
Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma
 0
0Abstract
Renal cell carcinoma (RCC), the most prevalent type of kidney cancer, is a significant cause of cancer morbidity and mortality worldwide. Antiangiogenic tyrosine kinase inhibitors (TKIs), in combination with immune checkpoint inhibitors (ICIs), are among the first-line treatment options for patients with advanced RCC. These therapies target the vascular endothelial growth factor receptor (VEGFR) tyrosine kinase pathway and other kinases crucial to cancer proliferation, survival, and metastasis. TKIs have yielded substantial improvements in progression-free survival (PFS) and overall survival (OS) for patients with advanced RCC. However, nearly all patients eventually progress on these drugs as resistance develops. This review provides an overview of TKI resistance in RCC and explores different mechanisms of resistance, including upregulation of alternative proangiogenic pathways, epithelial-mesenchymal transition (EMT), decreased intracellular drug concentrations due to efflux pumps and lysosomal sequestration, alterations in the tumor microenvironment including bone marrow-derived cells (BMDCs) and tumor-associated fibroblasts (TAFs), and genetic factors such as single nucleotide polymorphisms (SNPs). A comprehensive understanding of these mechanisms opens the door to the development of innovative therapeutic approaches that can effectively overcome TKI resistance, thereby improving outcomes for patients with advanced RCC.
Keywords
INTRODUCTION
Kidney cancer is the 6th most common cancer in men and the 9th most common cancer in women in the United States, and there will be an estimated 82,000 new cases and 15,000 deaths in 2023[1]. Renal cell carcinoma (RCC) is the most common type of kidney cancer, accounting for over 90%-95% of all cases[2]. With an annual incidence rate of 17.1 per 100,000 individuals and a mortality rate of 3.6%, RCC has become a significant public health concern, resulting in substantial morbidity and mortality[3]. RCC originates from renal tubular epithelial cells and comprises a diverse and heterogeneous group of pathologies[4]. RCC is categorized into multiple subtypes, the most common of which is clear cell RCC (ccRCC), accounting for approximately 75% of all cases and 85% of metastatic cases[5,6]. Other subtypes include papillary, chromophobe, collecting duct, and renal medullary carcinomas. Risk factors for RCC include age, male sex, tobacco use, hypertension, and genetic syndromes such as von Hippel-Lindau (VHL)[7,8].
Management of RCC varies based on disease stage and characteristics. For localized disease, treatment strategies encompass active surveillance, nephrectomy, radiofrequency ablation, and cryotherapy[9,10]. Adjuvant systemic therapies including antiangiogenic targeted therapies have thus far failed to demonstrate a benefit in overall survival, although there are several ongoing trials in this space[11]. The management of metastatic disease, predominantly ccRCC, is more complicated. Until the mid-2000s, systemic cytokines including interleukin-2 (IL-2) and interferon-alpha (IFN-α) were commonly used, but response rates to these therapies were poor[12]. The initial breakthrough in targeted therapy for RCC occurred in 2005 with the approval of sorafenib, a tyrosine kinase inhibitor (TKI) against vascular endothelial growth factor (VEGF). In 2009, bevacizumab, a monoclonal antibody against VEGF, was approved in combination with IFN-α. Subsequently, other multitargeted small molecule antiangiogenic TKIs emerged as treatment options and significantly improved progression-free survival (PFS) and overall survival (OS)[13]. Another class of agents, the mammalian target of rapamycin (mTOR) inhibitors, were introduced during the same period. In recent years, the addition of immune checkpoint inhibitors (ICIs) to TKIs in the frontline setting has reshaped the treatment landscape of advanced RCC[14].
TKIs are competitive inhibitors that bind to the kinase domain of their target receptor tyrosine kinase (RTK). RTKs are transmembrane proteins possessing an extracellular ligand-binding domain and an intracellular kinase domain, enabling them to transmit signals across the plasma membrane. Upon ligand binding, RTKs undergo dimerization and autophosphorylation, leading to kinase domain activation. This provides a docking site for proteins, enabling them to direct a cascade of intracellular events that regulate cellular proliferation, differentiation, survival, and migration [Figure 1][15]. Dysregulation of RTK signaling via constitutive autophosphorylation resulting in ligand-independent kinase activity has been implicated in many cancers, making RTKs attractive therapeutic targets[16]. In RCC, the signaling pathways initiated by overactive VEGF receptor (VEGFR) lead to enhanced endothelial cell migration, proliferation, permeability, survival, and lymphangiogenesis[17]. Antiangiogenic TKIs have been developed to mute this response by binding at or adjacent to the ATP-binding site, preventing autophosphorylation[18,19]. These drugs target various kinases, including VEGFR, platelet-derived growth factor receptor (PDGFR), c-Kit, FMS-related receptor tyrosine kinase 3 (FLT-3), and rearranged during transfection tyrosine-protein kinase (RET)[20]. The majority of TKIs are multitargeted and have activity against several kinases with varying potency. Importantly, all TKIs commonly used in the treatment of advanced RCC possess activity against VEGFR[21]. These include sunitinib, sorafenib, pazopanib, axitinib, cabozantinib, and lenvatinib.

Figure 1. Common receptor tyrosine kinases and their downstream signaling targets. ABC: ATP-binding cassette; ERK: extracellular signal-related kinases; FGF: fibroblast growth factors; FGFR: FGF receptor; HGF: hepatocyte growth factor; mTOR: mammalian target of rapamycin; PDGFR: platelet-derived growth factor receptor; PI3K: phosphoinositide-3-kinase; TKIs: tyrosine kinase inhibitors; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor.
Despite the success of these targeted agents, antiangiogenic TKI resistance is common and poses significant obstacles to achieving durable responses in the treatment of RCC. For instance, the majority of RCC patients who start on sunitinib will develop resistance within 6 months of treatment[22]. In this review, we aim to explore the molecular mechanisms underlying TKI resistance in RCC and shed light on potential strategies to overcome this phenomenon. By elucidating the complexities of TKI resistance, we hope to pave the way for the development of novel therapeutic approaches that can improve outcomes for patients with advanced RCC.
RATIONALE FOR ANTIANGIOGENIC TKIS IN RCC
The treatment of RCC stems from the underlying molecular and genetic alterations that drive tumorigenesis and progression. Almost all hereditary and most sporadic cases of RCC involve mutations leading to loss of tumor suppressor gene function. The most common mutation in ccRCC is the loss of the VHL via loss of chromosome 3p, seen in over 90% of sporadic cases[23]. This leads to the loss or impaired function of VHL tumor suppressor protein (pVHL). Normally, pVHL targets the alpha subunits of hypoxia-inducible factor (HIF) for proteasomal degradation through prolyl hydroxylases. In the absence of pVHL, HIF, particularly HIF-2α, accumulates and acts as a transcription factor to trigger the overexpression of hypoxia-inducible genes such as VEGF, platelet-derived growth factor-BB (PDGF-BB), transforming growth factor-alpha (TGF-α), c-Met, cyclin D1, and CXCR4[24]. These genes play a crucial role in angiogenesis, cell proliferation, and tumor growth. HIF-2α also upregulates the production of c-Myc, a transcription factor implicated in the progression of various cancers[25]. Antiangiogenic TKIs disrupt this aberrant molecular cascade by specifically targeting the dysregulated signaling pathways driven by HIF-2α and its downstream targets, thereby inhibiting tumor angiogenesis and growth in RCC.
TKI RESISTANCE
Resistance to a targeted agent develops when either the agent is no longer able to inhibit specific signaling pathways or when a tumor’s ability to survive becomes independent from those pathways. Various mechanisms contribute to drug resistance, such as alterations in drug targets, activation of alternative signaling pathways, enhancement of drug efflux, and evasion of apoptotic cell death[26].
Resistance to antiangiogenic targeted therapies can be categorized as intrinsic (primary) or acquired (secondary)[27]. Intrinsic resistance is characterized by an inherent insensitivity of cancer cells to TKIs and a lack of response to the drug. By contrast, acquired resistance arises when cancer cells initially respond to TKI treatment but eventually relapse as the drug loses efficacy over time due to the acquisition of various resistance mechanisms. Multiple in vitro and in vivo models involving cell lines, patient-derived xenografts (PDX), organoids, and genetically engineered mouse models (GEMMs) have been developed to assess the mechanisms, dynamics, and progression of drug resistance[28]. These models help elucidate the complex interplay between cancer cells and their microenvironment, offering valuable information for developing strategies to overcome resistance and improve treatment outcomes. In the following section, we summarize the numerous mechanisms of TKI resistance identified to date in RCC.
ACQUIRED MECHANISMS OF TKI RESISTANCE IN RCC
Alternative proangiogenic pathways
Successful antiangiogenic TKI therapy suppresses the production of proangiogenic factors such as VEGF and PDGF and inhibits angiogenesis. This eventually leads to tumor hypoxia, which then triggers the upregulation of alternative proangiogenic pathways, contributing to eventual drug resistance. This process is also known as angiogenic escape or angiogenic switch.
One such pathway involves the increased expression of interleukin-6 (IL-6). IL-6 activates the STAT3 pathway, leading to the upregulation of HIF-2α and subsequent increased production of VEGFR[29]. IL-8 is also upregulated as a response to hypoxia and contributes to angiogenesis by promoting endothelial cell proliferation, survival, and migration via VEGF mRNA transcription and autocrine VEGFR-2 activation[30,31]. High levels of IL-6 and IL-8 have been correlated with significantly shorter PFS and OS in metastatic RCC patients treated with sunitinib and pazopanib[32,33].
Angiopoietin 1 and 2 (Ang 1/2) are critical regulators of angiogenesis, acting as ligands for the Tie2 receptor tyrosine kinase on endothelial cells[34]. When Tie2 is activated, it promotes vessel stabilization, survival, and maturation, thereby boosting the VEGF pathway’s effectiveness in improving perfusion to RCC tumors[35]. Wang et al. followed Ang 2 levels as RCC patients were treated with sunitinib and found that Ang 2 decreased as patients responded to therapy, but then increased as patients became resistant and developed advanced disease[36].
C-Met, a tyrosine kinase encoded by the MET proto-oncogene, binds hepatocyte growth factor (HGF) and initiates an alternative proangiogenic pathway to VEGFR. Type 1 papillary RCC is commonly associated with activating MET alterations and has an unfavorable prognosis[37]. Inhibitors against c-Met have emerged as important treatment options for this disease in recent years. In the phase 2 CREATE trial by Schöffski
Other proangiogenic factors including fibroblast growth factors 1 and 2 (FGF 1/2) and ephrin A1 and A2 (EFNA 1/2) have also been found to be upregulated following VEGFR inhibition, likely as a result of tumor hypoxia[42]. The FGF receptor (FGFR) pathway regulates and activates alternative proangiogenic pathways such as mitogen-activated protein kinases/extracellular signal-related kinases (MAPK/ERK) and phosphoinositide-3-kinase/AK strain transforming signaling pathway (PI3K/Akt)[43].
Increased pericyte coverage
Pericytes are mural cells that surround the endothelial cells of blood vessels and promote vascular stability and angiogenesis[44]. PDGF-BB is secreted by vascular endothelial cells and binds to PDGFR-β on pericytes, activating signaling that leads to increased pericyte production of VEGF which promotes endothelial cell proliferation[45]. Increased pericyte activity in the tumor microenvironment has been associated with more aggressive ccRCC[46], and resistance to antiangiogenic TKIs has been observed in tumors with an increased number of pericytes[47]. A proposed mechanism of resistance is the inhibition of an important negative feedback loop. High VEGF levels lead to the formation of a VEGFR/PDGFR-β complex that suppresses PDGFR-β signaling, preventing the production of excessive VEGF[48]. In the setting of TKI therapy against VEGFR, this mechanism may be less effective, leading to overactive pericyte activity. Thus, all antiangiogenic TKIs induce resistance via this mechanism.
Multi-drug resistance via lysosomal sequestration and efflux transporters
Sunitib is a small, hydrophobic, weak base, which allows it to easily pass through the cellular membrane, accumulate intracellularly, and bind to the kinase domain of VEGFR[49]. However, this structure also lends itself to efficient passage into lysosomes, where a more acidic environment encourages protonation of the drug, conferring a positive charge and effectively sequestering it for degradation[50]. Although sunitinib is particularly susceptible due to its chemical properties, lysosomal sequestration has been identified as a contributor to multi-drug resistance (MDR) in cancer cells via a variety of mechanisms. Cancer cells have been found to upregulate PI3K, which promotes lysosomal activity and stability. Further, cancer cells have been found to abolish lysosomal membrane permeabilization (LMP), a process that normally induces apoptosis, via upregulation of cytosolic protease inhibitors and via increased translocation of Hsp70 to the lysosomal lumen, which stabilizes lysosomal membranes[51]. Following exposure to sunitinib, RCC cells exhibit a rise in lysosomal mass, facilitating enhanced sequestration[52]. Zhitomirsky et al. demonstrated that exposure of human carcinoma cells to sunitinib leads to an increase in the number of lysosomes per cell, as well as the number of lysosomes accumulating high levels of sunitinib[53].
Sunitinib exposure has also been associated with increased expression of ATP-binding cassette (ABC) transporters such as P-glycoprotein (P-GP), which are present both on the lysosomal and cell membranes and promote both lysosomal sequestration and extracellular efflux[54]. Thus, lysosomal sequestration and efflux transporter proteins work synergistically to promote the development of resistance[55].
Bone marrow-derived cell recruitment
Bone marrow-derived cells (BMDCs) are a pool of progenitor cells, including hematopoietic stem cells, endothelial progenitor cells, tumor-associated macrophages, mesenchymal stromal cells, VEGFR1-positive hemangiocytes, and more[56]. Myeloid-derived suppressor cells (MDSCs) are a subpopulation of BMDCs that most commonly express CD11b and serve an immunomodulatory role in the tumor microenvironment, suppressing the activity of infiltrating cytotoxic T lymphocytes[57]. MDSCs produce nitric oxide, which reacts with superoxide, generating peroxynitrite (PNT). PNT nitrates both T-cell receptors, reducing their responsiveness to antigen major histone compatibility (MHC) complexes, and
Tumor-associated fibroblasts
Tumor-associated fibroblasts (TAFs) are activated fibroblasts that have undergone phenotypic and functional changes in response to tumor-derived signals[63]. Unlike regular fibroblasts, TAFs remain chronically activated and continue to carry out their work indefinitely within the tumor microenvironment. They support tumor survival and growth by secreting various growth factors, cytokines, and chemokines, such as interleukins, HGF, and stromal cell-derived factor 1 alpha (SDF-1α)[64]. TAFs promote the deposition of a dense extracellular matrix, creating a physical barrier that hampers drug penetration into the tumor and supporting cell adhesion-mediated drug resistance[65,66]. Moreover, the role of TAFs in supporting cell adhesion-mediated drug resistance (CAM-DR) is well-defined. TAFs also play a part in reprograming tumor metabolism, making cancer cells less dependent on glucose, and increasing the lactate upload to drive anabolic pathways[64]. Platelet-derived growth factor-C (PDGF-C) mediates the angiogenic properties of TAFs and has been found to be upregulated in tumor cells resistant to anti-VEGF antibodies[67].
Epithelial-mesenchymal transition
Epithelial-mesenchymal transition (EMT) is an embryonic development process that can be hijacked by cancer cells to promote invasion, metastasis, and resistance to therapy. During EMT, epithelial cells lose characteristics such as cell-to-cell adhesion and apical-basal polarity, and acquire mesenchymal features including increased motility, invasiveness, and resistance to cell death[68]. This phenotypic switch is achieved by changes in gene expression, such as the downregulation of epithelial markers (E-cadherin) and upregulation of mesenchymal markers (N-cadherin, vimentin), as well as by epigenetic modifications[68]. EMT in cancer is largely driven by tumor hypoxia and resultant HIF-1α activation, as well as by inflammatory cytokines IL-6, IL-8, IL-15, and tumor necrosis factor-α (TNF-α)[20,69].
EMT is frequently discussed in the context of cancer stem cells (CSCs), which are subpopulations of cells within tumors that are capable of self-renewal and multi-lineage differentiation[70]. Because heterogeneous tumors contain many phenotypically distinct cells with varying degrees of response to chemotherapeutics, CSCs enhance drug resistance and tumor relapse. Processes associated with EMT, such as suppression of
Acquired resistance to sunitinib via EMT has been demonstrated in patient-derived xenograft models. In a study by Hammers et al., tumor tissue from skin metastases of a sunitinib-resistant ccRCC patient was implanted into nude mice, and these mice were treated with sunitinib or vehicle for 90 days. The average tumor volume was less than 200 mm3 in mice treated with sunitinib and more than 800 mm3 in mice treated with the vehicle, which suggests renewed sensitivity to sunitinib. The histology of the skin metastases indicated sarcomatous differentiation with a fibroblast-like appearance, indicating EMT. However, the cells in the xenograft returned to normal ccRCC histology[73].
Pharmacogenomic factors
Single nucleotide polymorphisms (SNPs) are variations in a single nucleotide within the DNA sequence, which achieve an allelic frequency of at least 1% in a population, and they can affect gene expression, protein function, and drug metabolism[3]. Specific SNPs in drug-metabolizing enzymes, drug transporters, or drug targets can impact the efficacy of TKIs.
CYP3A4 and CYP3A5 are two of the principal enzymes responsible for sunitinib metabolism, converting the drug to its active metabolite, N-desethyl sunitinib (SU12662)[74]. SU12662 has a longer half-life than sunitinib, resulting in increased drug exposure[75]. The CYP3A4 SNP rs4646437G>A has been associated with an increased incidence of hypertension in mRCC patients treated with sunitinib. This is presumably due to enhanced sunitinib metabolism leading to increased concentrations of SU12662[76]. The expression of CYP3A4 is negatively regulated by two ligand-activated nuclear receptors, NR112 and NR113[77]. SNPs present in the genes encoding NR112 and NR113 have been associated with decreased patient PFS and OS, likely due to enhanced NR112 and NR113 activity leading to decreased CYP3A4 expression and lower SU12662 concentrations[78]. SNPs in CYP3A5, such as the CYP3A5*1 allele, have been associated with sunitinib toxicity and the need for dose reduction, likely due to enhanced CYP3A5 activity leading to increased concentrations of SU12662[79].
Additionally, SNPs in genes coding for ABC transporters have been found to affect the uptake and efflux of TKIs, altering drug concentrations within tumor cells. The presence of CGT in the ABCB1 haplotype is associated with improved PFS, likely due to decreased clearance of sunitinib and its active metabolite SU12662[79]. On the other hand, certain variants such as the TT-genotype in ABCB1 rs1125803 or the TT/TA-variant in ABCB1 rs2032582 have been found to promote drug efflux, resulting in increased intracellular clearance of sunitinib, increased time-to-dose reduction, and decreased PFS[80].
MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNA molecules that play a significant regulatory role in gene expression. miRNAs are known to contribute to cancer progression by silencing tumor suppressor genes via destruction or decreased expression of mRNA[81]. Multiple miRNAs have been implicated in mediating TKI resistance in RCC by targeting key signaling pathways involved in cell proliferation, survival, and drug response. For example, miR-15b has been found to be overexpressed in sunitinib-resistant cell lines after incubation with the drug. In vivo models have produced similar results[82]. Other notable miRNAs overexpressed in sunitinib-resistant RCC cells include miRNA-575, miRNA-642b-3p, and miRNA-4430 (all studied in vitro), as well as miRNA-942, miRNA-133a, miRNA-628-5p, and miRNA-484 (all studied
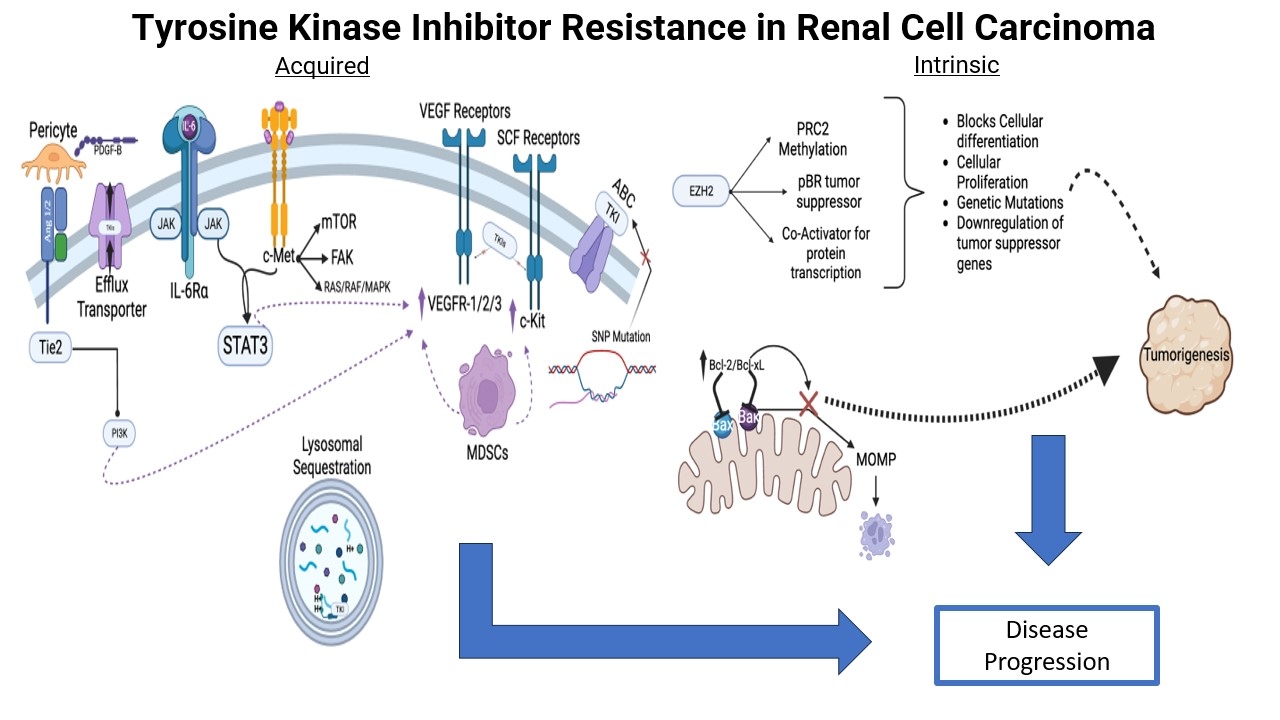
In contrast, miR-200b and miR-141 have been found to be downregulated in ccRCC compared to benign tissue, and their expression may represent an independent prognostic factor for increased PFS and OS[86]. Figure 2 illustrates the wide array of known acquired mechanisms of TKI resistance in RCC.

Figure 2. Common acquired mechanisms of resistance to tyrosine kinase inhibitors in RCC. ABC: ATP-binding cassette; IL-6: interleukin-6; MAPK: mitogen-activated protein kinases; MDSCs: myeloid-derived suppressor cells; mTOR: mammalian target of rapamycin; PDGF: platelet-derived growth factor; PI3K: phosphoinositide-3-kinase; RCC: renal cell carcinoma; SNP: single nucleotide polymorphism; TKIs: tyrosine kinase inhibitors; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor.
INTRINSIC MECHANISMS OF TKI RESISTANCE IN RCC
Methylation of tumor suppressor genes
Enhancer of zeste homolog 2 (EZH2) is an enzyme that functions as the catalytic subunit of the polycomb repressive complex 2 (PRC2). Its primary function is to methylate lysine 27 on histone H3, resulting in gene silencing and transcriptional repression[87]. Intrinsic increased expression of EZH2 can lead to aberrant methylation of specific tumor suppressor genes, rendering them inactive. Methylation of these genes can disrupt critical cellular pathways involved in growth control and DNA repair, leading to the promotion of tumorigenesis. The dysregulation of EZH2-mediated methylation can confer resistance to TKIs and other antineoplastic agents by circumventing the inhibitory effects of these drugs on oncogenic signaling pathways. The overexpression of EZH2 has been reported in RCC and is associated with poor prognosis[88,89].
Inhibition of apoptosis
B cell lymphoma-2 (Bcl-2) and B cell lymphoma-extra large (Bcl-xL) are proteins within the Bcl-2 family and play a key role in inhibiting apoptosis. These are known to be upregulated in many cancers, contributing to proliferation and metastasis[90]. There is limited evidence that overexpression of these proteins may confer intrinsic resistance to TKIs and other antineoplastic agents in RCC via inhibition of apoptosis, though further study is needed[91]. Figure 3 demonstrates known intrinsic mechanisms of resistance to TKIs in RCC.

Figure 3. Intrinsic resistance mechanisms affecting tyrosine kinase inhibitor activity in RCC. Bcl-2: B cell lymphoma-2; Bcl-xL: B cell lymphoma-extra large; EZH2: enhancer of zeste homolog 2; PRC2: polycomb repressive complex 2; RCC: renal cell carcinoma.
OVERCOMING TKI RESISTANCE IN RCC
Effective mitigation of TKI resistance plays a vital role in improving outcomes for patients with advanced RCC. Undoubtedly, this is a significant challenge due to the diverse range of mechanisms through which RCC can develop resistance. However, the multitude of mechanisms also offers numerous avenues for intervention, enabling the targeting of various processes to minimize resistance. Table 1 summarizes the many resistance mechanisms herein described, as well as potential approaches for overcoming these mechanisms.
Acquired and intrinsic mechanisms of tyrosine kinase inhibitor resistance in RCC
| Process | Mechanism | TKIs affected | Potential therapies | Ref. |
| Alternative proangiogenic pathways | Multiple; tumor hypoxia induces upregulation of interleukins, c-Met/HGF, angiopoietins, and growth factors | Sunitinib, pazopanib, sorafenib, axitinib, tivozanib | HIF-2α inhibitors; anti-IL-6 and IL-8 antibodies; TKIs against Ang 1 and 2, c-Met, and FGFR | [29-43,92-104] |
| Increased pericyte coverage | Increased tumor endothelial stability and production of VEGF | Sunitinib, pazopanib, sorafenib, axitinib, tivozanib | Unknown | [44-48] |
| Lysosomal sequestration | Drug sequestered; subtherapeutic intracellular concentrations; ineffective inhibition of receptor tyrosine kinases | Sunitinib | Alkalizing agents; drugs against lysosomal membrane proteins | [49-53,55,108] |
| Efflux pumps | Subtherapeutic intracellular concentrations | Sunitinib known, but other TKIs possible | P-GP inhibitors | [54,55,109-113] |
| Bone marrow-derived cell recruitment | Bone marrow-derived cells accumulate around tumor; immunomodulatory effects including suppression of cytotoxic T cells | Sunitinib, pazopanib, sorafenib, axitinib, tivozanib | Immune checkpoint inhibitors to rejuvenate immune response in the tumor microenvironment | [56-62] |
| Tumor-associated fibroblasts | Overactive fibroblasts secrete interleukins, hepatocyte growth factor, and more in tumor microenvironment; increased extracellular matrix deposition hampers drug penetration and supports cell adhesion | Unknown, but all TKIs possible | Histone deacetylase inhibitors | [63-67,105,107] |
| Epithelial-mesenchymal transition | Tumor hypoxia drives epithelial cells to acquire features such as increased motility, invasiveness, and resistance to cell death | Sunitinib, pazopanib, sorafenib, axitinib, tivozanib | Drugs with activity against EMT in combination with antineoplastic agents | [20,68-72,105,106] |
| Pharmacologic Factors | Single nucleotide polymorphisms in genes related to sunitinib metabolism lead to decreased concentrations of active metabolite | Sunitinib | Unknown | [3,74-80] |
| miRNAs | Silencing of tumor suppressor genes via destruction or decreased expression of mRNA | Sunitinib | Unknown | [81-86] |
| Methylation of tumor suppressor genes | EZH2 overexpression leads to aberrant methylation of tumor suppressor genes, rendering them inactive | All antineoplastic and targeted drugs | EZH2 inhibitors; hypomethylating agents | [87-89,114] |
| Inhibition of apoptosis | Bcl-2 and Bcl-xL overexpression inhibits tumor cell apoptosis | All antineoplastic and targeted drugs | Bcl-2 inhibitors | [90,91,115] |
One strategy is to target processes upstream from VEGF/VEGFR, such as HIF-2α. In hypoxic conditions, HIF-2α promotes the expression of multiple hypoxia-inducible genes, including VEGF and PDGF-BB. As previously discussed, in VHL syndrome, the negative regulatory protein pVHL is lost, leading to the accumulation of HIF-2α and excessive angiogenesis[24]. HIF-2α inhibitors act by inhibiting the dimerization of HIF-2α and its partner protein, ARNT1, thereby inhibiting HIF-2α mediated transcription[92]. The HIF-2α inhibitor, belzutifan, was approved by the FDA for the treatment of germline VHL-mutated RCC after a phase 2 trial demonstrated promising efficacy in this population[93]. Other studies examining belzutifan in sporadic (non-VHL associated) RCC are also encouraging[94]. In a phase 1 expansion cohort, belzutifan in combination with nivolumab was administered to patients with advanced ccRCC, and patients with therapeutic exposure to the drug had a median PFS of 10.0 months compared to just 4.7 months for patients with subtherapeutic exposure[95]. An ongoing trial evaluating the combination of belzutifan and cabozantinib is currently underway[96].
Directly targeting the alternative proangiogenic pathways upregulated during resistance development may also be effective. IL-6 and IL-8, both upregulated in the setting of tissue hypoxia following effective antiangiogenic therapy, are prime examples[47]. These cytokines can induce a cascade of immunologic changes that promote angiogenesis. Interestingly, co-administration of IL-8 neutralizing antibody has shown promise in re-sensitizing xenograft tumors to sunitinib treatment[97]. Administration of the anti-IL-6 antibody, tocilizumab, given in combination with interferon, has also slowed RCC xenograft proliferation[29]. The Ang/Tie signaling pathway, which augments the VEGFR pathway and promotes blood vessel stabilization, survival, and maturation, can be directly targeted with the TKI trebananib[98]. In xenograft mouse models, treatment with trebananib in combination with sunitinib slowed tumor progression compared to treatment with sunitinib plus control[36]. However, other results have been less promising. A phase 2 trial examined the efficacy of trebananib with or without continued anti-VEGF therapy in RCC patients who had previously progressed on anti-VEGF therapy and observed poor outcomes in both treatment arms[99]. Targeting c-Met in cells that have become resistant to TKI’s against VEGF may also be effective. Studies have shown that the growth of sunitinib-resistant tumors can be slowed with a combination of sunitinib and a c-Met inhibitor[39]. Likewise, Zhou et al. found that treatment with cabozantinib, a TKI against VEGFR and MET, can rescue acquired sunitinib resistance in xenograft mouse models[100]. Finally, fibroblast growth factor (FGF) overexpression, specifically FGF2, has been associated with a poor RCC prognosis and contributes to TKI resistance through suppression of antiangiogenic activity[42,101]. There are several multitargeted TKIs that inhibit FGFR, such as lenvatinib, but these drugs are themselves susceptible to resistance[102]. In vitro studies have suggested that combining FGFR inhibitors with other TKIs such as PI3K inhibitors may help mitigate this resistance and inhibit RCC cell metabolic activity[103]. PI3K inhibitors have also demonstrated strong in vitro anti-tumor activity when co-administered with sunitinib[104].
Targeting proangiogenic changes in the tumor microenvironment may be useful in combatting TKI resistance. The process of EMT, which promotes resistance to therapy and metastases, is associated with a host of metabolic changes. Although there are no direct inhibitors to EMT, multiple metabolism-inhibiting drugs have shown indirect activity against EMT changes through various complex biochemical mechanisms[105]. There are multiple ongoing clinical trials examining the combination of these drugs with antineoplastics agents, although none of these trials include RCC patients[106]. Additionally, TAFs may serve as a target for overcoming resistance. Histone deacetylase inhibitors offer a promising approach to diminish the activation of TAFs and eradicate their infiltration within the tumor stroma, as demonstrated by a study in patients with relapsed or refractory lymphoma or multiple myeloma[105,107].
Lysosomal sequestration of TKIs is another potentially reversible mechanism of resistance. Combination therapies incorporating alkalizing treatments have been explored, creating a toxic environment that counteracts the sequestration process[51]. Drugs targeting various lysosomal membrane proteins have shown promise in destabilizing the lysosomal membrane, leading to membrane permeabilization[55]. Interestingly, Gotink et al. showed that sunitinib efficacy in RCC cells that had developed resistance via lysosomal sequestration can be restored by culturing resistant cells with sunitinib-free media[108]. This supports lysosomal sequestration as an acquired, transient, and reversible resistance mechanism.
Additionally, RCC has been shown to overexpress the ABC transporter P-glycoprotein (P-GP) in response to hypoxia from antiangiogenic therapy. P-GP is predominantly found in the cell membrane and exports drugs, contributing to multi-drug resistance[109,110]. P-GP is also commonly found in enterocytes and contributes to resistance via drug export into the GI lumen, limiting drug oral bioavailability[111]. Several studies have examined the co-administration of inhibitors to P-GP with anticancer drugs in various malignancies, but this strategy has been limited by severe toxicity and unwanted side effects[112]. Despite evidence of in vitro anti-tumor activity, there are currently no P-GP inhibitors approved for cancer treatment[113].
Intrinsic mechanisms may also be targeted to overcome resistance. EZH2 overexpression in RCC contributes to hypermethylation, and ultimately inactivation, of tumor suppressor genes. The efficacy of EZH2 inhibitors and hypomethylating agents is currently not defined in many solid tumors, including RCC, but investigative studies are underway[114]. Antiapoptotic proteins such as Bcl-2 and Bcl-xL also confer intrinsic resistance. There is limited evidence regarding the use of Bcl-2 inhibitors in RCC. However, one preclinical study has demonstrated a potential synergetic effect of the Bcl-2 inhibitor, venetoclax, when given sequentially prior to sunitinib[115].
In recent years, ICIs targeting programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) have emerged as a cornerstone in the treatment of advanced RCC. These therapies block interactions between immune checkpoint proteins, thereby triggering immune activation against various cancers[116]. ICIs are frequently combined with TKIs to leverage their synergistic effects resulting from complementary mechanisms of action. Notably, all TKI resistance mechanisms share a common thread of promoting angiogenesis. Proangiogenic molecules like VEGF impede both the innate and adaptive immune systems by hindering precursor cell differentiation, upregulating PD-1, PD-L1, and CTLA-4 on immune cells, and recruiting MDSCs[117]. ICI therapy rejuvenates the immune response, counteracting these immunosuppressive effects and mitigating resistance.
CONCLUSION
TKIs have significantly improved the outcomes of patients with advanced RCC by targeting key pathways involved in cancer proliferation, survival, and metastasis. However, treatment with VEGF-targeted TKIs is almost always characterized by the eventual development of resistance and subsequent disease progression. Mitigating drug resistance is challenging due to the diverse mechanisms underlying TKI resistance, including upregulation of alternative proangiogenic pathways, EMT, efflux pumps reducing intracellular drug concentrations, lysosomal sequestration, alterations in the tumor microenvironment, and genetic factors.
Understanding these mechanisms is crucial for the development of innovative therapeutic approaches to overcome TKI resistance. Notably, the combination of TKIs with other agents, particularly ICIs, has made significant strides towards accomplishing this aim, solidifying ICIs as a cornerstone in the treatment of advanced RCC. Additionally, strategies to reverse or inhibit specific resistance mechanisms hold the potential to restore TKI efficacy. The continued exploration of combination therapies, comprehensive understanding of the tumor microenvironment, and identification of additional genetic biomarkers will pave the way for more effective treatments, ultimately improving outcomes for patients with advanced RCC.
DECLARATIONS
Authors’ contributions
Investigation, writing, reviewing and editing: Sweeney PL
Investigation, writing: Suri Y
Supervision, reviewing and editing: Basu A
Supervision, reviewing and editing: Koshkin VS
Project conceptualization, supervision, reviewing and editing: Desai A
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73:17-48.
2. Koul H, Huh JS, Rove KO, et al. Molecular aspects of renal cell carcinoma: a review. Am J Cancer Res 2011;1:240-54.
3. Jin J, Xie Y, Zhang JS, et al. Sunitinib resistance in renal cell carcinoma: from molecular mechanisms to predictive biomarkers. Drug Resist Updat 2023;67:100929.
4. Moch H, Amin MB, Berney DM, et al. The 2022 World Health Organization classification of tumours of the urinary system and male genital organs-part a: renal, penile, and testicular tumours. Eur Urol 2022;82:458-68.
5. Hsieh JJ, Purdue MP, Signoretti S, et al. Renal cell carcinoma. Nat Rev Dis Primers 2017;3:17009.
6. Escudier B, Porta C, Schmidinger M, et al. ESMO Guidelines Committee. Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27:v58-68.
7. Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011;19:617-23.
8. Capitanio U, Bensalah K, Bex A, et al. Epidemiology of renal cell carcinoma. Eur Urol 2019;75:74-84.
9. Ljungberg B, Albiges L, Abu-Ghanem Y, et al. European Association of Urology Guidelines on Renal Cell Carcinoma: the 2022 update. Eur Urol 2022;82:399-410.
10. Prins FM, Kerkmeijer LGW, Pronk AA, et al. Renal cell carcinoma: alternative nephron-sparing treatment options for small renal masses, a systematic review. J Endourol 2017;31:963-75.
11. Larroquette M, Peyraud F, Domblides C, et al. Adjuvant therapy in renal cell carcinoma: current knowledges and future perspectives. Cancer Treat Rev 2021;97:102207.
12. Rini BI, Battle D, Figlin RA, et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J Immunother Cancer 2019;7:354.
13. Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin 2017;67:507-24.
14. Liu YF, Zhang ZC, Wang SY, et al. Immune checkpoint inhibitor-based therapy for advanced clear cell renal cell carcinoma: a narrative review. Int Immunopharmacol 2022;110:108900.
16. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 2018;17:58.
17. Mabeta P, Steenkamp V. The VEGF/VEGFR axis revisited: implications for cancer therapy. Int J Mol Sci 2022;23:15585.
18. Ebrahimi N, Fardi E, Ghaderi H, et al. Receptor tyrosine kinase inhibitors in cancer. Cell Mol Life Sci 2023;80:104.
19. Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors - a review on pharmacology, metabolism and side effects. Curr Drug Metab 2009;10:470-81.
20. Sharma R, Kadife E, Myers M, Kannourakis G, Prithviraj P, Ahmed N. Determinants of resistance to VEGF-TKI and immune checkpoint inhibitors in metastatic renal cell carcinoma. J Exp Clin Cancer Res 2021;40:186.
21. Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 2010;13:1-14.
22. Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 2009;10:992-1000.
23. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43-9.
24. Kim H, Shim BY, Lee SJ, Lee JY, Lee HJ, Kim IH. Loss of von Hippel-Lindau (VHL) tumor suppressor gene function: VHL-HIF pathway and advances in treatments for metastatic renal cell carcinoma (RCC). Int J Mol Sci 2021;22:9795.
25. Meléndez-Rodríguez F, Roche O, Sanchez-Prieto R, Aragones J. Hypoxia-inducible factor 2-dependent pathways driving von Hippel-Lindau-deficient renal cancer. Front Oncol 2018;8:214.
26. Haider T, Pandey V, Banjare N, Gupta PN, Soni V. Drug resistance in cancer: mechanisms and tackling strategies. Pharmacol Rep 2020;72:1125-51.
27. Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol 2014;8:1132-9.
28. Xiang Y, Zheng G, Zhong J, Sheng J, Qin H. Advances in renal cell carcinoma drug resistance models. Front Oncol 2022;12:870396.
29. Ishibashi K, Koguchi T, Matsuoka K, et al. Interleukin-6 induces drug resistance in renal cell carcinoma. Fukushima J Med Sci 2018;64:103-10.
30. Rizzo M, Varnier L, Pezzicoli G, Pirovano M, Cosmai L, Porta C. IL-8 and its role as a potential biomarker of resistance to anti-angiogenic agents and immune checkpoint inhibitors in metastatic renal cell carcinoma. Front Oncol 2022;12:990568.
31. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem 2009;284:6038-42.
32. Harmon CS, DePrimo SE, Figlin RA, et al. Circulating proteins as potential biomarkers of sunitinib and interferon-α efficacy in treatment-naïve patients with metastatic renal cell carcinoma. Cancer Chemother Pharmacol 2014;73:151-61.
33. Tran HT, Liu Y, Zurita AJ, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol 2012;13:827-37.
34. Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells 2019;8:471.
35. He FF, Zhang D, Chen Q, et al. Angiopoietin-Tie signaling in kidney diseases: an updated review. FEBS Lett 2019;593:2706-15.
36. Wang X, Bullock AJ, Zhang L, et al. The role of angiopoietins as potential therapeutic targets in renal cell carcinoma. Transl Oncol 2014;7:188-95.
37. Deng J, Li L, Xia H, et al. A comparison of the prognosis of papillary and clear cell renal cell carcinoma: evidence from a meta-analysis. Medicine 2019;98:e16309.
38. Schöffski P, Wozniak A, Escudier B, et al. Crizotinib achieves long-lasting disease control in advanced papillary renal-cell carcinoma type 1 patients with MET mutations or amplification. EORTC 90101 CREATE trial. Eur J Cancer 2017;87:147-63.
39. Marona P, Górka J, Kotlinowski J, Majka M, Jura J, Miekus K. C-Met as a key factor responsible for sustaining undifferentiated phenotype and therapy resistance in renal carcinomas. Cells 2019;8:272.
40. Marona P, Górka J, Kwapisz O, et al. Resistance to tyrosine kinase inhibitors promotes renal cancer progression through MCPIP1 tumor-suppressor downregulation and c-Met activation. Cell Death Dis 2022;13:814.
41. Peltola KJ, Penttilä P, Rautiola J, et al. Correlation of c-Met expression and outcome in patients with renal cell carcinoma treated with sunitinib. Clin Genitourin Cancer 2017;15:487-94.
42. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8:299-309.
43. Tsimafeyeu I, Demidov L, Stepanova E, Wynn N, Ta H. Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma. Scand J Urol Nephrol 2011;45:190-5.
44. Dessalles CA, Babataheri A, Barakat AI. Pericyte mechanics and mechanobiology. J Cell Sci 2021;134:jcs240226.
45. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res 2005;97:512-23.
46. Cao Z, Shang B, Zhang G, et al. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim Biophys Acta 2013;1836:273-86.
47. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592-603.
48. Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 2008;456:809-13.
49. Mena AC, Pulido EG, Guillén-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs 2010;21 Suppl 1:S3-11.
50. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol Cancer Ther 2018;17:1355-64.
51. Halaby R. Influence of lysosomal sequestration on multidrug resistance in cancer cells. Cancer Drug Resist 2019;2:31-42.
52. Rausch M, Rutz A, Allard PM, et al. Molecular and functional analysis of sunitinib-resistance induction in human renal cell carcinoma cells. Int J Mol Sci 2021;22:6467.
53. Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015;6:1143-56.
54. Giuliano S, Cormerais Y, Dufies M, et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015;11:1891-904.
55. de Klerk DJ, Honeywell RJ, Jansen G, Peters GJ. Transporter and lysosomal mediated (Multi)drug resistance to tyrosine kinase inhibitors and potential strategies to overcome resistance. Cancers 2018;10:503.
56. Golle L, Gerth HU, Beul K, et al. Bone marrow-derived cells and their conditioned medium induce microvascular repair in uremic rats by stimulation of endogenous repair mechanisms. Sci Rep 2017;7:9444.
59. Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol 2011;11:856-61.
60. Susek KH, Karvouni M, Alici E, Lundqvist A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front Immunol 2018;9:2159.
61. Elkabets M, Ribeiro VS, Dinarello CA, et al. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol 2010;40:3347-57.
62. Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009;15:2148-57.
64. Errarte P, Larrinaga G, López JI. The role of cancer-associated fibroblasts in renal cell carcinoma. An example of tumor modulation through tumor/non-tumor cell interactions. J Adv Res 2020;21:103-8.
65. Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev 2010;21:21-6.
66. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 2009;9:665-74.
67. Crawford Y, Kasman I, Yu L, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009;15:21-34.
68. Piva F, Giulietti M, Santoni M, et al. Epithelial to mesenchymal transition in renal cell carcinoma: implications for cancer therapy. Mol Diagn Ther 2016;20:111-7.
69. He H, Magi-Galluzzi C. Epithelial-to-mesenchymal transition in renal neoplasms. Adv Anat Pathol 2014;21:174-80.
70. Babaei G, Aziz SG, Jaghi NZZ. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed Pharmacother 2021;133:110909.
71. Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008;3:e2888.
72. Yuan ZX, Mo J, Zhao G, Shu G, Fu HL, Zhao W. Targeting strategies for renal cell carcinoma: from renal cancer cells to renal cancer stem cells. Front Pharmacol 2016;7:423.
73. Hammers HJ, Verheul HM, Salumbides B, et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol Cancer Ther 2010;9:1525-35.
74. Amaya GM, Durandis R, Bourgeois DS, et al. Cytochromes P450 1A2 and 3A4 catalyze the metabolic activation of sunitinib. Chem Res Toxicol 2018;31:570-84.
75. Diekstra MH, Klümpen HJ, Lolkema MP, et al. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin Pharmacol Ther 2014;96:81-9.
76. Diekstra MH, Belaustegui A, Swen JJ, et al. Sunitinib-induced hypertension in CYP3A4 rs4646437 A-allele carriers with metastatic renal cell carcinoma. Pharmacogenomics J 2017;17:42-6.
77. van der Veldt AA, Eechoute K, Gelderblom H, et al. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin Cancer Res 2011;17:620-9.
78. Beuselinck B, Karadimou A, Lambrechts D, et al. Single-nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br J Cancer 2013;108:887-900.
79. Diekstra MH, Swen JJ, Boven E, et al. CYP3A5 and ABCB1 polymorphisms as predictors for sunitinib outcome in metastatic renal cell carcinoma. Eur Urol 2015;68:621-9.
80. Beuselinck B, Lambrechts D, Van Brussel T, et al. Efflux pump ABCB1 single nucleotide polymorphisms and dose reductions in patients with metastatic renal cell carcinoma treated with sunitinib. Acta Oncol 2014;53:1413-22.
81. Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ. Regulatory mechanism of microRNA expression in cancer. Int J Mol Sci 2020;21:1723.
82. Lu L, Li Y, Wen H, Feng C. Overexpression of miR-15b promotes resistance to sunitinib in renal cell carcinoma. J Cancer 2019;10:3389-96.
83. Yamaguchi N, Osaki M, Onuma K, et al. Identification of microRNAs involved in resistance to sunitinib in renal cell carcinoma cells. Anticancer Res 2017;37:2985-92.
84. Prior C, Perez-Gracia JL, Garcia-Donas J, et al. Identification of tissue microRNAs predictive of sunitinib activity in patients with metastatic renal cell carcinoma. PLoS One 2014;9:e86263.
85. Xiao W, Lou N, Ruan H, et al. Mir-144-3p promotes cell proliferation, metastasis, sunitinib resistance in clear cell renal cell carcinoma by downregulating ARID1A. Cell Physiol Biochem 2017;43:2420-33.
86. Saleeb R, Kim SS, Ding Q, et al. The miR-200 family as prognostic markers in clear cell renal cell carcinoma. Urol Oncol 2019;37:955-63.
88. Wang Y, Chen Y, Geng H, Qi C, Liu Y, Yue D. Overexpression of YB1 and EZH2 are associated with cancer metastasis and poor prognosis in renal cell carcinomas. Tumour Biol 2015;36:7159-66.
89. Adelaiye-Ogala R, Budka J, Damayanti NP, et al. EZH2 modifies sunitinib resistance in renal cell carcinoma by kinome reprogramming. Cancer Res 2017;77:6651-66.
90. Qian S, Wei Z, Yang W, Huang J, Yang Y, Wang J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front Oncol 2022;12:985363.
91. Gobé G, Rubin M, Williams G, Sawczuk I, Buttyan R. Apoptosis and expression of Bcl-2, Bcl-XL, and Bax in renal cell carcinomas. Cancer Invest 2002;20:324-32.
92. Cho H, Kaelin WG. Targeting HIF2 in clear cell renal cell carcinoma. Cold Spring Harb Symp Quant Biol 2016;81:113-21.
94. Ahmed R, Ornstein MC. Targeting HIF-2 alpha in renal cell carcinoma. Curr Treat Options Oncol 2023;24:1183-98.
95. Rini BI, Appleman LJ, Figlin RA, et al. Results from a phase I expansion cohort of the first-in-class oral HIF-2α inhibitor PT2385 in combination with nivolumab in patients with previously treated advanced RCC. JCO 2019;37:558.
96. Choueiri TK, Kaelin WG Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat Med 2020;26:1519-30.
97. Huang D, Ding Y, Zhou M, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res 2010;70:1063-71.
98. Eroglu Z, Stein CA, Pal SK. Targeting angiopoietin-2 signaling in cancer therapy. Expert Opin Investig Drugs 2013;22:813-25.
99. Semrad TJ, Groshen S, Luo C, et al. Randomized phase 2 study of trebananib (AMG 386) with or without continued anti-vascular endothelial growth factor therapy in patients with renal cell carcinoma who have progressed on bevacizumab, pazopanib, sorafenib, or sunitinib - results of NCI/CTEP protocol 9048. Kidney Cancer 2019;3:51-61.
100. Zhou L, Liu XD, Sun M, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016;35:2687-97.
101. Welti JC, Gourlaouen M, Powles T, et al. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene 2011;30:1183-93.
102. Yue S, Li Y, Chen X, et al. FGFR-TKI resistance in cancer: current status and perspectives. J Hematol Oncol 2021;14:23.
103. Rausch M, Weiss A, Achkhanian J, Rotari A, Nowak-Sliwinska P. Identification of low-dose multidrug combinations for sunitinib-naive and pre-treated renal cell carcinoma. Br J Cancer 2020;123:556-67.
104. Makhov PB, Golovine K, Kutikov A, et al. Modulation of Akt/mTOR signaling overcomes sunitinib resistance in renal and prostate cancer cells. Mol Cancer Ther 2012;11:1510-7.
105. Sekino Y, Teishima J, Liang G, Hinata N. Molecular mechanisms of resistance to tyrosine kinase inhibitor in clear cell renal cell carcinoma. Int J Urol 2022;29:1419-28.
106. Ramesh V, Brabletz T, Ceppi P. Targeting EMT in cancer with repurposed metabolic inhibitors. Trends Cancer 2020;6:942-50.
107. Younes A, Berdeja JG, Patel MR, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol 2016;17:622-31.
108. Gotink KJ, Broxterman HJ, Labots M, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res 2011;17:7337-46.
109. Mollazadeh S, Sahebkar A, Hadizadeh F, Behravan J, Arabzadeh S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci 2018;214:118-23.
110. Soto-Vega E, Arroyo C, Richaud-Patin Y, García-Carrasco M, Vázquez-Lavista LG, Llorente L. P-glycoprotein activity in renal clear cell carcinoma. Urol Oncol 2009;27:363-6.
111. Liu X. ABC family transporters. In: Liu X, Pan G, editors. Drug transporters in drug disposition, effects and toxicity. Singapore: Springer; 2019. pp. 13-100.
112. Callaghan R, Luk F, Bebawy M. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab Dispos 2014;42:623-31.
113. Heming C, Muriithi W, Wanjiku Macharia L, Niemeyer Filho P, Moura-Neto V, Aran V. P-glycoprotein and cancer: what do we currently know? Heliyon 2022;8:e11171.
114. Joosten SC, Smits KM, Aarts MJ, et al. Epigenetics in renal cell cancer: mechanisms and clinical applications. Nat Rev Urol 2018;15:430-51.
115. Tang Y, Song T, Gao L, Mao F. Venetoclax synergizes sunitinib in renal cell carcincoma through inhibition of Bcl-2. Anticancer Agents Med Chem 2023;23:2027-34.
116. Shiravand Y, Khodadadi F, Kashani SMA, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol 2022;29:3044-60.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sweeney PL, Suri Y, Basu A, Koshkin VS, Desai A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resist 2023;6:858-73. http://dx.doi.org/10.20517/cdr.2023.89
AMA Style
Sweeney PL, Suri Y, Basu A, Koshkin VS, Desai A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resistance. 2023; 6(4): 858-73. http://dx.doi.org/10.20517/cdr.2023.89
Chicago/Turabian Style
Sweeney, Patrick L., Yash Suri, Arnab Basu, Vadim S. Koshkin, Arpita Desai. 2023. "Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma" Cancer Drug Resistance. 6, no.4: 858-73. http://dx.doi.org/10.20517/cdr.2023.89
ACS Style
Sweeney, PL.; Suri Y.; Basu A.; Koshkin VS.; Desai A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resist. 2023, 6, 858-73. http://dx.doi.org/10.20517/cdr.2023.89
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 12
likes
Like This Article 12
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.