
Drug resistance in glioblastoma: from chemo- to immunotherapy
 0
0
Abstract
As the most common and aggressive type of primary brain tumor in adults, glioblastoma is estimated to end over 10,000 lives each year in the United States alone. Stand treatment for glioblastoma, including surgery followed by radiotherapy and chemotherapy (i.e., Temozolomide), has been largely unchanged since early 2000. Cancer immunotherapy has significantly shifted the paradigm of cancer management in the past decade with various degrees of success in treating many hematopoietic cancers and some solid tumors, such as melanoma and non-small cell lung cancer (NSCLC). However, little progress has been made in the field of neuro-oncology, especially in the application of immunotherapy to glioblastoma treatment. In this review, we attempted to summarize the common drug resistance mechanisms in glioblastoma from Temozolomide to immunotherapy. Our intent is not to repeat the well-known difficulty in the area of neuro-oncology, such as the blood-brain barrier, but to provide some fresh insights into the molecular mechanisms responsible for resistance by summarizing some of the most recent literature. Through this review, we also hope to share some new ideas for improving the immunotherapy outcome of glioblastoma treatment.
Keywords
INTRODUCTION
Brain tumors affect more than ~17,000 people in the United States each year, where gliomas are considered the most common type of primary brain tumor[1]. Glioblastoma is a grade IV astrocytoma that was initially categorized into four molecular subtypes, termed neural, proneural, classical, and mesenchymal subtype[2]. Transcriptional profiling and genetic modeling in mice showed that glioblastoma originated from neural stem cells (NSC), NSC-derived astrocytes, and oligodendrocyte precursor cells (OPCs)[3-5]. Besides the four molecular subtypes based on their transcription profiling, glioblastoma tumors can also be classified by the status of the isocitrate dehydrogenase gene (IDH) as IDH wild-type and IDH-mutant tumors. Similarly, epigenetics factors, such as CpG island methylation phenotype of O6-methylguanine-DNA methyltransferase (MGMT) promoter, are also commonly used for glioblastoma tumor stratification[6,7].
Since the approval of Temozolomide (TMZ) for newly diagnosed glioblastoma treatment by the FDA in early 2000, surgery followed by radiotherapy and TMZ treatment has remained the first-line glioblastoma treatment[8]. However, none of these therapies eliminate cancer cells entirely because of challenges marred by high infiltration rate, tumor heterogeneity, blood-brain barrier (BBB), and immunosuppressive environment factors[9,10]. The highly infiltrative nature of glioblastoma does not allow the removal of cancerous cells using resection; self-renewing cells followed by resection become more prone to radioresistance and chemoresistance. Similarly, cellular heterogeneity and BBB prevent targeted drug delivery in glioblastoma[11,12].
COMMON DRUG RESISTANCE MECHANISMS IN BRAIN CANCERS
Blood-brain barrier
Although the BBB in glioblastoma is compromised to some extent, tumor BBB still presents a great challenge for therapeutics to reach glioblastoma cells. As the intrinsic barrier for brain cancer, BBB is a microvasculature structure surrounding the central nervous system (CNS), tightly regulating the movement of molecules and cells between the CNS and blood. Normally, BBB maintains the homeostasis of CNS and prevents infiltration of toxins, pathogens, inflammation, and harmful metabolites[13-15]. Disruption of the neurovascular unit (NVU) is associated with blood-brain dysfunction in neurodegenerative disease and brain tumors[16]. The NVU consists of vascular cells (endothelial, pericytes, and vascular smooth muscle cells), glia (astrocytes, microglia, and oligodendroglia), and neurons, and it plays an important role in maintaining BBB functional integrity and regulating the volume of cerebral blood flow[17,18]. The endothelial cells in neurovascular parenchyma form capillary beds connected through tight junctions (TJs), surrounded by a specialized basal lamina shared with pericytes and astrocytic end feet. They are sparsely interconnected by neuronal endings and microglia[19,20]. Astrocytes and pericytes, an essential constituent of NVU, release Sonic Hedgehog and vitronectin and angiopoietin I, respectively, acting on endothelial cells for their survival and maintaining BBB.
Overexpression of efflux pumps
Efflux transporters on the BBB membrane also contribute to cerebrospinal fluid homeostasis by protecting it from potentially harmful endogenous and exogenous substances[21,22]. These transporters also pose challenges by blocking therapeutic compounds from entering the brain parenchyma. Efflux transporters on compartments of the BBB belong to either ATP-binding cassette (ABC) or the solute carrier (SLC) superfamilies[23,24]. Organic anion-transporting polypeptides (OATP) are a superfamily of solute carrier organic anionic (SLCO) transmembrane transporters that are known for cancer drug resistance[25,26]. These peptide transporters regulate a variety of xenobiotic and endogenous substrates, including endogenous hormones, their conjugates, and anticancer drugs[27]. OATP1A2 is a sodium-independent uptake transporter family member and is highly expressed on the luminal membrane of BBB in tumors and adjacent healthy tissues[28]. A study by Cooper et al. in glioblastoma patients showed significant over-expression of all the OATP isoforms (OATP1A2, 2B1, 1C1, and 4A1) in tumor tissues compared to non-neoplastic brain[29].
Enhanced DNA damage repair pathways (MGMT) and abnormal activation of survival signaling pathways
As part of the glioblastoma standard treatment regimen, TMZ is a potent DNA alkylating agent that leads to DNA damage in cancer cells and cell death[30]. However, TMZ treatment often results in drug resistance in ~50% of glioblastoma patients due to overexpression of MGMT, which reverses the methylation of the O6 position of guanine. In addition to upregulated MGMT expression, glioblastoma often exhibits enhanced DNA damage repair capacity through several related mechanisms. For instance, poly(ADP-ribose) polymerase (PARP) was shown to interact with MGMT and enhance MGMT function in the removal of O6-methylation of DNA[31]. Interestingly, even in MGMT-deficient glioblastoma, TMZ resistance may still arise due to the loss of mismatch repair (MMR) pathway in tumor cells. Recent work by Lin et al. developed a new class of compound (KL-50) to achieve MMR-independent glioblastoma cell killing. It demonstrated a promising strategy to exploit cancer-specific deficiencies in DNA repair pathways[32]. Glioblastoma tumors also have elevated levels of receptor tyrosine kinases, such as EGFR gene amplification or mutation (EGFRvIII), PDGFR and FGFR, and aberrant activation of PI3K/ATK signaling and other growth factors (e.g., IGF-1, CTGF, and TGFβ)[33-39], with a potential contribution to the drug resistance phenotype.
Role of glioma stem cells
Glioma stem cells (GSCs) represent a subpopulation of relatively undifferentiated cells capable of self-renewal while also generating clonal populations of differentiated tumor cells in glioblastoma. These cells are increasingly recognized as a driving force supporting glioma genesis, therapy resistance, and recurrence[40]. GSCs have high regenerative capacity and can differentiate into cells expressing several lineage markers such as CD133, SOX2, CD15, CD44, integrin α6, and CD36[41]. Along with heterogeneity, various factors contribute to the chemoresistance of GSCs. Intrinsic factors include upregulated MGMT, higher anabolic capacity, and autophagy-mediated clearance of ROS induced by chemotherapy. Extrinsic factor is mainly hypoxic tumor microenvironment (TME). Hypoxia promotes the expression of GSC markers and a cancer stem-like phenotype[42]. Hypoxia-response genes, such as hypoxia-inducible factor HIF-2α and VEGF, are highly expressed in GSCs. Intriguingly, two reports have demonstrated that hypoxia-associated transcriptional signatures can be used as prognostic markers for glioblastoma patients[43,44].
Epigenetic modulations
Epigenetic dysregulation has been increasingly recognized as one of the significant drivers of oncogenesis, and several subtypes of glioblastoma are associated with epigenetic alterations[45,46]. These epigenetic modifications may serve as valuable biomarkers for tumor stratification and prognostic prediction. For instance, the glioblastoma resistance to receptor tyrosine kinase (RTK) inhibitors has been found to involve both genetic and epigenetic mechanisms[47], resulting in subclones with a gain of copy number in the insulin receptor substrate-1(IRS1) and substrate-2 (IRS2) loci. Another study identified a long non-coding RNA (LINC00021) that promotes TMZ resistance through Notch signaling and epigenetically silenced p21 expression via recruiting EZH2[48], one of the methyltransferases responsible for histone methylation. Epigenetic modifications in glioblastoma are also exploited as drug targets. Among the promising epigenetic interventions for glioblastoma are the histone deacetylase (HDAC) inhibitors[49], which have been extensively tested in various cancers[50]. HDAC inhibitors can block cancer cell proliferation by inducing cell cycle arrest, cell differentiation, and/or apoptosis[51]. With a large amount of supportive preclinical data, various HDAC inhibitors in glioblastoma clinical trials are underway.
DRUG RESISTANCE TO IMMUNOTHERAPY IN GLIOBLASTOMA
Current status of immunotherapy trials in glioblastoma
Although immune checkpoint inhibitors have greatly improved cancer treatment today, the clinical trials in glioblastoma treatment have been largely unsuccessful.
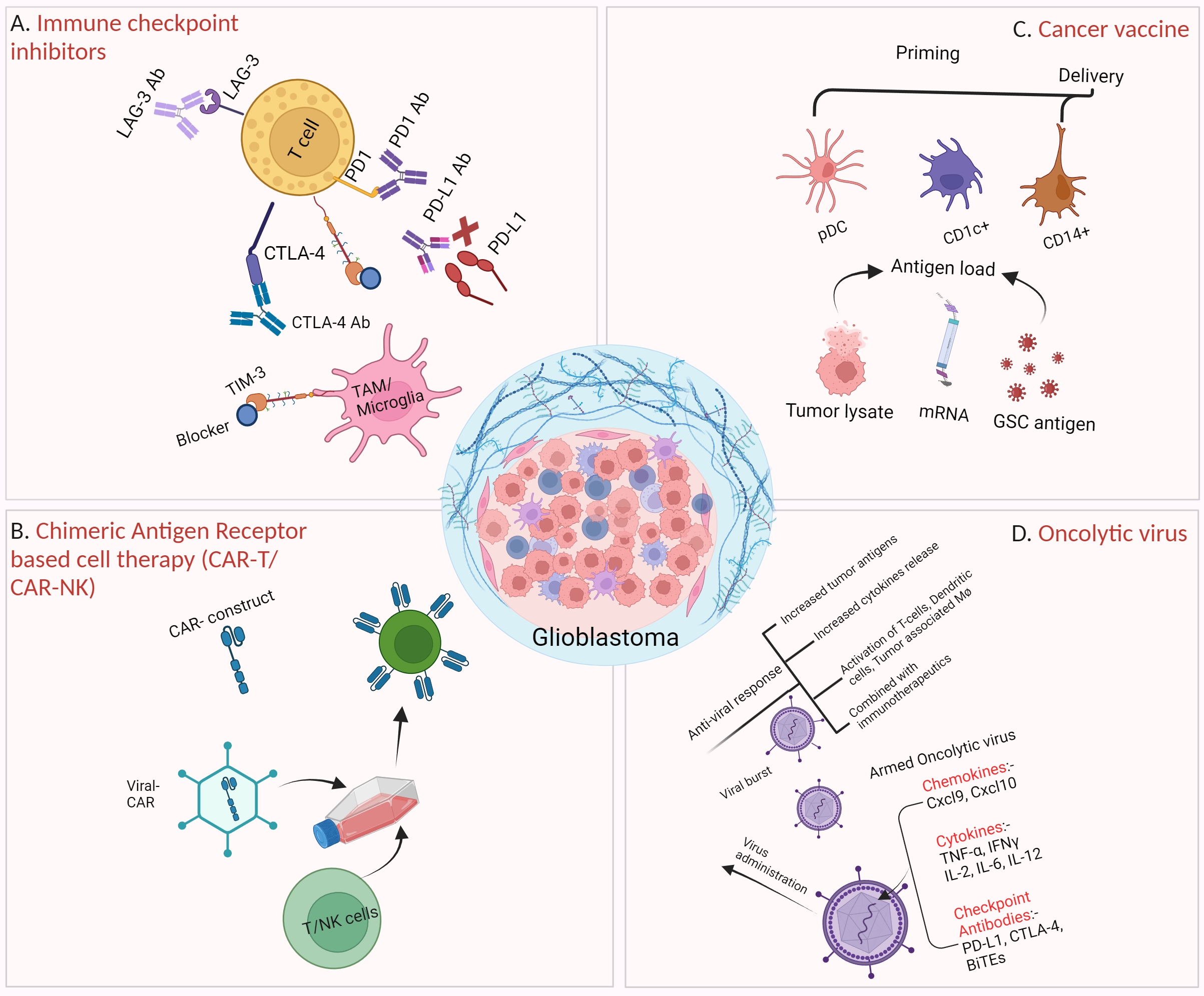
We summarized the most common immunotherapies that have been evaluated in glioblastoma in either preclinical or clinical trials [Figure 1]. The most widely tested immunotherapies in glioblastoma (like in all other cancers) are immune checkpoint inhibitors (ICIs). Immune checkpoint molecules are typically expressed on the surface of immune cells, and they play a crucial role in maintaining immune balance, preventing excessive immune activation, and avoiding auto-immune response. This function of immune regulation is achieved through the interaction of immune checkpoints with their corresponding ligands on other cells, and cancer cells often hijack this communication mechanism to suppress the anti-tumor immunity and evade immune surveillance[53-55]. A common working mechanism of ICIs is to block the inhibitory signal to the immune cells (usually from cancer cells) through an antibody binding to the checkpoint or its ligand to disengage their interaction. Since the discovery of the first immune checkpoint, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), more than a dozen of these checkpoint molecules have been identified to date, such as PD1 and its ligand PD-L1/L2, TIM3, lymphocyte activation gene-3 (LAG3), and TIGIT[54]. Among various ICIs, α-PD-1 has been widely studied as a monotherapy[56] or in a combination of either radiation or radiation plus TMZ in multiple trials (CheckMate 143, 498 and 548)[57-59]. Overall, the clinical outcome has been rather murky in both primary and recurrent glioblastoma due to multiple resistance mechanisms, including high tumor heterogeneity, low mutational burden, systemic immunosuppression, and local immune dysfunction[60].

Figure 1. Various forms of immunotherapy in preclinical and clinical trials for glioblastoma treatment. (A) Various checkpoint inhibitors, including α-PD1, α-PD-L1, α-CTLA-4, and α-TIM-3, have been studied in glioblastoma treatment; (B) CAR-based adoptive cell therapies have attained immense success against hematopoietic cancer, but have shown limited effects on glioblastoma; (C) Cancer vaccine has been tested in glioblastoma treatment by priming antigen-presenting cells (e.g., Dendritic Cells) with tumor antigens/lysate or synthetic antigen peptides, followed by infusion back to the patients; (D) An OV can lyse tumor cells through replication. OV can be armed with immunotherapy in which a virus is genetically modified to carry checkpoint inhibitors (e.g., α-PD-L1 and α-CTLA-4), therapeutic proteins, chemokine (Cxcl9, Cxcl10) or cytokines genes (IFNγ, IL-6, IL-12). Those armed OVs are more potent in killing cancer cells[52]. (Created with BioRender.com). CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; GSC: glioma stem cell; LAG-3: lymphocyte activation gene-3; OV: oncolytic virus; TAM: tumor-associated macrophage.
CAR-T therapy has been studied in glioblastoma[61]. The targets of these CARs in clinical trials span from growth signaling receptors (EGFR/EGFRvIII, Her2), cytokine receptors (IL13Rα2), immune checkpoint (B7-H3) to even matrix metalloproteinase (MMP2), and disialoganglioside (GD2)[62]. Besides very limited responders, including pediatric patients with diffuse intrinsic pontine glioma (DIPG)[63,64], most trials failed to demonstrate a sustained clinical benefit, mainly due to CAR-T-associated severe side effects, including cytokine release syndrome and high grade of neurotoxicity[65,66].
Cancer vaccines have also been explored in glioblastoma trials with minimal success. A peptide vaccine targeting EGFRvIII called rindopepimut has been tested in various trials, with only one trial (phase II) reporting a marginal increase in median overall survival of 12.0 months with rindopepimut plus bevacizumab compared to 8.8 months with bevacizumab plus vaccine placebo[67]. The main limitation of EGFRvIII vaccine is that the expression of EGFRvIII is only limited in some glioblastoma patients, and there is also an intra-tumoral heterogeneous pattern of EGFRvIII expression, which further hinders the overall immune response to the tumor. Another cancer vaccine strategy is to use patient-derived dendritic cells with ex vivo exposure to glioblastoma neoantigens. For instance, ICT-107 and DCVax-L both used patient autologous dendritic cells with pulse to either peptides designed based on patient tumors (ICT-107) or autologous tumor lysates (DCVax-L). Both trials have reached phase 3 and had an acceptable safety profile, though the efficacy was minimal[68,69].
Oncolytic virus (OV) can be viewed as a gene & immuno-hybrid therapy. Typically, an OV exerts its anti-tumor function through a dual mode of action - tumor cell killing (lysis) and induction of systemic anti-tumor immunity[70]. An OV can selectively infect and lyse cancer cells, and various viruses have been employed to develop oncolytic viruses[71]. Upon lysis of tumor cells due to OV replication, many tumor antigens will be released, leading to a local and systemic anti-tumor reaction[72]. One of the main issues associated with OV therapy is the host’s anti-viral immune response to the OV[73]. Currently, a modified herpes simplex virus type 1, named teserpaturev or G47Δ, is the only OV that received conditional approval (in Japan) for glioblastoma treatment[74], and many more oncolytic viruses are currently in clinical trials for glioblastoma treatment (reviewed by Suryawanshi & Schulze[75]). Among them, a retroviral OV called Toca511 reached phase III clinical trial, but was terminated due to its failure to improve survival and meet other endpoints[75].
Immunosuppressive TME
Glioblastoma tumors generally have a low to moderate mutation rate, especially compared to other solid tumors such as melanoma, non-small cell lung cancer, GI cancer, and head and neck cancer[76]. The tumor mutation burden was found to be correlated with immunotherapy treatment response[77]. In addition, glioblastoma also has a highly immune-suppressive microenvironment with a large amount of infiltrating myeloid cells, including bone marrow-derived macrophages (MΦ), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and neutrophils[78]. T lymphocyte dysfunction in the glioblastoma is very severe and was found to be mediated partially by IL-10 produced by the myeloid cells[79]. Additionally, within the TME, prolonged antigen exposure to T cells leads to the expression of LAG3, which in turn causes T cell exhaustion[80]. More strikingly, patients with glioblastoma also have systemic immune suppression. For instance, glioblastoma patients have lower numbers of circulating T cells due to the sequestration of T cells in the bone marrow, possibly due to loss of sphingosine-1-phosphate receptor 1 (S1P1) expression[81]. S1P1 is a GPCR that binds the lipid second messenger, sphingosine-1-phosphate (S1P), and the S1P-S1P1 axis plays a pivotal role in lymphocyte trafficking[82]. Typically, surface S1P1 affords T cell egress from the spleen, lymph node, and thymus. In a mouse glioblastoma model, the T cells from tumor-bearing mice were found to have lost surface expression of S1P1, leading to T cells sequestered mainly in bone marrow[81]. This may partially explain the T cell lymphopenia in glioblastoma patients. However, treatment (radiation and TMZ) associated T cell lymphopenia was also very common[83,84].
Glioblastoma tumors can produce IL-6 and drive myeloid immunosuppression by inducing PD-L1 expression on MDSCs[85]. Glioblastoma can also utilize the natural immune tolerance mechanisms to recruit regulatory T cells (Tregs) through the expression of indoleamine 2,3-dioxygenase (IDO)[86], as well as the tumor-associated macrophages (TAMs) expression of TIM4[87]. Besides soluble factors, extracellular vesicles containing various signaling molecules, including growth factors, non-coding RNAs, cytokines, and other functional proteins, have been found to play an important role in the regulation of glioblastoma TME[88]. Those mechanisms involve an extensive network of DCs, TAMs, MDSCs, and T lymphocytes with complex and dynamic crosstalk [Figure 2].

Figure 2. Main determinants of therapeutic failures in glioblastoma. (A) BBB can prevent the transport of most macromolecule therapeutics (e.g., immune checkpoint inhibitors), cell-based therapies, and most oncolytic viruses; (B) Within the glioblastoma, TME is a severely immunosuppressive local environment that can inhibit the function of most immunotherapies; (C) Clonal heterogeneity represents a complex problem for targeted therapeutics (e.g., receptor tyrosine kinase inhibitors and α-VEGF) to attack glioblastoma tumor cells effectively; (D) Various mechanisms for glioblastoma tumor cells to evade immune attack: tumor cells derived soluble factors (e.g., IL-4, IL-13, prostaglandin E2, and TGF-β) can suppress T cell proliferation; T cell exhaustion induced by prolonged antigen exposure can severely diminish CD8 CTL mediated cancer killing; FOXP3+ CD4 Tregs also block T cell activation. (Created with BioRender.com). BBB: Blood-brain barrier; CTL: cytotoxic T cell; DC: dendritic cell; ICIs: immune checkpoint inhibitors; MDSCs: myeloid-derived suppressor cells; TAMs: tumor-associated macrophages; TME: tumor microenvironment.
Heterogeneity in tumor microenvironment
Tumor heterogeneity has been well-known in glioblastoma biology at multiple levels[89], including genetics/epigenetics (molecular subtypes), molecular signaling (tumor driver mutations), cellular components (clonal and subclonal tumor cells vs. tumor microenvironment), and temporal (primary vs. secondary). scRNAseq analysis of infiltrating neoplastic cells in human glioblastoma revealed vast genomic and transcriptomic heterogeneity[90]. Another work in brain endothelial cells derived from human glioblastoma using a similar approach (scRNAseq) showed five distinct endothelial cell phenotypes representing different states of EC activation and BBB impairment and association with different anatomical locations within and around the tumor[91].
With the advancement of multi-omics platforms, tumor heterogeneity at both inter- and intra-tumoral levels has been much better depicted in glioblastoma[92-94]. The inter-tumoral heterogeneity can be readily appreciated by the molecular subtyping of human glioblastoma tumors by their transcriptional profile and phenotypical response to therapy[2,95,96]. Consistent with the four molecular subtypes of glioblastoma, a more recent scRNAseq analysis showed that glioblastoma cells can differentiate into four principal states, including astrocyte-like, oligodendrocyte progenitor-like, neural progenitor cell-like, and mesenchymal-like state[97]. These four cellular states are influenced by the tumor microenvironment and oncogenic drivers with certain plasticity[97].
The intra-tumoral heterogeneity in glioblastoma is characterized by the presence of clonal and subclonal differentiated tumor cells, glioma stem cells (GSCs), and various components of the tumor microenvironment (stromal, endothelial, and infiltrating immune cells). A recent study by Schaettler et al. using scRNAseq revealed the differences between primary and secondary glioblastoma in their genomic abnormality and neoantigen formation, as well as the spatially differential T cell clones within the glioblastoma[98]. The authors used TCR β-chain CDR3 sequences as unique barcodes of individual T cell clones, as TCR β-chain CDR3 is highly diverse with a significant role in antigen recognition[99]. Their results demonstrated a topological clonal diversity of T cells in glioblastoma[98]. Besides microglia, another representative cell population that further complicates glioblastoma heterogeneity is a large variety of myeloid cells in the TME[100]. They mainly comprise TAMs, MDSCs, DCs, neutrophils, and undifferentiated monocytes[78,101]. Another study using scRNAseq and multiplexing tissue-imaging techniques demonstrated a spatially differential tumor microenvironment characterized by inflammatory signaling and hypoxia in glioblastoma[102]. The authors revealed that CD73, a critical regulator of local purinergic signaling with an essential role in inflammatory response[103], was mainly expressed in glioblastoma cells with a positive correlation between levels of CD73 and HIF1α expression in the hypoxic tumor regions, where the CD73+ glioma cells co-localize with CD39+ microglia to form a spatially compartmentalized microenvironment to regulate the production of adenosine, a potent immunosuppressive metabolite[102].
Immune surveillance escape mechanisms
The crosstalk between glioblastoma and the TME through which glioblastoma tumors escape immune surveillance is very complex and highly dynamic, involving many signaling mechanisms, including both soluble factors and cell-cell interactions. Besides the BBB, which prevents drugs from reaching their target sites, these mechanisms include various immune-suppressive mechanisms, such as secretion of immunosuppressive cytokines (IL-10, TGF-β, and IL-6)[104,105], expression of immune checkpoints[106], and recruitment of regulatory T cells (Tregs)[107], induction of M2-like phenotype of tumor-associated MΦ and microglia[106], reduced tumor antigen presentation through downregulation of MHC expression, and the ability to evade immune through soluble ligands[108,109] [Figure 2].
T cell exhaustion
T cell exhaustion is exceptionally severe in glioblastoma[110], resulting in poor therapeutic efficacy of immunotherapy. Most immunotherapies focus on eliciting an anti-tumor T cell response that requires a collaboration of at least CD4 T Helper cells and CD8 cytotoxic T cells (CTLs). CD4 T Helpers can modulate antigen-specific immune response through their high plasticity and cytokine production, while CD8 CTLs exert cancer cell killing through direct cell-cell interaction and targeted release of effector molecules (perforin and granzymes)[111]. T cell exhaustion is mainly induced by persistent antigen exposure, and it is commonly seen in chronic infections and cancers. It is generally characterized by elevated expression of various immune checkpoints (PD-1, CTLA-4, LAG-3, and TIM-3). Remarkably, T cell exhaustion was also found to correlate with hypoxia in glioma, and both the number of exhausted T cells and the associated exhaustion markers (PD-L1, FOXO1, and PRDM1) correlated with HIF1α levels[112].
The presence of regulatory T cells (Tregs) is another contributing factor for the dysregulation of T cell function in glioma TME [Figure 2]. Tregs are a subset of CD4 T cells that usually prevent autoimmunity response via suppression of inflammation and maintenance of self-tolerance[113]. Tregs (CD4+ Foxp3+) naturally arise from thymic differentiation[114] or are induced in the already differentiated Foxp3- CD4+ T cells in the periphery[115]. A recent study showed that Tregs promote CD8 T cell exhaustion and restrict clonal diversity of tumor-infiltrating CD8 CTLs[116]. Therefore, strategies to eliminate Tregs have been developed to restore anti-tumor immunity in glioblastoma, including activation of glucocorticoid-induced tumor necrosis factor-related protein (GITR). GITR is an immune checkpoint constitutively expressed in Tregs, and its activation through ligand binding leads to the depletion of Tregs and reduced immuno-suppression. A preclinical study by Amoozgar et al. demonstrated that targeting Tregs with anti-GITR antibodies can relieve resistance to immunotherapy (e.g., anti-PD1) in mouse glioblastoma models[117].
Immunosuppression by myeloid cells
A large number of myeloid cells, such as monocytes, macrophages and MDSCs, in the glioblastoma TME impose another great challenge for immunotherapy to function [Figure 2]. Among the tumor-infiltrating myeloid populations in glioblastoma, TAMs play a pivotal role in tumor progression, immunosuppression, and therapy resistance. TAMs are usually found to exhibit a tumor-promoting phenotype by producing immune suppressive cytokines such as IL-6, IL-10, and TGF-β[104,105], and they represent a large population of cells with immunosuppressive function in TME. Various approaches have been proposed to target TAMs for glioblastoma treatment. For instance, by dual targeting IL-6 and CD40, Yang et al. showed that they could reverse TAMs-mediated tumor immunosuppression and sensitize the glioblastoma tumor to immune checkpoint inhibitors (anti-PD1 and anti-CTLA-4) in mouse tumor models[118]. In addition, the relatively undifferentiated monocytic MDSCs have been found to play a significant role in glioblastoma-associated immunosuppression. Domenis et al. demonstrate that CD14+ monocytic MDSCs were the primary mediators of the T cell suppression induced by the GSC-derived exosomes containing various immune suppressive cytokines[119].
Glioblastoma can also evade immune attack by down-regulating tumor antigen expression. Tumor antigen loss during immunotherapy treatment, especially by CAR-T therapy, has been frequently reported[120]. Migrating or invading glioblastoma cells were found to have reduced expression of major histocompatibility complex (MHC) class I and II genes, resulting in significant down-regulation of tumor antigen presentation[121]. Additionally, glioblastoma TME is quite a hypoxic and acidic environment. Both hypoxia and acidosis are essential environmental cues for maintaining GSCs, especially in a HIF1α-dependent manner[122,123]. GSCs are believed to be primarily responsible for tumor resistance to chemotherapy and radiotherapy[124,125]. More importantly, GSCs have also been shown to have a significant role in the evasion of immune function[126].
Resistance to ICIs
ICIs are currently the most prevalent immunotherapy for cancer treatment. Since the approval of the first ICI (α-CTLA-4) by the FDA in 2011, these antibodies have been studied in an increasingly growing number of clinical trials, including those cancers with low response rates, such as breast cancer, cervical cancer, and brain cancer[60,127,128]. Despite the success of ICIs in treating hematopoietic cancers, the clinical trials in glioblastoma have been underwhelming. Besides the BBB, several contributing factors that render ICIs ineffective in glioblastoma treatment have been identified.
Low tumor mutational burden in glioblastoma tumor
Glioblastoma is generally considered an immunologically “cold” tumor type with a relatively lower tumor mutational burden (TMB). Thus, the neoantigen levels are also lower[129,130]. Higher TMB often leads to the formation of a greater number of neoantigens and a greater potential for T-cell repertoire against tumor-specific antigens[131]. TMB has been found to be correlated with the clinical outcome of cancer immunotherapy[76]. Compared with the immunologically “hot” tumor types such as melanoma and NSCLC, glioblastoma shows a much lower neoantigen burden[132].
T cell dysfunction
Glioblastoma patients are often found to have T cell dysfunction in both CNS and peripheral blood, and T cell exhaustion is pervasive and severe in glioblastoma TME. CD8 T cell exhaustion usually starts with the loss of IL-2 production, a cytokine crucial for T cell proliferation, followed by loss or decreased production of TNF-α, IFN-γ, and granzyme B[133]. Tregs also make a significant contribution to the T cell dysfunction in glioma. Both natural and induced Tregs can suppress the cytotoxicity of CD8 CTLs. Tregs were found to be associated with worse prognosis in glioblastoma patients[134], and it seems that the natural Tregs are the dominant subpopulation of Tregs in glioblastoma. Besides dysregulated T cell function, surprisingly, neurons have been shown to play a role in the ICI therapy resistance in glioblastoma. A recent study reported neuronal calmodulin-dependent kinase kinase-2 (CaMKK2) as a driver for the resistance to ICIs in glioblastoma[56], in which CaMKK2 increased CD8 T cell exhaustion, reduced CD4 effector cell expansion, and played a role in the maintenance of immunosuppressive phenotype of tumor-associated microglia[135].
Deficits in antigen presentation by microglia
In glioblastoma TME, antigen presentation machinery is dysregulated in almost all types of antigen-presenting cells. The immunosuppressive microenvironment in glioblastoma leads to the downregulation of MHC expression in microglia[136,137]. The decreased MHC expression significantly impairs the ability of microglia to effectively present antigens, limiting the activation of other immune cells and undermining the immune response against the tumor. Similarly, TAMs were found to be deficient in antigen presentation, lacking costimulatory molecules CD86, CD80, and CD40 critical for T-cell activation[138]. In fact, although glioblastoma tumor-infiltrating dendritic cells seemed more efficient than both MΦ and microglia in priming T-cells with exogenous antigens[139], data from a preclinical study demonstrated that a better anti-tumor immunity is associated with both tumor-infiltrating dendritic cells and microglia[140].
TAMs
A new study using patient-derived recurrent glioblastoma tumors with neoadjuvant PD-1 antibody treatment showed that α-PD-1 activated T cells and dendritic cells, but was unable to reverse the immunosuppressive phenotype in TAMs[141]. Work by Chen et al. analyzed scRNAseq data from a combined of >19,000 individual macrophages from 66 human glioma cases (50 glioblastomas and 16 low-grade gliomas) and discovered a pro-tumor subset of bone marrow-derived macrophages with the expression of a scavenger receptor MARCO[142]. More interestingly, this subpopulation of MARCO+ TAMs was found almost exclusively in the IDH-WT glioblastoma, and they exhibited a completely oppositive dynamic in α-PD-1 responders vs. non- responders[142]. Park et al. studied the immune landscape of mouse glioblastoma with α-PD-1 treatment, and found that chemokine CCL5 induced by α-PD-1 treatment seemed to recruit the anti-inflammatory TAMs into the glioblastoma TME[143]. A CyToF-based high-plexing immune profiling approach revealed that ICI-sensitivity in both human and mouse tumors was associated with a higher number of T cells and dendritic cells (DCs) and a lower number of PD-L1 positive TAMs[144].
Anti-inflammatory glucocorticoids
Glucocorticoids have been used to control certain adverse effects associated with cancer immunotherapy. Interestingly, concurrent administration of dexamethasone, a potent corticosteroid frequently used in glioblastoma patients to decrease tumor-associated edema, has been shown to be detrimental to immunotherapy for patients with glioblastoma[145]. Though the clinical data in this study was limited to a subset of patients with wild-type IDH-1 glioblastoma under α-PD-L1 treatment, the concurrent dexamethasone diminished the response to α-PD-1 therapy in two different mouse glioma models[145]. It is worth mentioning that glioblastoma patients under standard (radiation plus TMZ) treatment who received dexamethasone treatment also showed a worse outcome[146]. However, this is likely because MGMT promoter contains two nonconsensus glucocorticoid-responsive elements and glucocorticoids can upregulate MGMT expression[147]. A comprehensive study of MGMT promoter activity in glioblastoma cell lines further clarified that dexamethasone, but not TMZ or irradiation, can induce the upregulation of MGMT expression via a SP-1 dependent fashion[148], while not through altering the epigenetic status (i.e., methylation) of the MGMT promoter.
Role of non-coding RNAs
Long non-coding RNAs (LncRNAs) have been increasingly recognized for their essential role in cell growth, survival, proliferation, pluripotency, and immune functions correlating to the malignant transformation of normal cells into cancerous cells[149-151]. MALAT1, NEAT1, and H19 are among the common LncRNAs that influence the response of glioblastoma/glioma to chemotherapeutics[152]. Another lncRNA, LINC00021, was significantly upregulated in glioblastoma, especially in the TMZ resistance cells or tissues, enhancing resistance to TMZ through Notch pathway and epigenetically silencing p21 expression[48]. A study also showed that LncRNA SNHG15 promotes pro-glioblastoma cytokines TGF-β and lL-6 in TMZ-resistance cells via M2-polarization of microglial cells[153].
Micro RNA (miRNA) also plays a role in the regulation of glioblastoma TME. One example is the
OPPORTUNITIES
Approaches to alter immuno-suppression in glioblastoma TME
Many great efforts have been made to overcome the difficulty of immunotherapy applications in neuro-oncology. For example, a clinical trial found that neoadjuvant PD-1 blockade resulted in significantly improved overall survival and progression-free survival in patients with recurrent glioblastoma[158]. In this study, patients received anti-PD1 treatment ~2 weeks before surgery, and the PD1 antibody was able to elicit both systemic and local anti-tumor immunity. Other attempts are primarily focused on modulating the immune suppression in the glioblastoma tumor microenvironment by targeting various components of the TME, such as TAMs and MDSCs (summarized in a recent review by Wang et al.[159]). In the meantime, new targets have been identified for future immunotherapy development. For instance, TAMs associated CD73 was found to be a promising target with potentially synergistic effects along with dual inhibition of PD1 and CTLA-4[160]. CD47/SIRPα axis is another exciting target to consider. SIRPα governs the phagocytosis activity of MΦ. When CD47 on the cancer cell surface engages with SIRPα on MΦ, it sends a “Don’t-eat-me” signal to prevent phagocytosis of cancer cells by MΦ. Treatment with anti-CD47 plus TMZ was shown to activate both innate and adaptive anti-tumor immunity in a preclinical study[161].
A single-cell RNA-seq study of patient glioma infiltrating T cells revealed CD161 (KLRB1) as a promising immunotherapy target. Depleting CD161 led to T cell activation and anti-tumor immunity both in vitro and in vivo[162]. An independent study using data from a large cohort of glioma patients confirmed that CD161 might play an important role in promoting glioma progression via inhibition of T cell function[163].
Besides checkpoint inhibition, a deeper understanding of the resistance mechanism to CAR-T therapy in solid tumors was achieved through a genome-wide CRISPR knockout screen in glioblastoma[164]. A recent study using a genome-wide CRISPR knockout screen in glioblastoma revealed a functional requirement of IFN-γ receptor in glioblastoma for sufficient adhesion of CAR-T cells to mediate productive cytotoxicity[164]. This study suggests that strategies to enhance the binding of CAR-T cells to the solid tumor will likely result in a better treatment response. Another strategy to enhance the infiltration of CAR-T cells into glioblastoma tumors by combining CAR-T with a CXCL11-armed oncolytic virus also demonstrated an improved anti-tumor immunity in a syngeneic mouse glioma model[165].
Combinatorial approaches and new forms of immunotherapies
Combination therapy has been extensively explored to improve glioblastoma treatment. For instance, resistance to α-VEGF monotherapy was common in glioblastoma. A new study reported that combined blockade of VEGF, Angiopoietin-2, and PD1 could reprogram glioblastoma endothelial cells into quasi-antigen-presenting cells and induced a durable anti-tumor T cell response[166]. A recent review has nicely summarized the current status of combinatorial approaches, including both chemo- and immunotherapies, for glioblastoma treatment[167]. Additionally, many new forms of immunotherapy are emerging with great hope to shift the paradigm of glioblastoma treatment. A recent study reported a nanoporter (NP)-hydrogel complex for local induction of CAR-macrophages (CAR-MΦ) targeting CD133+ glioblastoma stem cells in tumor resection cavity with promising results[168]. This nanomicelle complex consists of a self-assembled peptide-based hydrogel loaded with the CD133-targeting CAR construct and then was coated with a citraconic anhydride–modified dextran with the ability to bind to CD206, a typical surface marker of M2 macrophages. Different from the ex vivo engineering of CAR-MΦ developed by Klichinsky et al.[169], the nanoporter-hydrogel-based in situ induction of strategy CAR-MΦ largely simplified the process of
The CAR-NK cells have also been explored to treat glioblastoma either by Her2 targeting monotherapy[170] or in combination therapy. For instance, the Off-the-Shelf EGFR-targeting CAR-NK cells have been tested in combination with an oncolytic virus expressing the IL15/IL15Ralpha complex and the combinatorial therapy demonstrates a strong anti-tumor immunity[171]. A significant problem associated with CAR-NK cell therapy is the shedding or down-regulation of the ligands in cancer cells that bind natural killer group 2D (NKG2D) receptors on the natural killer (NK) cells. NKG2D is an activating receptor widely expressed in NK cells as well as in some subsets of T cells[172]. To overcome the limitation of NKG2DL heterogeneity in the tumor, a recent study using a bispecific antibody with two ScFv fragments (linked with a IgG4-Fc) that target Her2 (tumor) and NKG2D (NK cells), respectively, in combination with human NK-92 cells, showed synergistic tumor cell killing effects in both in vitro and in vivo conditions[173]. Although the syngeneic tumor model they used represents a situation of a heterogenous expression of NKG2DLs in tumor cells, the flank tumors they used did not address the difficulty in delivery of the combination therapy across the BBB[173].
Another interesting phenomenon is the sex difference in response to immunotherapy in glioma. The sex disparity in brain cancer has been reported by several groups[174-177]. A recent meta-analysis revealed that female patients with glioblastoma treated with immunotherapy had a statistically significant survival advantage in overall survival over their male counterparts[178]. They also found that female patients exhibited a more robust survival advantage with cancer vaccine treatment. Another study by Bayik et al. discovered that two subsets of myeloid-derived suppressor cells (MDSCs) have a sex-specific tumor-promoting phenotype in both mouse and human glioblastoma[179]. All these data suggest that a more personalized approach, which at least considers sex differences in glioblastoma treatment, will more accurately evaluate the efficacy of immunotherapy.
New drug delivery technologies to overcome BBB limitation and activate glioblastoma TME
Various new technologies have demonstrated promising progress in overcoming BBB, and we summarized a few new approaches with great potential to improve the glioblastoma treatment outcome [Figure 3]. Among those new approaches, the use of ultrasound to open BBB for glioblastoma treatment has been applied in several areas, including immunotherapy delivery. Using low-intensity pulsed ultrasound to temporarily disrupt BBB, Sabbagh et al. demonstrated a significantly improved BBB penetration of both anti-PD1 antibody and EGFRvIII targeting CAR-T cells, as well as significantly improved survival in mouse glioblastoma models[180]. Another study by Sheybani et al. applied MRI-guided focused ultrasound with systemic injection of microbubbles and studied the impact of this approach on temporary BBB disruption in a mouse glioma model[181]. This approach caused a transient local inflammatory phenotype in the mouse glioblastoma, with an increased number of dendritic cells and the upregulated maturation marker. However, they did not see a significant increase in CD8 T cells in the TME[181].

Figure 3. Potential new approaches to improve glioblastoma treatment. (A) The focused ultrasound in combination with micro-bubbles and photodynamic therapy (PDT) can temporarily open BBB to allow therapeutics crossing. PDT can also activate local immunity in TME; (B) New approaches to modulate glioblastoma TME by targeting hypoxia, activating suppressed local immunity, or enhancing cancer neoantigen formation in tumor cells; (C) Novel nanodrug delivery technologies in combination with CRISPR/Cas9-based gene editing and immune checkpoint inhibitors; (D) Various forms of adoptive cell therapies; (E) Better strategies for tumor stratification, prognostic prediction and personalized medicine would enhance the clinical outcome of glioblastoma treatment. (Created with BioRender.com). BBB: Blood-brain barrier; DC: dendritic cell; TAMs: tumor-associated macrophages; TME: tumor microenvironment.
Another technology to modulate BBB function is photodynamic therapy (PDT). Conventionally, PDT relies on a photosensitizer, such as 5-aminolevulinic acid (5-ALA)[182], that can accumulate in tumor tissue, plus a laser that can stimulate the photosensitizer, followed by energy transfer to generate reactive oxygen species, leading to damages to the cancer cells[183]. It is noteworthy that PDT has shown promise in temporary opening of BBB, possibly through modulating certain components of TJs[184]. Interestingly, PDT can also induce an acute inflammatory response in which both innate and adaptive immune systems are activated[185]. Recently, BBB opening was shown to affect the meningeal lymphatic system characterized by an anti-tumor effect of talaporfin sodium (TS)-PDT as well as its synergy with the immune checkpoint inhibitor[186]. In vitro studies have demonstrated that targeted TS-PDT triggers various forms of cell death, including apoptosis, necrosis, and autophagy-associated cell death. Furthermore, TS-PDT induces the acute activation of lymphatic drainage in the brain and the clearance of unwanted molecules from the CNS[187,188]. The approval of 5-ALA by the FDA for fluorescence-guided glioblastoma resection has sparked a renewed interest in its potential application for PDT[182].
Nanotechnology has also made significant advancements in the field of glioblastoma treatment. Various forms of nanomedicines have exploited the features of the glioblastoma tumor microenvironment for efficient BBB crossing and release of payloads[189-191]. Fan et al. engineered an MMP-2-activated nanoparticle to carry anti-CD276 & CD3 bispecific antibodies and demonstrated that this strategy enhanced IFN-γ-induced tumor cell ferroptosis[192]. A polylactic-co-glycolic acid (PLGA) nanoparticle encapsulated disulfiram was used to block hypoxia-induced NF-κB signaling and glioma stem cells[193]. Zou et al. devised a polymer-based CRISPR-Cas9 nano-capsule for systemic gene therapy delivery to glioblastoma[194]. This nano-capsule has both the BBB crossing and tumor targeting functions mediated through an angiopep-2 peptide[195]. By targeting polo-like kinase (PLK-1) via a sgRNA, the strategy demonstrated a significant survival advantage over the control mice[194].
CONCLUSION
Despite advances in surgical technologies and therapeutics development, there has been limited improvement in the long-term survival rate of glioblastoma patients, with a 5-year survival still around 5%-10%. Many lessons have been learned in glioblastoma drug resistance mechanisms, especially with cutting-edge scRNAseq, spatial biology, and other-omics platforms. Efforts are needed to overcome BBB and tumor heterogeneity, targeting glioma stem cells and their niches, enhancing T cell trafficking and preventing their exhaustion, and modulating the immunosuppressive TME in glioblastoma. A complex disease, such as glioblastoma, would require a complex solution. Multidisciplinary approaches involving nanodrug carriers, focused ultrasound, plus temporary BBB permeability enhancement technologies (micro-bubbles, phototherapy) in combination with gene and immuno-therapy will likely lead to an improved outcome [Figure 3]. In addition, a much less traveled path is to enhance glioblastoma neoantigen formation. Glioblastoma tumors have a relatively lower TMB, which was shown to correlate with immunotherapy outcomes in solid tumors[76,196]. Lower TMB results in lower neoantigen generation, which enables a stealth mode of glioblastoma cells. Therefore, increasing the formation of neoantigens may significantly promote tumor recognition and clearance by the immune system[197]. Besides T cells, strategies to activate other infiltrating immune cells (TAMs, microglia, and MDSCs) that reside in the glioma TME in large abundance may effectively reverse the local immunosuppression. Finally, a more precise tumor stratification approach and improved prognostic biomarkers will help determine the most effective combinatorial therapies for glioblastoma treatment.
DECLARATIONS
Authors’ contributionsConceptualization, investigation, writing: Sharma S, Chepurna O
Conceptualization, supervision, writing: Sun T
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Tran B, Rosenthal MA. Survival comparison between glioblastoma multiforme and other incurable cancers. J Clin Neurosci 2010;17:417-21.
2. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98-110.
3. Verdugo E, Puerto I, Medina MÁ. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun 2022;42:1083-111.
4. Wen PY, Weller M, Lee EQ, et al. Glioblastoma in adults: a society for neuro-oncology (SNO) and European society of neuro-oncology (EANO) consensus review on current management and future directions. Neuro Oncol 2020;22:1073-113.
5. Drakulic D, Schwirtlich M, Petrovic I, et al. Current opportunities for targeting dysregulated neurodevelopmental signaling pathways in glioblastoma. Cells 2022;11:2530.
6. Perez A, Huse JT. The evolving classification of diffuse gliomas: World Health Organization updates for 2021. Curr Neurol Neurosci Rep 2021;21:67.
7. Grochans S, Cybulska AM, Simińska D, et al. Epidemiology of glioblastoma multiforme-literature review. Cancers 2022;14:2412.
8. Fisher JP, Adamson DC. Current FDA-approved therapies for high-grade malignant gliomas. Biomedicines 2021;9:324.
9. Xue J, Wu Y, Liu N. Ultrasound enhanced anti-tumor effect of temozolomide in glioblastoma cells and glioblastoma mouse model. Cell Mol Bioeng 2019;12:99-106.
10. Barker CA, Chang M, Chou JF, et al. Radiotherapy and concomitant temozolomide may improve survival of elderly patients with glioblastoma. J Neurooncol 2012;109:391-7.
11. Noch EK, Ramakrishna R, Magge R. Challenges in the treatment of glioblastoma: multisystem mechanisms of therapeutic resistance. World Neurosurg 2018;116:505-17.
12. Nicholas MK. Glioblastoma multiforme: evidence-based approach to therapy. Expert Rev Anticancer Ther 2007;7:S23-7.
13. Whelan R, Hargaden GC, Knox AJS. Modulating the blood-brain barrier: a comprehensive review. Pharmaceutics 2021;13:1980.
14. Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020;17:69.
15. Hladky SB, Barrand MA. Elimination of substances from the brain parenchyma: efflux via perivascular pathways and via the blood-brain barrier. Fluids Barriers CNS 2018;15:30.
16. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell 2015;163:1064-78.
17. Simon MJ, Iliff JJ. Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim Biophys Acta 2016;1862:442-51.
18. Wolburg H, Wolburg-Buchholz K, Liebner S, Engelhardt B. Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci Lett 2001;307:77-80.
19. Bhalerao A, Sivandzade F, Archie SR, Chowdhury EA, Noorani B, Cucullo L. In vitro modeling of the neurovascular unit: advances in the field. Fluids Barriers CNS 2020;17:22.
21. de Gooijer MC, Kemper EM, Buil LCM, et al. ATP-binding cassette transporters restrict drug delivery and efficacy against brain tumors even when blood-brain barrier integrity is lost. Cell Rep Med 2021;2:100184.
22. Qosa H, Miller DS, Pasinelli P, Trotti D. Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res 2015;1628:298-316.
23. Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005;2:86-98.
24. Liu H, Yu N, Lu S, et al. Solute carrier family of the organic anion-transporting polypeptides 1A2- Madin-Darby Canine Kidney II: a promising in vitro system to understand the role of organic anion-transporting polypeptide 1A2 in blood-brain barrier drug penetration. Drug Metab Dispos 2015;43:1008-18.
25. He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum Genomics 2009;3:195-206.
26. Hagenbuch B, Gui C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica 2008;38:778-801.
27. Thakkar N, Lockhart AC, Lee W. Role of organic anion-transporting polypeptides (OATPs) in cancer therapy. AAPS J 2015;17:535-45.
28. Bronger H, König J, Kopplow K, et al. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res 2005;65:11419-28.
29. Cooper E, Woolf Z, Swanson MEV, et al. Single-cell image analysis reveals over-expression of organic anion transporting polypeptides (OATPs) in human glioblastoma tissue. Neurooncol Adv 2022;4:vdac166.
30. Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol 2012;5:102-14.
31. Wu S, Li X, Gao F, de Groot JF, Koul D, Yung WKA. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol 2021;23:920-31.
32. Lin K, Gueble SE, Sundaram RK, Huseman ED, Bindra RS, Herzon SB. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022;377:502-11.
33. Tirrò E, Massimino M, Romano C, et al. Prognostic and therapeutic roles of the insulin growth factor system in glioblastoma. Front Oncol 2020;10:612385.
35. Jimenez-Pascual A, Siebzehnrubl FA. Fibroblast growth factor receptor functions in glioblastoma. Cells 2019;8:715.
36. Li X, Wu C, Chen N, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016;7:33440-50.
37. Snuderl M, Fazlollahi L, Le LP, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011;20:810-7.
38. Yin D, Chen W, O’Kelly J, et al. Connective tissue growth factor associated with oncogenic activities and drug resistance in glioblastoma multiforme. Int J Cancer 2010;127:2257-67.
39. Nie E, Jin X, Miao F, et al. TGF-β1 modulates temozolomide resistance in glioblastoma via altered microRNA processing and elevated MGMT. Neuro Oncol 2021;23:435-46.
40. Huang T, Song X, Xu D, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020;10:8721-43.
41. Mattei V, Santilli F, Martellucci S, et al. The importance of tumor stem cells in glioblastoma resistance to therapy. Int J Mol Sci 2021;22:3863.
42. Fidoamore A, Cristiano L, Antonosante A, et al. Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int 2016;2016:6809105.
43. Qin Y, Zhang X, Chen Y, Zhang W, Du S, Ren C. Prognostic analysis of a hypoxia-associated lncRNA signature in glioblastoma and its pan-cancer landscape. J Neurol Surg A Cent Eur Neurosurg 2023.
44. Ren P, Wang JY, Zeng ZR, et al. A novel hypoxia-driven gene signature that can predict the prognosis and drug resistance of gliomas. Front Genet 2022;13:976356.
45. Wu Y, Fletcher M, Gu Z, et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat Commun 2020;11:6434.
46. Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22:425-37.
47. Eyler CE, Matsunaga H, Hovestadt V, Vantine SJ, van Galen P, Bernstein BE. Single-cell lineage analysis reveals genetic and epigenetic interplay in glioblastoma drug resistance. Genome Biol 2020;21:174.
48. Zhang S, Guo S, Liang C, Lian M. Long intergenic noncoding RNA 00021 promotes glioblastoma temozolomide resistance by epigenetically silencing p21 through Notch pathway. IUBMB Life 2020;72:1747-56.
49. Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol 2014;31:985.
50. Ramaiah MJ, Tangutur AD, Manyam RR. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci 2021;277:119504.
51. Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett 2008;269:7-17.
52. Zeng J, Li X, Sander M, Zhang H, Yan G, Lin Y. Oncolytic viro-immunotherapy: an emerging option in the treatment of gliomas. Front Immunol 2021;12:721830.
53. Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol 2022;86:137-50.
55. Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015;35 Suppl:S185-98.
56. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol 2020;6:1003-10.
57. Omuro A, Reardon DA, Sampson JH, et al. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: results from exploratory phase I cohorts of checkmate 143. Neurooncol Adv 2022;4:vdac025.
58. Weller M, Lim M, Idbaih A, et al. CTIM-25. A randomized phase 3 study of nivolumab or placebo combined with radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma with methylated mgmt promoter: checkmate 548. Neuro-Oncology 2021;23:vi55-6.
59. Sampson JH, Omuro AMP, Preusser M, et al. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498. J Clin Oncol 2016;34:TPS2079.
60. Medikonda R, Dunn G, Rahman M, Fecci P, Lim M. A review of glioblastoma immunotherapy. J Neurooncol 2021;151:41-53.
61. Lin YJ, Mashouf LA, Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol 2022;13:817296.
62. Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S. CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci 2021;15:662064.
63. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9.
64. Majzner RG, Ramakrishna S, Yeom KW, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022;603:934-41.
65. Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol 2020;11:1973.
66. Schubert ML, Schmitt M, Wang L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol 2021;32:34-48.
67. Reardon DA, Schuster J, Tran DD, et al. ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol 2015;33:2009.
68. Wen PY, Reardon DA, Phuphanich S, et al. A randomized, double-blind, placebo-controlled phase 2 trial of dendritic cell (DC) vaccination with ICT-107 in newly diagnosed glioblastoma (GBM) patients. J Clin Oncol 2014;32:2005.
69. Liau LM, Ashkan K, Tran DD, et al. Correction to: first results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med 2018;16:179.
70. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2016;15:660.
71. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol 2020;13:84.
72. Engeland CE, Bell JC. Introduction to oncolytic virotherapy. In: Engeland CE, editor. Oncolytic Viruses. New York: Springer; 2020. p. 1-6.
73. Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol 2017;7:106.
74. Sugawara K, Iwai M, Ito H, Tanaka M, Seto Y, Todo T. Oncolytic herpes virus G47Δ works synergistically with CTLA-4 inhibition via dynamic intratumoral immune modulation. Mol Ther Oncolytics 2021;22:129-42.
75. Suryawanshi YR, Schulze AJ. Oncolytic viruses for malignant glioma: on the verge of success? Viruses 2021;13:1294.
76. Sha D, Jin Z, Budczies J, Kluck K, Stenzinger A, Sinicrope FA. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov 2020;10:1808-25.
77. Kao C, Powers E, Datto MB, et al. Tumor mutational burden (TMB) as a predictive biomarker of immune checkpoint blockade (ICB) in metastatic solid tumors. J Clin Oncol 2020;38:80.
79. Ravi VM, Neidert N, Will P, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun 2022;13:925.
80. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev 2017;276:80-96.
81. Chongsathidkiet P, Jackson C, Koyama S, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 2018;24:1459-68.
82. Garris CS, Blaho VA, Hla T, Han MH. Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology 2014;142:347-53.
83. Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol 2012;2012:831090.
84. Mapelli R, Julita C, Bianchi SP, et al. Association between treatment-related lymphopenia and survival in glioblastoma patients following postoperative chemoradiotherapy. Strahlenther Onkol 2022;198:448-57.
85. Lamano JB, Lamano JB, Li YD, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res 2019;25:3643-57.
86. Wainwright DA, Balyasnikova IV, Chang AL, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res 2012;18:6110-21.
87. Xu L, Xiao H, Xu M, Zhou C, Yi L, Liang H. Glioma-derived T cell immunoglobulin- and mucin domain-containing molecule-4 (TIM4) contributes to tumor tolerance. J Biol Chem 2011;286:36694-9.
88. Yekula A, Yekula A, Muralidharan K, Kang K, Carter BS, Balaj L. Extracellular vesicles in glioblastoma tumor microenvironment. Front Immunol 2019;10:3137.
89. Becker AP, Sells BE, Haque SJ, Chakravarti A. Tumor heterogeneity in glioblastomas: from light microscopy to molecular pathology. Cancers 2021;13:761.
90. Darmanis S, Sloan SA, Croote D, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep 2017;21:1399-410.
91. Xie Y, He L, Lugano R, et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021;6:e150861.
92. Gularyan SK, Gulin AA, Anufrieva KS, et al. Investigation of inter- and intratumoral heterogeneity of glioblastoma using TOF-SIMS. Mol Cell Proteomics 2020;19:960-70.
93. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344:1396-401.
94. Martínez A, Madurga R, García-Romero N, Ayuso-Sacido Á. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett 2022;527:66-79.
95. Zhang P, Xia Q, Liu L, Li S, Dong L. Current opinion on molecular characterization for GBM classification in guiding clinical diagnosis, prognosis, and therapy. Front Mol Biosci 2020;7:562798.
96. Chen X, Fan X, Zhao C, et al. Molecular subtyping of glioblastoma based on immune-related genes for prognosis. Sci Rep 2020;10:15495.
97. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019;178:835-49.e21.
98. Schaettler MO, Richters MM, Wang AZ, et al. Characterization of the genomic and immunologic diversity of malignant brain tumors through multisector analysis. Cancer Discov 2022;12:154-71.
99. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17:61.
100. Fu W, Wang W, Li H, et al. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front Immunol 2020;11:835.
101. Pinton L, Masetto E, Vettore M, et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J Immunother Cancer 2019;7:58.
102. Coy S, Wang S, Stopka SA, et al. Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat Commun 2022;13:4814.
103. Verkhratsky A, Burnstock G. Biology of purinergic signalling: its ancient evolutionary roots, its omnipresence and its multiple functional significance. Bioessays 2014;36:697-705.
104. Rodrigues JC, Gonzalez GC, Zhang L, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol 2010;12:351-65.
105. Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin Dev Immunol 2011;2011:732413.
106. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013;19:3165-75.
107. Jacobs JF, Idema AJ, Bol KF, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol 2010;225:195-9.
108. Chitadze G, Kabelitz D. Immune surveillance in glioblastoma: role of the NKG2D system and novel cell-based therapeutic approaches. Scand J Immunol 2022;96:e13201.
109. Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol 2012;14:584-95.
110. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24:4175-86.
111. Mohme M, Neidert MC. Tumor-specific T cell activation in malignant brain tumors. Front Immunol 2020;11:205.
112. Liu S, Liu X, Zhang C, Shan W, Qiu X. T-cell exhaustion status under high and low levels of hypoxia-inducible factor 1α expression in glioma. Front Pharmacol 2021;12:711772.
113. Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev 2001;182:18-32.
114. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 2006;25:249-59.
115. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol 2012;30:733-58.
116. Noyes D, Bag A, Oseni S, et al. Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J Immunother Cancer 2022;10:e004605.
117. Amoozgar Z, Kloepper J, Ren J, et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun 2021;12:2582.
118. Yang F, He Z, Duan H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun 2021;12:3424.
119. Domenis R, Cesselli D, Toffoletto B, et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS One 2017;12:e0169932.
120. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984.
121. Zagzag D, Salnikow K, Chiriboga L, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 2005;85:328-41.
122. Pistollato F, Chen HL, Rood BR, et al. Hypoxia and HIF1α repress the differentiative effects of BMPs in high-grade glioma. Stem Cells 2009;27:7-17.
123. Filatova A, Seidel S, Böğürcü N, Gräf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res 2016;76:5845-56.
124. Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008;26:3015-24.
125. Goffart N, Lombard A, Lallemand F, et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro Oncol 2017;19:66-77.
126. Sesé B, Íñiguez-Muñoz S, Ensenyat-Mendez M, et al. Glioblastoma embryonic-like stem cells exhibit immune-evasive phenotype. Cancer 2022;14:2070.
127. Gaynor N, Crown J, Collins DM. Immune checkpoint inhibitors: key trials and an emerging role in breast cancer. Semin Cancer Biol 2022;79:44-57.
128. De Felice F, Marchetti C, Palaia I, et al. Immune check-point in cervical cancer. Crit Rev Oncol Hematol 2018;129:40-3.
129. Zhao X, Pan X, Wang Y, Zhang Y. Targeting neoantigens for cancer immunotherapy. Biomark Res 2021;9:61.
131. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275-87.
132. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-21.
133. Watowich MB, Gilbert MR, Larion M. T cell exhaustion in malignant gliomas. Trends Cancer 2023;9:270-92.
134. Yue Q, Zhang X, Ye HX, et al. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J Neurooncol 2014;116:251-9.
135. Tomaszewski WH, Waibl-Polania J, Chakraborty M, et al. Neuronal CaMKK2 promotes immunosuppression and checkpoint blockade resistance in glioblastoma. Nat Commun 2022;13:6483.
136. Mieczkowski J, Kocyk M, Nauman P, et al. Down-regulation of IKKβ expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget 2015;6:33077-90.
137. Schartner JM, Hagar AR, Van Handel M, Zhang L, Nadkarni N, Badie B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005;51:279-85.
138. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol 2006;8:261-79.
139. Beauvillain C, Donnou S, Jarry U, et al. Neonatal and adult microglia cross-present exogenous antigens. Glia 2008;56:69-77.
140. Jarry U, Jeannin P, Pineau L, Donnou S, Delneste Y, Couez D. Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur J Immunol 2013;43:1173-84.
141. Lee AH, Sun L, Mochizuki AY, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun 2021;12:6938.
142. Chen AX, Gartrell RD, Zhao J, et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med 2021;13:88.
143. Park JH, Kang I, Lee HK. The immune landscape of high-grade brain tumor after treatment with immune checkpoint blockade. Front Immunol 2022;13:1044544.
144. Simonds EF, Lu ED, Badillo O, et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer 2021;9:e002181.
145. Iorgulescu JB, Gokhale PC, Speranza MC, et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin Cancer Res 2021;27:276-87.
146. Shields LB, Shelton BJ, Shearer AJ, et al. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat Oncol 2015;10:222.
147. Ueda S, Mineta T, Nakahara Y, Okamoto H, Shiraishi T, Tabuchi K. Induction of the DNA repair gene O6-methylguanine-DNA methyltransferase by dexamethasone in glioblastomas. J Neurosurg 2004;101:659-63.
148. Aasland D, Reich TR, Tomicic MT, Switzeny OJ, Kaina B, Christmann M. Repair gene O6 -methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J Neurochem 2018;144:139-51.
152. Ghafouri-Fard S, Agabalazadeh A, Abak A, et al. Role of long non-coding RNAs in conferring resistance in tumors of the nervous system. Front Oncol 2021;11:670917.
153. Li Z, Zhang J, Zheng H, et al. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J Exp Clin Cancer Res 2019;38:380.
154. Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 2008;14:1271-7.
155. Ye X, Wei W, Zhang Z, et al. Identification of microRNAs associated with glioma diagnosis and prognosis. Oncotarget 2017;8:26394-403.
156. Yang J, Liu R, Deng Y, et al. MiR-15a/16 deficiency enhances anti-tumor immunity of glioma-infiltrating CD8+ T cells through targeting mTOR. Int J Cancer 2017;141:2082-92.
157. Hübner M, Moellhoff N, Effinger D, et al. MicroRNA-93 acts as an “anti-inflammatory tumor suppressor” in glioblastoma. Neurooncol Adv 2020;2:vdaa047.
158. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019;25:477-86.
159. Wang EJ, Chen JS, Jain S, et al. Immunotherapy resistance in glioblastoma. Front Genet 2021;12:750675.
160. Goswami S, Walle T, Cornish AE, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med 2020;26:39-46.
161. von Roemeling CA, Wang Y, Qie Y, et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat Commun 2020;11:1508.
162. Mathewson ND, Ashenberg O, Tirosh I, et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021;184:1281-98.e26.
163. Di W, Fan W, Wu F, et al. Clinical characterization and immunosuppressive regulation of CD161 (KLRB1) in glioma through 916 samples. Cancer Sci 2022;113:756-69.
164. Larson RC, Kann MC, Bailey SR, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature 2022;604:563-70.
165. Wang G, Zhang Z, Zhong K, et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol Ther 2023;31:134-53.
166. Amoozgar Z, Ren J, Wang N, et al. Combined blockade of VEGF, Angiopoietin-2, and PD1 reprograms glioblastoma endothelial cells into quasi-antigen-presenting cells. bioRxiv 2022.
167. Bausart M, Préat V, Malfanti A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exp Clin Cancer Res 2022;41:35.
168. Chen C, Jing W, Chen Y, et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Transl Med 2022;14:eabn1128.
169. Klichinsky M, Ruella M, Shestova O, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol 2020;38:947-53.
170. Zhang C, Burger MC, Jennewein L, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst 2016;108:djv375.
171. Ma R, Lu T, Li Z, et al. An oncolytic virus expressing IL15/IL15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res 2021;81:3635-48.
172. Siemaszko J, Marzec-Przyszlak A, Bogunia-Kubik K. NKG2D natural killer cell receptor-a short description and potential clinical applications. Cells 2021;10:1420.
173. Zhang C, Röder J, Scherer A, et al. Bispecific antibody-mediated redirection of NKG2D-CAR natural killer cells facilitates dual targeting and enhances antitumor activity. J Immunother Cancer 2021;9:e002980.
174. Sun T, Warrington NM, Luo J, et al. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J Clin Invest 2014;124:4123-33.
175. Sun T, Plutynski A, Ward S, Rubin JB. An integrative view on sex differences in brain tumors. Cell Mol Life Sci 2015;72:3323-42.
176. Ostrom QT, Kinnersley B, Wrensch MR, et al. Sex-specific glioma genome-wide association study identifies new risk locus at 3p21.31 in females, and finds sex-differences in risk at 8q24.21. Sci Rep 2018;8:7352.
177. Zhang H, Liao J, Zhang X, et al. Sex difference of mutation clonality in diffuse glioma evolution. Neuro Oncol 2019;21:201-13.
178. Shireman JM, Ammanuel S, Eickhoff JC, Dey M. Sexual dimorphism of the immune system predicts clinical outcomes in glioblastoma immunotherapy: a systematic review and meta-analysis. Neurooncol Adv 2022;4:vdac082.
179. Bayik D, Zhou Y, Park C, et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov 2020;10:1210-25.
180. Sabbagh A, Beccaria K, Ling X, et al. Opening of the blood-brain barrier using low-intensity pulsed ultrasound enhances responses to immunotherapy in preclinical glioma models. Clin Cancer Res 2021;27:4325-37.
181. Sheybani ND, Witter AR, Garrison WJ, Miller GW, Price RJ, Bullock TNJ. Profiling of the immune landscape in murine glioblastoma following blood brain/tumor barrier disruption with MR image-guided focused ultrasound. J Neurooncol 2022;156:109-22.
182. Hadjipanayis CG, Stummer W. 5-ALA and FDA approval for glioma surgery. J Neurooncol 2019;141:479-86.
183. Henderson BW, Dougherty TJ. How does photodynamic therapy work? Photochem Photobiol 1992;55:145-57.
184. Zhang C, Feng W, Vodovozova E, et al. Photodynamic opening of the blood-brain barrier to high weight molecules and liposomes through an optical clearing skull window. Biomed Opt Express 2018;9:4850-62.
185. Mroz P, Vatansever F, Muchowicz A, Hamblin MR. Photodynamic therapy of murine mastocytoma induces specific immune responses against the cancer/testis antigen P1A. Cancer Res 2013;73:6462-70.
186. Sasaki M, Tanaka M, Kojima Y, et al. Anti-tumor immunity enhancement by photodynamic therapy with talaporfin sodium and anti-programmed death 1 antibody. Mol Ther Oncolytics 2023;28:118-31.
187. Semyachkina-glushkovskaya O, Khorovodov A, Fedosov I, et al. A novel method to stimulate lymphatic clearance of beta-amyloid from mouse brain using noninvasive music-induced opening of the blood-brain barrier with EEG markers. Appl Sci 2021;11:10287.
188. Semyachkina-Glushkovskaya O, Chehonin V, Borisova E, et al. Photodynamic opening of the blood-brain barrier and pathways of brain clearing. J Biophotonics 2018;11:e201700287.
189. Tian T, Liang R, Erel-Akbaba G, et al. Immune checkpoint inhibition in GBM primed with radiation by engineered extracellular vesicles. ACS Nano 2022;16:1940-53.
190. Sun T, Patil R, Galstyan A, et al. Blockade of a laminin-411-notch axis with CRISPR/Cas9 or a nanobioconjugate inhibits glioblastoma growth through tumor-microenvironment cross-talk. Cancer Res 2019;79:1239-51.
191. Quader S, Liu X, Toh K, et al. Supramolecularly enabled pH- triggered drug action at tumor microenvironment potentiates nanomedicine efficacy against glioblastoma. Biomaterials 2021;267:120463.
192. Fan R, Chen C, Mu M, et al. Engineering MMP-2 activated nanoparticles carrying B7-H3 bispecific antibodies for ferroptosis-enhanced glioblastoma immunotherapy. ACS Nano 2023;17:9126-39.
193. Kannappan V, Liu Y, Wang Z, et al. PLGA-nano-encapsulated disulfiram inhibits hypoxia-induced NF-κB, cancer stem cells, and targets glioblastoma in vitro and in vivo. Mol Cancer Ther 2022;21:1273-84.
194. Zou Y, Sun X, Yang Q, et al. Blood-brain barrier-penetrating single CRISPR-Cas9 nanocapsules for effective and safe glioblastoma gene therapy. Sci Adv 2022;8:eabm8011.
195. Jiang Y, Zhang J, Meng F, Zhong Z. Apolipoprotein E peptide-directed chimeric polymersomes mediate an ultrahigh-efficiency targeted protein therapy for glioblastoma. ACS Nano 2018;12:11070-9.
196. Okada M, Shimizu K, Fujii SI. Identification of neoantigens in cancer cells as targets for immunotherapy. Int J Mol Sci 2022;23:2594.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sharma S, Chepurna O, Sun T. Drug resistance in glioblastoma: from chemo- to immunotherapy. Cancer Drug Resist 2023;6:688-708. http://dx.doi.org/10.20517/cdr.2023.82
AMA Style
Sharma S, Chepurna O, Sun T. Drug resistance in glioblastoma: from chemo- to immunotherapy. Cancer Drug Resistance. 2023; 6(4): 688-708. http://dx.doi.org/10.20517/cdr.2023.82
Chicago/Turabian Style
Sharma, Sachin, Oksana Chepurna, Tao Sun. 2023. "Drug resistance in glioblastoma: from chemo- to immunotherapy" Cancer Drug Resistance. 6, no.4: 688-708. http://dx.doi.org/10.20517/cdr.2023.82
ACS Style
Sharma, S.; Chepurna O.; Sun T. Drug resistance in glioblastoma: from chemo- to immunotherapy. Cancer Drug Resist. 2023, 6, 688-708. http://dx.doi.org/10.20517/cdr.2023.82
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 6
likes
Like This Article 6
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.