
Mechanisms of resistance to anti-angiogenic treatments

Abstract
Hailed as the cancer treatment to end all the resistance to treatment, anti-angiogenic therapy turned out to be not quite what was promised. The hope that this therapeutic approach would not have suffered by the phenomenon of resistance was based on the fact that was targeting normal vessels rather than tumour cells prone to mutation and subject to drug induced selection. However, reality turned out to be more complex and since 1997, several mechanisms of resistance have been described to the point that the study of resistance to these drugs is now a very large field. Far from being exhaustive, this paper presents the main mechanisms discovered trough some examples.
Keywords
Introduction
Solid tumours need a blood supply and a large body of evidence has previously suggested that they can only grow behind a few millimetres in diameter if they induce the development of new blood vessels, a process known as angiogenesis. Based on this hypothesis, it was proposed that anti-angiogenic drugs should be able to suppress the growth of all solid tumours by killing off the blood supply[1]. However, after many clinical trials with anti-angiogenic agents, we now know that this is not always the case. Unfortunately, hope for a “pan-cancer” anti-angiogenic therapy[2] had been greatly diminished with the finding that the efficacy of vascular endothelial growth factor (VEGF) signaling pathway inhibitors is modest and is restricted to certain advanced-stage cancers only. Crucially, overshadowed until now, there is an increasing body of research, spanning now 25 years, demonstrating that not all the tumours are angiogenic. There are also non-angiogenic tumours that grow by exploiting pre-existing blood vessels of the surrounding nonmalignant tissue[3] using a process called vessel co-option[4]. This is a frequently overlooked mechanism of tumour vascularization that can mediate disease progression and metastasis. Its identification led also, for the first time, to raise the hypothesis that resistance to anti-angiogenic treatments can occur[5]. Until than this type of cancer treatment was regarded as unable to meet or induce any type resistance[6].
Clinical findings
In 1971, Folkman[1] proposed that new blood vessel formation (angiogenesis) is necessary for the progression of solid tumours beyond a size of a few millimetres cube and that blocking angiogenesis in tumours might be an effective means of maintaining or inducing dormancy and preventing metastasis. The premise of this hypothesis was that cancers cells chances of survival are dependent on their ability to induce formation of new vessels to deliver oxygen as all the pre-existing vessels were assumed to be destroyed by the neoplastic process. In the following years, many studies seemed to support this hypothesis. The direct association between the microvascular density observed in the neoplastic tissue and tumour aggressiveness[7,8], the identification of the angiogenic factors of the VEGF family[9,10] and the discovery that they are widely upregulated in human tumours[11-14] seemed to provide more evidences. The report in the mid 90s that angiostatin could induces astonishing levels of tumour regression in mouse models[15,16] further pushed the idea that antiangiogenic agents could treat patients with both early and advanced malignancies. Based in large part on these data, induction of angiogenesis has been considered as a hallmark of cancer since 2000[17,18]. It also emerged at the time the idea that, as the targeted vessels were not neoplastic, this type of cancer treatment could be immune from resistance[6].
In spite of all the high expectations, clinical trials produced disappointing results. For example, in high grade glioma, a Cochrane Review concluded that anti-angiogenic drugs do not improve significantly overall survival and there are no evidences to support this therapeutic approach[19]. However, in these patients, bevacizumab treatment can relieve symptoms by reducing the severity of intracranial oedema[20]. In advanced breast cancer improvement in disease free but crucially not overall survival has been seen while the results in early breast tumours are inconclusive[21]. Anti-angiogenic treatment has been instead effective in improving the outcome of patients with metastatic[22] colorectal cancer, although the improvements achieved are in the range of months rather than years. No benefit has been instead found for patients with early colorectal cancer[23]. Disappointing the results in Small Cell Lung cancer[24] while positive results have been reported in non-Small Cell Lung cancer, although the improvements in progression free and overall survival observed are, again, modest[25].
Mechanisms of resistance
The first evidence that cancer could be resistant to anti-angiogenic treatment was published in 1997 when non-angiogenic tumours were recognised and formally described for the first time in the lung[5]. In the following years, a number of mechanisms of resistance have been discovered. Resistance to antiangiogenic therapies can be intrinsic, when it is observed at the beginning of the treatment, or acquired, i.e., that it affects the relapsing disease after an initial response to therapy[26,27]. Here are illustrated the main mechanisms.
Non angiogenic growth by vascular co-option
Non-angiogenic cancers are early or advanced symptomatic tumours which grow by exploiting pre-existing vessels by means of vascular co-option. These neoplasms can be very aggressive but as they are lacking angiogenesis, it was immediately evident that anti-angiogenic treatment could have been completely ineffective in these patients and, for the first time, was suggested that this type of therapeutic approach could face resistance, as all the others do[5]. That non angiogenic growth, by vascular co-option is a mechanism of resistance to anti agiogenic treatmentrs has been now proved by a large numebr of clinical and pre-clinical studies. The rational behind this mechanism is illustrated in Figure 1.
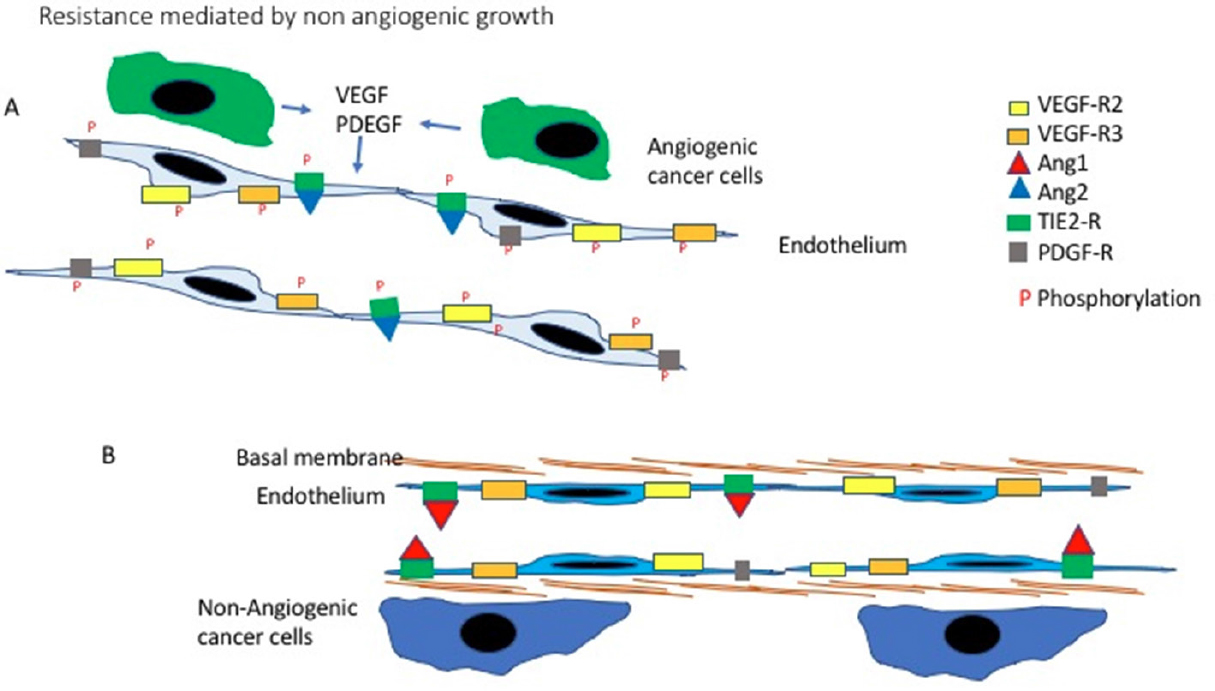
Figure 1. Non-angiogenic tumours: rational for resistance to anti-angiogenic treatment. A: Newly formed vessel. When cancer cells induce angiogenesis, new vessels sprout from the pre-existing one. These new vessels are lined up by newly formed proliferating endothelial cells. Under the influence of the angiogenic factors released by the cancer cells, like VEGF and PDEGF, the endothelial cells express higher levels of the relevant receptors. Following binding with their ligands, the intracellular portion of the receptors is phosphorylated activating the downstream intracellular pathways. A third angiogenic mechanisms is also present in this example: Angiopoietin 2 compete with Angiopoietin 1 and binds with the Tie receptor. Angiopoietin 1 maintains the endothelial cell quiescent, by taking its places Angiopoietin 2 trigger endothelial proliferation. In this situation compounds blocking VEGF or inhibitors of the Tyrosine kinases, blocking the activation of receptors like VEGFR2, can be effective; B: A co-opted pre-existing normal vessel. In non-angiogenic tumours the cells do not trigger angiogenesis but grow by exploiting the pre-existing vessels. These are lined by quiescent mature endothelial cells those few receptors for angiogenic factors remain inactive as no angiogenic stimuli are present. The Tie2 receptors remain linked to Angiopoietin 1 which maintain the vessel quiescent. Much is still to be learned about the biology of this system but is it is possible to appreciate how antibodies blocking VEGF or tyrosine kinases inhibiting angiogenesis, will not have any effects as the pathways they target are not contributing to the growth of these tumours. VEGF: vascular endothelial growth factor; PDEGF: platelets derived endothelial growth factor
Comparative clinco pathological investigations have confirmed that intrinsec and extrinsec resistance can be modulated by non angiogenic growth. The secondary location of breast carcinomas to the lung[28-30] liver[31,32], lymph nodes[33], skin[34] and brain[35-37] is frequently due to non-angiogenic growth. This conclusion can explain why phase III clinical trials in patients with advanced breast cancer involving sunitinib or bevacizumab as anti-angiogenic agent, either alone or with chemotherapy, have not been as successful as expected.
Similar observations have been provided by the study of brain tumours. Post mortem studies in patients treated with Cediranib, an inhibitor of VEGF receptor 2 (VEGFR2) tyrosine kinases, or Bevacizumab regimen[38,39] showed that the glioma cells were growing around pre-existing vessels. Contrast-enhanced MRI found, having administered bevacizumab, that spreading gliomas had a non-enhancing pastern consistent with invasive perivascular malignant progression[40,20]. Again, patients with metastatic colorectal carcinoma to the liver have a higher rate of response to bevacizumab plus chemotherapy when they have angiogenic metastases, while the poor responders have mostly non angiogenic secondaries[30]. The responders have also a significantly improved OS compared to the poor responder with non-angiogenic lesions (HR 3.50, 95% CI 1.49-8.20; P = 0.0022) indicating an association between vessel co-option and a poor response to antiangiogenic therapy.
Frentzas et al.[32] have provided evidences in a mouse model that co-option is effectively the cause for the resistance described in the above clinical examples. As vascular co-option requires for the cancer cells to be motile, the authors scrutinised the role of ARP2/3 complex. The actin-related protein 2 (ARP2; encoded by ACTR2) and ARP3 complex (known as the ARP2/3 complex) is involved with the nucleation-promoting factor into the nucleation of Actin filaments, leading to cell motility. In cancer, the ARP2/3 complex is well known as one of the key factors promoting invasion and metastases[41]. First Frentzas et al.[32] analysed the expression of the ARPC3 component of the complex by immunohistochemistry in the liver metastases proving higher expression in the non-angiogenic lesions compared to the angiogenic once. Than they moved to investigate the role of ARP2/3 in co-option using a mouse model of metastases in which the human colorectal cancer cell line HT29 is injected into the liver [Figure 2]. Metastases are than produced with both angiogenic and non-angiogenic areas. The authors than successfully knocked down ARP2/3 in this cell line using two different short hairpin mRNA. Depleted of ARP2/3 the cell motility was impaired but not its proliferation. Once injected into the liver these cells still produced metastatic lesions but prevalently angiogenic.
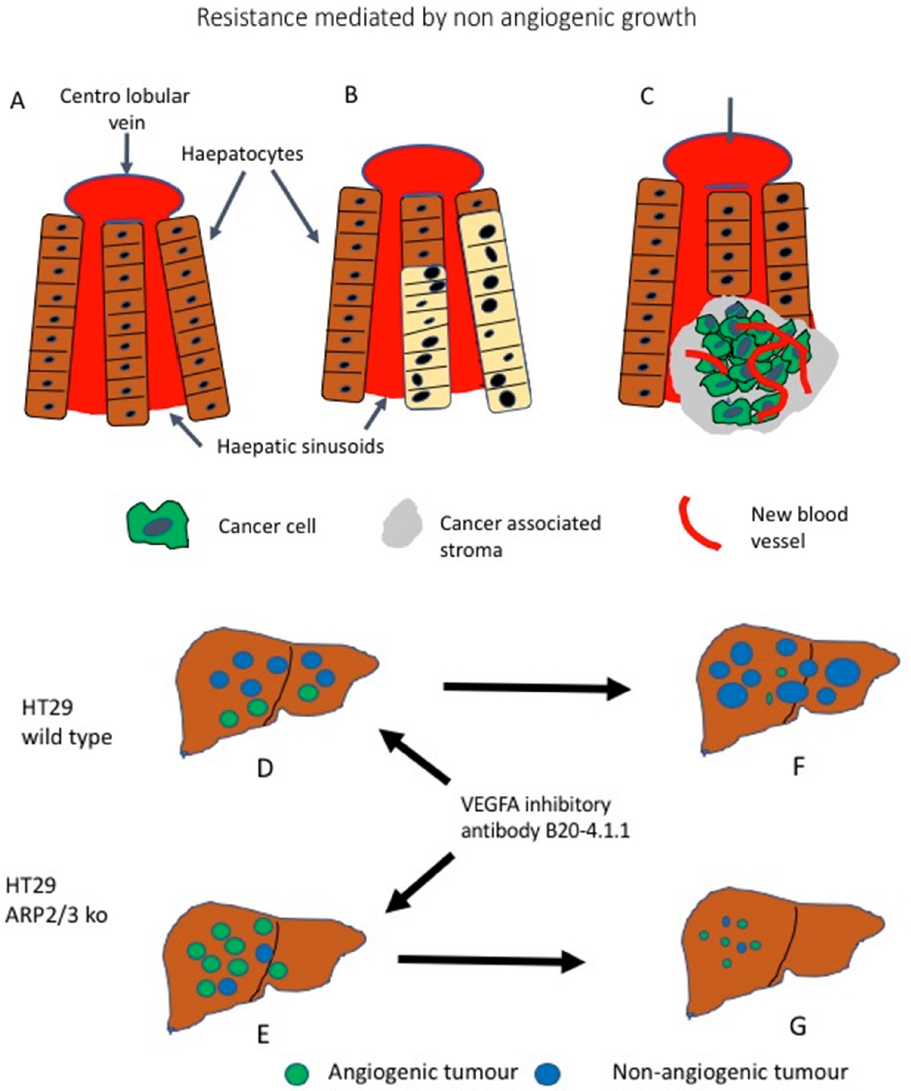
Figure 2. Non-angiogenic tumours are a cause for resistance to anti angiogenic treatment. A: Schematic representation of normal liver; B: a non-angiogenic liver metastases in which the liver architecture is preserved, the metastatic cells (beige) take the place of the hepatocytes exploiting the liver sinusoidal vascular system; C: an angiogenic metastases (green) grows by destroying the liver tissue, new vessels (in red) are sprouting providing the blood flow; D: HT29 Wild Type colorectal cancer cells produces metastases when injected into the mouse liver and the majority of them are non-angiogenic (blue). If HT29 cells with silenced ARP2/3 complex are injected; E: the metastatic growth will be predominantly angiogenic (green). When treatment with an anti VEGF antibody is performed, the angiogenic metastases of wild type tumours regresses, but the non-angiogenic lesions progresses; F: However, in tumours made up by HT29 cells with silenced ARP2/3 complexes the angiogenic metastases respond; G: Because of the silencing of the ARP2/3 complexes, the non-angiogenic metastases do not develop further. (Based on[32])
The mice with HT29 wild type metastatic disease, mostly non angiogenic, did not responded well to treatment with the VEGFA inhibitory antibody B20-4.1.1, however this antibody was effective in significantly reducing the neoplastic bulk in mice injected with ARP2/3 negative HT29 cells. This indicate that abrogation of motility and subsequent co-option, drive the cells to use angiogenesis and the lesion is therefore sensible to anti angiogenic compounds. It can be concluded that is the non-angiogenic nature of the tumour causing resistance and this can be reversed by preventing co-option happening[32]. Association between non-angiogenic tumours and resistance has been also illustrated in a different mouse model in which an orthotopic model of hepatocellular carcinoma is initially sensitive to Sorafenib, another Tyrosine Kinase inhibitor blocking the VEGF induced signalling, only to develop resistance after one month, by switching to an invasive phenotype, with upregulation of EMT-associated genes and co-option of sinusoidal and portal-tract vessels[42].
A first step in overcoming these problems would be an assessment of anti-angiogenic drugs in clinical trials where patients are selected according to predictive biomarkers (e.g., the vascular pattern). So far there have been no randomized phase III clinical trial of an antiangiogenic drug guided by biomarkers reflecting the type of vascularization present (e.g., newly formed vessels versus vascular co-option)[1,43]. This stands in marked contrast to what has happened in different situations in which clinical trials were designed according to predictive biomarkers such as, e.g., trastuzumab in HER2-positive breast cancer patients. A second approach under inquiry is the combination of treatment against angiogenesis and against vascular co-option. This follow the observations that vascular co-option is a mechanism of resistance[32] but the angiogenic status of a tumour can change during progression in both ways[3,29]. Therefore it is emerging that, as an angiogenic tumour treated with anti-angiogenic drugs can “escape” by turning non-angiogenic, also a non-angiogenic tumour treated with anti-co-option drugs could “escape” acquiring an angiogenic phenotype. The combined approach has been also suggested by animal model studies. Seaman et al.[44] described that CD276, a highly conserved cell-surface protein, is overexpressed in many different types of cancer both on cancer cells and endothelium. The most interesting finding was that CD276 was expressed on both newly formed and pre-existing vessels inside the tumours but not in normal vessels outside the malignant lesion and not during physiological angiogenesis. In a mouse model, a drug conjugated with anti CD276 antibody, eradicated both large established tumours and metastases and improved long-term overall survival likely because of the targeting of both angiogenic and non-angiogenic intra-tumour blood vessels[44].
Hypoxia mediated resistance
Some of the first, among other mechanisms of resistance discovered, have been those mediated by hypoxia[45]. It is a heterogeneous group, as different can be the causes of the better adaptation of a cell to hypoxia, but the common factor is that cells more equipped to survive in hypoxia, are more likely to remain vital and able to growth after treatment-induced reduction of the vascularity. In one of the first studies published, the cause is a genetic damage. Following the observation that p53 negative neoplastic cells are more resistant to apoptosis induced by hypoxia[46], Yu et al.[47] investigated in a mouse model whether the increased hypoxia which follow the targeting of blood vessels, could select the growth of p53 negative cancer cells [Figure 3A]. Mice were injected with human colorectal cancer cell lines HCT116 wild type, p53 positive, and HCT116 p53-/p53-. Once the neoplasms had developed, the mice were treated with low dose vinblastine and the antibody DC101, against the anti-murine VEGFR2, or with the DC101 antibody alone. The HCT116 p53-/53- xenograft were slower to respond to both treatment schedules. In a second set of experiments xenografts were induced by injecting a mixture of an equal amount of HCT116 p53-/53- and Wild Type (p53+/p53+) cells. Following treatment with Vinblastim and DC101, a slowdown of the tumour growth compared to the negative control was found. However, these residual tumours were still growing and the percentage of p53-/53- cells in the tumour had increased as they demonstrated to have a selective advantage over the p53+/p53+ [Figure 4]. Finally, the authors showed that in an untreated xenograft induced by injecting 50%/50% mixture of p53 positive and negative cells, the less hypoxic areas, closer to the vessels, were richer in p53+ positive cells than the sections of tumours more distant from the blood supply. This was due to a higher rate of apoptosis among the p53+/p53+ cells distant from the blood vessels. The author concluded that the genetic imprinting of the neoplastic cell can therefore be a cause of resistance to at least some types of antiangiogenic treatments[47].

Figure 3. Resistance by Hypoxia. Following anti-angiogenic treatment, the decrease in number of vessels frequently leads to increased hypoxia levels. A: Mechanism driven by p53 mutation. In the model investigated by Yu et al.[47] the human colorectal cancer HCT116 cell line is used in its wild type and p53-/p53- versions. When a mouse carrying a xenograft produced by a mixture of HCTT16 WT and HCT116 p53-/p53- is treated with an anti VEGF antibody an initial shrinkage of the tumour occurs as most of the WT-hypoxia sensitive cells undergo apoptosis. However, the p53-/p53- cells, hypoxia resistant, continuous to growth producing eventually an even larger mass. (based on Yu et al.[47]); B: Mechanism driven by Metabolic Symbiosis. As the number of hypoxic cells increases, following treatment-induced vascular disappearance, the hypoxic cells by releasing LDH allows the normoxic cells, which internalise it and turn it into pyruvate, to improve their Oxidative Phosphorylation, decrease their consume of glucose and therefore leave more glucose available. for the hypoxic cells which have therefore the possibility to produce more energy and survive. The same phenomenon occurs for the tumour-associated stroma. (Based on [51]and [52])

Figure 4. Redundant angiogenic pathways. Binding of VEGFA with is receptor VEGFR2 lead to the activation of four main pathways. Two, the Ras/MAPK and the PI3K/AKT promotes proliferation of the endothelial cell, while the other two, Fak and Cdc42 pathways induces migration of the same cells. Proliferation and migration of the endothelial cells is necessary for angiogenesis to happen. However, angiogenic stimuli leading to these two events in the endothelium can also be independent from the VEFG action. As illustrated in this picture, PDEGF and FGF2 also lead to activation of the Ras/MAPK pathways and therefore proliferation and on activation of migration. Angiopoietin 1 can activate motility as the Wnt Canonical pathway can trigger proliferation. (Based on data from[81]). HGF: hepatocyte growth factor; Mek: methyl ethyl ketone; eNOS: endothelial Nitric Oxide Synthase 3; MAPK: mitogen activated protein kinase; Erk: extracellular signal regulated kinase; PI3K/AKT: phosphoinositide 3-kinase/protein kinase B.
A second way in which hypoxia is believed to mediate resistance is after treatment with anti VEGF therapy. One example is that reported in a study by Paez-Ribes et al.[48]. In murine models of neuro endocrine pancreatic cancer and of glioblastoma, treatment with the anti-murine VEGRF2 antibody DC101 is followed by reduction in volume of the tumours but, at the same time, the residual malignancy has a more invasive phenotype which persisted also after stopping treatment. In tumour cells with the VEGF-A gene deleted, a similar aggressive phenotype was found. Anatomical examination also revealed an increased number of metastatic lesions occurring. Similar results were found when, instead of the DC101 antibody, an anti-angiogenic tyrosine kinase inhibitor, Sunitinib, was employed to treat the tumours. As in the previous model, also in this one an increase in the number of hypoxic cells has been found suggesting again a link between post treatment development of hypoxia and escape from drugs targeting the new vessels[48,49].
In a third study where treatment with Bevacizumab is followed again by hypoxia and increased invasiveness of glioma cells[50] the authors dissect the mechanism by which hypoxia leads to invasiveness. In the neoplastic cell investigated, the higher levels of Hif1 which follows hypoxia induces, among others, the transcription of ZEB2. ZEB2 protein overexpression leads to down regulation of Ephrin B2 and, in this model the authors demonstrated that, following loss of Ephrin B2 expression, the glioma cells become more invasive[50].
A final example of how the increased levels of hypoxia caused by anti-angiogenic treatment can lead to resistance and tumour growth is the one which relay on metabolic symbiosis[51]. This is a process in which both cancer cells and tumour associated stromal cells located in the best oxygenated areas, collaborate with the more hypoxic cells in order to optimise their respective metabolisms and energy production [Figure 3B]. The more hypoxic the cell, the more glycolysis is used leading to production of pyruvate. As oxygen is scarce, most of the pyruvate is converted to lactate, instead of entering the tricarboxylic acid cycle (TAC). This excess of lactate is secreted in the extra cellular environment where it diffuses and is picked up by the more oxygenated cells which internalise the lactate in the cytoplasm, revert it to pyruvate and use it to further enhance the efficiency of their TAC and, consequently, of their oxidative phosphorylation. As the better oxygenate cells improve the use of their respiratory chain, their need for glucose decreases. As a consequence, more glucose is left available in extracellular compartment for the more hypoxic areas of the tumour[51]. Following anti-angiogenic treatment therefore, it has been observed that the establishment of this positive symbiotic loop allows the cells to remain viable and proliferate also in face of the dramatic increases of hypoxia which follow angiogenesis inhibition and collapse of part of the vasculature[52].
How to overcome hypoxia mediate resistance has been the object of investigation for many years[53] and its discussion would be too long for this review. Several the approaches currently under scrutiny. Some of the most interesting are those looking at targeting the associated metabolic changes[54,55], targeting epigenetic changes, using drug associated nano particles[56], use hypoxia imaging as predictive factor[57] or use of small molecules inhibiting protein-protein interaction to target Hif1[58].
Redundancy of the angiogenic signals
The main angiogenic pathway is the VEGF one[59] however it is not the only and, in its complexity, interacts with several other pathways. Recently in a very thorough review Gacche and Assaraf[60] identified three main mechanisms of resistance due to redundancy of the angiogenic signals: 1) activation of pathways involving angiogenic factors other than VEGF; 2) replacement production of VEGF by non- neoplastic, stromal cells or 3) pericytes-driven angiogenesis. There are several angiogenic factors other than VEGF (for review by Gacche et al.[60] and Ribatti[49]) and the main one, with their correspondent pathways, are illustrated in Figure 4. Among these pathways an important VEGF- independent angiogenic activity is provided by the interplay of fibroblastic growth factor (FGF2) and PDGF-BB[61]. PDGF-BB is a factor with strong chemotactic and mitogenic action on the pericytes while the FGF2 induces proliferation mostly on the endothelium. In this study the authors demonstrate that the angiogenic factor FGF2 has two actions on the endothelial cells: triggers proliferation, through the Ras/MAPK pathway and also induces higher levels of two PDEGF receptors: the alpha and the beta. In this way, the sensitivity of the endothelial cell to PDEGF is increased and, as consequence, also its motility is stepped up[61]. Instead while the presence of FGF2 recruits and maintain pericytes, the additional expression of PDEGF inhibits their recruitment, making the vessels very leaky[61]. As this pathway is VEGF independent, it can maintain angiogenesis in presence of anti VEGF pathways drugs.
These factors providing redundant angiogenic signals have also other roles in cancer and many approaches to target them are being investigated. FGF2 is widely involved in many types of cancer trough activation of the Ras/MAPK and PI3K pathways causing not only angiogenesis but also increased proliferation and metastatic spread[62]. Targeting FGF2 is therefore one of the ways to overcome resistance to anti VEGF therapy[63,64]. PDGF also is involved in many aspects of the cancer cell biology and both pharmacological compounds and inhibitors of tyrosine kinases specific for this pathway are being investigated[65,66]. Finally, because of the emerging role of pericytes in maintaining intra tumour vessels viable, targeting these cells has become the latest approach investigated to overcome resistance to anti VEGF treatment[67,68].
Vascular heterogeneity
Endothelial cells sensitive to anti VEGF therapy rely on this pathway. However not all the endothelial cells are the same. In the human body differences exists according to the anatomical location and the type of vessels[69,70]. Inside cancer lesions vascular heterogeneity is even more pronounced, intra tumour vessels have a heterogeneous anatomical structure and endothelial phenotype[71]. Anatomically the main features causing heterogeneity are a variable degree of leakage and variable coverage by pericytes. The endothelial cells themselves are than heterogeneous as far as the genotype and the phenotype is concerned. Endothelial genotypic differences in mice models are due to the occurrence of aneuploidy and presence of abnormal centromeres, and the genetic defects, in this model, are not due to contamination form the tumour. Variable patterns of mRNA transcriptions have also been found which lead to a variable protein phenotype resulting in differences in behaviour and response to drugs and growth factors[71]. One case of heterogeneity is due the up regulation in endothelial cells, associated with tumours, of the PI3K/AKT pathway which has been reported in some intra tumour endothelial cells[72], one of the consequences is the variable response to angiogenic factors like VEGF and to their blockage[73].
One example that demonstrates both the effect of angiogenesis redundancy and vascular heterogeneity is the resistance to Bevacizumab observed in ovarian cancer. In human tissue samples of this malignancy, heterogeneity of AKT phosphorylation in the intra tumour endothelial cells has been reported by Guerrouahen et al.[74]; they show how within one single vessels, a mixture of heterogeneous endothelial cells is present, as exemplified in Figure 5. To investigate the role of endothelial AKT phosphorylation as a mechanism or resistance “in vitro” the authors first selected, trough continuous exposure to Bevacizumab, HUVECs (Human umbilical vein endothelial cells) resistant to Bevacizumab. These cells showed resistance to Bevacizumab “in vitro” and also had higher levels of AKT phosphorylation. Treatment with the pan-PI3K inhibitor LY294002 blocked AKT phosphorylation but did not harmed the cells, however abolished the resistance to Bevacizumab as its addiction lead to endothelial cell death indicating that AKT phosphorylation was mediating the resistance to anti VEGF treatment. These resistant cells have also higher levels of FGF2 compared to the normal HUVAC, and following incubation with Bevacizumab, a further increase in FGF2 transcription and translation was observed alongside an increase in levels of FGFR1 and its phosphorylation. The authors than demonstrated that these higher levels of FGF2 leads to yet more phosphorylation of AKT alongside activating Src and the pro-angiogenic pathway ERK1/STAT3[Figure 5]. As selective inhibition of FGF2 reverse this process, the authors conclude demonstrating that in these cells, Bevacizumab treatment leads to an autocrine loop supporting the viability of the endothelial cell[74]. This intra cellular mechanism can be further enhanced by the recruitment, following Bevacizumab treatment, of marrow-derived fibrocyte-like cells which also produce FGF2[75,76].

Figure 5. Angiogenic pathways redundancy and endothelial heterogeneity. Intra-tumour endothelial cells, despite being normal, non-neoplastic cells, can be still heterogeneous as far as their biology is concern. Guerrouahen et al.[74] have demonstrated that endothelial cells from vessels in ovarian carcinoma can considerably differ as far as phosphorylation of AKT is concerned. A:They describe that, following blockage of VEGF, the endothelium with low levels of AKT phosphorylation undergo apoptosis; B: However, in the endothelial cells with high level of phosphor AKT, following the block of VEGF stimulation, the AKT pathway remains active inducing transcription of FGF, an angiogenic factor, which on one side promotes endothelium proliferation and on the other further phosphorylates AKT producing a positive loop. (Based on [74]). FGFR: fibroblast growth factor receptor
Increased knowledge of the phenotype of intratumor endothelial cells is now allowing to plan new approaches to overcome the heterogeneity-linked problems met so far[73]. One way is to exploit the expression of markers which are more diffusely expressed but are not, by themselves, targets of treatments. By developing petide-ligand motives, and conjugating these peptides to drugs or drug-containing liposome, a broader range of endothelial cells can be targeted[77,78,73]. A second approach is to deliver with liposome siRNA which can “switch off” the transcription of factors inducing resistance[73]. A third approach is to target the cytoskeleton and the cell-cell junctions of the endothelial cells as these are all structures fairly uniform in endothelial cells[79]. Some phase III trials with anti-endothelial compounds have been done but the results have not been encouraging indicating that this could eventually be a rather difficult problem to solve[80].
Conclusions
The main conclusion that most of these studies share is that resistance to anti angiogenic treatment is very frequent and mediated by several different mechanisms. Therefore if therapies targeting new vessels are going to stay, it will be as part of combined treatments. An emerging new way to use these drugs is in a dual approach, targeting both angiogenic and non-angiogenic growth patterns. Some possible ways to achieve this are emerging. For example, CD276, a highly conserved cell-surface protein, is found broadly overexpressed by multiple malignancies on both cancer cells and blood vessels[44]. Notably, CD276 was expressed on both newly formed and pre-existing vessels present inside tumours but not in normal vessels outside the neoplastic mass and not during physiological angiogenesis (e.g., regenerating liver tissue)[44]. In a mouse model, an antibody-drug conjugate pyrrobenzodiazepine-conjugated CD276, eradicated both large established tumours and metastases and improved long-term overall survival potentially as a result of targeting both angiogenic and non-angiogenic tumour blood vessels[44]. However, as these observations are based on animal studies, work is necessary to confirm whether this would be an effective therapy for angiogenic and non-angiogenic human cancers. Progresses on the study of the biology of non-angiogenic tumours and how they co-opt vessels is therefore essential to develop new approaches to cancer treatment but they are likely to be effective only as a part of multi drugs therapeutic protocols.
Declarations
Authors’ contributionsWriting-original draft preparation and editing: Pezzella F
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Kolata G. Hope in the lab: A special report; Tow drugs eradicate tumors in mice. The New York Times 1998.
3. Donnem T, Reynolds AR, Kuczynski EA, Gatter K, Vermeulen PB, et al. Non-angiogenic tumours and their influence on cancer biology. Nat Rev Cancer 2018;18:323-36.
4. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol 2019; doi: 10.1038/s41571-019-0181-9. [Epub ahead of print]
5. Pezzella F, Pastorino U, Tagliabue E, Andreola S, Sozzi G, et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am J Pathol 1997;151:1417-23.
6. Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays 1991;13:31-6.
7. Weidner N, Folkman J, Pozza F, Bevilacqua P, Allred EN, et al. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst 1992;84:1875-87.
8. Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med 1991;324:1-8.
9. Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989;161:851-8.
10. Senger DR, Connolly DT, Van de Water L, Feder J, Dvorak HF. Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res 1990;50:1774-8.
11. Mise M, Arii S, Higashituji H, Furutani M, Niwano M, et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996;23:455-64.
12. Obermair A, Kucera E, Mayerhofer K, Speiser P, Seifert M, et al. Vascular endothelial growth factor (VEGF) in human breast cancer: correlation with disease-free survival. Int J Cancer 1997;74:455-8.
13. Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res 1995;55:3964-8.
14. Toi M, Inada K, Hoshina S, Suzuki H, Kondo S, et al. Vascular endothelial growth factor and platelet-derived endothelial cell growth factor are frequently coexpressed in highly vascularized human breast cancer. Clin Cancer Res 1995;1:961-4.
15. O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994;79:315-28.
16. O’Reilly MS, Holmgren L, Chen C, Folkman J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med 1996;2:689-92.
19. Ameratunga M, Pavlakis N, Wheeler H, Grant R, Simes J, et al. Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst Rev 2018;11:CD008218.
20. Verhoeff JJ, van Tellingen O, Claes A, Stalpers LJ, van Linde ME, et al. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 2009;9:444.
21. Mackey JR, Kerbel RS, Gelmon KA, McLeod DM, Chia SK, et al. Controlling angiogenesis in breast cancer: a systematic review of anti-angiogenic trials. Cancer Treat Rev 2012;38:673-88.
22. Lopez A, Harada K, Vasilakopoulou M, Shanbhag N, Ajani JA. Targeting Angiogenesis in Colorectal Carcinoma. Drugs 2019;79:63-74.
23. Kerr RS, Love S, Segelov E, Johnstone E, Falcon B, et al. Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): an open-label, randomised phase 3 trial. Lancet Oncol 2016;17:1543-57.
24. Stratigos M, Matikas A, Voutsina A, Mavroudis D, V. G. Targeting angiogenesis in small cell lung cancer. Transl Lung Cancer Res 2016;5:389-400.
25. Manzo A, Montanino A, Carillio G, Costanzo R, Sandomenico C, et al. Angiogenesis Inhibitors in NSCLC. Int J Mol Sci 2017;18:20121.
26. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis 2014;17:471-94.
27. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592-603.
28. Pezzella F, Di Bacco A, Andreola S, Nicholson AG, Pastorino U, et al. Angiogenesis in primary lung cancer and lung secondaries. Eur J Cancer 1996;32A:2494-500.
29. Breast Cancer Progression Working Party. Evidence for novel non-angiogenic pathway in breast-cancer metastasis. Breast Cancer Progression Working Party. Lancet 2000;355:1787-8.
30. Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol 2017;241:362-74.
31. Stessels F, Van den Eynden G, Van der Auwera I, Salgado R, Van den Heuvel E, et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer 2004;90:1429-36.
32. Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med 2016;22:1294-302.
33. Jeong HS, Jones D, Liao S, Wattson DA, Cui CH, et al. Investigation of the Lack of Angiogenesis in the Formation of Lymph Node Metastases. J Natl Cancer Inst 2015;107:djv155.
34. Colpaert CG, Vermeulen PB, Van Beest P, Soubry A, Goovaerts G, et al. Cutaneous breast cancer deposits show distinct growth patterns with different degrees of angiogenesis, hypoxia and fibrin deposition. Histopathology 2003;42:530-40.
35. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156:1002-16.
36. Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS One 2009;4:e5857.
37. Bugyik E, Dezso K, Reiniger L, László V, Tóvári J, et al. Lack of angiogenesis in experimental brain metastases. J Neuropathol Exp Neurol 2011;70:979-91.
38. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 2010;12:233-42.
39. di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of “rebound” revascularization as mode of escape. Cancer Res 2011;71:19-28.
40. Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol 2009;5:610-20.
42. Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J Natl Cancer Inst 2016;108.
43. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990;82:4-6.
44. Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer cell 2017;31:501-15.
46. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996;379:88-91.
47. Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science 2002;295:1526-8.
48. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15:220-31.
49. Ribatti D. Novel angiogenesis inhibitors: addressing the issue of redundancy in the angiogenic signaling pathway. Cancer Treat Rev 2011;37:344-52.
50. Depner C, Zum Buttel H, Böğürcü N, Cuesta AM, Aburto MR, et al. EphrinB2 repression through ZEB2 mediates tumour invasion and anti-angiogenic resistance. Nat Commun 2016;7:12329.
51. Nakajima EC, Van Houten B. Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog 2013;52:329-37.
52. Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, et al. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Rep 2016;15:1161-74.
53. Poon E, Harris AL, Ashcroft M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med 2009;11:e26.
55. Ward C, Langdon SP, Mullen P, Harris AL, Harrison DJ, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171-9.
56. Rey S, Schito L, Koritzinsky M, Wouters BG. Molecular targeting of hypoxia in radiotherapy. Adv Drug Deliv Rev 2017;109:45-62.
57. Suh YE, Lawler K, Henley-Smith R, Pike L, Leek R, et al. Association between hypoxic volume and underlying hypoxia-induced gene expression in oropharyngeal squamous cell carcinoma. Br J Cancer 2017;116:1057-64.
58. Li J, Xi W, Li X, Sun H, Li Y. Advances in inhibition of protein-protein interactions targeting hypoxia-inducible factor-1 for cancer therapy. Bioorg Med Chem 2019;27:1145-58.
59. Adams RH, K. A. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007;8:464-78.
60. Gacche RN, YG. A. Redundant angiogenic signaling and tumor drug resistance. Drug Resist Updat 2018;52:1350-62.
61. Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, et al. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest 2007;117:2766-77.
62. Akl MR, Nagpal P, Ayoub NM, Tai B, Prabhu SA, et al. Molecular and clinical significance of fibroblast growth factor 2 (FGF2 /bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016;7:44735-62.
63. Alessi P, Leali D, Camozzi M, Cantelmo A, Albini A, et al. Anti-FGF2 approaches as a strategy to compensate resistance to anti-VEGF therapy: long-pentraxin 3 as a novel antiangiogenic FGF2-antagonist. Eur Cytokine Netw 2009;20:225-34.
64. Ma H, Qiu P, Xu H, Xu X, Xin M, et al. The Inhibitory Effect of Propylene Glycol Alginate Sodium Sulfate on Fibroblast Growth Factor 2-Mediated Angiogenesis and Invasion in Murine Melanoma B16-F10 Cells In Vitro. Mar Drugs 2019;17.
65. Papadopoulos N, Lennartsson J. The PDGF/PDGFR pathway as a drug target. Mol Aspects Med 2018;62:75-88.
66. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal 2013;11:97.
67. Meng MB, Zaorsky NG, Deng L, Wang HH, Chao J, et al. Pericytes: a double-edged sword in cancer therapy. Future Oncol 2015;11:169-79.
68. Chen Z, Xu XH, Hu J. Role of pericytes in angiogenesis: focus on cancer angiogenesis and anti-angiogenic therapy. Neoplasma 2016;63:173-82.
69. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 2007;100:174-90.
70. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 2007;100:158-73.
71. Hida K, Ohga N, Akiyama K, Maishi N, Hida Y. Heterogeneity of tumor endothelial cells. Cancer Sci 2013;99:1391-5.
72. Bussolati B, Assenzio B, Deregibus MC, Camussi G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J Mol Med (Berl) 2006;84:852-63.
73. Hida K MN, Sakurai Y, Hida Y, Harashima H. Heterogeneity of tumor endothelial cells and drug delivery. Adv Drug Deliv Rev 2016;99:140-7.
74. Guerrouahen BS, Pasquier J, Kaoud NA, Maleki M, Beauchamp MC, et al. Akt-activated endothelium constitutes the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol Cancer Ther 2014;13:3123-36.
75. Mitsuhashi A, Goto H, Saijo A, Trung VT, Aono Y, et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat Commun 2015;6:8792.
76. Ma S, Pradeep S, Hu W, Zhang D, Coleman R, Sood A. The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Res 2018;7:326.
77. Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 2000;60:722-7.
78. Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, et al. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res 2003;63:7400-9.
79. Hinnen P, Eskens FA. Vascular disrupting agents in clinical development. Br J Cancer 2007;96:1159-65.
80. Lorusso PM, Boerner SA, Hunsberger S. Clinical development of vascular disrupting agents: what lessons can we learn from ASA404? J Clin Oncol 2011;29:2952-5.
81. KEGG PATHWAY. (Accessed March April 2019, 2019, at https://www.kegg.jp/kegg/pathway.html.).
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pezzella F. Mechanisms of resistance to anti-angiogenic treatments. Cancer Drug Resist 2019;2:595-607. http://dx.doi.org/10.20517/cdr.2019.39
AMA Style
Pezzella F. Mechanisms of resistance to anti-angiogenic treatments. Cancer Drug Resistance. 2019; 2(3): 595-607. http://dx.doi.org/10.20517/cdr.2019.39
Chicago/Turabian Style
Pezzella, Francesco. 2019. "Mechanisms of resistance to anti-angiogenic treatments" Cancer Drug Resistance. 2, no.3: 595-607. http://dx.doi.org/10.20517/cdr.2019.39
ACS Style
Pezzella, F. Mechanisms of resistance to anti-angiogenic treatments. Cancer Drug Resist. 2019, 2, 595-607. http://dx.doi.org/10.20517/cdr.2019.39
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 0
likes
Like This Article 0
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.