
Overcome cancer drug resistance by targeting epigenetic modifications of centrosome


Abstract
The centrosome is an organelle that serves as the microtubule- and actin-organizing center of human cells. Although the centrosome is small of size, it is great important on cellular function that regulates cytoskeletal organization and governs precise spindle orientation/positioning ensuring equal distribution of cellular components in cell division. Epigenetic modifications to centrosome proteins can lead to centrosome aberrations, such as disorganized spindles and centrosome amplification causing aneuploidy and genomic instability. Epigenetic disturbances are associated not only with carcinogenesis and cancer progression, but also with drug resistance to chemotherapy. In this review, we discuss mechanisms of epigenetic alteration during the centrosome biogenesis in cancer. We provide an update on the current status of clinical trials that aim to target epigenetic modifications in centrosome aberrations and to thwart drug resistance.
Keywords
Introduction
It has long been considered that the accumulation of genetic mutations in tumor suppressors and/or oncogenes causes cancer[1]. However, mounting evidences have emerged that alterations of every component in the epigenetic regulatory machinery also participate in carcinogenesis[2,3]. Ultimately both genetic and epigenetic changes determine abnormal gene expression. Centrosomes play a key role in establishment and maintenance of the bipolar mitotic spindle that require to accurately divide genetic material (chromosomes) into daughter cells during cell division. Centrosome aberrations are either numerical or structural aberrations since they arise when centrosome structure, duplication or segregation are deregulated. So far, it has not been shown whether structural centrosome aberrations directly trigger drug resistance. In this review, we only focus on numerical aberrations of centrosome. Acquisition of ≥ 3 centrosomes in the centrosome cycle was termed centrosome amplification. Failure to properly control centrosome number leads to aneuploidy, which is frequently found in cancer cells. Mechanistically centrosome amplification may cause multipolar spindles or monopolar aster resulting in chromosome missegregation[4]. Thus, centrosome amplification is a hallmark of human tumors[5-10]. The BRCA1 E3 ligase specifically ubiquitinated γ-tubulin at lysine-48 (K48) and the expression of a mutant γ-tubulin protein in which K48 was mutated to arginine induces centrosome amplification[11]. However, centrosome amplification does not necessarily require DNA damages, epigenetic changes is one potential mechanistic link in dysregulation of centrosome function. Here we discuss in detail on centrosome structure, aberrations of centrosome in cancer, the relationship of cancer drug resistance to centrosome amplification and new drug development.
Centrosome aberrations, cancer and cancer drug resistance
In this section, we briefly summarize the structure and biogenesis of centrosomes (for more detail reviews, please refer to[12]. Morphologically, centrosomes are non-membranous organelles. Each centrosome consists of a pair of centrioles surrounded by the pericentriolar material (PCM) [Figure 1]. Although molecular compositions of the centrosome remain poorly defined, hundreds of proteins have been detected using proteomics[13,14]. As illustrated in Figure 2[15], cycling cells begin the cell cycle with one centrosome in G1, and centriole duplication occurs once per cell cycle, paralleled with DNA replication during the S phase. The duplication of the centrosome is initiated (formation of the procentriole) with Plk4 as the dominant kinase in centriole biogenesis[16,17]. Depletion of Plk4 induces the loss of centrioles and overexpression of Plk4 conversely causes the formation of multi-daughter centrioles[18].
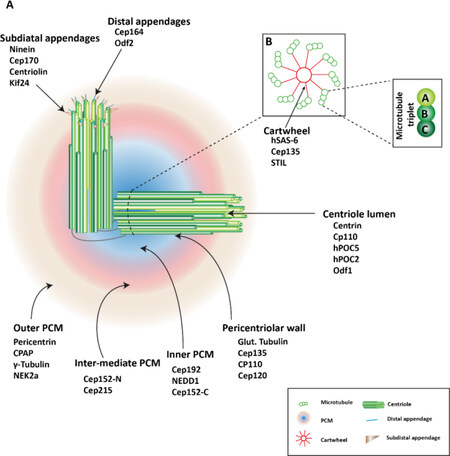
Figure 1. Centrosome structure. A: the centrosome consists a pair of orthogonal centrioles, surrounded by mass proteins, referred to as the pericentriolar material (PCM). PCM proteins subdivide the PCM to three layers depending on diameter of the ring-like structures. Mother centrioles possess subdistal appendages and distal appendages; B: each centriole is a cylindrical structure with nine-fold microtubule organized like a central cartwheel. The triplet microtubules are composed of internal A, middle B, and external C microtubules. The figure is adapted and modified[9]
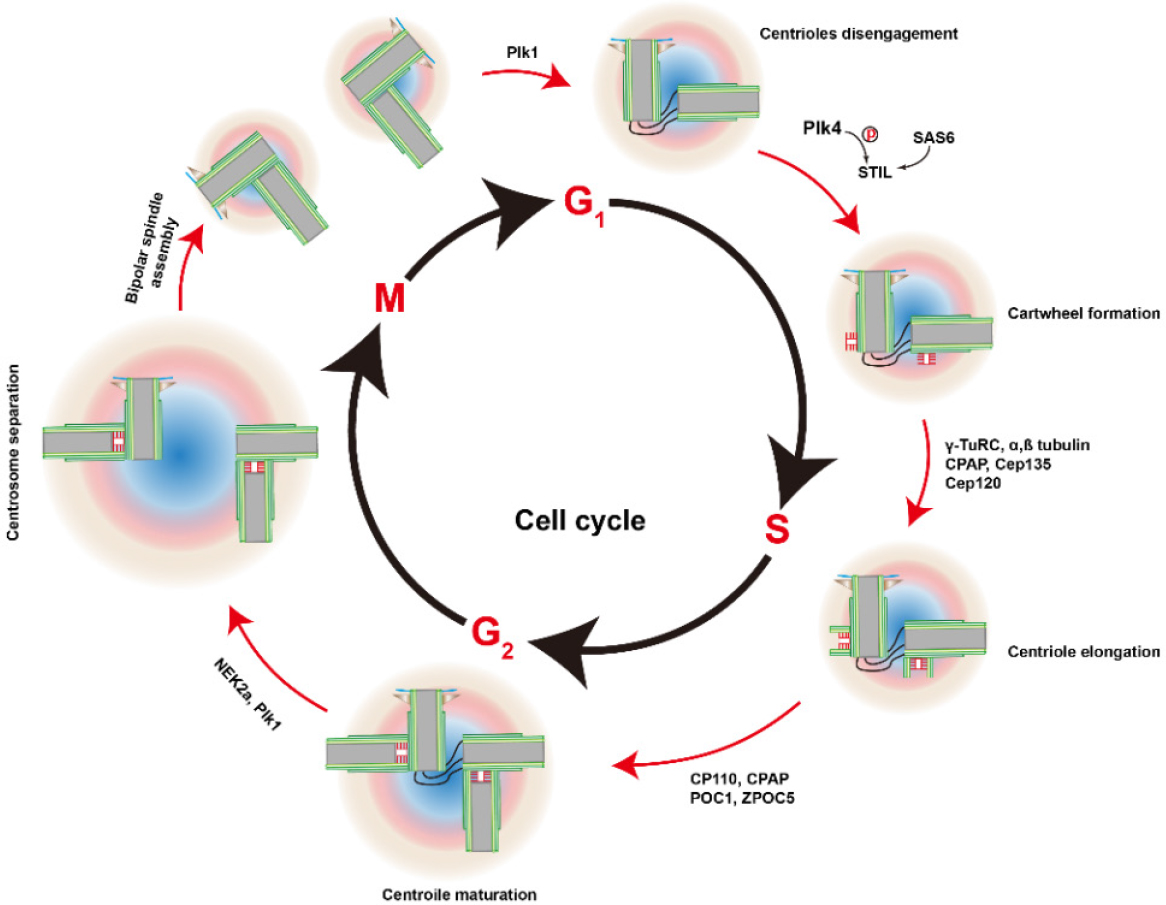
Figure 2. Centrosome duplication cycle. Centrosome duplication begins at the G1-S transition, with the disengagement of the pair of centrioles. Plk4 binds and phosphorylates STIL and associates with SAS-6. Thus, the cartwheel forms the proximal wall of the mother centriole. Other proteins are recruited to the cartwheel and the new daughter centriole (procentriole) begins to grow from the existing centrioles during S and reaches the full length at the G2 phase. Duplicated centrosomes separate at the beginning of the M phase, helped by kinases NEK2a and Plk1. PCM mature and the cartwheels disassemble in the early M phase. After the separation of two daughter cells, each cell inherits one centrosome. The figure is adapted and modified[15]
Once centrioles are assembled, PCM proteins will be recruited, nucleating more microtubules during interphase. These proteins are not only important for centrosome biogenesis but also contribute to the maintenance of cell polarity, cell vesicle transport, cell adhesion, cell signal transduction (Reviewed[19]). Duplicated centrosomes separate at the onset of mitosis for bipolar spindle formation, which equally segregate sister chromatids to two daughter cells. Centrosome disjunction is modulated by NEK2a to remove centrosomal linkers, such as C-Nap1 (also known as CEP250) and rootletin[20]). Over-expression of NEK2a induces immature centrosome separation[21,22].
It is worth to note that majority of centrosome proteins have multiple locations. Approximately 77% (n = 370) of the centrosome and microtubule-organizing center (MTOC) proteins detected in the cell atlas also localize to other cellular compartments[23]. The network plot shows that the most common locations shared with centrosome and MTOC are the cytoplasm, nucleus and vesicles. For example, CTCF is associated with the centrosome in metaphase to anaphase of the cell cycle. At telophase, CTCF dissociates from the centrosome and localizes to the midbody and the newly formed nuclei.
Functionally the centrosome in human cells acts as the MTOC, which has been studied widely ever since first described by Theodor Bovery in 1900[24], and the actin-organizing center[25]. Importantly precisely duplicated and matured centrosomes ensure faithful chromosome segregation into two daughter cells via the formation of the bipolar mitotic spindle[26]. Thus, when centrosome structure, duplication or segregation are deregulated, centrosome aberrations with either numerical or structural aberrations arise.
Many studies established a link of centrosome aberrations and solid tumors or hematological malignancies; the correlations are present not only in pre-invasive lesions but also with tumor progression[27,28]. Although bipolar spindles could be detected in centrosome depletion or centrosome over-duplicated cells by clustering mechanisms[29], the numerical centrosome aberrations are still the most common cause for chromosomal segregation errors[30]. Centrosome amplification thus can be as a novel biomarker for personalized treatment of cancers[31]. Several oncogenic and tumor suppressor proteins, such as BRCA1 and p53, are the best-known centrosome proteins[32,33]. Either overexpression or downregulation of centrosome proteins evoke centrosome abnormalities resulting tumorigenesis [Table 1].
Association of centrosome proteins with epigenetic disturbances in different types of tumors
As discussed above, NEK2 is an important centrosome protein, which regulates centrosome separation and bipolar spindle formation in mitotic cells. High expression of NEK2 also mediates drug resistance to cisplatin or lipo-doxorubicin in myeloma[48], breast cancer[49], ovarian cancer[50] and liver cancer[51]. Nlp (ninein-like protein) is involved in centrosome maturation and spindle formation. Nlp overexpression was detected in human breast and lung cancers[52]. By examining 55 breast cancer samples, a study found that the breast cancer patients with high expression of Nlp were likely resistant to the treatment of paclitaxel. KIFC1 is a nonessential minus end-directed motor of the kinesin-14 family and it functions as a centrosome clustering molecule[53,54]. In breast cancer cells, overexpression of KIFC1 and KIFC3 confer docetaxel resistance[55]. These studies indicate that centrosome aberrations not only produce aneuploidy, chromosome instability leading to tumorigenesis but also promote cancer drug resistance.
Mechanisms of cancer drug resistance involving centrosome
The mechanism underlying chemo-resistance (mitotic drug resistance) is not yet clear. Several studies provide insights into the molecular basis of centrosome abnormalities that produce drug resistance, either directly by dysregulation of centrosome protein levels or indirectly by regulating gene expression of other proteins.
Gene dosage
As discussed previously, gene dosage may affect centrosome duplication through a balance of the relative abundance of one or more proteins essential for the assembly of new centrioles and the availability of assembly sites. Thus, dysregulation of gene dosage for centrosome proteins produce aneuploidy and chromosome instability. Moreover, there are a close relationship between aneuploidy, chromosome instability and chemotherapy resistance.
The Aurora A kinase regulates centrosome maturation and separation and thereby play important roles in spindle assembly and stability. Overexpression of Aurora-A kinase induces centrosome amplification and chromosomal instability that create tumor cell heterogeneity, thus is associated with acquired drug resistance[56].
PLK4 is a key component of the centrosome. Dysregulation of PLK4 activity causes loss of centrosome numeral integrity. Its overexpression is responsible centrosome amplification and contributes to resistance to tamoxifen and trastuzumab[57].
Mitotic slippage
Chemotherapy is commonly used in order to induce cell death or to prevent proliferation of cancer cells by impairing spindle function and chromosome segregation. However sometimes cancer cells evade cell death for those that are arrested in mitosis[58]. Instead these cells leave mitosis without completing a normal cell division and become tetraploid. This phenomenon is called mitotic slippage. The examples can be seen in the drugs that target microtubule assembly (nocodazole, vincristine) or the disassembly (taxol or paclitaxel).
Avoiding apoptosis through mitotic slippage in cancer cells is thought to be a major mechanism contributing to cancer drug resistance. An interesting recent study provides insight into the mechanism of mitotic slippage. BH3-only pro-apoptotic proteins are necessary to initiate the molecular process of apoptosis in cells undergoing perturbed mitosis[59]. NEK2 conferred drug resistance is associated with decreased apoptosis[60]. Furthermore, overexpression of NEK2 suppressed the expression of the BH3-only genes BAD and PUMA and upregulated the expression of pro-survival genes BCL-xL and MCL-1, indicating a possible role of NEK2 in cancer drug resistance via mitotic slippage.
Phosphorylation at S69 of BIM, which is also a BH3-only protein, leads to its ubiquitin-dependent degradation[61]. During mitotic arrest, BIM is known to be heavily by Aurora A kinase, which could result in mitotic slippage.
Centrosome protein CEP55 was found to have a role in promoting mitotic slippage, which again is mediated by the Bcl2 family proteins in breast cancer[62]. In breast cancer patients, high-level expression of CEP55 associates with chemotherapeutic resistance, particularly to docetaxel. Similarly, docetaxel induces spindle multipolarity, higher KIFC1 expression might counteract this effect to prevent cell death and enable bipolar spindle formation through centrosome clustering[55].
These studies demonstrated that centrosome proteins regulate gene expression of apoptotic/anti-apoptotic genes to induce cancer drug resistance.
Regulation of drug transporters by centrosome proteins
A recent study demonstrates that overexpression of NEK2 and drug resistance are closely correlated in other cancers through activation of efflux drug pumps[57,60]. Overexpression of NEK2 upregulated ABC transporter family members, including ABCB1 (p-glycoprotein, MDR1), the multidrug resistance protein ABCC1 (MRP1), and the breast cancer resistant protein ABCG2 (BCRP). High expression of NEK2 promoted a higher efflux of the hydrophilic eFluxx-ID gold fluorescent dye from cancer cells. Verapamil, an ABC transporter inhibitor, was able to abrogate part of the NEK2-induced drug resistance by showing a decrease in colony formation. Downregulation of NEK2 by shRNA decreased the expression of phosphorylated PP1, AKT, nuclear β-catenin, and ABC transporters.
However, it is also worth to note that drug resistance can also be secondary since chemotherapy or radiation may induced centrosome abnormalities. This is associated with tumor cell heterogeneity. Accumulations of centrosome aberrations after nilotinib and imatinib treatment in vitro are associated with mitotic spindle defects and genetic instability[63].
Epigenetic regulation of centrosome protein expression
Epigenetic mechanisms that alter functional gene dosage through hyper- or hypo-methylation, and consequently the abundance of key centrosome precursor molecules, may result in centrosome abnormalities, spindle defects, aneuploidy and polyploidy.
Although any genetic aberrations of the centrosome proteins contributes to tumorigenesis, alterations of epigenetic gene regulation are found more frequently as cancer drivers, which include widespread alterations of CpG island methylation, histone modifications, and dysregulation DNA binding proteins disrupt normal patterns of gene expression.
Phosphorylation
Many kinases (e.g., CDKs, Aurora A, polo-like kinases, etc.) participate in the regulation of centrosome duplication even they themselves are also controlled by phosphorylation and dephosphorylation. For instance, the phosphorylation status of CKAP2 during mitosis is critical for controlling both centrosome biogenesis and bipolar spindle formation[64]. It has been shown that the Cyclin-Dependent Kinase (CDK)-activating phosphatase (CDC25B) localizes to centrosome and involves in the centrosome duplication cycle and in microtubule nucleation[65]. The activity of CDC25B is positively or negatively regulated by several kinases including Aurora A and CHK1[66,67]. The phosphorylation of CDC25B by Aurora-A locally participate in the control of the onset of mitosis[68]. Abnormal expression of CDC25B in numerous human tumors might have a critical role in centrosome amplification and genomic instability[69].
Activities of Aurora-A (AurA) for its cellular function are regulated by different protein-protein interactions and posttranslational modifications. It has been established that Twist1 has a critical role in promoting EMT and drug resistance. AURKA phosphorylates Twist1 at three positions (S123, T148 and S184). AURKA-mediated phosphorylation of Twist1 is crucial for EMT, the cancer stem cell phenotype and drug resistance togemcitabine[70]. On the other hands, activation of AurA at centrosomes occurs through autophosphorylation at the critical activating residue Thr288[71]. The autophosphorylation is regulated by PLK1[72] and TPX2[73].
PLK4 contains an N-terminal kinase domain (residues 12-284) a C-terminal localization domain (residues 596-898) and 3 polo box domains, which facilitates oligomerization, targeting, and promotes trans-autophosphorylation. PLK4 can be directly phosphorylated and activated by stress-activated protein kinase kinase kinases[74]. Indeed, tumor-derived SAPKK1/MKK4 mutants induced centrosome amplification under genotoxic stress (only in p53-negative cells)[74], which lead to increased resistance to apoptosis, chemotherapy and radiotherapy[75].
PLK4 is also a substrate of itself (via autophosphorylation). Autophosphorylation of PLK4 results in ubiquitination and subsequent destruction by the proteasome[76-78]. It has been shown that mutagenesis of ASP-154 in the catalytic domain that causes centrosome amplification above background levels when overexpressed[16]. This may be due to the loss of self-destruction of PLK4.
Acetylation
Acetylation and deacetylation are highly common posttranslational modifications. Several studies demonstrate that acetylation/deacetylation play a role in the regulation of centrosome duplication and induction of abnormal amplification of centrosomes. KAT2A/KAT2B function as histone acetyltransferase or lysine acetyltransferases. Fournier and her colleagues showed that KAT2A/2B acetylate the PLK4 kinase domain on residues K45 and K46[79]. Impairing KAT2A/2B-acetyltransferase activity results in diminished phosphorylation of PLK4 and in excess centrosome numbers in cells. Therefore KAT2A/2B acetylation of PLK4 prevents centrosome amplification. On the other hands, through focusing on the deacetylases, Fukasawa’s group found that the deacetylation event negatively controls centrosome duplication and amplification. Of the 18 total known deacetylases (HDAC1-11, SIRT1-7), ten deacetylases possess the activity to suppress centrosome amplification, and their centrosome amplification suppressing activities are strongly associated with their abilities to localize to centrosomes. Among them, HDAC1, HDAC5 and SIRT1 show the highest suppressing activities, but each of them suppresses centrosome duplication and/or amplification with its unique mechanism[80].
Methylation
G9a is a histone methyltransferase enzyme, also known as euchromatic histone-lysine N-methyltransferase 2 (EHMT2)[81]. G9a catalyzes the mono- and di-methylated states of histone H3 at lysine residue 9 (i.e., H3K9me1 and H3K9me2) and lysine residue 27 (H3K27me1 and HeK27me2). G9a plays a critical role in regulating centrosome duplication. Knockdown of G9a significantly reduces di- and trimethylation of H3K9, resulting in disruptions in centrosome amplification and chromosome instability in cancer cells[82]. Furthermore, silencing G9a leads to down-modulation of gene expressions, including that of p16INK4A. It has been shown that cells lacking p16 (INK4A) activity exhibit phenotypes associated with malignancy[83]. p16INK4A is the CDK2, Cdk4 and Cdk6-specific inhibitor[84]. The observations of the effects on G9a silencing are in support of the studies linking cyclin D1/Cdk4 with centrosome amplification[85,86]. Initiation of tumorigenesis was found in the loss of p16INK4A through hypermethylation of its promoter[87-89]. Thus, it has been postulated that loss of p16 expression coupled with increased γ-tubulin contributes to centrosome amplification and breast cancer progression[90].
14-3-3 proteins are associated with centrosomes[91]. 14-3-3γ prevents centrosome amplification and neoplastic progression[92]. Inactivation of the 14-3-3 sigma gene is associated with 5’ CpG island hypermethylation in human cancers[93,94]. Promoter hypermethylation of p53 genes is detected in many cancers[95-97].
Promoter
There is a functional link between centrosome and transcription factors. NF-κB can induce abnormal centrosome amplification by upregulation of CDK2[98]. A functional NF-κB binding site was located in the CDK2 promoter.
Methyl-CpG binding protein 2 (MeCP2) localizes at the centrosome. Its loss causes deficient spindle morphology and microtubule nucleation. In addition, MECP2 binds to histone deacetylases and represses gene transcription[99].
E2Fs affect the expression of proteins, including Nek2 and Plk4, thereby deregulation of E2Fs induces centrosome amplification in breast cancer[100]. A further example showed that arsenic induced centrosome amplification via SUV39H2-mediated epigenetic modification of E2F1[101].
DDX3 regulates epigenetic transcriptional and translational activation of p53 and colocalizes with p53 at centrosome during mitosis to ensure proper mitotic progression and genome stability, which supports the tumor-suppressive role of DDX3[102]. DDX3 knockdown suppressed p53 transcription through activation of DNA methyltransferases along with hypermethylation of p53 promoter and promoting the binding of repressive histone marks to p53 promoter.
During tumor development, especially to most solid tumors, cancer cells are often subjected to hypoxia[103]. A recent study showed that via upregulation of HIF1, proteins whose overexpression drives centrosome amplification (such as Cyclin E, Aurora A, and PLK4) were upregulated[104].
MiRNA
Recently gene expression of centrosome proteins was found as miRNA targets. MiR-129-3p is identified as a novel metastatic microRNA. CP110 expression was repressed by miR-129-3p[105].
PLK1 is one of targets of miRNA-210-3p, and lnc-RI regulates PLK1 mRNA stability by competing with the PLK1 mRNA 3’UTR for binding to miRNA-210-3p[106].
MiR-128 inhibited NEK2 expression and miR-128 was silenced by DNA methylation. Up-regulation of NEK2 by MicroRNA-128 methylation is associated with poor prognosis in colorectal cancer[107].
Ubiquitination
Dysfunction in the ubiquitin-proteasome degradation has implicated in several cancer drug resistance. MDM2 is an E3 ubiquitin-protein ligase that mediates ubiquitination and degradation of p53[108,109]. Increased levels of MDM2 would inactivate the functions of p53 to similar extent that do in deletion or mutation of p53 and found in a variety of human tumors[110]. Several studies demonstrated that MDM2 overexpression increases cancer drug resistance of tumors[111,112]. Mind bomb (Mib1) was identified as the E3 ubiquitin ligase of PLK4[113]. Recently we found that HECTD1, a HECT-type E3 ubiquitin ligase is a novel centrosome protein whose deficiency induces centrosome amplification and promotes epithelial-mesenchymale transition[114,115]. These results indicate ubiquitination is one of the important epigenetic modifications for centrosome.
New drugs in trial
Abnormalities in size, number and microtubule nucleation capacity of centrosome are resulted from genetic disorders or epigenetic disturbances of gene expression. Epigenetic modifications are temporally dynamic and reversible changes. Development of small molecules targeting epigenetic regulators are promising anticancer strategies, involving elimination of cancer cells with chromosome instability and aneuploid in combination with targeting centrosome proteins to overcome mitotic slippage and to induce apoptosis in cancer cells. The drugs that focused on the centrosome amplification may provide possibilities to treat cancer or overcome some forms of drug resistance. Recently clinical trials of inhibitors targeting kinases that function as centrosome regulators are under way for hematologic malignancies and solid tumors. We summarize the development of therapies targeting these mechanisms.
Although a lot of progresses have been made in treating cancer, the most cancer chemotherapeutics develop drug resistant (secondary) that limits the efficacy of treatments. This happened even for the newly approved NTRK inhibitors[116]. Thus, there is a significant need to target drug resistance for improved therapeutics for cancer. Several clinical trials, in single or combination of drugs, have been focused on blocking centrosome clustering to combat drug resistance [Table 2].
Ongoing trials with known inhibitory activity to centrosome aberrations
| NCT number | Start date | Drug | Targets | Cancers | Phases | Patient number |
|---|---|---|---|---|---|---|
| NCT03654716 | 01 2018 | ALRN-6924 | MDM2 and MDMX | Solid tumor | 1 | 69 |
| NCT03634228 | 08 2018 | DS-3032b | MDM2 | Refractory Acute Myeloid Leukemia | 2 | 52 |
| NCT02098967 | 03 2014 | RO6839921 | MDM2 | Neoplasms, Myelogenous Leukemia, Acute | 1 | 68 |
| NCT03671564 | 09 2018 | Milademetan | MDM2 | Acute Myeloid Leukemia | 1 | 24 |
| NCT03787602 | 12 2018 | KRT-232 | MDM2 | Merkel Cell Carcinoma | 2 | 27 |
| NCT03781986 | 03 2019 | APG-115 | MDM2 | Malignant Salivary Gland Cancer | 2 | 62 |
| NCT03566485 | 06 2018 | Atezolizumab (an Anti-PD-L1 Monoclonal Antibody) With Idasanutlin | PD-L1/MDM2 | Metastatic ER + Breast Cancer | 1+2 | 92 |
| NCT01014429 | 11 2009 | NMS-1286937 | PLK1 | Advanced/Metastatic Solid Tumors | 1 | 21 |
| NCT01954316 | 10 2013 | CFI-400945 | PLK4 | Advanced Cancer | 1 | 48 |
| NCT02187783 | 11 2014 | LEE011 | CDK4/6 Pathway | CDK4/6 Pathway Activated Tumors | 2 | 106 |
| NCT03242382 | 08 2017 | Palbociclib | CDK4 Overexpression | Advanced Sarcomas | 2 | 38 |
| NCT00536835 | 09 2007 | GSK461364 | PLK1 | Non-Hodgkins Lymphoma | 1 | 40 |
| NCT03555877 | 06 2018 | Ribociclib | CDK4/6 | Breast Cancer Metastatic | 2 | 150 |
| NCT01676753 | 06 2016 | Dinacicli + pembrolizumab | Cyclin-dependent Kinase | advanced breast cance | 1 | 32 |
| NCT01037790 | 2009 | PD-0332991 | CDK4 mutation | Solid Tumor | 2 | 205 |
| NCT03242382 | 08 2017 | Palbociclib | CDK4 Overexpression | Sarcoma | 2 | 38 |
| NCT03024489 | 01 2017 | Palbociclib + cetuximab | CDK4 Overexpression | Head and Neck Cancer | 2 | 33 |
| NCT03310879 | 11 2017 | Abemaciclib | Abnormality in one of the following genes: CCND1, CCND2, CCND3, CDK4, or CDK6 | Breast | 2 | 38 |
| NCT03050398 | 06 2017 | Ribociclib+ letrozole | CDK4/6 | breast cancer | 3 | 140 |
| NCT03096912 | 06 2016 | Ribociclib | CDK4/6 | Liposarcoma | 2 | 30 |
| NCT03054363 | 11 2017 | Palbociclib + letrozole | CDK4 overexpression | Hormone Receptor Positive and HER2-positive Metastatic Breast Cancer | 1 + 2 | 40 |
Since increased levels of MDM2 inactivate the functions of p53 thereby induce centrosome amplification and drug resistance. Inhibition of MDM2 is obviously a good strategy to fight cancer drug resistance. Several clinical trials have been setting up to a variety of human tumors, such as Leukemia, Myeloma, Brain Tumor, Solid Tumor and Lymphoma [Table 2].
As mentioned above, PLK4 is important in centrosome biogenesis and regulates mitotic progression. PLK4 has, therefore been identified as a candidate anticancer target. With directed virtual screening using a ligand-based focused library, several leads have been identified. CFI-400945 was generated through further optimization[117]. CFI-400945 is a potent and selective small molecule inhibitor of PLK4[118].
Monopolar spindle 1 (Mps1/TTK) kinase is essential for safeguarding proper chromosome alignment and segregation during mitosis[119]. Its overexpression contributes to more aggressive and drug resistant breast tumors but the reduction of the Mps1 level can sensitize several tumor cells to paclitaxel[120]. In the two ongoing clinical trials, the Mps1 inhibitors are tested along with paclitaxel in triple negative breast cancer patients [Table 2, NCT02138812 and NCT03328494].
Plk1, known as polo-like kinase 1, supports the functional maturation of the centrosome and establishment of the bipolar spindle. Overexpression of Plk1 is often observed in cancer cells[121]. This protein therefore is a potential drug target in cancer[122]. Several inhibitors of PLK1 have been developed and the promising results have been obtained in clinical trials[123]. For example, as compared to administration of cytarabine (a chemotherapy medication used to treat acute myeloid leukemia ) alone, a combination of BI 6727 with cytaribine increased the response with 31% total remission from 13%[124].
Cyclin-dependent kinases (Cdks) are a family of protein kinases that regulate the centrosome cycle but deregulation of those Cdks by oncogenes and tumor supressors results to centrosome amplification[125]. More than 30 small-molecule inhibitors developed[126]. Many of them have been used in clinical trials studies for the treatment of various cancers [Table 2][127]. Flavopiridol is the first Cdk inhibitor used in clinical trials[128]. Flavopiridol has been successful in the treatment of AML and chronic lymphocytic leukemia[129,130].
The Aurora kinases (AURKs) are involved in different aspects of mitotic control during cell cycle. Importantly, PLK1 is activated by AURKA/B. Therefore AURKs are potential targets against centrosome for cancer therapy. More than 30 AURK inhibitors have been developed and used in clinical studies[131]. For example, the inhibitor MLN8237 (alisertib), which targets AURKA, showed promising efficacy in several solid tumors[132]. AZD1152 (barasertib) is a selective inhibitor of AURKB and has been effective in AML patients with an overall response rate of 25%, but with no effective results in patients with solid tumors. In addition, AURKB/AURKC kinase inhibitor GSK1070916A is actually being tested in patients with solid tumors and phase I in the clinical trial has been completed.
Conclusion
The consequences of numerical aberrations/centrosome amplification leading to tumorigenesis have been studied extensively. In contrast, studies on mechanisms of cancer drug resistance in relation to centrosome aberrations have received little attention. Epigenetic modifications in centrosome biogenesis have important implications for the origin of some malignant tumors and play a role in cancer drug resistance. The current review discussed the connection of epigenetic changes causing centrosome aberrations to cancer drug resistance. For clinical, the ultimate goal is to identify effective cancer therapy. So far, most of clinical trials targeting possible drug resistance, which were registered to ClinicalTrials.gov at NIH are still monotherapies and in early stages of development. One important factor we should not forget is that regulation pathways to epigenetic modifications of centrosome, such as positive or negative feedback signaling circuits involved in cancer drug resistance are more complex than once thought. The selection of drugs, together with other treatment like immunotherapy, for combination therapy may lead to improve efficacy and to thwart drug resistance. In the future, one need to address molecular mechanisms how the trafficking of centrosome proteins between centrosome and nucleus determine expression/subcellular localization of downstream signaling molecules, such as the Bcl2 family proteins and ABC transporters. Further understanding of centrosome biology including basic cell biology or pathobiology of epigenetic controls in centrosome will provide potential to establish translatable strategies for cancer treatment and to prevent drug resistance.
Declarations
Authors’ contributionsConception: Jia ZH, Wang XG, Zhang H
Design of the study: Jia ZH, Wang XG, Zhang H
Wrote: Jia ZH, Wang XG, Zhang H
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis research was funded by university of Basel, Switzerland. Jia ZH was supported by CSC, China.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Baylin SB, Jones PA. A decade of exploring the cancer epigenome-biological and translational implications. Nat Rev Cancer 2011;11:726-34.
4. Salisbury JL. The contribution of epigenetic changes to abnormal centrosomes and genomic instability in breast cancer. J Mammary Gland Biol Neoplasia 2001;6:203-12.
5. Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res 1998;58:3974-85.
6. Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, et al. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016;16:47.
7. Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol 1999;155:1941-51.
8. Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A 1998;95:2950-5.
9. Krämer A, Neben K, Ho AD. Centrosome aberrations in hematological malignancies. Cell Biol Int 2005;29:375-83.
10. Giehl M, Fabarius A, Frank O, Hochhaus A, Hafner M, et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005;19:1192-7.
11. Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, et al. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 2004;24:8457-66.
12. Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 2015;16:611-24.
13. Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 2011;30:1520-35.
14. Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003;426:570-4.
15. Loncarek J, Bettencourt-Dias M. Building the right centriole for each cell type. J Cell Biol 2018;217:823-35.
16. O’Connell KF, Caron C, Kopish KR, Hurd DD, Kemphues KJ, et al. The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell 2001;105:547-58.
17. Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 2005;7:1140-6.
18. Arquint C, Nigg EA. The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem Soc Trans 2016;44:1253-63.
19. Pihan GA. Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol 2013;3:277.
20. Yang J, Adamian M, Li T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol Biol Cell 2006;17:1033-40.
21. Mardin BR, Lange C, Baxter JE, Hardy T, Scholz SR, et al. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat Cell Biol 2010;12:1166-76.
22. Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J 1998;17:470-81.
23. Centrosome. Available from: https://www.proteinatlas.org/humanproteome/cell/centrosome. [Last accessed on 22 Apr 2019].
24. Boveri T. 1900. Ueber die Natur der Centrosomen. Zellen-Studien 4. Jena, Germany: G. Fischer; .
25. Farina F, Gaillard J, Guérin C, Couté Y, Sillibourne J, et al. The centrosome is an actin-organizing centre. Nat Cell Biol 2016;18:65-75.
26. Nam HJ, Naylor RM, van Deursen JM. Centrosome dynamics as a source of chromosomal instability. Trends Cell Biol 2015;25:65-73.
27. Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res 2003;63:1398-1404.
28. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 2011;7:1122-44.
29. Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 2008;22:2189-203.
30. Neben K, Giesecke C, Schweizer S, Ho AD, Krämer A. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood 2003;101:289-91.
31. Mahathre MM, Rida PC, Aneja R. The more the messier: centrosome amplification as a novel biomarker for personalized treatment of colorectal cancers. J Biomed Res 2015;30:441-51.
32. Kais Z, Parvin JD. Regulation of centrosomes by the BRCA1-dependent ubiquitin ligase. Cancer Biol Ther 2008;7:1540-3.
33. Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science 1996;271:1744-7.
34. Li X, Song N, Liu L, Liu X, Ding X, et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat Commun 2017;8:14866.
35. Xie S, Qin J, Liu S, Zhang Y, Wang J, et al. Cep70 overexpression stimulates pancreatic cancer by inducing centrosome abnormality and microtubule disorganization. Sci Rep 2016;6:21263.
36. Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, et al. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016;16:47.
37. Miyachika Y, Yamamoto Y, Matsumoto H, Nishijima J, Kawai Y, et al. Centrosome amplification in bladder washing cytology specimens is a useful prognostic biomarker for non-muscle invasive bladder cancer. Cancer Genet 2013;206:12-8.
38. Zeng YR, Han ZD, Wang C, Cai C, Huang YQ, et al. Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol 2015;15:90.
39. Ogden A, Rida PC, Aneja R. Prognostic value of CA20, a score based on centrosome amplification-associated genes, in breast tumors. Sci Rep 2017;7:262.
40. Huang J, Sun SG, Hou S. Aberrant NEK2 expression might be an independent predictor for poor recurrence-free survival and overall survival of skin cutaneous melanoma. Eur Rev Med Pharmacol Sci 2018;22:3694-3702.
41. Yun M, Rong J, Lin ZR, He YL, Zhang JX, et al. High expression of transforming acidic coiled coil-containing protein 3 strongly correlates with aggressive characteristics and poor prognosis of gastric cancer. Oncol Rep 2015;34:1397-1405.
42. Karkera JD, Cardona GM, Bell K, Gaffney D, Portale JC, et al. Oncogenic characterization and pharmacologic sensitivity of activating fibroblast growth factor receptor (FGFR) genetic alterations to the selective FGFR inhibitor erdafitinib. Mol Cancer Ther 2017;16:1717-26.
43. Lauffart B, Vaughan MM, Eddy R, Chervinsky D, DiCioccio RA, et al. Aberrations of TACC1 and TACC3 are associated with ovarian cancer. BMC Women’s Health 2005;5:8.
44. Fan G, Sun L, Shan P, Zhang X, Huan J, et al. Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun 2015;6:8450.
45. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251-63.
46. Tamotsu K, Okumura H, Uchikado Y, Kita Y, Sasaki K, et al. Correlation of Aurora-A expression with the effect of chemoradiation therapy on esophageal squamous cell carcinoma. BMC Cancer 2015;15:323.
47. Rakha EA, Pinder SE, Paish CE, Ellis IO. Expression of the transcription factor CTCF in invasive breast cancer: a candidate gene located at 16q22.1. Br J Cancer 2004;91:1591-6.
48. Lee J, Gollahon L. Nek2-targeted ASO or siRNA pretreatment enhances anticancer drug sensitivity in triplenegative breast cancer cells. Int J Oncol 2013;42:839-47.
49. Zhou W, Yang Y, Xia J, Wang H, Salama ME, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013;23:48-62.
50. Liu X, Gao Y, Lu Y, Zhang J, Li L, et al. Upregulation of NEK2 is associated with drug resistance in ovarian cancer. Oncol Rep 2014;31:745-54.
51. Wu SM, Lin SL, Lee KY, Chuang HC, Feng PH, et al. Hepatoma cell functions modulated by NEK2 are associated with liver cancer progression. Int J Cancer 2017;140:1581-96.
52. Zhao W, Song Y, Xu B, Zhan Q. Overexpression of centrosomal protein Nlp confers breast carcinoma resistance to paclitaxel. Cancer Biol Ther 2012;13:156-63.
53. Kleylein-Sohn J, Pöllinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings, et al. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J Cell Sci 2012;125:5391-402.
54. Kwon M, Bagonis M, Danuser G, Pellman D. Direct microtubule-binding by myosin-10 orients centrosomes toward retraction fibers and subcortical actin clouds. Dev Cell 2015;34:323-37.
55. De S, Cipriano R, Jackson MW, Stark GR. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res 2009;69:8035-42.
56. Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, et al. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci USA 2004;101:18123-8.
57. Marina M, Saavedra HI. Nek2 and Plk4: prognostic markers, drivers of breast tumorigenesis and drug resistance. Front Biosci (Landmark Ed) 2014;19:352-65.
58. Jusino S, Fernández-Padín FM, Saavedra HI. Centrosome aberrations and chromosome instability contribute to tumorigenesis and intra-tumor heterogeneity. J Cancer Metastasis Treat 2018;4:pii:43.
59. Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 2008;27:S2-19.
60. Zhou W, Yang Y, Xia J, Wang H, Salama ME, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013;23:48-62.
61. Moustafa-Kamal M, Gamache I, Lu Y, Li S, Teodoro JG, et al. BimEL is phosphorylated at mitosis by Aurora A and targeted for degradation by βTrCP1. Cell Death Differ 2013;20:1393-1403.
62. Kalimutho M, Sinha D, Jeffery J, Nones K, Srihari S, et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol Med 2018;10:e8566.
63. Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, et al. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res 2004;64:7370-6.
64. Yoo BH, Kang DS, Park CH, Kang K, Bae CD. CKAP2 phosphorylation by CDK1/cyclin B1 is crucial for maintaining centrosome integrity. Exp Mol Med 2017;49:e354.
65. Boutros R, Ducommun B. Asymmetric localization of the CDC25B phosphatase to the mother centrosome during interphase. Cell Cycle 2008;7:401-6.
66. Cazales M, Schmitt E, Montembault E, Dozier C, Prigent C, et al. CDC25B phosphorylation by Aurora-A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle 2005;4:1233-8.
67. Bouché JP, Froment C, Dozier C, Esmenjaud-Mailhat C, Lemaire M, et al. NanoLC-MS/MS analysis provides new insights into the phosphorylation pattern of Cdc25B in vivo: full overlap with sites of phosphorylation by Chk1 and Cdk1/cycB kinases in vitro. J Proteome Res 2008;7:1264-73.
68. Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, et al. Phosphorylation of cdc25b by aurora-a at the centrosome contributes to the g2-m transition. J Cell Sci 2004;117:2523-31.
69. Boutros R, Lobjois V, Ducommun B. CDC25B involvement in the centrosome duplication cycle and in microtubule nucleation. Cancer Res 2007;67:11557-64.
70. Wang J, Nikhil K, Viccaro K, Chang L, Jacobsen M, et al. The Aurora-A-Twist1 axis promotes highly aggressive phenotypes in pancreatic carcinoma. J Cell Sci 2017;130:1078-93.
71. Joukov V, De Nicolo A, Rodriguez A, Walter JC, Livingston DM. Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc Natl Acad Sci USA 2010;107:21022-7.
72. Scutt PJ, Chu ML, Sloane DA, Cherry M, Bignell CR, et al. Discovery and exploitation of inhibitor-resistant aurora and polo kinase mutants for the analysis of mitotic networks. J Biol Chem 2009;284:15880-93.
73. Zorba A, Buosi V, Kutter S, Kern N, Pontiggia F, et al. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. ELife 2014;3:e02667.
74. Nakamura T, Saito H, Takekawa M. SAPK pathways and p53 cooperatively regulate PLK4 activity and centrosome integrity under stress. Nat Commun 2013;4:1775.
75. Rauch N, Rukhlenko OS, Kolch W, Kholodenko BN. MAPK kinase signaling dynamics regulate cell fate decisions and drug resistance. Current Opin Struct Biol 2016;41:151-8.
76. Cunha-Ferreira I, Bento I, Pimenta-Marques A, Jana SC, Lince-Faria M, et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Current Biology 2013;23:2245-54.
77. Guderian G, Westendorf J, Uldschmid A, Nigg EA. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J Cell Sci 2010;123:2163-9.
78. Slevin LK, Nye J, Pinkerton DC, Buster DW, Rogers GC, et al. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure 2012;20:1905-17.
79. Fournier M, Orpinell M, Grauffel C, Scheer E, Garnier JM, et al. KAT2A/KAT2B-targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat Commun 2016;7:13227.
80. Ling H, Peng L, Seto E, Fukasawa K. Suppression of centrosome duplication and amplification by deacetylases. Cell Cycle 2012;11:3779-91.
81. Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem 2001;276:25309-17.
82. Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, et al. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 2008;3:e2037.
83. Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, et al. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001;409:633.
84. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16INK4a inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol 1999;19:1981-9.
85. Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A 1999;96:2817-22.
86. Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol 1999;9:429-32.
87. Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol 1998;18:1793-1801.
88. Holst CR, Nuovo GJ, Esteller M, Chew K, Baylin SB, et al. Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res 2003;63:1596-601.
89. McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, et al. p16(INK4a) prevents centrosome dysfunction and genomic instability in primary cells. PLoS Biol 2006;4:e51.
90. Berman H, Zhang J, Crawford YG, Gauthier ML, Fordyce CA, et al. Genetic and epigenetic changes in mammary epithelial cells identify a subpopulation of cells involved in early carcinogenesis. Cold Spring Harb Symp Quant Biol 2005;70:317-27.
91. Pietromonaco SF, Seluja GA, Aitken A, Elias L. Association of 14-3-3 proteins with centrosomes. Blood Cells Mol Dis 1996;22:225-37.
92. Mukhopadhyay A, Sehgal L, Bose A, Gulvady A, Senapati P, et al. 14-3-3γ prevents centrosome amplification and neoplastic progression. Sci Rep 2016;6:26580.
93. Suzuki H, Itoh F, Toyota M, Kikuchi T, Kakiuchi H, et al. Inactivation of the 14-3-3 sigma gene is associated with 5’ CpG island hypermethylation in human cancers. Cancer Res 2000;60:4353-7.
94. Iwata N, Yamamoto H, Sasaki S, Itoh F, Suzuki H, et al. Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3 sigma gene in human hepatocellular. Oncogene 2000;19:5298-302.
95. Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, et al. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci 2006;89:431-7.
96. Jha AK, Nikbakht M, Jain V, Sehgal A, Capalash N, et al. Promoter hypermethylation of p73 and p53 genes in cervical cancer patients among north Indian population. Mol Biol Rep 2012;39:9145-57.
97. Adrian Jarzynski, Katarzyna Papiernik, Malgorzata Polz-Dacewicz. Analysis of mutation and promoter methylation of TP53 gene in tumors of the head and neck. Current Issues in Pharmacy and Medical Sciences 2016;29:53-6.
98. Liu JL, Ma HP, Lu XL, Sun SH, Guo X, et al. NF-κB induces abnormal centrosome amplification by upregulation of CDK2 in laryngeal squamous cell cancer. Int J Oncol 2011;39:915-24.
99. Yu F, Thiesen J, Strätling WH. Histone deacetylase-independent transcriptional repression by methyl-CpG-binding protein 2. Nucleic Acids Res 2000;28:2201-6.
100. Lee MY, Moreno CS, Saavedra HI. E2F activators signal and maintain centrosome amplification in breast cancer cells. Mol Cell Biol 2014;34:2581-99.
101. Liao WT, Lu JH, Lee CH, Lan CE, Chang JG, et al. An interaction between arsenic-induced epigenetic modification and inflammatory promotion in a skin equivalent during arsenic carcinogenesis. J Invest Dermatol 2017;137:187-96.
102. Chen WJ, Wang WT, Tsai TY, Li HK, Lee YW. DDX3 localizes to the centrosome and prevents multipolar mitosis by epigenetically and translationally modulating p53 expression. Sci Rep 2017;7:9411.
103. Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006;441:437-43.
104. Mittal K, Choi DH, Ogden A, Donthamsetty S, Melton BD, et al. Amplified centrosomes and mitotic index display poor concordance between patient tumors and cultured cancer cells. Sci Rep 2017;7:43984.
105. Bijnsdorp IV, Hodzic J, Lagerweij T, Westerman B, Krijgsman O, et al. miR-129-3p controls centrosome number in metastatic prostate cancer cells by repressing CP110. Oncotarget 2016;7:16676-87.
106. Wang ZD, Shen LP, Chang C, Zhang XQ, Chen ZM, et al. Long noncoding RNA lnc-RI is a new regulator of mitosis via targeting miRNA-210-3p to release PLK1 mRNA activity. Sci Rep 2016;6:25385.
107. Takahashi Y, Iwaya T, Sawada G, Kurashige J, Matsumura T, et al. Up-regulation of NEK2 by microRNA-128 methylation is associated with poor prognosis in colorectal cancer. Ann Surg Oncol 2014;21:205-12.
108. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296-9.
109. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387:299-303.
110. Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, et al. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res 1993;53:2231-4.
111. Akio Suzuki, Masakazu Toi, Yutaka Yamamoto, Shigehira Saji, Mariko Muta, et al. Role of MDM2 overexpression in doxorubicin resistance of breast carcinoma,. Jpn J Cancer Res 1998;89:221-7.
112. Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res 2001;61:6185-93.
113. Čajánek L, Glatter T, Nigg EA. The E3 ubiquitin ligase Mib1 regulates Plk4 and centriole biogenesis. J Cell Sci 2015;128:1674-82.
114. Shen X, Jia Z, D‘Alonzo D, Wang X, Bruder E, et al. HECTD1 controls the protein level of IQGAP1 to regulate the dynamics of adhesive structures. Cell Commun Signal 2017;15:2.
115. Wang XG, De Geyter C, Jia ZH, Peng Y, Zhang H. HECTD1 regulates expression of SNAIL: implications for epithelial-mesenchymal transition (EMT). Forthcoming 2019.
116. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018;378:731-9.
117. Sampson PB, Liu Y, Patel NK, Feher M, Forrest B, et al. The discovery of polo-like kinase 4 inhibitors: design and optimization of spiro[cyclopropane-1,3ʹ[3H]indol]-2ʹ(1ʹH)-ones as orally bioavailable antitumor agents. J Med Chem 2015;58:130-46.
118. Mason JM, Lin DC, Wei X, Che Y, Yao Y, et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014;26:163-76.
120. Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A 2009;106:19108-13.
121. Jeong SB, Im JH, Yoon JH, Bui QT, Lim SC, et al. Essential role of polo-like kinase 1 (Plk1) oncogene in tumor growth and metastasis of tamoxifen-resistant breast cancer. Mol Cancer Ther 2018;17:825-37.
122. Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, et al. Targeting mitosis in cancer: emerging strategies. Mol Cell 2015;60:524-36.
123. Yim H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs 2013;24:999-1006.
124. Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015;29:11-9.
125. Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol 2018;13:21-38.
126. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153-66.
127. Cicenas J, Kalyan K, Sorokinas A, Jatulyte A, Valiunas D, et al. Highlights of the latest advances in research on CDK inhibitors. Cancers (Basel) 2014;6:2224-42.
128. Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res 1996;56:2973-8.
129. Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res 2015;39:1312-8.
130. Blachly JS, Byrd JC, Grever M. Cyclin-dependent kinase inhibitors for the treatment of chronic lymphocytic leukemia. Semin Oncol 2016;43:265-73.
131. Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, et al. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs 2012;30:2411-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jia ZH, Wang XG, Zhang H. Overcome cancer drug resistance by targeting epigenetic modifications of centrosome. Cancer Drug Resist 2019;2:210-24. http://dx.doi.org/10.20517/cdr.2018.010
AMA Style
Jia ZH, Wang XG, Zhang H. Overcome cancer drug resistance by targeting epigenetic modifications of centrosome. Cancer Drug Resistance. 2019; 2(2): 210-24. http://dx.doi.org/10.20517/cdr.2018.010
Chicago/Turabian Style
Jia, Zan-Hui, Xing-Gang Wang, Hong Zhang. 2019. "Overcome cancer drug resistance by targeting epigenetic modifications of centrosome" Cancer Drug Resistance. 2, no.2: 210-24. http://dx.doi.org/10.20517/cdr.2018.010
ACS Style
Jia, Z.H.; Wang X.G.; Zhang H. Overcome cancer drug resistance by targeting epigenetic modifications of centrosome. Cancer Drug Resist. 2019, 2, 210-24. http://dx.doi.org/10.20517/cdr.2018.010
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 4
likes
Like This Article 4
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.