
Resistance to immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer

Abstract
Non-small cell lung cancer (NSCLC) patients with Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation are associated with significant clinical heterogeneity and a poor prognosis to standard NSCLC therapies such as surgical resection, radiotherapy, chemotherapies, and targeted medicines. However, the application of immune checkpoints inhibitors (ICIs) has dramatically altered the therapeutic pattern of NSCLC management. Clinical studies have indicated that some KRAS-mutant NSCLC patients could benefit from ICIs; however, the responses in some patients are still poor. This review intends to elucidate the mechanisms of resistance to immunotherapy in KRAS-driven NSCLC and highlight the TME functions altered by immunoinhibitors, immunostimulators, and cancer metabolism. These metabolic pathways could potentially be promising approaches to overcome immunotherapy resistance.
Keywords
INTRODUCTION
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer-related deaths worldwide[1]. According to Global Cancer Statistics 2020, lung cancer is estimated to represent 11.4% of new cancer cases and 18.0% of cancer-related deaths[1]. There are two main histological types of lung cancer: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC)[2]. About 85% of lung cancer is NSCLC, which comprises lung adenocarcinoma (LUAD), lung squamous cell carcinoma, and large cell neuroendocrine carcinoma. SCLC accounts for the remaining 15%[3]. Despite inducements such as smoking, genetic mutations are the leading causes of lung cancer. The most common driver mutations in NSCLC involve Kirsten rat sarcoma viral oncogene homolog (KRAS), epidermal growth factor receptor (EGFR), and anaplastic lymphoma kinase[4].
KRAS, a member of the RAS family, was one of the first oncogenes identified in NSCLC[5]. Since KRAS mutation is reported to be associated with resistance to multiple therapies and poor prognosis in NSCLC, several preclinical and clinical studies have investigated effective therapies, including immunotherapy and targeted therapy[6,7]. In 2021, AMG510, specifically targeting KRASG12C, was approved by the FDA as the orphan drug to treat the NSCLC with KRASG12C[8]. Other inhibitors, peptides, and tumor vaccines are under preclinical and clinical studies, including MRTX849 targeting KRASG12C, MRTX1133 targeting KRASG12D, 12VC1 targeting KRASG12C/V, mRNA-5671 targeting KRASG12C/D/V, etc.[9]. In addition to targeted therapy, immunotherapies have also remarkably changed the management of NSCLC[10]. The increased expression of programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) has also been demonstrated to be closely related to KRAS status[11]. Controversial results have been reported in multiple clinical studies on the efficacy of immune checkpoints inhibitors (ICIs) in NSCLC with KRAS mutation[12-15]. Jeanson et al.[16] and Arbour et al.[13] demonstrated no difference in response to ICIs among NSCLC patients with or without KRAS mutations. However, other studies have demonstrated more benefits from ICIs for NSCLC patients with KRAS mutation than those without[14,15]. In this review, we summarize and analyze the possible mechanisms underlying the resistance to ICIs in NSCLC with KRAS mutation and mainly focus on the role of the tumor microenvironment (TME) and metabolism, therapeutic implications, and potential targets for overcoming the resistance to or improving the efficacy of ICIs treatments in KRAS-mutant NSCLC.
ICIS TREATMENTS IN NSCLC WITH KRAS MUTATION
Even though the status of KRAS mutation as being able to alter the responses to ICIs in NSCLC has been verified in multiple fundamental studies, solid clinical evidence is still lacking. In this section, we review the clinical trials of ICIs in KRAS-mutant NSCLC patients.
Checkmate-057 is a random and double-blind phase 3 clinical trial with a size of 582 cases, which is designed to compare the efficacy of monotherapy of nivolumab or docetaxel in advanced non-squamous NSCLC patients. Comparing to docetaxel, nivolumab significantly elongates the overall survival (OS) (medium OS = 12.2 months vs. 9.4 months; HR = 0.73; 95%CI: 0.59-0.89; P = 0.0002). The subgroup analysis showed that KRAS-mutant patients have better survival benefits than KRAS-wildtype patients (HR = 0.52, 95%CI: 0.29-0.95; HR = 0.98, 95%CI: 0.29-0.95)[17]. Another phase 3 clinical trial, KEYNOTE-042, investigated the efficacy and safety of monotherapy of pembrolizumab or platinum-based chemotherapy in locally advanced or metastatic NSCLC harboring wildtype EGFR/anaplastic lymphoma kinase (ALK) and PD-L1+ (TPS ≥ 1%). The results show that the monotherapy of pembrolizumab can prolong the OS compared to platinum-based chemotherapy. Further analysis revealed that KRAS-mutant patients show higher PD-L1 expression and tumor mutational burden (TMB) and have significantly longer progression-free survival (PFS) in the pembrolizumab treatment group than those in the platinum treatment group[14,18]. KEYNOTE-189 evaluated the efficacy and safety of pemetrexed and platinum-based therapy or combined with pembrolizumab in metastatic non-squamous NSCLC patients. The results demonstrate that a combination of pembrolizumab and chemotherapy can significantly prolong the OS than chemotherapy regardless of the PD-L1 expression level. Both KRAS-wildtype and -mutant NSCLC patients benefit from chemotherapy combined with pembrolizumab. The pooled analysis of KEYNOTE-189 and KEYNOTE-042 indicated that the non-squamous NSCLC patients with wildtype EGFR/ALK or KRAS show a better prognosis to the combination of pembrolizumab and chemotherapy (HR = 0.55) compared to pembrolizumab monotherapy (HR = 0.86). However, for KRAS-mutant non-squamous patients, the monotherapy of pembrolizumab shows a better prognosis (HR = 0.42 and HR = 0.28) than combination treatment of pembrolizumab and chemotherapy (HR = 0.79 and HR = 1.14)[19]. Altogether, the clinical outcomes above indicate that KRAS-mutant NSCLC patients can benefit from immunotherapy or combined treatment of immunotherapy and chemotherapy, whereas the conclusion is not supported by some meta-analyses with more solid evidence. Two meta-analyses and some retrospective analyses have reported that the status of KRAS mutation has no negative association with the survival outcome of immunotherapy in advanced NSCLC patients[7,16,20,21].
One possible illustration is that the co-mutations besides KRAS mutation might play the predictive and prognostic role in response to ICIs in KRAS-mutant NSCLC. Skoulidis et al.[22] defined three distinct co-mutation subgroups of the early stage and advanced KRAS-mutant tumors: KRASmut/serine/threonine kinase 11 (STK11)-/-, KRASmut/tumor protein 53 (TP53)-/- (KP), and KRASmut/cyclin dependent kinase inhibitor 2A, 2B (CDKN2A, 2B)-/-/thyroid transcription factor 1low (KC)[22]. The retrospective analysis revealed that the KL patients show less responses to immunotherapy and shorter PFS and OS than KP patients[23]. Other investigations have revealed that KP patients show significant benefits from PD-1 blockade monotherapy[24]. Collectively, PD-L1 blockade might be more beneficial to KP patients than KL patients.
Taken together, not all the KRAS-mutant NSCLC patients could benefit from ICIs. Co-mutations increase the TMB in patients and might contribute to the poor response to ICIs. The mechanisms of resistance to ICIs in NSCLC with KRAS mutation are sophisticated and not fully investigated, which we discuss in the following sections.
THE RESISTANT MECHANISMS OF ICIS TREATMENTS IN NSCLC WITH KRAS MUTATION
KRAS regulates TME through immunomodulatory molecules
Growing evidence has implicated that the intrinsic and extrinsic resistance mechanisms to ICIs might result from the immunosuppressive TME caused by alterations in disparate signaling pathways in tumor cells[25]. Previous studies have clearly indicated that the immunosuppressive TME can be regulated by alterations of immunoinhibitors and immunostimulators in tumor and stromal cells[26].
Immune checkpoints exert crucial roles in preventing overreaction and minimize the duration and expansion of immune responses[27]. PD-1 (CD279), coded by pdcd1, is ubiquitously expressed on various immune cells, including activated T cells, B cells, monocytes, NK cells, and dendritic cells (DCs)[28]. PD-1 can be recognized by the ligands, PD-L1 and PD-L2, on normal tissue cells and cancer cells, which maintains the immune homeostasis but might also revoke the anti-tumor immunity[29]. After PD-1 binds to PD-L1, PD-1 undergoes a conformational change, which leads to the inactivation of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3K)-serine/threonine kinase (AKT) and extracellular signal-regulated kinase 2 (ERK) pathways and, consequently, dysfunction of T cells[30]. In addition, studies have reported that tumor cells can secrete exosomes carrying PD-L1, which impede the function of CD8+ T cells, CD4+ T cells, and Tregs before infiltration[31]. It has been widely reported that oncogenic KRAS can increase PD-L1 from different aspects, including transcription, stabilization, and recycling. In LUAD cell lines, KRASmut has been found to increase the transcriptional level of PD-L1 through the mitogen-activated protein kinase kinase 1 (MEK) pathway[32]. In human lung and colorectal cancer, Coelho et al.[33] demonstrated that the adenylate-uridylate-rich element-binding protein tristetraprolin (TTP) can bind to the 3’ untranslated region of PD-L1 mRNA and consequently induce the degradation of PD-L1. The oncogenic KRAS activates MEK signaling and further phosphorylates and inhibits the TTP by MK2, which leads to the stabilization of PD-L1 mRNA[33]. In addition, it has been reported that mutant KRAS suppresses programmed cell death 4 and then promotes the translation of ADP ribosylation factor 6 and MYCBP associated protein expression, which facilitates PD-L1 recycling driven by platelet-derived growth factor and eventually the immune evasion[34].
As another factor, cytokine-modulated immune responses have been investigated for decades. Cytokines are a type of small proteins secreted by almost all types of cells and can be classified into seven families: interleukin (IL), colony stimulating factor (CSF), interferon (IFN), tumor necrosis factor, tumor growth factor-beta (TGF-β) family, growth factor (GF), and chemokine family[35]. Cytokines deliver messages in a paracrine, autocrine, or endocrine manner and can coordinate the recruitment of immune cells, spatial organization, and cellular interactions[36,37]. Plentiful cytokines and chemokines have both pro- and anti-inflammatory functions and exert different functions in particular scenarios[36]. It has been reported that mutant KRAS can remodel the TME by regulating the production and secretion of pro- or anti-inflammatory cytokines and chemokines[38]. The oncogenic mutant KRAS represses the expression of interferon regulatory factor 2 (IRF2) and results in increased production of C-X-C motif chemokine ligand 3 (CXCL3), which binds to
Immunomodulators are composed of all molecules that can regulate the immune response, including receptors/ligands on both tumor cells and stromal cells in TME and cytokines and chemokines secreted by cells. The studies cited above have uncovered that KRAS could mediate the rearrangement of immunomodulators through different signaling pathway in multiple cancers, which might contribute to the resistance to ICIs in NSCLC. A schematic figure of KRAS-driven signaling pathways in the regulation of immunomodulators is presented in Figure 1.

Figure 1. A schematic figure of KRAS-driven signaling pathways in the regulation of immunomodulators. PI3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; AKT: serine/threonine kinase; MAPK: mitogen-activated protein kinase 1; ERK: extracellular signal-regulated kinase 2; PFK: phosphofructokinase; HIF1α: hypoxia inducible factor 1 subunit alpha; JNK: mitogen-activated protein kinase 8; mTOR: mechanistic target of rapamycin kinase; LDHA/B: lactate dehydrogenase A/B; YAP: yes1 associated transcriptional regulator; IRF2: interferon regulatory factor 2; STAT3: signal transducer and activator of transcription 3; NF-κB: transcription factor P65; MHCs: major histocompatibility complex.
KRAS regulates TME through metabolic alteration
For the past decades, many studies have revealed adjustments of energy metabolism in cancers, which are involved in almost every stage of cancer development[43,44]. Oncogenic KRAS mutation has been reported to regulate diverse metabolic networks to fulfill the excessive requirement of distinct nutrients to support tumor growth and metastasis[45,46]. In this section, we discuss the mechanisms underlying how KRAS orchestrates the TME through metabolic networks.
Glycolysis
In normal cells, glucose can be consumed either through oxidative phosphorylation (in mitochondria) or anaerobic glycolysis (in cytosol) to produce energy for cell activities. Glucose is mainly transported into cells with the help of glucose transporters (GLUTs) in an energy-consuming-free way. In cytosol, glucose is catalyzed to glucose-6-phosphate (G6P) by hexokinase (HK). G6P can enter the pentose-phosphorylation pathway with catalyzation of G6P dehydrogenase and be processed to ribulose-5-phosphate for the synthesis of purine. G6P can also be isomerized to fructose-6-phosphate (F6P) and further phosphorylated to fructose-1,6-biphosphate (FBP), which enters the glycolysis pathway. Under the aerobic condition, FBP is catalyzed to pyruvate and then converted to acetyl-CoA by pyruvate dehydrogenase (PDH), which participates in the TCA cycle. Under the anaerobic condition, pyruvate does not enter the TCA cycle but is converted to lactate by the lactate hydrogenase (LDH). Under normal condition, a small portion of F6P also enters the hexosamine pathway (HBP) with the help of glutamine--fructose-6-phosphate aminotransferase (GFPT) and provides glycans for protein glycosylation[47,48].
Back in 1985, it was reported that rat-1 cell maintains a higher level of glycolysis in the presence of ras transfection[49]. Accumulating evidence shows that oncogenic KRAS mutation modulates glycolysis through promoting the uptake of glucose and increasing glycolysis flux. In pancreatic ductal adenocarcinoma (PDAC) with KRASG12D, metabolomic and RT-qPCR analyses have revealed increasing glucose transporter solute carrier family 2-member 1 (GLUT1) expression and enhanced glycolysis flux. KRASG12D was found to upregulate the expression of several enzymes, including HK, PFK, enolase, etc.[50]. Many studies have explored in more detail about how mutant KRAS interacts with the enzymes involved in the carbon metabolism pathways. In KRAS-driven PDAC, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 is phosphorylated by p38γ induced by KRAS mutation and forms a ternary complex with solute carrier family 2 member 2 (GLUT2), which promotes the uptake of glucose[51]. In addition, GLUT1 has been found significantly upregulated in NSCLC with KRAS mutation[52]. KRAS4A, a shorter isoform of KRAS, has been found to bind to the N-terminal of HK1 and block the allosteric inhibition feedback, which promotes the transition of glucose to G6P and increases the glycolysis flux[53]. Based on both an in vivo mice model and an in vitro H23 cell line study, Wang et al.[54] reported that KRAS activation is required for HK2 expression. In a KRASmut/STK11-/- co-mutated lung cancer model, KRAS mutation induces the expression of GLUT. Meanwhile, the loss of STK11 impedes the phosphorylation and activation of protein kinase AMP-activated catalytic subunit alpha 1 (AMPK), thereby leading to the activation of GFPT enzyme. The increasing activity of GLUT and GFPT promotes HBP[55]. Glucose is also the primary energy source for the proliferation and differentiation of immune cells[56,57]. As a result of increasing glycolysis in tumor cells, the access to glucose for immune cells is limited in TME. A cruel competition between tumor cells stromal cells consequently impairs the glycolytic metabolism and normal functions of immune cells[58,59]. More specifically, the limited glycolysis causes the weakened activity of the AKT/mammalian target of rapamycin (mTOR) pathway in T cells, which impedes the production of
One important metabolite of glycolysis is lactate. In KRAS-driven NSCLC, the high expressions of LDH-A and LDH-B are reported to be associated with tumorigenesis and disease progression[63,64]. One study found that phosphoglycerate kinase 1 (PGK1) can be phosphorylated by ERK1/2, which is the downstream of KRAS. The phosphorylated PGK1 phosphorylates PDHK1, the latter further phosphorylating and inhibiting PDH, which blocks the conversion of pyruvate to acetyl-CoA and subsequently promotes the conversion of pyruvate to lactate[65]. In PDAC, hypoxia and active KRAS can induce the expression of hypoxia inducible factor 1 subunit alpha (HIF1α) and inhibit the degradation of HIF1α, respectively. HIF1α then transcriptionally enhances the expression of the lactate transporter, solute carrier family 16 member 4 (MCT4), which transports lactate out of cells[66]. The accumulation of lactate from the excessive glycolysis in tumor cells is transported by MCT4 out of cells, which generates a low-pH environment that discourages the normal proliferation and function of immune cells[67]. The accumulated lactate promotes the polarization of macrophages toward anti-inflammatory M2 phenotype and leads to a downregulation of interferon gamma (IFN-γ) secreted by the infiltration of T cells and NK cells[68,69]. Besides, the accumulated lactate directly blocks the glycolytic flux in T cells[70].
Taken together, the studies described above suggest that KRAS mutation can promote glycolysis by promoting various signaling pathways and molecules, such as related enzymes and transporters. The outrageous requirement of glucose and excessive excretion of lactate impair the normal proliferation and function of different immune cells, which ultimately diminishes the anti-tumor activity.
Lipid metabolism
Lipid metabolism is constantly in homeostasis due to the balance of fatty acids synthesis (FAS) and fatty acids oxidation (FAO) in normal cells. In the process of de novo lipogenesis, citrate is catalyzed to acetyl-CoA in assistance with ATP-citrate lyase (ACLY) and then transformed to palmitate by FA synthetase (FASN), phosphatidic acids, and finally triacylglycerol. Triacylglycerol can be stored in the lipid droplets (LDs) in the cytoplasm[71]. Phospholipids are another source of lipogenesis and can be converted to arachidonic acid (ARA) by phospholipases (PLA2)[72]. ARA can be catalyzed by cyclooxygenase 1 and 2 (COX-1 and COX-2) and transformed to prostaglandins 2 (PGE2), which has been shown to play a vital role in tumorigenesis and immune response[73]. The FAO process can be classified into harmless β-oxidation and fatal lipid peroxidation. In β-oxidation, FAs are catalyzed to produce acetyl-CoA, NADH, and FAD. Besides, lipid peroxidation is believed to start from polyunsaturated fatty acid chains, induced by reactive oxygen species (ROS), and results in ferroptosis[74]. In the mouse model of KRAS-driven LUAD, the loss of stress-induced metabolic regulators, regulated in development and DNA damage responses 1, can induce a HIF-dependent lipid storage pathway, which induces the FA transporter CD36 and thereby uptake of exogenous fatty acid[75]. KRAS mutation has been shown to promote FA synthesis through inducing FASN and stearoyl-CoA desaturase mediated by ERK2[76]. Besides, the FA metabolism has been reported to be upregulated by the PI3K-mTOR pathway[77], which has already been demonstrated to be regulated by KRAS[78]. The active AKT can facilitate both the ACLY and mTORc1. ACLY promotes the transformation of citrate to acetyl-CoA, which subsequently boosts lipogenesis. Active mTORc1 can activate slicing factor serine/threonine-protein kinase (SRPK2) and transcriptional factor sterol regulatory element binding protein-1c in the nucleus, which ensures the expression of a series of lipogenesis enzymes, including ACLY, FASN, etc.[79-81]. Besides promoting FAS, KRAS mutation has also been reported to suppress the oxidation of lipids. The suppression of hormone-sensitive lipase (HSL) by KRAS mutation leads to the decreased
Previous studies have revealed that the accumulated FAs provides sufficient raw material for PGE2 production[90,91], and KRAS mutation can also enhance the production of PGE2 via upregulation of
Cholesterol is normally synthesized from acetyl-CoA. Recent studies have found that cholesterol exerts an immunosuppressive TME by promoting the expression of immune checkpoint on CD8+ T cells, which induces ER stress and consequently exhaustion of those T cells[96,97]. In KRAS-driven NSCLC, the synthesis of cholesterol has been found to be induced by Myc proto-oncogene protein (MYC) activation. The activation of MYC leads to an accumulation of cholesteryl esters (CE) stored in LDs and the increased influx and the decreased efflux of cholesterol in tumor cells. Of note, the deactivation of MYC following activation gives rise to an additional increase of CEs[98].
In conclusion, there is supporting evidence indicating that FA metabolism can be induced by KRAS mutation through regulating various molecules. The excessive synthesis of FA and associated metabolites impedes the normal function of immune cells.
Glutamine and tryptophan metabolism
In normal cells, amino acid metabolism facilitates cells with energy and material for the biosynthesis of macromolecules, such as proteins and nucleotides. Amino acid metabolism reprogramming is required for hyperactive tumor cells to meet the high demand of energy and proteins required for cell proliferation[99]. Among the 21 amino acids within the human body, glutamine and tryptophan have been broadly reported to be rewired in various tumor cells and build an immune suppressive TME, which facilitates tumor cells to evade immune surveillance.
Glutamine can be successively transformed to glutamate, α-ketoglutarate, oxaloacetate, and aspartate by the catalyzation of glutaminase (GLS1), glutamate dehydrogenase 1, and glutamic-oxaloacetic transaminase 1/2, respectively[100]. KRAS mutation in NSCLC enhances the glutamine metabolism by inducing glutamine uptake, which results in a glutamine deficiency in TME[101]. KRAS mutation has been found to induce multiple glutamine transporters, such as solute carrier family 1 member 5 (SLC1A5) in NSCLC[102,103], solute carrier family 7 member 5 (SLC7A5)[104], and solute carrier family 38 member 2 (SLC38A2) via activation of hippo effector yes1 associated transcriptional regulator (YAP1)[103]. The upregulated transporter SLC1 increases the influx of glutamine, and the increased antiporter SLC7A effluxes glutamines and intakes other essential amino acids. The essential amino acids transported by SLC7A induce mTORC1-ribosomal protein S6 kinase B1 signaling and further promote protein synthesis[103,104]. It has also been shown that the mRNA level of GLS is higher in KRAS-mutated NSCLC, which indicates a higher consumption of glutamine in NSCLC[102]. Additionally, the KRASmut/STK11-/-/KEAP1-/- co-mutant NSCLC was found dependent on glutaminolysis for fuel and was specifically sensitive to GLS inhibitors[85]. The high uptake of glutamine in tumor cells leads to a glutamine deficiency in TME, which causes immune suppression through blocking active T cells or producing immune suppressive cells. One study illustrated that the lack of glutamine results in the increase of HIF and then the rising secretion of IL-23 by macrophages, which promotes the proliferation of Tregs through the STAT3 signaling pathway. Tregs subsequently secretes TGF-β and IL-10 and suppresses the cytotoxicity of T cells[105]. Besides the shortage of glutamine in the TME, other conditions can also result in glutamine deprivation in tumor infiltrated immune cells. One study showed that the shortage of glucose causes defective N-linked glycosylation and endoplasmic reticulum stress and accordingly activates the endoplasmic reticulum to nucleus signaling 1 (IRE1) - X-box binding protein 1 (XBP1) axis. Activated XBP1 represses the glutamine transporters through post-translational regulation, which blocks glutamine uptake. The low level of glucose and glutamine causes the dysfunction of mitochondria, which impedes the production of IFN-γ by active T cells[106]. Moreover, glutamine deprivation increases the secretion of G-CSF and GM-CSF through activating the IRE1α/c-Jun N-terminal kinase pathway in cancer cells, which manipulates the formation of immunosuppressive MDSCs in TME[107]. However, the results from one study also indicate that high glutamine in TME tends to polarize the macrophages to pro-tumorigenic M2 type and inhibit the differentiation of the M1 macrophage[108], which is contrary to the conclusion from the above research. The contradictory conclusion of deprived glutamine indicates the complexity of glutamine in regulating TME.
Tryptophan can either be catalyzed to 5-hydrooxytryptophan, serotonin, and 5-hydroxyindoleacetic acid by tryptophan hydroxylase 1 and 2 (TPH1, TPH2), dopa decarboxylase, and monoamine oxidase A and B, respectively, or be catalyzed to kynurenine, kynurenic acid by indoleamine 2,3-dioxygenase 1 and 2 (IDO1/2), and tryptophan 2,3-dioxygenase (TDO2). In NSCLC, the KRAS mutation has been shown to reprogram tryptophan metabolism through upregulating the expression of immune checkpoint markers IDO1 according to microarray data[109]. The detailed mechanism remains to be discovered. The tryptophan metabolism has a pivotal role in promoting immune evasion. In tumor cells, tryptophan is transformed into kynurenine by IDO1. The upregulated IDO1 level induces excess kynurenine, which could be transferred to TME to promote Tregs and suppress effector T cells[110]. It has been illustrated that the activity of IDO can reflect the advanced disease, tumor metastasis, and immunotherapy responses to PD-1 inhibitors. The high expression of IDO can recruit immunosuppressive MDSCs through Tregs, which is Foxp3 dependent[111,112].
In summary, cancer cells can rely on glutamine as the main source of fuel and the precursor of other biomacromolecules. The deficiency of glutamine and increase of metabolite from tryptophan metabolism in TME give rise to an immunosuppressive phenotype. KRAS has been found to modulate several signaling pathways involved in glutamine metabolism, whereas further work is required to illustrate the more detailed mechanisms.
Besides the metabolism mentioned above, mutant KRAS has also been reported to regulate additional metabolic signaling pathways, including asparagine metabolism[113], authophagy[46,114], mcropinocytosis[115], etc. More detailed reviews of the metabolic rewiring driven by KRAS mutation can be found elsewhere[46,116-118]. Nutrient scavenging is a practical method for both tumor cells and immune cells to grab necessities from TME. Tumor cells and immune cells often outcompete stromal cells for nutrients or metabolites in TME, which leads to a nutrient-deprived environment[43]. However, most nutrients and some metabolites are also essential for the biosynthesis of macromolecules and production of energy craved by immunocytes[56,57,61,119,120]. Studies have also asserted that excess metabolites, such as lactate, alter the environment and affect the functions of immune cells[60,86,87,89,121]. A more comprehensive description of cancer metabolic reprogramming and immune response can be found in other reviews[122-125]. Collectively, the limited nutrients and redundant metabolites arouse the dysfunction in immune cells, which is one of the leading causes of resistance to ICIs. Taken together, KRAS mutation rewires the metabolic network in TME and thereby might cause a poor response to ICIs. A schematic figure of KRAS-driven metabolic signaling pathways in cancer cells is shown in Figure 2.

Figure 2. A schematic figure of KRAS-driven signaling pathways in the regulation of metabolic signaling pathways. GLUT: Glucose transporters; HK: hexokinase; PFK: phosphofructokinase; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; AKT: serine/threonine kinase; MAPK: mitogen-activated protein kinase 1; ERK: extracellular signal-regulated kinase 2; PDH: pyruvate dehydrogenase; PGK: phosphoglycerate kinase 1; HIF1α: hypoxia inducible factor 1 subunit alpha; JNK: mitogen-activated protein kinase 8; mTOR: mechanistic target of rapamycin kinase; LDHA/B: lactate dehydrogenase A/B; YAP: yes1 associated transcriptional regulator; IRF2: interferon regulatory factor 2; STAT3: signal transducer and activator of transcription 3; NF-κB: transcription factor P65; HSL: hormone-sensitive lipase; COX: cyclooxygenase; SCD: stearoyl-CoA desaturase; FASN: FA synthetase; ACLY: ATP-citrate lyase; GLS: glutaminase.
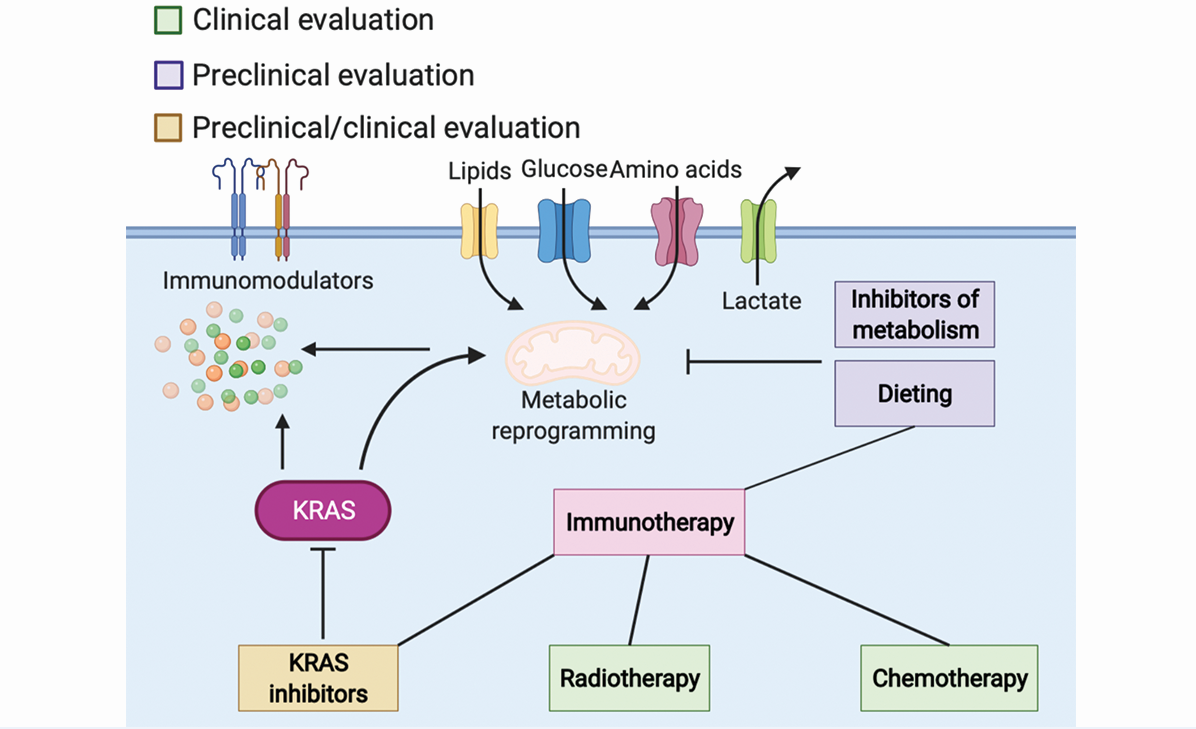
PROSPECTIVE STRATEGIES FOR OVERCOMING RESISTANCE TO ICIS
According to the ASCO guidelines, five main therapeutic strategies are applied in NSCLC: surgery, radiation therapy, chemotherapy, target therapy, and immunotherapy. The therapeutic schedule depends mostly on the stage and mutation type. As illustrated above, the efficacy of monotreatment of immunotherapy is controversial in NSCLC patients; thus, researchers have begun investigating the combinational strategy of immunotherapy and other therapies. In this section, we discuss the promising combined treatments of immunotherapy and other therapies in preclinical and clinical studies.
It is discussed above that PD-1/-L1 expression plays an essential role in response to ICIs. Therefore, targeting pathways modulating PD-1/-L1 expression might show potential for overcoming the resistance to ICIs, which has been demonstrated in several studies. In the KRAS-mutant lung cancer model, blockade of both PD-1 and helix-loop-helix transcription factor inhibitor of differentiation 1 knock out significantly enhances the amount of CD8+ T cells as well as the expression of PD-L1, which impairs the tumor growth and increases the survival[126]. In other studies of KRASmut lung cancer, anti-PD-1 combined with inhibition of the AKT-mTOR pathway by mTOR inhibitor or the STAT3 pathway by natural compound luteolin can remarkably decreased the expression of PD-1 or PD-L1, respectively, which consequently surmounts the resistance to anti-PD-1[127,128].
Other potential strategies to overcome the ICIs resistance include promoting the proliferation and activation of cytotoxic T cells and thwarting immunosuppressive immune cells, among others. The cholesterol-lowering drug statin has been reported to inhibit prenylation of KRAS and cause ER stress and immunogenic cell death, which cross-primes DCs and provokes CD8+ T cells. Combinational treatment of anti-PD-1, statin, and oxaliplatin has shown the potential to overcome resistance to anti-PD-1[129]. In the KRASmut/TP53-/- lung cancer mouse model, chemokine CCL7 has also been found to recruit CD11c+ or Zbtb46+ DCs and promote T cell expansion, which increases the immune responses. When combined with anti-PD-1, CCL7 profoundly increases the survival of the NSCLC mouse model[130]. The BET bromodomain inhibitor has also been demonstrated to increase the number of infiltrated T-helper type 1 cells and reduce the population of Tregs, which augments the anti-tumor response to anti-PD-1[131]. Utilization of antibodies or natural compounds to block immunosuppressive cytokines or chemokines has been reported to hamper the resistance to ICIs in KRAS-mutant NSCLC mouse models[41,132].
Due to the approval of the KRASG12C inhibitor AMG 510 in KRASG12C NSCLC by the FDA, studies have been investigating the effect of the combination of KRAS inhibitors and ICIs at the bench and in clinics. In the KRASG12C CT26 syngeneic mouse model, AMG510 treatment has been found to induce a pro-inflammatory microenvironment by enhancing the tumor infiltrated CD8+ T cells, DCs, NKs, etc., and it exerted extended anti-tumor efficacy when combined with anti-PD-1[133]. A phase 1 clinical study designed to evaluate the safety and efficacy of AMG 510 and immunotherapy is in process now (NCT03600883). In the KRASG12C CT26 syngeneic mouse model, KRAS inhibitor MRTX849 combined with anti-PD-1 has also been shown to increase major histocompatibility complex I expression and decrease immunosuppressive factors, which increased PFS compared to monotherapy[134]. Phase 1/2 studies evaluating the safety, tolerability, PK, and clinical activity of MRTX849 in combination with pembrolizumab in patients with NSCLC are now recruiting (NCT04613596 and NCT03785249).
Other attempts have been made to combine ICIs with inhibitors targeting KRAS downstream. A phase 1/2 clinical trial has investigated the efficacy of the PLK inhibitor rigosertib combined with nivolumab in NSCLC patients with KRAS mutation (NCT04263090). The early results indicate that 29% (2/7) of patients achieved partial responses, most of whom harbor a KRASG12C or KRASG12V mutation; 43% (3/7) of patients had a stable disease; and 57% (4/7) of patients had progressive disease[135]. Studies on the combinational efficacy of other KRASG12C inhibitors and ICIs, such as GDC-6036 (NCT04449874) and JDQ-443 (NCT04699188), are now recruiting. The efficacy of ICIs combined with tumor vaccine mRNA-5671, which specifically target KRASG12C/D/V and KRASG13D, is now under investigation (NCT03948763).
Combination treatments which are not specifically for KRAS-mutant NSCLC have also received positive results in the NSCLC group. Multiple ICI treatment has shown promising results in NSCLC patients. In a phase 3 trial, patients were designed to receive mono-nivolumab, nivolumab combined with ipilimumab, nivolumab combined platinum-based chemotherapy, or mono-chemotherapy. The nivolumab combined with ipilimumab treatment group (medium OS = 17.1 months; 95%CI: 15.0-20.1) has shown longer median duration of overall survival compared to the chemotherapy treatment group (medium OS = 14.9 months; 95%CI: 12.7-16.7) among patients with PD-L1 (TPS ≥ 1%). The two-year overall survival rates were 40% and 32.8% and the median duration responses were 23.2 and 6.2 months, in the nivolumab combined with ipilimumab treatment group and chemotherapy treatment group, respectively. Patients with a PD-L1 (TPS < 1%) also benefit from nivolumab combined with ipilimumab (medium OS = 17.2; 95%CI: 12.8-22.0) compared with chemotherapy treatment (medium OS = 12.2 months; 95%CI: 9.2-14.3)[136]. In a phase 2 clinical trial, 97 patients were enrolled to study whether stereotactic body radiotherapy (SBRT) enhances the tumor responses to pembrolizumab. The results show that the ORR at 12 weeks was 36% in experimental arm (SBRT plus pembrolizumab) vs. 18% in control arm (pembrolizumab), the median PFS was 6.6 months (SBRT plus pembrolizumab, 95%CI: 4.0-14.6) vs. 1.9 months (pembrolizumab, 95%CI: 1.7-6.9), and the median OS was 15.9 months (SBRT plus pembrolizumab, 95%CI: 7.1 to not reached) vs. 7.6 months (pembrolizumab, 95%CI: 6.0-13.9)[137]. A phase 2 trial enrolled 96 patients to study the efficacy of combinational anti-PD-1 (SHR-1210) and anti-VEGFR (Apatinib) in NSCLC patients with wild-type EGFR and ALK (NCT03083041). The ORR was 29.7% and DCR was 81.3%. Further analysis showed that the ORR reached 50% in the patients with high TMB, indicating that this combo-treatment is acceptable in NSCLC patients with high TMB[138].
All the clinical trials mentioned above have illustrated good outcomes comparing with mono-immunotherapy in NSCLC, regardless of KRAS status. Further umbrella analyses are still needed to elucidate the combo-treatment efficacy on the KRAS-mutant NSCLC groups. All clinical trials are listed in Table 1.
Immune checkpoint inhibitors active in clinical trials
| Immune checkpoint inhibitors | NCT identifier numbers | Combined therapy | Target | Allocation | Phase | Size | Primary ends | Status |
| Anti-PD-1/-L1 | NCT03600883 | AMG 510 | KRASG12C | Randomized | Phase 1 | 733 | AEs, DLTs, significant clinical changes | Recruiting |
| Atezolizumab | NCT04449874 | GDC-6036 bevacizumab, cetuximab, rrlotinib | KRASG12C VGFR EGFR TKI | Non-randomized | Phase 1 | 236 | AEs, DLTs | Recruiting |
| Pembrolizumab | NCT04429542 | BCA101 | EGFR/TGF-β | Non-randomized | Phase 1 | 292 | Safety, Cmax, PFS, AEs, ORR, OS, DLTs, MTD | Recruiting |
| NCT04613596 | MRTX849 | KRASG12D | N/A | Phase 2 | 120 | Clinical activity | Recruiting | |
| NCT03785249 | MRTX849 | KRASG12D | N/A | Phase 1 | 565 | Safety, tolerability, drug levels, molecular effects, and clinical activity | Recruiting | |
| NCT03299088 | Trametinib | MEK | Non-randomized | Phase 1 | 15 | DLTs, ORR, PFS | Active, not recruiting | |
| NCT02779751 | Abemaciclib Anastrozole | CDK Estrogen synthesis | Non-randomized | Phase 1 | 100 | PFS, AEs, ORR, OS, DCR, PK | Active, not recruiting | |
| NCT03948763 | mRNA-5671/V941 | Vaccine | Non-randomized | Phase 1 | 100 | DLTs, AEs, ORR | Recruiting | |
| NCT04340882 | Docetaxel Ramucirumab | Chemotherapy VEGFR | N/A | Phase 2 | 41 | PFS, AEs, ORR, OS | Recruiting | |
| NCT03225664 | Trametinib | MEK | N/A | Phase 1/2 | 37 | ORR | Active, not recruiting | |
| NCT02492568 | Radiation | N/A | Phase 2 | 96 | ORR, toxicity | Complete | ||
| Nivolumab | NCT04263090 | Rigosertib | PLK1 | N/A | Phase 1/2 | 30 | MTD, ORR, PFS, OS | Recruiting |
| NCT02852083 | Pioglitazone Clarithromycin | PPARγ Bacteria proteins | Randomized | Phase 2 | 86 | PFS, AEs, ORR, OS | Unknown status | |
| NCT02492568 | Ipilimumab | Anti-CTLA-4 | N/A | Phase 3 | 1980 | OS, ORR, disease related symptom | Recruiting | |
| SHR-1210 | NCT03083041 | Apatinib | Anti-VEGFR | N/A | Phase 2 | 117 | AEs, ORR | N/A |
The application of dietary modifications to supplement other cancer therapies is drawing more attention currently. The intake of different nutrients might alter the nutrient availability in the plasma, thus the TME[139]. Preclinical and retrospective studies have assumed that cancer development and prognosis are negatively correlated to unhealthy diets, such as diets high in sodium and fat[121,140]. Growing research has found that restriction or supplementation of specific metabolites might ameliorate therapeutic responses. The manipulation of nutrient accessibility remarkably reprograms the metabolic activity and therefore leads to alterations in cell activities and sensitivity to therapies, which is mainly modulated by nutrient-sensing pathways[141-144]. The dietary modifications enhance cancer therapy through sophisticated mechanisms, which are well summarized in other reviews[145-147]. As mentioned above, glucose is the primary source for not only energy but also metabolic intermediates for macromolecule synthesis for both tumor cells and immune cells. A diet low in glucose but normal in calories can reduce the blood glucose and decelerate tumor growth in some tumor models[148]. However, the glucose restriction might also affect the normal function of immune effect cells. During the process of proliferation, differentiation, and activation of immune cells, the requirement for glucose might differ. Studies have found that inhibition of glycolysis restrains the differentiation of CD4+ T cells to Treg cells and promotes the differentiation of activated CD8+ T cells to long-lived memory CD8+ T cells[119,149]. Accordingly, we presume that timing and personalized diet should be taken into account when applying glucose restriction to supplement ICI treatment. Likewise, the lipid and amino acid metabolisms function differently at different stages of differentiation and activation of immune cells, thus the need for nutrients varies[120,150-152]. Therefore, it is critical to understand the metabolic network and landscape in TME in KRAS-driven NSCLC and how they affect the behavior of tumor cells and the function of stromal cells. Moreover, the impact of dietary modification on ICI response might be influenced by patient-specific variables. Many studies are currently investigating the molecular mechanisms and evaluating the effects of dietary interventions in enhancing cancer therapies. Collectively, dietary modification is a promising strategy for overcoming resistance to or improving the efficacy of ICI treatment in KRAS-mutant NSCLC.
CONCLUSION
Further understanding of the resistance mechanisms to ICIs mediated by KRAS mutation in NSCLC could provide implications on prospective therapeutic interventions to overcome the resistance or improve the efficacy. This will need further investigations to unearth metabolic pathways regulated by specific KRAS mutations, as well the modulatory effect on shaping TME directly and indirectly. Additional attempts to identify metabolic signaling pathways that promote immunosuppressive TME and resistance to ICIs will help discover the targetable metabolic vulnerabilities to improve the efficacy or overcome the resistance to ICIs through dietary modifications.
DECLARATIONS
AcknowledgmentsFigure 2 was illustrated using BioRender.com.
Authors’ contributionsResearch, image illustration and contributed to writing of review manuscript: Li Y, Hu L
Performed data acquisition for tables: Li Y, Peng X, Xu H, Tang B
Substantial contributions to conception and planning of review and editing: Hu L, Xu C
Conception, writing and editing of review manuscript: Xu C
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by research grants from the National Natural Science Foundation of China (No. 81873048) and Sichuan Provincial Science Fund for Distinguished Young Scholars of China (2020JDJQ0065).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209-49.
2. Chen J, Liu A, Wang Z, et al. LINC00173.v1 promotes angiogenesis and progression of lung squamous cell carcinoma by sponging miR-511-5p to regulate VEGFA expression. Mol Cancer 2020;19:98.
3. Gridelli C, Rossi A, Carbone DP, et al. Non-small-cell lung cancer. Nat Rev Dis Primers 2015;1:15009.
4. Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 2010;28:4953-60.
5. Shepherd FA, Lacas B, Le Teuff G, et al. LACE-Bio Collaborative Group. Pooled analysis of the prognostic and predictive effects of TP53 comutation status combined with KRAS or EGFR mutation in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 2017;35:2018-27.
6. Garassino MC, Marabese M, Rusconi P, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol 2011;22:235-7.
7. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thorac Oncol 2017;12:403-7.
9. Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann Oncol 2021;32:1101-10.
10. Wang M, Herbst RS, Boshoff C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat Med 2021;27:1345-56.
11. Ghimessy A, Radeczky P, Laszlo V, et al. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev 2020;39:1159-77.
12. Galland L, Le Page AL, Lecuelle J, et al. Prognostic value of thyroid transcription factor-1 expression in lung adenocarcinoma in patients treated with anti PD-1/PD-L1. Oncoimmunology 2021;10:1957603.
13. Arbour KC, Rizvi H, Plodkowski AJ, et al. Treatment outcomes and clinical characteristics of patients with KRAS-G12C-mutant non-small cell lung cancer. Clin Cancer Res 2021;27:2209-15.
14. Sun L, Hsu M, Cohen RB, Langer CJ, Mamtani R, Aggarwal C. Association between KRAS variant status and outcomes with first-line immune checkpoint inhibitor-based therapy in patients with advanced non-small-cell lung cancer. JAMA Oncol 2021;7:937-9.
15. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol 2019;30:1321-8.
16. Jeanson A, Tomasini P, Souquet-Bressand M, et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC). J Thorac Oncol 2019;14:1095-101.
17. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015;373:1627-39.
18. Mok TSK, Wu Y, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 2019;393:1819-30.
19. Gadgeel S, Rodríguez-Abreu D, Speranza G, et al. Updated analysis from KEYNOTE-189: pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol 2020;38:1505-17.
20. Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: a meta-analysis and review. Oncotarget 2017;8:48248-52.
21. Passiglia F, Cappuzzo F, Alabiso O, et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br J Cancer 2019;120:57-62.
22. Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860-77.
23. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822-35.
24. Dong ZY, Zhong WZ, Zhang XC, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res 2017;23:3012-24.
25. Bai R, Chen N, Li L, et al. Mechanisms of cancer resistance to immunotherapy. Front Oncol 2020;10:1290.
26. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol 2019;20:1100-9.
27. Blank CU, Haining WN, Held W, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol 2019;19:665-74.
28. Bai J, Gao Z, Li X, Dong L, Han W, Nie J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017;8:110693-707.
29. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv 2020;6:eabd2712.
30. Wu X, Gu Z, Chen Y, et al. Application of PD-1 blockade in cancer immunotherapy. Comput Struct Biotechnol J 2019;17:661-74.
31. Chen G, Huang AC, Zhang W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018;560:382-6.
32. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell 2019;76:359-70.
33. Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 2017;47:1083-99.e6.
34. Hashimoto S, Furukawa S, Hashimoto A, et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc Natl Acad Sci U S A 2019;116:17450-9.
37. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity 2021;54:859-74.
38. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014;1843:2563-82.
39. Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019;35:559-72.e7.
40. Yang Z, Xu G, Wang B, et al. USP12 downregulation orchestrates a protumourigenic microenvironment and enhances lung tumour resistance to PD-1 blockade. Nat Commun 2021;12:4852.
41. Peng DH, Rodriguez BL, Diao L, et al. Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers. Nat Commun 2021;12:2606.
42. Hamarsheh S, Groß O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun 2020;11:5439.
44. McGuirk S, Audet-Delage Y, St-Pierre J. Metabolic fitness and plasticity in cancer progression. Trends Cancer 2020;6:49-61.
46. Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer 2021;21:510-25.
47. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011;27:441-64.
48. Coussement P, Bauwens D, Peters G, Maertens J, De Mey M. Mapping and refactoring pathway control through metabolic and protein engineering: The hexosamine biosynthesis pathway. Biotechnol Adv 2020;40:107512.
49. Racker E, Resnick RJ, Feldman R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci U S A 1985;82:3535-8.
50. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656-70.
51. Wang F, Qi XM, Wertz R, et al. p38γ MAPK is essential for aerobic glycolysis and pancreatic tumorigenesis. Cancer Res 2020;80:3251-64.
52. Sasaki H, Shitara M, Yokota K, et al. Overexpression of GLUT1 correlates with Kras mutations in lung carcinomas. Mol Med Rep 2012;5:599-602.
53. Amendola CR, Mahaffey JP, Parker SJ, et al. KRAS4A directly regulates hexokinase 1. Nature 2019;576:482-6.
54. Wang H, Wang L, Zhang Y, Wang J, Deng Y, Lin D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int 2016;16:9.
55. Kim J, Lee HM, Cai F, et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat Metab 2020;2:1401-12.
56. Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002;16:769-77.
57. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 2005;5:844-52.
58. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol 2008;38:2438-50.
59. Chang CH, Qiu J, O'Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015;162:1229-41.
60. Ho PC, Bihuniak JD, Macintyre AN, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015;162:1217-28.
61. Cong J, Wang X, Zheng X, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab 2018;28:243-55.e5.
62. Guak H, Al Habyan S, Ma EH, et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun 2018;9:2463.
63. Xie H, Hanai J, Ren JG, et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 2014;19:795-809.
64. McCleland ML, Adler AS, Deming L, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res 2013;19:773-84.
65. Li X, Jiang Y, Meisenhelder J, et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell 2016;61:705-19.
66. McDonald PC, Chafe SC, Brown WS, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology 2019;157:823-37.
67. Certo M, Tsai CH, Pucino V, Ho PC, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol 2021;21:151-61.
68. Brand A, Singer K, Koehl GE, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab 2016;24:657-71.
69. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014;513:559-63.
70. Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007;109:3812-9.
71. Walther TC, Chung J, Farese RV Jr. Lipid droplet biogenesis. Annu Rev Cell Dev Biol 2017;33:491-510.
73. Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta 2015;1851:414-21.
74. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021;22:266-82.
75. Qiao S, Koh SB, Vivekanandan V, et al. REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors. Genes Dev 2020;34:751-66.
76. Gouw AM, Eberlin LS, Margulis K, et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U S A 2017;114:4300-5.
77. Biffo S, Manfrini N, Ricciardi S. Crosstalks between translation and metabolism in cancer. Curr Opin Genet Dev 2018;48:75-81.
78. Uras IZ, Moll HP, Casanova E. Targeting KRAS mutant non-small-cell lung cancer: past, present and future. Int J Mol Sci 2020;21:4325.
79. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer 2020;122:4-22.
80. Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med 2008;14:461-9.
81. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016;35:1250-60.
82. Man J, Pajic M, Joshua AM. Fats and mets, KRAS-driven lipid dysregulation affects metastatic potential in pancreatic cancer. Cancer Res 2020;80:4886-7.
83. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012;485:661-5.
84. Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci 2020;14:267.
85. Galan-Cobo A, Sitthideatphaiboon P, Qu X, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 2019;79:3251-67.
86. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res 2020;80:101055.
87. Manzo T, Prentice BM, Anderson KG, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med 2020;217:e20191920.
88. Gao A, Liu X, Lin W, et al. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J Immunother Cancer 2021;9:e001536.
89. Yin X, Zeng W, Wu B, et al. PPARα inhibition overcomes tumor-derived exosomal lipid-induced dendritic cell dysfunction. Cell Rep 2020;33:108278.
90. Goor SA, Dijck-Brouwer DA, Fokkema MR, van der Iest TH, Muskiet FA. Maternal and fetal brain contents of docosahexaenoic acid (DHA) and arachidonic acid (AA) at various essential fatty acid (EFA), DHA and AA dietary intakes during pregnancy in mice. Prostaglandins Leukot Essent Fatty Acids 2008;78:159-69.
91. Qari HA, Oves M. Fatty acid synthesis by Chlamydomonas reinhardtii in phosphorus limitation. J Bioenerg Biomembr 2020;52:27-38.
92. Backlund MG, Mann JR, Wang D, Dubois RN. . Ras up-regulation of cyclooxygenase-2. Regulators and effectors of small GTPases: ras family. Elsevier; 2006. p. 401-10.
93. Sharma S, Yang SC, Zhu L, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 2005;65:5211-20.
94. Basingab FS, Ahmadi M, Morgan DJ. IFNγ-dependent interactions between ICAM-1 and LFA-1 counteract prostaglandin E2-mediated inhibition of antitumor CTL responses. Cancer Immunol Res 2016;4:400-11.
95. Böttcher JP, Bonavita E, Chakravarty P, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018;172:1022-37.e14.
96. Raccosta L, Fontana R, Corna G, Maggioni D, Moresco M, Russo V. Cholesterol metabolites and tumor microenvironment: the road towards clinical translation. Cancer Immunol Immunother 2016;65:111-7.
97. Ma X, Bi E, Lu Y, et al. Cholesterol Induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab 2019;30:143-56.e5.
98. Hall Z, Wilson CH, Burkhart DL, Ashmore T, Evan GI, Griffin JL. Myc linked to dysregulation of cholesterol transport and storage in nonsmall cell lung cancer. J Lipid Res 2020;61:1390-9.
99. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol 2020;8:603837.
100. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients 2018;10:1564.
101. Bernfeld E, Foster DA. Glutamine as an essential amino acid for KRas-driven cancer cells. Trends Endocrinol Metab 2019;30:357-68.
102. Meijer TWH, Looijen-Salamon MG, Lok J, et al. Glucose and glutamine metabolism in relation to mutational status in NSCLC histological subtypes. Thorac Cancer 2019;10:2289-99.
103. Kandasamy P, Zlobec I, Nydegger DT, et al. Oncogenic KRAS mutations enhance amino acid uptake by colorectal cancer cells via the hippo signaling effector YAP1. Mol Oncol 2021;15:2782-800.
104. Najumudeen AK, Ceteci F, Fey SK, et al. CRUK Rosetta Grand Challenge Consortium. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat Genet 2021;53:16-26.
105. Fu Q, Xu L, Wang Y, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol 2019;75:752-63.
106. Song M, Sandoval TA, Chae CS, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018;562:423-8.
107. Sun HW, Wu WC, Chen HT, et al. Glutamine deprivation promotes the generation and mobilization of MDSCs by enhancing expression of G-CSF and GM-CSF. Front Immunol 2020;11:616367.
108. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;18:985-94.
109. Parra ER, Villalobos P, Zhang J, et al. Immunohistochemical and image analysis-based study shows that several immune checkpoints are co-expressed in non-small cell lung carcinoma tumors. J Thorac Oncol 2018;13:779-91.
110. Pranzini E, Pardella E, Paoli P, Fendt SM, Taddei ML. Metabolic reprogramming in anticancer drug resistance: a focus on amino acids. Trends Cancer 2021;7:682-99.
111. Holmgaard RB, Zamarin D, Li Y, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep 2015;13:412-24.
112. Botticelli A, Cerbelli B, Lionetto L, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J Transl Med 2018;16:219.
113. Doubleday PF, Fornelli L, Ntai I, Kelleher NL. Oncogenic KRAS creates an aspartate metabolism signature in colorectal cancer cells. FEBS J 2021;288:6683-99.
114. Moldogazieva NT, Mokhosoev IM, Terentiev AA. Metabolic heterogeneity of cancer cells: an interplay between HIF-1, GLUTs, and AMPK. Cancers (Basel) 2020;12:862.
115. Baek G, Tse YF, Hu Z, et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep 2014;9:2233-49.
117. Kawada K, Toda K, Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol 2017;22:651-9.
118. Pupo E, Avanzato D, Middonti E, Bussolino F, Lanzetti L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front Oncol 2019;9:848.
119. Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011;186:3299-303.
120. Reilly NA, Lutgens E, Kuiper J, Heijmans BT, Wouter Jukema J. Effects of fatty acids on T cell function: role in atherosclerosis. Nat Rev Cardiol 2021;18:824-37.
121. Ringel AE, Drijvers JM, Baker GJ, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 2020;183:1848-66.e26.
122. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015;43:435-49.
123. Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov 2019;18:669-88.
124. Cerezo M, Rocchi S. Cancer cell metabolic reprogramming: a keystone for the response to immunotherapy. Cell Death Dis 2020;11:964.
125. Xia L, Oyang L, Lin J, et al. The cancer metabolic reprogramming and immune response. Mol Cancer 2021;20:28.
126. Baraibar I, Roman M, Rodríguez-Remírez M, et al. Id1 and PD-1 combined blockade impairs tumor growth and survival of KRAS-mutant lung cancer by stimulating PD-L1 expression and tumor infiltrating CD8+ T cells. Cancers (Basel) 2020;12:3169.
127. Lastwika KJ, Wilson W 3rd, Li QK, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res 2016;76:227-38.
128. Jiang ZB, Wang WJ, Xu C, et al. Luteolin and its derivative apigenin suppress the inducible PD-L1 expression to improve anti-tumor immunity in KRAS-mutant lung cancer. Cancer Lett 2021;515:36-48.
129. Nam GH, Kwon M, Jung H, et al. Statin-mediated inhibition of RAS prenylation activates ER stress to enhance the immunogenicity of KRAS mutant cancer. J Immunother Cancer 2021;9:e002474.
130. Zhang M, Yang W, Wang P, et al. CCL7 recruits cDC1 to promote antitumor immunity and facilitate checkpoint immunotherapy to non-small cell lung cancer. Nat Commun 2020;11:6119.
131. Adeegbe DO, Liu S, Hattersley MM, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in Kras-mutant non-small cell lung cancer. Cancer Immunol Res 2018;6:1234-45.
132. Li R, Salehi-Rad R, Crosson W, et al. Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in LKB1-deficient non-small cell lung cancer. Cancer Res 2021;81:3295-308.
133. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217-23.
134. Briere DM, Li S, Calinisan A, et al. The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther 2021;20:975-85.
135. Onconova Therapeutics initiates phase 1/2a study of rigosertib plus nivolumab to treat KRAS+ lung adenocarcinoma. Available from: http://www.pharmabiz.com/NewsDetails.aspx?aid=129076&sid=2 [Last accessed on 17 Jan 2022].
136. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med 2019;381:2020-31.
137. Theelen WSME, Peulen HMU, Lalezari F, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol 2019;5:1276-82.
138. Zhou CC, Gao G, Wang YN, et al. Efficacy of PD-1 monoclonal antibody SHR-1210 plus apatinib in patients with advanced nonsquamous NSCLC with wild-type EGFR and ALK. J Clin Oncol 2019;37:9112.
139. Sullivan MR, Danai LV, Lewis CA, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019;8:e44235.
140. Barbi J, Patnaik SK, Pabla S, et al. Visceral obesity promotes lung cancer progression-toward resolution of the obesity paradox in lung cancer. J Thorac Oncol 2021;16:1333-48.
141. Marsh J, Mukherjee P, Seyfried TN. Drug/diet synergy for managing malignant astrocytoma in mice: 2-deoxy-D-glucose and the restricted ketogenic diet. Nutr Metab (Lond) 2008;5:33.
142. Yang J, Yang X, Pan W, et al. Fucoidan-supplemented diet potentiates immune checkpoint blockage by enhancing antitumor immunity. Front Cell Dev Biol 2021;9:733246.
143. Malczewski AB, Ketheesan N, Coward JIG, Navarro S. Enhancing checkpoint inhibitor therapy in solid tissue cancers: the role of diet, the microbiome & microbiome-derived metabolites. Front Immunol 2021;12:624434.
145. Zitvogel L, Pietrocola F, Kroemer G. Nutrition, inflammation and cancer. Nat Immunol 2017;18:843-50.
146. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature 2017;542:177-85.
147. Soldati L, Di Renzo L, Jirillo E, Ascierto PA, Marincola FM, De Lorenzo A. The influence of diet on anti-cancer immune responsiveness. J Transl Med 2018;16:75.
148. Allen BG, Bhatia SK, Anderson CM, et al. Ketogenic diets as an adjuvant cancer therapy: history and potential mechanism. Redox Biol 2014;2:963-70.
149. Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 2013;123:4479-88.
150. Almeida L, Dhillon-LaBrooy A, Carriche G, Berod L, Sparwasser T. CD4+ T-cell differentiation and function: unifying glycolysis, fatty acid oxidation, polyamines NAD mitochondria. J Allergy Clin Immunol 2021;148:16-32.
151. Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol 2019;40:699-718.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Li Y, Hu L, Peng X, Xu H, Tang B, Xu C. Resistance to immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer. Cancer Drug Resist 2022;5:129-46. http://dx.doi.org/10.20517/cdr.2021.102
AMA Style
Li Y, Hu L, Peng X, Xu H, Tang B, Xu C. Resistance to immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer. Cancer Drug Resistance. 2022; 5(1): 129-46. http://dx.doi.org/10.20517/cdr.2021.102
Chicago/Turabian Style
Li, Yunchang, Lanlin Hu, Xinhao Peng, Huasheng Xu, Bo Tang, Chuan Xu. 2022. "Resistance to immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer" Cancer Drug Resistance. 5, no.1: 129-46. http://dx.doi.org/10.20517/cdr.2021.102
ACS Style
Li, Y.; Hu L.; Peng X.; Xu H.; Tang B.; Xu C. Resistance to immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer. Cancer Drug Resist. 2022, 5, 129-46. http://dx.doi.org/10.20517/cdr.2021.102
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 20
likes
Like This Article 20
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.