL2-SEVs from endothelial cells and this release of ANGPTL2-SEVs was required for primary human AML cell maintenance. These findings demonstrate that SEVs have a role in cancer development and suggest that targeting the release of ANGPTL2-SEVs from endothelial cells would be an effective strategy in the treatment of some types of AML[134]. While IO-108, an antibody targeting LILRB2, is in Phase 1 clinical development for solid tumors[135]; LILRB4 antibody (IO-202) is in phase 1 cohort expansion clinical trial [NCT0437243] in combination with azacitidine and venetoclax, for the treatment of AML and likely to be effective in monocytic AML[136,137].
Cytarabine and decitabine treatment resulted in significantly higher intracellular levels of cholesterol and HMGCR (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase), the rate-limiting enzyme in the cholesterol synthesizing mevalonate pathway, in cultured AML and non-malignant cells. Concomitantly, these cells produced higher levels of SEVs[138]. Interestingly, there was an increase in HMGCR levels in SEVs in response to chemotherapy. These HMGCR+ SEVs promoted the growth of AML cells by upregulating intracellular cholesterol levels. This chemotherapy-induced enhancement of SEV secretion in AML cells was prevented by HMGCR inhibition[138]. Thus, HMGCR blockade could potentially provide a therapeutic alternative in AML by inhibiting cholesterol-driven chemoresistance caused by SEV signaling[138]. Another study reported the secretion of exosomes containing vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) by AML cells. These exosomes induced glycolysis in human umbilical vein endothelial cells (HUVECs), which resulted in vascular remodeling and chemoresistance[139].
BM stromal cells exposed to AML-derived exosomes exhibit decreased expression of genes supporting normal hematopoiesis (CXCL12, KITL, IL-7, IGF1) and osteogenesis (OCN, Col1A1, IGF1) and increased expression of genes supporting AML growth (DKK1, IL-6, CCL3)[140]. When BM is preconditioned with AML-derived exosomes, AML cell engraftment and growth are significantly accelerated, whereas disruption of exosome secretion in AML cells significantly reduced these cells’ ability to engraft and grow in vivo[140]. In coculture, serum and leukemic cells from patients with NPM1-mutated AML impair CD8+ T cells’ immune function[141]. Through SEVs, leukemic cells secrete miR-19a-3p into the tumor microenvironment. This process was mediated by the NPM1-mutated protein/CCCTC-binding factor (CTCF)/poly (A)-binding protein cytoplasmic 1 (PABPC1). MiR-19a-3p released from SEV was found to be internalized by CD8+
AML utilizes SEV miRNAs to target the mTOR pathway which suppresses protein synthesis in the LT‐HSC to elicit their quiescence[143]. While LT-HSCs are functional after the recovery, their DNA is damaged for a long time. Hematopoietic stem and progenitor cells (HSPCs) are generally susceptible to SEV entry and use the mTOR pathway, but HSCs selectively enter quiescence because of their unique sensitivity to protein synthesis disruption[143].
TECHNIQUES TO ASSESS MITOCHONDRIAL FUNCTION AND METABOLISM
A tightly coordinated flux of metabolites occurs in mitochondria, integrating bioenergetics, redox homeostasis, and anabolic metabolism. By assessing mitochondrial function, researchers have gained a better understanding of metabolism in cellular physiology, disease pathology, and etiology. The reduction of oxygen into water is essential to oxidative phosphorylation and measuring oxygen consumption is therefore one way to assess mitochondrial function[144]. The mitostress test using a high-throughput real-time mode approach in Agilent Seahorse XF analyzers is the best assay for measuring basal cell respiratory rate, ATP production, proton leak, maximum respiration, and spare respiratory capacity in an intact cell[145]. This single assay yields a wide range of parameters, providing insight into mitochondrial dysfunction and reveals the functional differences between cell types, cellular metabolic state (quiescent versus active), effect of drug candidates, impact of genetic modifications, or biochemical interactions[146].
Mitochondria are known to generate approximately 90% of cellular reactive oxygen species (ROS)[147]. As a result of excessive production of ROS and/or decreased antioxidant defense activity, mitochondrial reactive oxygen species (mtROS) accumulate, causing oxidative stress (OS). It leads to oxidative damage that affects several cellular components, including lipids, DNA, and proteins[147]. To detect mitochondrial superoxide selectively, MitoSOX Red and Green are used, which are fluorogenic reagents designed for highly selective detection of superoxide in the mitochondria of viable cells[148]. A notable feature of these probes is that they are readily and specifically oxidized by superoxide alone and not by other ROS or reactive nitrogen species (RNS) generating systems at optimum concentration. However, the user should be aware of the cytotoxic or mitotoxic effect of these probes and their potential impact on mitochondrial morphology at higher concentrations[149]. Furthermore, oxidation of these probes is inhibited by superoxide dismutase. Small, cell-permeant dyes such as TMRM (tetramethylrhodamine, methyl ester) and TMRE (tetramethylrhodamine, ethyl ester) accumulate in active mitochondria and may be used to quantify mitochondrial membrane potential[150]. They appear bright in healthy cells with functioning mitochondria, but are dimmed or undetectable upon loss of mitochondrial membrane potential[151].
Metabolome consists of all the molecules with low molecular weight (metabolites) within a cell, tissue or organism. Analyzing the metabolites within a cell, tissue or organism following a genetic change or physiological stimulus is known as metabolomics[152]. The metabolomics approach can be divided into two distinct types: untargeted metabolomics and targeted metabolomics. Targeted approaches focus on identifying and quantifying a small number of known metabolites, such as those found in clinical analyses. In untargeted approaches or hypothesis-generating approaches, data are obtained for as many species as possible, metabolites are annotated, and metabolic changes are reviewed regardless of whether they are known or unknown[153]. However, measuring metabolite concentrations by metabolomics only tells half the story. The accumulation of metabolites can be caused not only by increased production, but also by decreased consumption. The metabolic flux, which can be quantified as material flow per unit time, is equally important for understanding pathway activity[153].
Unlike metabolites, fluxes are not physical entities that can be detected by mass spectrometry. But they can be inferred by using isotope tracers. Radioactive tracer studies laid the groundwork for modern metabolism research[154]. Today, similar studies can be performed using stable non-radioactive isotope tracers, which can be tracked quantitatively and broadly using MS or NMR[155]. Furthermore, an approach called metabolic activity screening integrates the metabolomics data with metabolic pathways and systems biology data, including proteomics and transcriptomics, in order to identify endogenous metabolites that may alter phenotypic characteristics and functionality[156].
CONCLUDING REMARKS
Nine therapeutic agents for AML have been developed and approved since 2017. Treatment improvements have increased the overall survival rate of AML patients, from 13% to 55% in those aged younger than 60 years and 8% to 17% in patients older than 60 years[157]. Further improvements in our understanding of the disease would further improve outcomes. The genetic makeup of AML, as well as its metabolic requirements for survival and proliferation, are heterogeneous. Diverse types of metabolic vulnerabilities, including dependence on OXPHOS, amino acid metabolism, nucleotide metabolism, and fatty acid metabolism, were reported. In addition, metabolic changes occurring in the tumor microenvironment have also been used as therapeutic targets. However, a thorough understanding of biomarkers of response to these therapies and mechanisms of resistance and combinatorial therapies would pave the way for better clinical management of AML. Understanding how the genetic architecture works with the tumor microenvironment is critical to uncovering new vulnerabilities. In addition, a thorough understanding of the interactions between different cell populations within the BM microenvironment in response to transient stress due to therapy which leads to changes in nutrient availability and metabolic adaptations, will increase the chances of a true cure.
LSCs have metabolically distinct subpopulations and relapsed/refractory LSCs switch to alternative metabolic pathways to fuel OXPHOS, rendering them therapeutically resistant to conventional treatments. Developing treatment strategies that target these metabolic pathways is key to eliminating relapsed/refractory LSC populations and improving treatment outcomes.
DECLARATIONS
AcknowledgmentsWe thank Ann Sutton, scientific editor at Research Medical Library, The University of Texas MD Anderson Cancer Center, for editing this article.
Authors’ contributionsDrafted the article: Sharma P
Revised the article critically for important intellectual content: Sharma P, Borthakur G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Zhang N, Wu J, Wang Q, et al. Global burden of hematologic malignancies and evolution patterns over the past 30 years. Blood Cancer J 2023;13:82.
2. de Kouchkovsky I, Abdul-Hay M. 'Acute myeloid leukemia: a comprehensive review and 2016 update'. Blood Cancer J 2016;6:e441.
3. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2012;2:881-98.
4. Papa L, Djedaini M, Hoffman R. Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem Cells Int 2019;2019:4067162.
5. Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006;12:446-51.
6. Katajisto P, Döhla J, Chaffer CL, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015;348:340-3.
7. Khacho M, Clark A, Svoboda DS, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 2016;19:232-47.
8. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329-41.
9. Sriskanthadevan S, Jeyaraju DV, Chung TE, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015;125:2120-30.
10. Tan AS, Baty JW, Dong LF, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015;21:81-94.
11. Dong LF, Kovarova J, Bajzikova M, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017;6:e22187.
12. Moschoi R, Imbert V, Nebout M, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016;128:253-64.
13. Strakova A, Nicholls TJ, Baez-Ortega A, et al. Recurrent horizontal transfer identifies mitochondrial positive selection in a transmissible cancer. Nat Commun 2020;11:3059.
14. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015;162:540-51.
15. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552-63.
16. Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036-46.
17. Janku F, Beom SH, Moon YW, et al. First-in-human study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. Invest New Drugs 2022;40:1001-10.
18. Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med 2013;2:611-24.
19. Bendell JC, Patel MR, Infante JR, et al. Phase 1, open-label, dose escalation, safety, and pharmacokinetics study of ME-344 as a single agent in patients with refractory solid tumors. Cancer 2015;121:1056-63.
20. Diamond JR, Goff B, Forster MD, et al. Phase Ib study of the mitochondrial inhibitor ME-344 plus topotecan in patients with previously treated, locally advanced or metastatic small cell lung, ovarian and cervical cancers. Invest New Drugs 2017;35:627-33.
21. Ishizawa J, Zarabi SF, Davis RE, et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell 2019;35:721-37.e9.
22. Baccelli I, Gareau Y, Lehnertz B, et al. Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell 2019;36:84-99.e8.
23. Huang Z, Shen Y, Liu W, et al. Berberine targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia with IDH1 mutation. Chin J Nat Med 2023;21:136-45.
24. Pardee TS, Anderson RG, Pladna KM, et al. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin Cancer Res 2018;24:2060-73.
25. Stuart SD, Schauble A, Gupta S, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab 2014;2:4.
26. You R, Wang B, Chen P, et al. Metformin sensitizes AML cells to chemotherapy through blocking mitochondrial transfer from stromal cells to AML cells. Cancer Lett 2022;532:215582.
27. Yuan F, Cheng C, Xiao F, Liu H, Cao S, Zhou G. Inhibition of mTORC1/P70S6K pathway by Metformin synergistically sensitizes Acute Myeloid Leukemia to Ara-C. Life Sci 2020;243:117276.
28. Wang F, Liu Z, Zeng J, et al. Metformin synergistically sensitizes FLT3-ITD-positive acute myeloid leukemia to sorafenib by promoting mTOR-mediated apoptosis and autophagy. Leuk Res 2015;39:1421-7.
29. Velez J, Pan R, Lee JT, et al. Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Oncotarget 2016;7:51435-49.
30. Zhou FJ, Zeng CX, Kuang W, et al. Metformin exerts a synergistic effect with venetoclax by downregulating Mcl-1 protein in acute myeloid leukemia. J Cancer 2021;12:6727-39.
31. Valiulienė G, Vitkevičienė A, Skliutė G, Borutinskaitė V, Navakauskienė R. Pharmaceutical drug metformin and MCL1 inhibitor S63845 exhibit anticancer activity in myeloid leukemia cells via redox remodeling. Molecules 2021;26:2303.
32. Rha SY, Beom S, Shin YG, et al. Phase I study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. J Clin Oncol 2020;38:3590.
33. Farge T, Saland E, de Toni F, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 2017;7:716-35.
34. Ma J, Liu B, Yu D, et al. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br J Haematol 2019;187:49-64.
35. Zhang Y, Shen Y, Wei W, et al. Dysregulation of SIRT3 SUMOylation confers AML chemoresistance via controlling HES1-dependent fatty acid oxidation. Int J Mol Sci 2022;23:8282.
36. Aroua N, Boet E, Ghisi M, et al. Extracellular ATP and CD39 activate cAMP-mediated mitochondrial stress response to promote cytarabine resistance in acute myeloid leukemia. Cancer Discov 2020;10:1544-65.
37. Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010;28:2348-55.
38. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol 2015;90:732-6.
39. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225-34.
40. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17-30.
41. Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013;493:561-4.
42. Montalban-Bravo G, DiNardo CD. The role of IDH mutations in acute myeloid leukemia. Future Oncol 2018;14:979-93.
43. Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013;340:622-6.
45. Norsworthy KJ, Luo L, Hsu V, et al. FDA approval summary: ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin Cancer Res 2019;25:3205-9.
46. Chaturvedi A, Herbst L, Pusch S, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017;31:2020-8.
47. Chaturvedi A, Gupta C, Gabdoulline R, et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica 2021;106:565-73.
48. Shih AH, Meydan C, Shank K, et al. Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in IDH2- and TET2-mutant acute myeloid leukemia. Cancer Discov 2017;7:494-505.
49. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018;559:125-9.
50. Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 2015;21:178-84.
51. Bewersdorf JP, Shallis RM, Derkach A, et al. Venetoclax-based salvage therapy in patients with relapsed/refractory acute myeloid leukemia previously treated with FLT3 or IDH1/2 inhibitors. Leuk Lymphoma 2023;64:188-96.
52. DiNardo CD, Hochhaus A, Frattini MG, et al. A phase 1 study of IDH305 in patients with IDH1R132-mutant acute myeloid leukemia or myelodysplastic syndrome. J Cancer Res Clin Oncol 2023;149:1145-58.
53. Sule A, Van Doorn J, Sundaram RK, Ganesa S, Vasquez JC, Bindra RS. Targeting IDH1/2 mutant cancers with combinations of ATR and PARP inhibitors. NAR Cancer 2021;3:zcab018.
54. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017;9:eaal2463.
55. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res 2017;77:1709-18.
56. Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:796-807.
57. Mugoni V, Panella R, Cheloni G, et al. Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res 2019;29:446-59.
58. Molenaar RJ, Radivoyevitch T, Nagata Y, et al.
60. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 2016;6:1106-17.
61. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020;383:617-29.
62. DiNardo CD, Rausch CR, Benton C, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol 2018;93:401-7.
63. Wang F, Morita K, DiNardo CD, et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat Commun 2021;12:2607.
64. Tateishi K, Wakimoto H, Iafrate AJ, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 2015;28:773-84.
66. Jacque N, Ronchetti AM, Larrue C, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015;126:1346-56.
67. Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol 2014;42:247-51.
68. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016;16:619-34.
69. Gregory MA, Nemkov T, Park HJ, et al. Targeting glutamine metabolism and redox state for leukemia therapy. Clin Cancer Res 2019;25:4079-90.
70. Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009;136:521-34.
71. Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol 2005;15:254-66.
72. Willems L, Jacque N, Jacquel A, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013;122:3521-32.
73. Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 2001;15:2852-64.
74. Beckett A, Gervais D. What makes a good new therapeutic L-asparaginase? World J Microbiol Biotechnol 2019;35:152.
75. Emadi A, Kapadia B, Bollino D, et al. Venetoclax and pegcrisantaspase for complex karyotype acute myeloid leukemia. Leukemia 2021;35:1907-24.
76. Gallipoli P, Giotopoulos G, Tzelepis K, et al. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018;131:1639-53.
77. Delpuech O, Griffiths B, East P, et al. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol Cell Biol 2007;27:4917-30.
78. Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004;23:3338-49.
79. Jones CL, Stevens BM, D’Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018;34:724-740.e4.
80. Jones CL, Stevens BM, D’Alessandro A, et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019;134:389-94.
81. Chen Q, Xu H, Xu A, et al. Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS One 2015;10:e0118834.
82. Mussai F, Egan S, Higginbotham-Jones J, et al. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood 2015;125:2386-96.
83. Miraki-Moud F, Ghazaly E, Ariza-McNaughton L, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood 2015;125:4060-8.
84. Zhang W, Trachootham D, Liu J, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol 2012;14:276-86.
85. Taya Y, Ota Y, Wilkinson AC, et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 2016;354:1152-5.
86. Wilkinson AC, Morita M, Nakauchi H, Yamazaki S. Branched-chain amino acid depletion conditions bone marrow for hematopoietic stem cell transplantation avoiding amino acid imbalance-associated toxicity. Exp Hematol 2018;63:12-6.e1.
87. Kreitz J, Schönfeld C, Seibert M, et al. Metabolic plasticity of acute myeloid leukemia. Cells 2019;8:805.
88. Raffel S, Falcone M, Kneisel N, et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017;551:384-8.
89. Tönjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013;19:901-8.
90. Sainas S, Pippione AC, Lupino E, et al. Targeting myeloid differentiation using potent 2-hydroxypyrazolo[1,5-
91. Sykes DB, Kfoury YS, Mercier FE, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 2016;167:171-86.e15.
92. Wu D, Wang W, Chen W, et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica 2018;103:1472-83.
93. Christian S, Merz C, Evans L, et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 2019;33:2403-15.
94. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021;593:586-90.
95. Noe DA, Rowinsky EK, Shen HS, et al. Phase I and pharmacokinetic study of brequinar sodium (NSC 368390). Cancer Res 1990;50:4595-9.
96. Martínez-Reyes I, Cardona LR, Kong H, et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020;585:288-92.
97. Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 2006;27:639-45.
99. Rohlena J, Dong LF, Ralph SJ, Neuzil J. Anticancer drugs targeting the mitochondrial electron transport chain. Antioxid Redox Sign 2011;15:2951-74.
100. Bajzikova M, Kovarova J, Coelho AR, et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab 2019;29:399-416.e10.
101. Jones CL, Stevens BM, Pollyea DA, et al. Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell 2020;27:748-64.e4.
102. Ito K, Carracedo A, Weiss D, et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 2012;18:1350-8.
103. Vriens K, Christen S, Parik S, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019;566:403-6.
104. Pan R, Ruvolo V, Mu H, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell 2017;32:748-60.e6.
105. Escudero S, Zaganjor E, Lee S, et al. Dynamic regulation of long-chain fatty acid oxidation by a noncanonical interaction between the MCL-1 BH3 helix and VLCAD. Mol Cell 2018;69:729-43.e7.
106. Perciavalle RM, Stewart DP, Koss B, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012;14:575-83.
107. Danial NN, Walensky LD, Zhang CY, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med 2008;14:144-53.
108. Ponnusamy S, Meyers-Needham M, Senkal CE, et al. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol 2010;6:1603-24.
109. Kao LP, Morad SAF, Davis TS, et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL-60 cells. J Lipid Res 2019;60:1590-602.
110. Dany M, Gencer S, Nganga R, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016;128:1944-58.
111. Li HY, Appelbaum FR, Willman CL, Zager RA, Banker DE. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003;101:3628-34.
112. Karakitsou E, Foguet C, Contreras Mostazo MG, et al. Genome-scale integration of transcriptome and metabolome unveils squalene synthase and dihydrofolate reductase as targets against AML cells resistant to chemotherapy. Comput Struct Biotechnol J 2021;19:4059-66.
113. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab 2020;31:62-76.
114. Humbert M, Seiler K, Mosimann S, et al. Reducing FASN expression sensitizes acute myeloid leukemia cells to differentiation therapy. Cell Death Differ 2021;28:2465-81.
115. Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol 2002;92:2475-82.
116. Ito H, Nakamae I, Kato JY, Yoneda-Kato N. Stabilization of fatty acid synthesis enzyme acetyl-CoA carboxylase 1 suppresses acute myeloid leukemia development. J Clin Invest 2021;131:e141529.
117. Yoon H, Spinelli JB, Zaganjor E, et al. PHD3 loss promotes exercise capacity and fat oxidation in skeletal muscle. Cell Metab 2020;32:215-28.e7.
118. German NJ, Yoon H, Yusuf RZ, et al. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol Cell 2016;63:1006-20.
120. Stevens BM, Jones CL, Pollyea DA, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer 2020;1:1176-87.
121. Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol 2018;16:e2003782.
122. Jones CL, Stevens BM, Culp-hill R, et al. Inhibition of fatty acid metabolism re-sensitizes resistant leukemia stem cells to venetoclax with azacitidine. Blood 2019;134:1272.
123. Samudio I, Harmancey R, Fiegl M, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 2010;120:142-56.
124. Salunkhe S, Mishra SV, Ghorai A, et al. Metabolic rewiring in drug resistant cells exhibit higher OXPHOS and fatty acids as preferred major source to cellular energetics. Biochim Biophys Acta Bioenerg 2020;1861:148300.
125. Abdul-Aziz AM, Shafat MS, Mehta TK, et al. MIF-induced stromal PKCβ/IL8 is essential in human acute myeloid leukemia. Cancer Res 2017;77:303-11.
126. Marlein CR, Zaitseva L, Piddock RE, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017;130:1649-60.
127. Hou D, Wang B, You R, et al. Stromal cells promote chemoresistance of acute myeloid leukemia cells via activation of the IL-6/STAT3/OXPHOS axis. Ann Transl Med 2020;8:1346.
128. Méndez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010;466:829-34.
129. Forte D, García-Fernández M, Sánchez-Aguilera A, et al. Bone marrow mesenchymal stem cells support acute myeloid leukemia bioenergetics and enhance antioxidant defense and escape from chemotherapy. Cell Metab 2020;32:829-43.e9.
130. Stuani L, Sarry JE. Microenvironmental aspartate preserves leukemic cells from therapy-induced metabolic collapse. Cell Metab 2020;32:321-3.
131. van Gastel N, Spinelli JB, Sharda A, et al. Induction of a timed metabolic collapse to overcome cancer chemoresistance. Cell Metab 2020;32:391-403.e6.
132. Ye H, Adane B, Khan N, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 2016;19:23-37.
133. Javidi-Sharifi N, Martinez J, English I, et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. Elife 2019;8:e40033.
134. Huang D, Sun G, Hao X, et al. ANGPTL2-containing small extracellular vesicles from vascular endothelial cells accelerate leukemia progression. J Clin Invest 2021;131:e138986.
135. Powderly J, Patel M, Woodard P, et al. Abstract CT209: A first-in-human phase 1 trial of IO-108, an antagonist antibody targeting LILRB2 (ILT4), as monotherapy and in combination with pembrolizumab in adult patients with advanced relapsed or refractory solid tumors (NCT05054348). Cancer Res 2022;82:CT209.
136. Anami Y, Deng M, Gui X, et al. LILRB4-targeting antibody-drug conjugates for the treatment of acute myeloid leukemia. Mol Cancer Ther 2020;19:2330-9.
137. Dinardo CD, Pollyea DA, Konopleva M, et al. A first-in-human (FIH) phase 1 study of the anti-LILRB4 antibody IO-202 in relapsed/refractory (R/R) myelomonocytic and monocytic acute myeloid leukemia (AML) and R/R chronic myelomonocytic leukemia (CMML). Blood 2020;136:19-20.
138. Hong CS, Jeong E, Boyiadzis M, Whiteside TL. Increased small extracellular vesicle secretion after chemotherapy via upregulation of cholesterol metabolism in acute myeloid leukaemia. J Extracell Vesicles 2020;9:1800979.
139. Wang B, Wang X, Hou D, et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J Cell Physiol 2019;234:10602-14.
140. Kumar B, Garcia M, Weng L, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018;32:575-87.
141. Peng M, Ren J, Jing Y, et al. Tumour-derived small extracellular vesicles suppress CD8+ T cell immune function by inhibiting SLC6A8-mediated creatine import in NPM1-mutated acute myeloid leukaemia. J Extracell Vesicles 2021;10:e12168.
142. Hong CS, Danet-Desnoyers G, Shan X, Sharma P, Whiteside TL, Boyiadzis M. Human acute myeloid leukemia blast-derived exosomes in patient-derived xenograft mice mediate immune suppression. Exp Hematol 2019;76:60-6.e2.
143. Abdelhamed S, Butler JT, Doron B, et al. Extracellular vesicles impose quiescence on residual hematopoietic stem cells in the leukemic niche. EMBO Rep 2019;20:e47546.
144. Divakaruni AS, Jastroch M. A practical guide for the analysis, standardization and interpretation of oxygen consumption measurements. Nat Metab 2022;4:978-94.
145. Pfleger J. Measurements of mitochondrial respiration in intact cells, permeabilized cells, and isolated tissue mitochondria using the seahorse XF analyzer. Methods Mol Biol 2022;2497:185-206.
146. Leung DTH, Chu S. Measurement of oxidative stress: mitochondrial function using the seahorse system. Methods Mol Biol 2018;1710:285-93.
147. Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol 2021;12:627837.
148. Puleston D. Detection of mitochondrial mass, damage, and reactive oxygen species by flow cytometry. Cold Spring Harb Protoc 2015;2015:pdb.prot086298.
149. Wojtala A, Bonora M, Malinska D, Pinton P, Duszynski J, Wieckowski MR. Chapter thirteen - methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol 2014;542:243-62.
152. Nicholson JK, Lindon JC, Holmes E. 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999;29:1181-9.
154. Umpleby AM, Sönksen PH. 1 Measurement of the turnover of substrates of carbohydrate and protein metabolism using radioactive isotopes. Baillieres Clin Endocrinol Metab 1987;1:773-96.
155. Klapa MI, Aon JC, Stephanopoulos G. Systematic quantification of complex metabolic flux networks using stable isotopes and mass spectrometry. Eur J Biochem 2003;270:3525-42.
156. Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol 2018;36:316-20.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sharma P, Borthakur G. Targeting metabolic vulnerabilities to overcome resistance to therapy in acute myeloid leukemia. Cancer Drug Resist 2023;6:567-89. http://dx.doi.org/10.20517/cdr.2023.12
AMA Style
Sharma P, Borthakur G. Targeting metabolic vulnerabilities to overcome resistance to therapy in acute myeloid leukemia. Cancer Drug Resistance. 2023; 6(3): 567-89. http://dx.doi.org/10.20517/cdr.2023.12
Chicago/Turabian Style
Sharma, Priyanka, Gautam Borthakur. 2023. "Targeting metabolic vulnerabilities to overcome resistance to therapy in acute myeloid leukemia" Cancer Drug Resistance. 6, no.3: 567-89. http://dx.doi.org/10.20517/cdr.2023.12
ACS Style
Sharma, P.; Borthakur G. Targeting metabolic vulnerabilities to overcome resistance to therapy in acute myeloid leukemia. Cancer Drug Resist. 2023, 6, 567-89. http://dx.doi.org/10.20517/cdr.2023.12
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 8
likes
Like This Article 8
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.