
How acute myeloid leukemia (AML) escapes from FMS-related tyrosine kinase 3 (FLT3) inhibitors? Still an overrated complication?

 , ...
, ... Abstract
FMS-related tyrosine kinase 3 (FLT3) mutations, present in about 25%-30% of acute myeloid leukemia (AML) patients, constitute one of the most frequently detected mutations in these patients. The binding of FLT3L to FLT3 activates the phosphatidylinositol 3-kinase (PI3K) and RAS pathways, producing increased cell proliferation and the inhibition of apoptosis. Two types of FLT3 mutations exist: FLT3-ITD and FLT3-TKD (point mutations in D835 and I836 or deletion of codon I836). A class of drugs, tyrosine-kinase inhibitors (TKI), targeting mutated FLT3, is already available with 1st and 2nd generation molecules, but only midostaurin and gilteritinib are currently approved. However, the emergence of resistance or the selection of clones not responding to FLT3 inhibitors has become an important clinical dilemma, as the duration of clinical responses is generally limited to a few months. This review analyzes the insights into mechanisms of resistance to TKI and poses a particular view on the clinical relevance of this phenomenon. Has resistance been overlooked? Indeed, FLT3 inhibitors have significantly contributed to reducing the negative impact of FLT3 mutations on the prognosis of AML patients who are no longer considered at high risk by the European LeukemiaNet (ELN) 2022. Finally, several ongoing efforts to overcome resistance to FLT3-inhibitors will be presented: new generation FLT3 inhibitors in monotherapy or combined with standard chemotherapy, hypomethylating drugs, or IDH1/2 inhibitors, Bcl2 inhibitors; novel anti-human FLT3 monoclonal antibodies (e.g., FLT3/CD3 bispecific antibodies); FLT3-CAR T-cells; CDK4/6 kinase inhibitor (e.g., palbociclib).
Keywords
INTRODUCTION
In 1996, the first unexpectedly longer transcripts of FMS-like tyrosine kinase 3 (FLT3) were reported in the transmembrane domain through the juxtamembrane domain of the gene[1]. This represented a cornerstone discovery in acute myeloid leukemia (AML), since internal tandem duplications (ITD)-FLT3 mutations represent one of the most recurrent alterations, occurring in 22-32% and the remaining 8% including the tyrosine kinase domain (TKD), de novo AML[2,3]. Moreover, FLT3-ITD mutations, particularly those carrying a high FLT3-ITD mutant to wild-type allelic ratio when detected at initial diagnosis, seem to confer a short duration of remission and globally a worse prognosis[4]. The presence of FLT3-ITD appears to be strongly associated with hyperleukocytosis and high blast percentages in both peripheral blood and bone marrow at presentation[5]. From a morphologic standpoint, it has been suggested that AML with cup-like nuclei is associated with co-occurring mutations of both NPM1 and ITD- or TKD- FLT3[6]. Most importantly, we have an interesting class of drugs targeting FLT3, tyrosine-kinase inhibitors (TKI), which have an already long history in the treatment of this cancer. With the availability of 1st and 2nd generation inhibitors, the emergence of resistance or the selection of clones not responding to FLT3 inhibitors has become an important clinical dilemma, probably recalling the challenge of TKI in CML and ALL BCR: ABL positive. This review will try to summarize the evidence, from a clinical viewpoint, regarding the development of resistance to FLT3-inhibitors, their real clinical impact in AML patients, and strategies to obviate the development of resistance.
FLT3 GENE AND ITS MOLECULAR FUNCTION
FLT3 (Fms-like tyrosine kinase 3) has strong similarities in its sequence with other members of the class III receptor tyrosine kinase (RTKIII) receptor family (FMS, platelet-derived growth factor receptor (PDGFR) α and β, and KIT)[7]. FLT3 is composed of an extracellular domain consisting of 5 immunoglobulin-like (Ig-like) domains, and by a cytoplasmic domain with a juxtamembrane domain and two intracellular tyrosine-kinase domains (TKDs)[8]. Physiologically, FLT3 is expressed over the cytoplasmic membrane of immature myeloid and lymphoid progenitor cells[9] and is activated by its ligand FLT3L, which can be found in the cytoplasm of bone marrow stromal fibroblasts, T-cells, B-cells, and progenitors CD34+ [Figure 1][10]. Noteworthy, FLT3 (CD135) is expressed on the surface of more than half of AML and seems associated with a worse outcome[11]. The binding of FLT3L to FLT3 activates the phosphatidylinositol 3-kinase (PI3K) and RAS pathways, producing increased proliferation and impaired apoptosis in cells. Moreover, activated PI3K stimulates downstream proteins like 3-phosphoinositide-dependent protein kinase 1 (PDK1), protein kinase B (PKB/AKT) and the mammalian target of rapamycin (mTOR), and PI3K activation hampers apoptosis through phosphorylation of the pro-apoptotic protein BAD, member of the BCL2-family. Moreover, the activation of RAS stimulates other downstream effectors like RAF, MAPK/ERK kinases (MEKs), extracellular-signal-regulated kinase (ERK), and the 90-kDa ribosomal protein S6 kinase (RSK). These downstream effectors can activate signal transducers and activators of transcription (STATs), which lead to the transcription of genes involved in cellular proliferation[7]. On the other hand, activating genetic mutations of FLT3 can produce the abnormal expression of a constitutively activated tyrosine kinase receptor in AML blasts, which is independent of the physiological FLT3L stimulus[12].
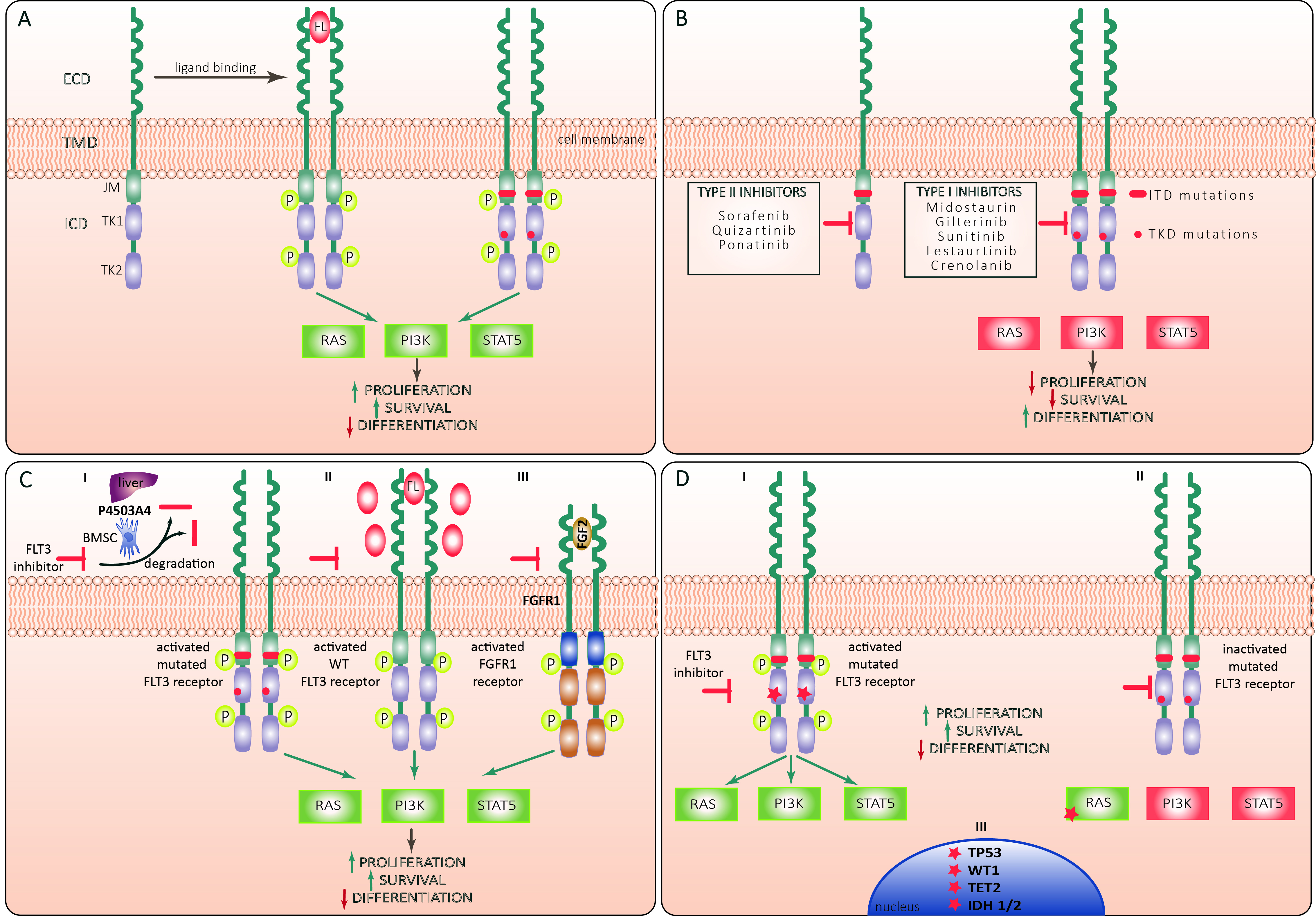
Figure 1. (A) Structure and activation of FLT3 receptor. FLT3 molecule is a tyrosine kinase receptor composed of an extracellular domain (ECD) that comprises 5 immunoglobuline-like repeats, a short transmembrane domain (TMD) and an intracellular domain (ICD) that includes a juxtramembrane (JM) domain followed by two tyrosine kinase (TK) domains (TK1 and TK2). Binding of the FLT3 ligand (FL) to ECD leads to formation of a ternary complex between FL and two receptor subunits with consequent conformational changes and trans-phosphorylation of TK and JM domains. The activated receptor exerts a tyrosine kinase activity against a series of adaptor and effector molecules leading to activation of Ras/Raf/MAPK (RAS) PI3K/Akt (PI3K) and Jak/STAT5 (STAT5) pathways promoting leukemic cell proliferation and survival and suppressing differentiation. Both FLT3-ITD and FLT3-TKD mutations lead to ligand-independent activation of FLT3 signaling; (B) types of FLT3 inhibitors. Type I FLT3 inhibitors interact with the ATP-binding site of the intracellular TK domain (TKD) when the receptor is in active conformation, whereas Type II inhibitors bind to the inactive form of the receptor, thus preventing its activation. Type I inhibitors inhibit FLT3 signaling in AML cells harboring ITD or TKD mutations, whereas Type II inhibitors are active against AML with ITD but not TKD mutations; (C) mechanisms of primary resistance to FLT3 inhibitors: Increased proliferation and survival and decreased differentiation of AML cells may be sustained through (I) Cytochrome P450 3A4-mediated degradation of FLT3-inhibitors by hepatocytes (liver symbol) and BM stromal cells (BMSC); (II) Restoration of FL/FLT3 signaling through compensatory overexpression of wild-type (WT) FLT3 receptor and FL; (III) Activation of common FLT3 and fibroblast growth factor receptor type 1 (FGFR1) downstream target pathways through the increased fibroblast growth factor 2 (FGF2)/FGFR1 signaling; (D) mechanisms of secondary resistance to FLT3 inhibitors: (I) Secondary resistance of FLT3 receptor to pharmacological inhibition may be conferred through newly acquired activating mutations of FLT3 TKD (red asterisk), and upregulation of pro-survival mechanisms through acquired activating mutations in RAS/MAPK pathway genes (II) and epigenetic modifiers like DNMT3A, TET2, IDH2 or transcriptional regulators like WT1 and TP53 (III). Green and red rectangles correspond to activated and inactivated FLT3 downstream signaling pathways, respectively. Yellow “P” circles indicate phosphorylated receptor domains. Green and red arrows indicate positive and negative regulation, respectively.
Two kinds of FLT3 mutations have been individuated: FLT3-ITD and FLT3-TKD (including point mutations in D835 and I836 or deletion of codon I836)[13]. FLT3-ITD takes place between exons 14 and 15. When AML cells carry FLT3-ITD, PCR products generate a wild-type band and a larger ITD band after electrophoresis. D835 and I836 codons are encoded by the nucleotide GATATC, which forms the Eco-RV restriction site. The amplified products of wild-type FLT3 are digested into two bands by the Eco-RV enzyme, while amplified products with D835-mutations (FLT3-TKD) result in uncut bands, and this permits a traditional diagnosis[14]. Nowadays, several methods have been developed or adapted to help identify mutations of FLT3 and aberrant karyotypes (e.g., Multiplex-targeted next-generation sequencing)[15], and moreover, next-generation sequencing (NGS) techniques for Measurable residual disease (MRD) detection are under development for clinical practice[16]. Especially in the pre-FLT3 inhibitors era, patients harboring FLT3-ITD had a worse prognosis in terms of OS and relapse-free survival[17]. Moreover, some studies suggested that a high Mutant-to-wild-type allelic ratio of FLT3-ITD was associated with a worse prognosis[4,18]. Other studies, however, found no increased risk of relapse in patients with FLT3-ITDhigh[19]. Other important determinants of prognosis in patients with FLT3-ITD are the karyotype status[20] and NPM1mut which confers a more favorable outcome[21,22]. Indeed, patients harboring NPM1mut and FLT3-ITDhigh are classified as intermediate risk in ELN2017[23]. Different is the case for AML with FLT3-TKD; for these patients, a clear impact on prognosis is not established due to conflicting results[24-30]. However, in the new ELN2022 classification, probably due to the therapeutic benefit of FLT3 inhibitors in the new AML scenario, all patients with FLT3 mutation are classified as intermediate risk regardless of whether the mutation is detected alone or with other co-mutations. However, after the introduction of FLT3-inhibitors, this prognostic disadvantage of FLT3-mutations seems abrogated, improving levels of MRD[31,32]. MRD techniques to monitor FLT3 mutations are still being explored. In the next future, the improvement and the standardization of next-generation sequencing analysis could strongly increase and refine the opportunities for monitoring FLT3 mutations during treatment.
CLASS OF DRUGS: FLT3-INHIBITORS
The class of drugs of FLT3-inhibitors can be categorized according to chronological order in 1st generation (sorafenib and midostaurin) and 2nd generation (gilteritinib, quizartinib, crenolanib). Another classification is according to the capacity of the TKI to link the active and inactive status of the mutated FLT3 (Type 1: midostaurin, gilteritinib) or the inactivated only status of FLT3 (Type 2: quizartinib, sorafenib)[33]. An important clinical implication is that Type 2 inhibitors at therapeutic concentrations are unable to inhibit FLT3-TKD, and, especially for the D835 mutation, this favors the active conformation of FLT3 and alters the binding of TKIs[34].
Midostaurin is a first-generation TKI of class 1 and was the first drug to be approved for clinical use in FLT3-mutated AML in 2017[35]. Midostaurin has moderate activity as a single agent, as shown in 92 patients who achieved a reduction of bone marrow blasts in 71% of cases[36]. This evidence paved the way for a phase III trial of the association of 7 + 3 and midostaurin. From 2008 to 2011, the RATIFY trial enrolled a total of 717 patients harboring a mutated FLT3 (22.6% had the TKD mutation). The 4-year overall survival (OS) rate was 51.4% in the midostaurin group and 44.3% in the placebo group and the Median event-free survival (EFS) was 8.2 months in the midostaurin group vs. 3.0 months[31]. In a post-hoc analysis in patients with FLT3-TKD, the 5-year EFS rate was significantly extended in patients treated with midostaurin than in the placebo arm (45.2% vs. 30.1%; P = 0.044)[37].
Gilteritinib, which belongs to the 2nd generation of TKI, has more profound single-agent activity than 1st generation drugs and has activity against both FLT3-ITD and TKD mutations[38]. Indeed, in phase I/II study with gilteritinib, 40% of patients achieved a response, with 19 (8%) reaching complete remission[39]. This fueled interest in a randomized phase 3 trial comparing gilteritinib against any chemotherapy (selected by investigators) in relapsed/refractory (R/R) AML with mutated FLT3. In the ADMIRAL trial, the median OS was significantly longer in patients treated with gilteritinib than among those receiving chemotherapy
Sorafenib, a multitargeted TKI, is approved for hepatocellular carcinoma and renal cell carcinoma, but its off-label use in AML, especially after HSCT, is performed in many centers. Sorafenib is active only in
Quizartinib, a second-generation TKI, was specifically selected from a library of drugs to deeply inhibit FLT3-ITD, while it is inactive against FLT3-TKD[50]. It has clinical activity as a single agent in the relapsed setting, where 56% of FLT3-ITD-positive patients achieved a composite CR[51]. However, the FDA expressed concerns about the lack of improvement in EFS, so quizartinib was initially rejected. Recently, the FDA granted quizartinib Priority Review for newly diagnosed (ND) FLT3-ITD+ AML based on data from the QuANTUM-First study. QuANTUM-First was a pivotal trial randomizing quizartinib vs. placebo added to standard 7 + 3 backbone in FLT3-ITD+ AML patients. The OS was significantly prolonged in the quizartinib arm compared to the placebo arm. The median OS was of about 32 months with quizartinib vs. 15 months with placebo, while CR rates were 71.6% and CRi rates 64.9%[52,53].
Crenolanib, compared to other drugs of the same class, demonstrates some attractive characteristics to target FLT3 mutations in AML. Being a potent type I pan-FLT3 inhibitor, crenolanib is also active in FLT3 TKD mutations[54]. As a single agent, in R/R AML Crenolanib showed 39% of CRi and 11% of PR among the 18 patients treated[55], and in another study in 34 evaluable patients, 12% achieved CRi[56]. However, these responses to crenolanib were transient [Table 1].
Clinical trials including targeted FLT3 Inhibitors in adult patients with AML
| Type of treatment | Therapeutic combinations including FLT3 inhibitors | Trial Phase | ClinicalTrials.gov identifier | AML1 setting |
| Chemotherapy + FLT3 inhibitors | “7 + 3” + gilteritinib vs. “7 + 3” | 3 | NCT02236013 | de novo AML1 |
| “7 + 3” + quizartinib vs. “7 + 3” | 3 | NCT02668653 | de novo AML1 with mutated FLT3-ITD2 | |
| “7 + 3” + crenolanib | 2 | NCT02283177 | de novo AML with mutated FLT3 | |
| CPX-351 + quizartinib | 2 | NCT04209725 | R/R3 AML1 with mutated FLT3-ITD2 | |
| “7 + 3” + crenolanib vs. “7 + 3” + midostaurin | 3 | NCT03258931 | de novo AML1 with mutated FLT3 | |
| “7 + 3” + crenolanib vs. “7 + 3” | 3 | NCT03250338 | R/R3 AML1 with mutated FLT3 | |
| HD-Ara-c5 + Mito6 + quizartinib | 2 | NCT03989713 | R/R3 AML1 with mutated FLT3-ITD2 | |
| CdA7 + ida8 + Ara-c9 + quizartinib | 2 | NCT04047641 | de novo and R/R3 AML1 | |
| HMAs + FLT3 inhibitors | Aza10 or LD-Ara-c11 + quizartinib | 1/2 | NCT01892371 | Phase I cohort: R/R3 AML1 Phase II cohort: de novo and R/R3 AML1 with mutated FLT3 |
| Aza10 + gilteritinib vs. aza10 | 3 | NCT02752035 | de novo AML1 with mutated FLT3 | |
| Maintenance with FLT3 inhibitors | Crenolanib | 2 | NCT02400255 | AML1 with mutated FLT3 post-transplant |
| Gilteritinib vs. placebo | 3 | NCT02997202 | AML1 with mutated FLT3 post-transplant | |
| Gilteritinib | 2 | NCT02927262 | AML1 with mutated FLT3 post CR14 achievement | |
| New molecules + FLT3 inhibitors | Dec12 + ven13 + quizartinib | 1/2 | NCT03661307 | de novo and R/R3 AML1 with mutated FLT3 |
| Ven13 + gilteritinib | 1 | NCT03625505 | R/R3 AML | |
| Ven13 + quizartinib | 1/2 | NCT03735875 | R/R3 AML with mutated FLT3 | |
| Aza10 + ven13 + gilteritinib | 1/2 | NCT04140487 | Phase I cohort: R/R3 AML with mutated FLT3 Phase IIa cohort: de novo AML1 | |
| Atezolizumab + gilteritinib | 1/2 | NCT03730012 | R/R3 AML1 with mutated FLT3 | |
| MTOR inhibitor (RAD001) + FLT3 inhibitor (PKC412) | 1 | NCT00819546 | R/R3 AML1 | |
| Milademetan (MDM2inh.) + quizartinib | 1 | NCT03552029 | de novo and R/R3 AML with mutated FLT3 | |
| “7+3” + GO14 + midostaurin | 1 | NCT03900949 | de novo AML1 with mutated FLT3 |
MOLECULAR MECHANISMS OF RESISTANCE
The heterogeneity of FLT3‐ITD mutations such as length, sequence, and duplication site is unique for every AML patient[57,58], and this molecular diversity increases the complex biology of FLT3-ITD. As previously reported, FLT3-ITD length mutations are found in the JM domain of the gene in 70% of mutated AML cases, while in the remaining 30%, the duplication region can involve the TKD1 with a different prognostic impact[30,59]. However, the molecular mechanisms responsible for the differential signaling based on
Proposed mechanisms of resistance to FLT3 Inhibitors in AML
| Mechanism | Description | References |
| Primary resistance | ||
| Cytochrome P450 3A4-mediated degradation of FLT3-inhibitors | Plasma levels of FLT3 inhibitors can be decreased through their enhanced degradation by hepatocytes and BMSC in Cytochrome P450 3A4-mediated manner | [64] |
| Restoration of FL/FLT3 signaling through compensatory overexpression of wild-type (WT) FLT3 receptor and FL | FLT3-mutated AML increases the expression of FL binding to WT FLT3 receptor and restores the downstream FLT3-MAPK signaling in WT and FLT3-ITD co-mutated cells | PMID: 27331411 PMID: 21263155 |
| Activation of common FLT3 and fibroblast growth factor receptor type 1 (FGFR1) downstream target pathways through the increased fibroblast growth factor 2 (FGF2)/FGFR1 signaling | FGF2 ligand highly expressed in BMSC binds to FGFR1 receptor in FLT3-ITD mutated cells and activates the downstream MAPK signaling shared by both receptor tyrosine kinases | [63] |
| Secondary resistance | ||
| Newly acquired activating mutations of FLT3 TKD | Acquired mutations (D835, N676, F691 and Y862, A627 and others) within FLT3 TK domain confer resistance to different TKI and sustain activation of the downstream effectors | [66,71] PMID: 22858906 PMID: 33780043 |
| Acquired activating mutations in RAS/MAPK pathway genes | Activating mutations in RAS/MAPK pathway genes upregulate pro-survival and pro-proliferative mechanisms in leukemic cells | [66,67,69,74] |
| Acquired mutations in epigenetic regulators and transcriptional regulators | Acquired mutations in epigenetic modifiers like DNMT3A, TET2, IDH2 or transcriptional regulators like WT1 and TP53 upregulate pro-survival mechanisms and favor leukemic cells survival | [60,67,76] |
| GM-CSF and IL-3 maintain cell survival without rescuing proliferation | Cytokine-mediated resistance through GM-CSF and IL-3 is dependent on JAK kinase, STAT5, and pro-viral integration site of Moloney murine leukemia virus (PIM) but not MAPK or mammalian target of rapamycin signaling | PMID: 30944098 |
| Novel ATM/mTOR pathway regulating oxidative phosphorylation | Marrow-mediated activation of ATM, in turn, upregulates oxidative phosphorylation via mTOR signaling. mTOR is required for the bone marrow stroma-dependent maintenance of protein translation | PMID: 36259537 |
Acquired mutation on FLT3 gene
Several studies have demonstrated that TKIs drugs may have an impact on the clonal evolution of FLT3-ITD mutated AML by downregulating certain clones[68,69]. These findings emphasize the importance of repeated mutation analysis for FLT3-ITD to discriminate between patients in whom TKI may induce long-lasting remission, and from those in whom relapse may originate from subclones, which may carry a FLT3-wild type at diagnosis. As previously discussed, secondary TKD mutations, mainly reported at residues D835/F691 in FLT3-ITD mutated patients treated with TKIs, confer therapy resistance and poor outcomes[70]. In particular, at AML onset, type II of FLT3-TKIs have an effect on FLT3-TKD mutations, but secondary alterations in the TK domain during the disease progression may confer treatment resistance interfering with the inhibitory activity on FLT3-ITD mutated clones. Acquired alterations at D835, F691 and Y842 residues have been reported in patients who developed resistance to sorafenib or quizartinib, both type II-TKIs[71]. Although the acquisition of mutations in the TK domain of FLT3 is rarely reported in patients treated with gilteritinib and crenolanib (a type I-TKIs), the gaining of F691L alteration, defined as gatekeeper mutation of FLT3 gene, was frequently observed. This mutation confers substantial resistance to both type I and type II-TKIs[65]. The latter mutations occur in residues that directly interact with the TKIs and become prevalent in AML clones almost exclusively after treatment with the inhibitors[72]. Although in FLT3-mutated AML, the addition of midostaurin to standard “7 + 3” treatment regimen has been widely used in clinical practice, only 60% of patients achieved a CR, and almost half of these cases developed a relapse. Schmalbrock and colleagues[16] provided novel biological features into the clonal evolution and mechanisms of resistance of FLT3-ITD-mutated AML exposed to midostaurin. They demonstrated that during the disease progression, almost 46% became FLT3-ITD negative and acquired mutations in MAPK pathways, conferring an additional proliferative advantage. By contrast, in AML cases that relapse with FLT3-ITD persistence, they showed a clonal selection of driver mutations in 11% of cases. FLT3-ITD clones persist in the remaining patients during relapse disease, indicating a failure of midostaurin inhibition activity.
Intracellular pathways alterations
The phosphorylation of FLT3 receptor activates several downstream intracellular signaling pathways, such as RAS/MAPK, PI3K/Akt/mTOR, and JAK/STAT5, which are mainly implicated in the survival, proliferation, and differentiation of hematopoietic cells[73]. During the TKI treatment, clonal selection of cells characterized by activating gene mutations involved in RAS/MAPK pathway is often detected during progression in AML patients who received frontline midostaurin combination therapy[69] or gilteritinib as monotherapy for relapsed/refractory disease in 2nd line[67,74,75]. These results suggest that RAS mutations may drive the clonal evolution in relapsed/refractory AML that occurs independently from TKI type administration. Other mechanisms of off-target resistance during FLT3-TKI therapy have been recently described. Alotaibi and colleagues[67] analyzed the mutational status of patients who relapsed after different FLT3 inhibitors and demonstrated that the most common gene alterations involved alterations in IDHs, NRAS, WT1, and TP53 genes. In particular, they identified in responding patients a higher occurrence of IDH2 alterations at diagnosis as compared to non-responder cases. This data suggests an advantage of combining targeted therapies in AML patients who harbored concomitant FLT3 and IDH2 mutations[76]. Instead, mutations of NRAS and IDHs genes often occur in leukemia subclones; TET2 alterations are described in FLT3 mutated clones and mainly enriched in crenolanib poor-responders AML. In particular, Zhang et al. reported that the mutation type of TET2 gene correlates with a different prognosis in patients treated with crenolanib, suggesting that TET2 truncation mutations may contribute to TKI resistance as compared to missense mutations that did not show a correlation with an unfavorable response to crenolanib[60].
CLINICAL RELEVANCE OF TKI RESISTANCE
It has been demonstrated that FLT3 inhibitor therapy promotes drug-resistant clonal populations that harbor secondary, on-target FLT3-mutations and that are prone to resistance to numerous TKIs in patients with relapsed AML who possess FLT3-ITD mutations[77]. Prolonged therapy with FLT3-TKIs can exert clonal pressure for the selection of drug-resistant sub-clones with additional mutations that facilitate leukemic proliferation regardless of the FLT3 kinase’s activation state, as well as for sub-clones that are completely lacking FLT3 mutations; this is particularly relevant because relapsed AML is demonstrably a polyclonal disease[69].
Sorafenib and midostaurin, FLT3 TKIs of 1st-generation, have in proportion a low selectivity for FLT3 mutations. Nevertheless, the rates of response are augmented with midostaurin and relapse rates show a significant reduction when either drug is administered in association with frontline chemotherapy, like 3 + 7 induction therapy in patients with newly diagnosed (ND-AML), FLT3-mut AML[31]. FLT3-ITDs are patient-specific, and due to the unique positioning and variable extension of the duplicated genomic sequence, there is a wide range of variability. This translates into unique peptide motifs derived from duplicate aminoacidic sequences within the FLT3 gene. Therefore, many studies have tried to answer the question if diversity of FLT3-ITD exerts any effect on the clinical outcome of AML patients[30,78-81]. In a study conducted on 151 elderly AML patients who had received standard chemotherapy, Stirewalt et al. showed that the length of the ITD (40 vs. > 40 base pairs) had an effect on the patients’ long-term survival[78]. According to research by Kayser and colleagues, patients with AML who have FLT3-ITD placed into the first tyrosine kinase domain (TKD1) of the FLT3-gene present a poorer outcome than those who have FLT3-ITD in the juxtamembrane domain (JMD)[30]. However, a retrospective analysis in 260 patients with AML FLT3-ITD positive, which divided FLT3-ITD into 3 groups based on its position, was unable to confirm this finding, and just showed a statistical trend towards a link between a more C-terminal location of FLT3-ITD and worse survival, but without impacting on EFS[79]. However, Fisher et al. reported data favorable to the assumption that FLT3-ITDs located closer to the C-terminus of the FLT3 gene correlate with an adverse prognosis[80]. They showed that the localization of the ITD affected the percentage of remission following AML first-line chemotherapy, independently of the allelic burden[80]. Rucker et al. assessed the prognostic and predictive role of FLT3-ITD insertion site (IS) considering 452 subjects treated in the RATIFY trial, identifying 265 IS in the tyrosine kinase domain-1 (TKD1) and 43 IS in the juxtamembrane domain (JMD) by NGS[81]. Four-year OS probabilities significantly differed between JMDsole, JMD/TKD1, and TKD1sole, respectively; specifically, multivariate Cox models for OS and cumulative occurrence of relapse after HSCT identified TKD1sole as a negative prognostic factor[81].
Gilteritinib and quizartinib, members of the second-generation FLT3 TKIs, show higher selectivity for FLT3 mutations. Indeed, when they are used as single agents in patients with FLT3-mutated R/R AML, both molecules exhibit clinical activity and have shown survival advantages over salvage approaches[17,61]. In comparison with quizartinib, gilteritinib showed activity for FLT3-ITD and FLT3-TKD mutations[82]. Nevertheless, secondary resistance to gilteritinib can arise via off-target mechanisms, such as the appearance of NRAS or similar mutations that activate MAPK signaling downstream of FLT3, as well as on-target FLT3 mutations at a gatekeeper FLT3 residue (F691L)[40,74]. As already discussed, gilteritinib has been approved as single-agent therapy for patients with FLT3-mutated R/R AML after positive results of ADMIRAL trial[61,79]. Patients randomized to receive 120 mg of gilteritinib showed a significantly longer median OS than those who received salvage chemotherapies and higher rates of CR[17]. Furthermore, Perl et al.[39] confronted post-hoc the clinical outcomes in patients with R/R FLT3-mutated AML enrolled in CHRYSALIS and ADMIRAL trials[17,39], with a view on those who were previously exposed to midostaurin or sorafenib against naive patients. Similar to those pre-treated, high rates of overall response emerged among patients treated with a FLT3 TKI before gilteritinib (CHRYSALIS, 42%; ADMIRAL, 52%) and patients without previous FLT3 TKI therapy (CHRYSALIS, 43%; ADMIRAL, 55%). Furthermore, in ADMIRAL study, a higher rate of response and a trend toward longer median OS was observed in the arm with gilteritinib vs. salvage chemotherapies in patients who had previously received a FLT3 TKI[83]. Therefore, these results encouraged the use of gilteritinib in FLT3-mutated R/R AML also after prior exposure to sorafenib or midostaurin.
POSSIBLE STRATEGIES TO OVERCOME RESISTANCE
New therapeutic combinations are now being tested in order to potentially reduce the development of resistances to FLT3 inhibitors or the possible occurrence of new point mutations within the FLT3 gene during the therapeutic approaches; this would allow an increased rate of response to treatment and a lower cumulative incidence of relapse in FLT3 mutated patients.
A phase 1 study (NCT02236013) assessed the safety, tolerability, and efficacy of gilteritinib plus 7 + 3 induction, followed by consolidation chemotherapy with high-dose ARA-C, and then a period of maintenance with gilteritinib in adults with ND-AML. The study included 80 patients; the rate of composite CR was 81.8% for all the four doses and 81.6% for patients who received 120 mg of gilteritinib (that represented the maximum tolerated dose). The median follow-up for OS was 35.8 months, with a mOS for patients with FLT3-mutated disease that was not reached. Serious treatment-related adverse events (AE) took place in 10 patients; the most common nonhematologic AE of grade ≥ 3 were increased liver enzymes, pneumonia, and sepsis/bacteremia[84]. Recently, the MD Anderson group presented preliminary data from a phase II trial that evaluated the combination of gilteritinib with intensive chemotherapy (CLIA = cladribine, cytarabine and idarubicin) in ND-AML with FLT3 mutation. Twenty-four patients were enrolled; 13 patients (54%) achieved a CR and underwent an allogeneic SCT[85]. Furthermore, treatment results for those patients with ND-AML, FLT3-mutated considered ineligible for a course of intensive chemotherapy are strongly disappointing. In a pooled analysis of the phase IB and phase III VIALE-A study (HMA + venetoclax), the median OS in FLT3 mutated patients was only 12.5 months, lower than what the 15 months achieved in FLT3 unmutated patients with this regimen[86]. Moreover, a phase III trial randomized (with a 2:1 ratio) untreated adults with FLT3 mutated AML unfit for intensive induction chemotherapy to gilteritinib (120 mg/day orally) and azacitidine (AZA + GIL) or azacitidine (AZA) alone. In the interim analysis, 123 patients were randomized (AZA + GIL, n = 74; AZA, n = 49). The median OS was not significantly different between the two arms (9.82 vs. 8.87 months, respectively; P = 0.753)[87]. As the median OS was the primary endpoint of the trial, the study was closed. Moreover, combination therapies to overcome resistance with FLT3 inhibitors and alternative resistance factors like apoptosis (BCL2) have been developed using venetoclax[88]. Preclinical data suggest that gilteritinib, inhibited the expression of BCL2A1 through the inactivation of STAT5 and alleviated TKI resistance of FLT3-mutated cell lines. Combining gilteritinib and venetoclax that suppresses BCL2A1 could improve the prognosis of AML with FLT3-ITD/D835 mutations[89].
Some patients harbor IDH and FLT3 mutations at the time point of diagnosis or experience a second mutation while undergoing treatment. Particularly FLT3 mutations are linked to low IDH inhibitors response rates, and treatment-emergent FLT3 mutations also seem to impart therapeutic resistance to these drugs. As a result, there is increased interest in researching combined therapies that target multiple pathways; however, there is currently no information available regarding the clinical experience of combined IDH inhibitors and FLT3 inhibitors therapy. A retrospective analysis identified 12 patients who received concurrent IDH and FLT3 inhibitors therapy, 11 of whom had R/R AML. The composite remission rate was 33%, and the ORR (CR + CRi + MLFS) was 42%, while the treatment combination was well tolerated[76]. Therefore, a notable proportion of R/R AML patients benefitted from concurrent FLT3 and IDH inhibitors therapy and studies are ongoing to investigate this promising combination.
In FLT3-ITD AML preclinical models, FLT3-ITD inhibition in combination with venetoclax exhibits impressive anti-tumor effectiveness and offers a solid molecular basis for clinical trials. However, the use of selective BCL-2 family inhibitors revealed a new function for BCL-2, BCL-XL, and MCL-1 in FLT3-ITD positive cells' in vivo survival, underscoring the necessity of targeting all three proteins for the most effective anti-tumor effect[88]. An open-label, phase 1b trial (NCT03625505) evaluated the combination of venetoclax and gilteritinib in R/R AML. Among the 54 patients enrolled, 38 patients (74.5%) achieved a response, with a median OS and a median duration of response of 10.5 and 5.6 months, respectively. In a post hoc analysis of the 30 analyzable patients who had achieved a CR with at least one follow-up MRD assessment, 17 (56.7%) achieved molecular clearance defined as FLT3 allelic burden < 10-2[90]. The MD Anderson group hypothesized that triplet therapy combining FLT3 inhibitors, venetoclax, and hypomethylating agents (HMAs) would further improve outcomes of FLT3 mutated patients. Therefore, they added FLT3 inhibitors to a regimen of 10-day decitabine with venetoclax in newly diagnosed (ND) and R/R FLT3 mutated patients. In ND patients, the composite complete remission (CRc) rate was 92%, with MRD negativity by flow cytometry in 56% and by next-generation sequencing (NGS) in 91% of responders. In R/R AML, the CRc rate was 62%, with MRD negativity rate by flow cytometry in 63% and by NGS in 100% of responders[91]. Therefore, triplet therapy with FLT3 inhibitors, venetoclax, and decitabine seems safe and an excellent frontline option for older patients with ND FLT3 mutated AML, and effective for R/R AML. A transition to allogenic transplant and post-transplant maintenance with FLT3 inhibitors would offer further improvement in long-term outcomes.
Although midostaurin and gilteritinib have been given FDA approval to treat patients with FLT3-mutations, the effectiveness of these treatments is still constrained by the fact that half of patients die during the first five years from diagnosis. Our actual understandings of the limits of TKIs highlight the demand for alternative therapeutic approaches. Although FLT3 antigen density on AML blasts is substantially lower than - for example, CD20 on lymphoma cells - and is not thought to be sufficient for producing powerful antibody-mediated effector functions, the membrane receptor FLT3 has also been studied as a target for immunotherapy using monoclonal antibodies. AML blasts and normal hematopoietic stem cells (HSCs), to a lesser extent, are selectively bound by the mouse anti-human FLT3 monoclonal antibody (mAb) 4G8. In preclinical models, 4G8 conferred selective reactivity against AML blasts with high FLT3 antigen density after Fc tuning[92]. Recently, T-cells engineered to express a FLT3-specific chimeric antigen receptor (CAR) were produced and it was shown that they confer robust reactivity against both AML cell lines and primary AML blasts, that express either wild-type FLT3 or FLT3-ITD. Furthermore, it was observed that treatment with crenolanib produced an increased surface expression of FLT3, particularly on FLT3-ITD + AML cells. Therefore, it enhanced the recognition by FLT3-CAR T-cells in vitro and in vivo[93]. Sommer et al. reported the preclinical evaluation of another off-the-shelf CAR-T cell construct targeting FLT3[94]. This CAR construct with single-chain variable fragments (scFvs) was directed against multiple FLT3 extracellular epitopes and was investigated for its capacity to direct T-cell selectivity and effector function to FLT3-mut AML cells. AML initial blasts are eliminated by allogeneic FLT3 CAR T cells made from a group of voluntary donors of T-cells; however, these cells are also effective against mouse and human hematopoietic stem and progenitor cells, raising concerns about myelotoxicity. Authors demonstrated that rituximab-mediated reduction of FLT3 CAR T cells following AML eradication permitted bone marrow regeneration without compromising leukemia remission by using a surrogate CAR with an affinity for murine FLT3[94].
Moreover, bispecific antibody cross-linking of FLT3 and CD3 demonstrated potent anti-leukemia activity against FLT3-mut cells. An anti-FLT3-CD3 immunoglobulin G (IgG)-based bispecific antibody (7,370) with a high affinity for FLT3 and a long half-life was developed to target FLT3-expressing AML blasts regardless of FLT3 mutational status. In vitro and in vivo testing revealed that 7,370 exhibits picomolar potency against AML cell lines. Additionally, 7,370 was able to stimulate T cells from AML patients, refocusing their cytotoxic activity at low effector-to-target ratios on autologous blasts[95].
Finally, cyclin-dependent kinase 6 (CDK6) is a protein that works as a transcriptional factor, regulating FLT3 and the serine-threonine kinase PIM1, a different step in the process of leukemogenesis[96]. Uras et al. found that FLT3-mutant AML cells are very responsive to palbociclib, one of several available inhibitors of CDK4/6 kinase[97]. They showed that the cell cycle kinase CDK6 is required for the viability of FLT3-dependent blastic cells and that FLT3-ITD could induce leukemogenesis [Figure 2][97].
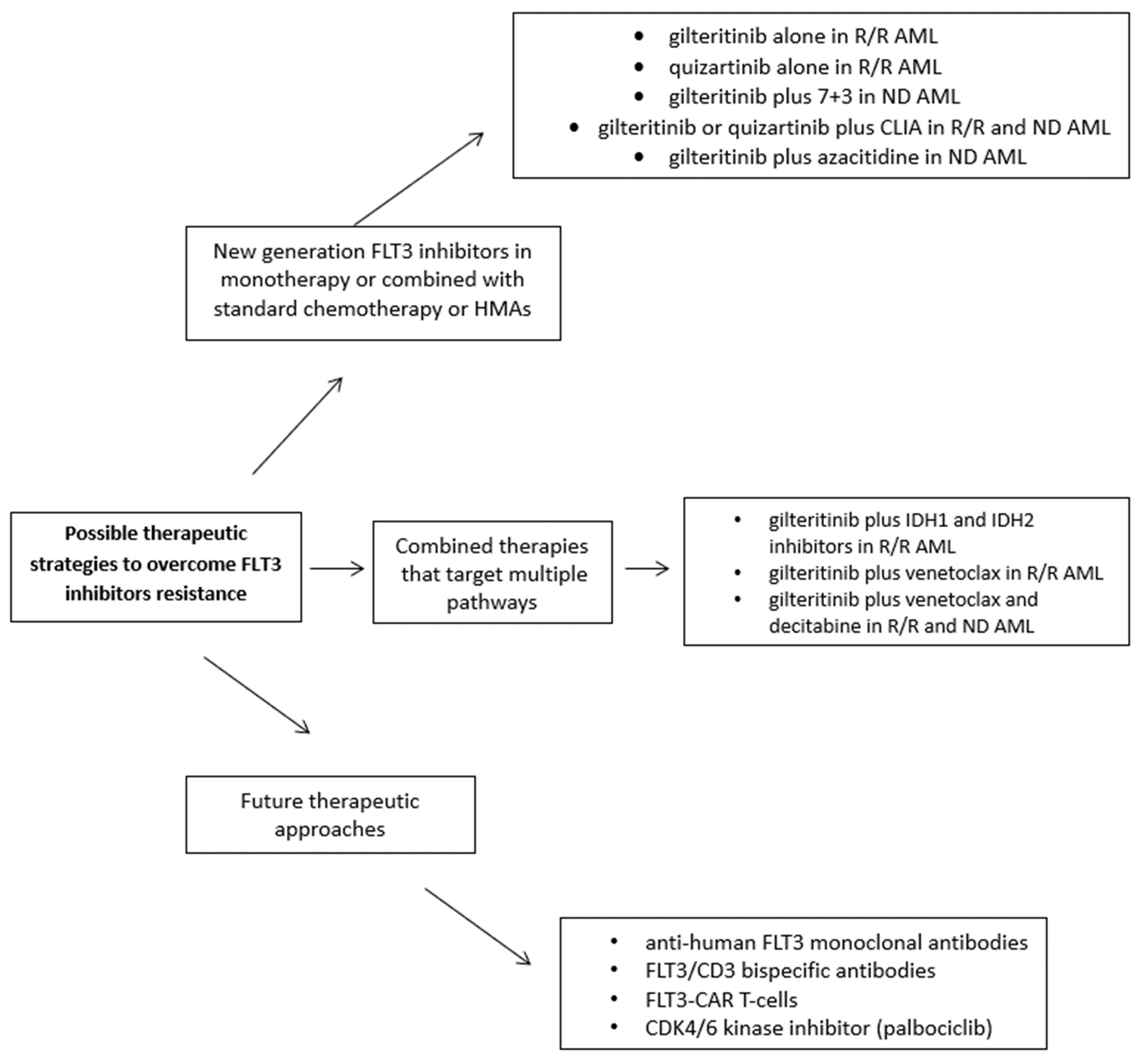
Figure 2. Possible strategies to overcome FLT3-inhibitor resistance.
CONCLUSIONS
One of the most important modifications introduced with the current ELN2022 is that the FLT3-ITD allelic ratio is no more included in the ELN risk classification; as a consequence, AML with FLT3-ITD (without other adverse-risk genetic lesions) are now comprised into the group of the intermediate-risk and not the high-risk group, this independently from the allelic-ratio or the concurrent presence of NPM1-mut. The reasons for this modification are related to methodological difficulties in standardizing the assay adopted to measure the FLT3-ITD allelic ratio in different labs, and the improved effect of midostaurin-based chemotherapy on AML patients with FLT3-ITD without NPM1-mut[55,98]. Several mechanisms of resistance to available FLT3-inhibitors have been partly elucidated, but it is a process that acts underneath many biologic fields (e.g., the emergence of antibiotic-resistant strains in many bacteria), also with a clonal selection of resistant sub-clones of AML after protracted biological pressure from TKI-inhibitors. Nevertheless, in the ADMIRAL trial, patients who achieved a CR experienced a median duration of this response of 23.0 months, suggesting that the effect is protracted in a subset of patients. However, the cumulative effect of these drugs on patients with AML harboring FLT3-mutations has been demonstrated to be largely beneficial in clinical trials and current clinical practice. In Europe, we have only gilteritinib and midostaurin approved in clinical practice, so an exchange for a different TKI is not possible yet.
As a result, we reasoned, is resistance “Much Ado about nothing?” Probably, from the actual clinical perspective, mechanisms of resistance to FLT3-inhibitors are somehow overlooked. For patients obtaining a CR after therapy with gilteritinib, if they are transplant eligible, considering an early Allogeneic transplant seems appropriate because the duration of a CR is generally short-lived and gilteritinib may represent a bridge to transplant[99]. When gilteritinib is given after Allo-transplantation, it results in a significant improvement in overall survival. In brief, exposure to gilteritinib for a limited interval after allo-SCT enhances Graft-vs.-Leukemia effects against FLT3-ITD+ leukemia without exacerbating GvHD[100].
Poor results have been reported in patients with R/R FLT3-ITD AML, who were treated with standard salvage chemotherapy and had a median OS of 5.5 months and 1- and 5-year OS rates of 25% and 7%, respectively[101]. On the contrary, the use of single-agent FLT3 inhibitors has improved survival over chemotherapy in the relapsed setting[17]. The path of FLT3 inhibitors is marked by possible strategies to combine different drugs to overcome the emergence of resistance[91,93]. We hope that these ongoing studies will benefit many patients in the future.
DECLARATIONS
Authors’ contributionsDesigned the paper: Perrone S, Molica M
Prepared Figure 1, graphical abstract, and wrote legends: Zhdanovskaya N
Wrote the paper and critically revised it: Perrone S, Molica M, Ottone T
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996;10:1911-8.
2. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209-21.
3. Fröhling S, Schlenk RF, Breitruck J, et al. Acute myeloid leukemia. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 2002;100:4372-80.
4. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002;99:4326-35.
5. Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood 2010;116:5089-102.
6. Park BG, Chi HS, Jang S, et al. Association of cup-like nuclei in blasts with FLT3 and NPM1 mutations in acute myeloid leukemia. Ann Hematol 2013;92:451-7.
7. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 2003;3:650-65.
8. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002;100:1532-42.
9. Rusten L, Lyman S, Veiby O, Jacobsen S. The FLT3 ligand is a direct and potent stimulator of the growth of primitive and committed human CD34+ bone marrow progenitor cells in vitro. Blood 1996;87:1317-25.
10. Wodnar-Filipowicz A. Flt3 ligand: role in control of hematopoietic and immune functions of the bone marrow. News Physiol Sci 2003;18:247-51.
11. Kandeel EZ, El Sayed G, Elsharkawy N, et al. Impact of FLT3 Receptor (CD135) Detection by Flow Cytometry on Clinical Outcome of Adult Acute Myeloid Leukemia Patients. Clin Lymphoma Myeloma Leuk 2018;18:541-7.
12. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev 2012;6:e8.
13. Fröhling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell 2007;12:501-13.
14. Murphy KM, Levis M, Hafez MJ, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn 2003;5:96-102.
15. Patnaik MM. The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma 2018;59:2273-86.
16. Blätte TJ, Schmalbrock LK, Skambraks S, et al. getITD for FLT3-ITD-based MRD monitoring in AML. Leukemia 2019;33:2535-9.
17. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med 2019;381:1728-40.
18. Schlenk RF, Kayser S, Bullinger L, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 2014;124:3441-9.
19. Linch DC, Hills RK, Burnett AK, Khwaja A, Gale RE. Impact of FLT3(ITD) mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood 2014;124:273-6.
20. Angenendt L, Röllig C, Montesinos P, et al. Chromosomal abnormalities and prognosis in NPM1-mutated acute myeloid leukemia: a pooled analysis of individual patient data from nine international cohorts. J Clin Oncol 2019;37:2632-42.
21. Pratcorona M, Brunet S, Nomdedéu J, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood 2013;121:2734-8.
22. Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008;111:2776-84.
23. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424-47.
24. Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood 2008;111:2527-37.
25. Whitman SP, Ruppert AS, Radmacher MD, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 2008;111:1552-9.
26. Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia 2005;19:1345-9.
27. Boddu P, Kantarjian H, Borthakur G, et al. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv 2017;1:1546-50.
28. Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007;110:1262-70.
29. Mead AJ, Gale RE, Hills RK, et al. Conflicting data on the prognostic significance of FLT3/TKD mutations in acute myeloid leukemia might be related to the incidence of biallelic disease. Blood 2008;112:444-5; author reply 445.
30. Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009;114:2386-92.
31. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454-64.
32. Levis M, Shi W, Chang K, et al. FLT3 inhibitors added to induction therapy induce deeper remissions. Blood 2020;135:75-8.
33. Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia 2020;34:682-96.
34. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015;29:2390-2.
36. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 2010;28:4339-45.
37. Voso MT, Larson RA, Jones D, et al. Midostaurin in patients with acute myeloid leukemia and FLT3-TKD mutations: a subanalysis from the RATIFY trial. Blood Adv 2020;4:4945-54.
38. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol 2001;11:130-5.
39. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol 2017;18:1061-75.
41. Perl AE, Larson RA, Podoltsev NA, et al. Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood 2022;139:3366-75.
42. Zhang W, Konopleva M, Shi YX, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst 2008;100:184-98.
43. Metzelder S, Wang Y, Wollmer E, et al. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood 2009;113:6567-71.
44. Metzelder SK, Schroeder T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia 2012;26:2353-9.
45. Sharma M, Ravandi F, Bayraktar UD, et al. Treatment of FLT3-ITD-positive acute myeloid leukemia relapsing after allogeneic stem cell transplantation with sorafenib. Biol Blood Marrow Transplant 2011;17:1874-7.
46. Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol 2020;38:2993-3002.
47. Ravandi F, Arana Yi C, Cortes JE, et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia 2014;28:1543-5.
48. Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol 2015;16:1691-9.
49. Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol 2013;31:3110-8.
51. Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol 2018;19:889-903.
52. Erba H, Montesinos P, Vrhovac R, et al. S100: quizartinib prolonged survival vs placebo plus intensive induction and consolidation therapy followed by single-agent continuation in patients aged 18-75 years with newly diagnosed FLT3-ITD+ AML. HemaSphere 2022;6:1-2.
53. Molica M, Perrone S. Molecular targets for the treatment of AML in the forthcoming 5th World Health Organization Classification of Haematolymphoid Tumours. Expert Rev Hematol 2022;15:973-86.
54. Smith CC, Lasater EA, Lin KC, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A 2014;111:5319-24.
55. Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. JCO 2016;34:7008-7008.
56. Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood 2014;124:389-389.
57. Döhner K, Thiede C, Jahn N, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 2020;135:371-80.
58. Desikan SP, Daver N, DiNardo C, Kadia T, Konopleva M, Ravandi F. Resistance to targeted therapies: delving into FLT3 and IDH. Blood Cancer J 2022;12:91.
59. Breitenbuecher F, Schnittger S, Grundler R, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009;113:4074-7.
60. Zhang H, Savage S, Schultz AR, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun 2019;10:244.
61. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2019;20:984-97.
62. Ghiaur G, Levis M. Mechanisms of resistance to FLT3 inhibitors and the role of the bone marrow microenvironment. Hematol Oncol Clin North Am 2017;31:681-92.
63. Javidi-Sharifi N, Martinez J, English I, et al. FGF2-FGFR1 signaling regulates release of leukemia-protective exosomes from bone marrow stromal cells. Elife 2019:8.
64. Chang YT, Hernandez D, Alonso S, et al. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv 2019;3:908-16.
65. Eguchi M, Minami Y, Kuzume A, Chi SG. Mechanisms underlying resistance to FLT3 inhibitors in acute myeloid leukemia. Biomedicines 2020;8:245.
66. Smith CC, Levis MJ, Perl AE, Hill JE, Rosales M, Bahceci E. Molecular profile of FLT3-mutated relapsed/refractory patients with AML in the phase 3 ADMIRAL study of gilteritinib. Blood Adv 2022;6:2144-55.
67. Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Discov 2021;2:125-34.
68. Travaglini S, Angelini DF, Alfonso V, et al. Characterization of FLT3-ITD(mut) acute myeloid leukemia: molecular profiling of leukemic precursor cells. Blood Cancer J 2020;10:85.
69. Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021;137:3093-104.
70. Alvarado Y, Kantarjian HM, Luthra R, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer 2014;120:2142-9.
71. Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012;485:260-3.
72. Friedman R. The molecular mechanisms behind activation of FLT3 in acute myeloid leukemia and resistance to therapy by selective inhibitors. Biochim Biophys Acta Rev Cancer 2022;1877:188666.
73. Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000;96:3907-14.
74. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov 2019;9:1050-63.
75. Tarver TC, Hill JE, Rahmat L, et al. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv 2020;4:514-24.
76. Fathi AT, Perl AE, Levis M, et al. Concurrent FLT3 inhibitor and IDH inhibitor therapy in patients with acute myeloid leukemia (AML). Blood 2020;136:11-2.
77. Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012;119:5133-43.
78. Stirewalt DL, Kopecky KJ, Meshinchi S, et al. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood 2006;107:3724-6.
79. Schnittger S, Bacher U, Haferlach C, Alpermann T, Kern W, Haferlach T. Diversity of the juxtamembrane and TKD1 mutations (exons 13-15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Gene Chromosome Canc 2012;51:910-24.
80. Fischer M, Schnetzke U, Spies-Weisshart B, et al. Impact of FLT3-ITD diversity on response to induction chemotherapy in patients with acute myeloid leukemia. Haematologica 2017;102:e129-31.
81. Rücker FG, Du L, Luck TJ, et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2022;36:90-9.
82. Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017;129:257-60.
83. Perl AE, Hosono N, Montesinos P, et al. Clinical outcomes in patients with relapsed/refractory FLT3-mutated acute myeloid leukemia treated with gilteritinib who received prior midostaurin or sorafenib. Blood Cancer J 2022;12:84.
84. Pratz KW, Cherry M, Podoltsev NA, et al. AML-256 a phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia: final study results. Cl Lymph Myelom Leuk 2022;22:S230.
85. Abuasab T, Kantarjian HM, Garcia-manero G, et al. Phase II study of cladribine, idarubicin, cytarabine (CLIA) plus gilteritinib in patients (pts) with FLT3 mutated acute myeloid leukemia (AML). JCO 2022;40:e19036-e19036.
86. Konopleva M, Thirman MJ, Pratz KW, et al. Impact of FLT3 mutation on outcomes after venetoclax and azacitidine for patients with treatment-naïve acute myeloid leukemia. Clin Cancer Res 2022;28:2744-52.
87. Wang ES, Montesinos P, Minden MD, et al. Phase 3 trial of gilteritinib plus azacitidine vs azacitidine for newly diagnosed FLT3mut+ AML ineligible for intensive chemotherapy. Blood 2022;140:1845-57.
88. Singh Mali R, Zhang Q, DeFilippis RA, et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021;106:1034-46.
89. Yamatani K, Ai T, Saito K, et al. Inhibition of BCL2A1 by STAT5 inactivation overcomes resistance to targeted therapies of FLT3-ITD/D835 mutant AML. Transl Oncol 2022;18:101354.
90. Daver N, Perl AE, Maly J, et al. Venetoclax in combination with gilteritinib demonstrates molecular clearance of
91. Maiti A, DiNardo CD, Daver NG, et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J 2021;11:25.
92. Hofmann M, Große-Hovest L, Nübling T, et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012;26:1228-37.
93. Jetani H, Garcia-Cadenas I, Nerreter T, et al. CAR T-cells targeting FLT3 have potent activity against FLT3-ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018;32:1168-79.
94. Sommer C, Cheng HY, Nguyen D, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol Ther 2020;28:2237-51.
95. Yeung YA, Krishnamoorthy V, Dettling D, et al. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol Ther 2020;28:889-900.
96. Otto T, Sicinski P. The kinase-independent, second life of CDK6 in transcription. Cancer Cell 2013;24:141-3.
97. Uras IZ, Walter GJ, Scheicher R, et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016;127:2890-902.
98. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022;140:1345-77.
99. Day JW, Fox TA, Gupta R, Khwaja A, Wilson AJ, Kottaridis PD. Gilteritinib monotherapy as a transplant bridging option for high risk FLT3-mutated AML with t(6;9)(p23;q34.1);DEK-NUP214 in morphological but not cytogenetic or molecular remission following standard induction chemotherapy. Leuk Res Rep 2022;17:100291.
100. Zhang Z, Hasegawa Y, Hashimoto D, et al. Gilteritinib enhances graft-versus-leukemia effects against FLT3-ITD mutant leukemia after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2022;57:775-80.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Perrone S, Ottone T, Zhdanovskaya N, Molica M. How acute myeloid leukemia (AML) escapes from FMS-related tyrosine kinase 3 (FLT3) inhibitors? Still an overrated complication?. Cancer Drug Resist 2023;6:223-38. http://dx.doi.org/10.20517/cdr.2022.130
AMA Style
Perrone S, Ottone T, Zhdanovskaya N, Molica M. How acute myeloid leukemia (AML) escapes from FMS-related tyrosine kinase 3 (FLT3) inhibitors? Still an overrated complication?. Cancer Drug Resistance. 2023; 6(2): 223-38. http://dx.doi.org/10.20517/cdr.2022.130
Chicago/Turabian Style
Perrone, Salvatore, Tiziana Ottone, Nadezda Zhdanovskaya, Matteo Molica. 2023. "How acute myeloid leukemia (AML) escapes from FMS-related tyrosine kinase 3 (FLT3) inhibitors? Still an overrated complication?" Cancer Drug Resistance. 6, no.2: 223-38. http://dx.doi.org/10.20517/cdr.2022.130
ACS Style
Perrone, S.; Ottone T.; Zhdanovskaya N.; Molica M. How acute myeloid leukemia (AML) escapes from FMS-related tyrosine kinase 3 (FLT3) inhibitors? Still an overrated complication?. Cancer Drug Resist. 2023, 6, 223-38. http://dx.doi.org/10.20517/cdr.2022.130
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 5
likes
Like This Article 5
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.