
Venetoclax resistance: mechanistic insights and future strategies

Abstract
Acute myeloid leukemia (AML) is historically associated with poor prognosis, especially in older AML patients unfit for intensive chemotherapy. The development of Venetoclax, a potent oral BH3 (BCL-2 homology domain 3) mimetic, has transformed the AML treatment. However, the short duration of response and development of resistance remain major concerns. Understanding mechanisms of resistance is pivotal to devising new strategies and designing rational drug combination regimens. In this review, we will provide a comprehensive summary of the known mechanisms of resistance to Venetoclax and discuss Venetoclax-based combination therapies. Key contributing factors to Venetoclax resistance include dependencies on alternative anti-apoptotic BCL-2 family proteins and selection of the activating kinase mutations. Mutational landscape governing response to Venetoclax and strategic approaches developed considering current knowledge of mechanisms of resistance will be addressed.
Keywords
INTRODUCTION
Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults[1]. The disease often affects older adults with a median age of 68 years at diagnosis[1]. Conventional treatments using intensive chemotherapy are less beneficial for older patients with poor tolerance and modest outcome[2,3]. Introduction of Venetoclax (ABT-199), a selective inhibitor of BCL-2, has advanced the treatment options for AML patients, especially older patients[4]. Venetoclax in combination with hypomethylating agents (HMA) or low-dose cytarabine (LDAC) was approved by US FDA for newly diagnosed AML patients who are either unable to receive intensive chemotherapy or older than 75 years old. Nowadays, this has become the standard therapy for such a population[5]. However, the prevalent use of Venetoclax comes with a new challenge of resistance, particularly in the relapsed/refractory setting[6,7]. Here, we review the mechanisms of Venetoclax resistance in AML and discuss strategies to overcome resistance.
MECHANISM OF ACTION OF VENETOCLAX
BCL-2 family proteins and mechanism of action of Venetoclax
Venetoclax (ABT-199) is an oral, selective antagonist of the B-cell lymphoma 2 (BCL-2), a key protein modulating intrinsic (mitochondrial) apoptosis[8]. Apoptosis is regulated and balanced by protein-protein interactions among BCL-2 family members[9]. Different members of the BCL-2 family share BCL-2 homology motifs (BH1 to BH4)[9]. Anti-apoptotic proteins (BCL-2, BCL2A2, MCL-1, and BCL2L1 (BCL-xL), BCL-w, BFL-1/A1) sequester pro-apoptotic proteins by binding to its BH3 motifs[9-13]. Pro-apoptotic proteins consist of BH3-only proteins and effector proteins, BAK and BAX, which have BH1-4 motifs. BH3-only proteins act as sensitizers (BAD, BIK, HRK, NOXA) or activators (BIM, BID, PUMA) of apoptosis[10,11,13]. BH3-only sensitizer proteins are unable to activate downstream effector proteins (BAX, BAK) directly. However, they are able to “sensitize” cells toward apoptosis by binding to BCL-2 anti-apoptotic protein, releasing bound BAX or BAK or BH3-only activator protein[14]. Upon activation by bound BH3-only activator proteins, effector protein oligomerizes, leading to increase mitochondrial outer membrane permeabilization (MOMP) and initiation of cytochrome c mediated intrinsic apoptosis[9-13].
Dysregulation and imbalance of BCL-2 family members controlling apoptosis are commonly found in multiple hematologic malignancies[12]. Likewise, the BCL-2 family has an essential role in mediating AML survival and chemoresistance. Previous studies have demonstrated that an increase in the level of anti-apoptotic proteins, including BCL-2, is associated with chemotherapy resistance[15-17]. BCL-2 also supports the survival of leukemic stem cells in AML, and its inhibition induces the death of quiescent leukemic stem cells[18]. Hence, BCL-2 is an important therapeutic target. Venetoclax, a BH3 mimetic, binds to a BH3-binding groove of BCL-2 protein with high selectivity[8]. This relieves inhibition of BCL-2 toward BAX and BAK, resulting in cell death[9]. Preclinical studies demonstrated high anti-tumor activity of Venetoclax in AML, which facilitated further clinical studies[16,19]. In an initial phase 2 study, Venetoclax monotherapy produced modest responses in heavily treated relapsed or refractory (R/R) AML patients with an overall response rate (ORR) of 19%[4]. As AML may not depend on BCL-2 for its survival or the dependency may evolve during tumor progression and after therapy[20], a method to assess BCL-2 family dependency is critically needed to predict sensitivity to Venetoclax[4,13,21,22]. BH3 profiling is performed by exposing mitochondria to a specific BH3 peptide, followed by measurement of cytochrome c release and MOMP[23]. Addiction to a specific BCL-2 anti-apoptotic protein is inferred from cellular apoptotic sensitivity when exposed to different BH3 peptides[23].
Genomic biomarkers associated with Venetoclax sensitivity
Early phase 2 study on Venetoclax monotherapy suggested spliceosomal mutation in SRSF2/ZRS2 and IDH1/2 as predictors for Venetoclax sensitivity[4]. Ten out of 11 patients with baseline SRSF2 or ZRSR2 mutation had a measurable reduction in bone marrow (BM) blast after treatment with Venetoclax[24]. SRSF2 genes were found to induce alternative splicing of apoptosis regulating genes and modulate the expression of BCL-2 family proteins, which may increase sensitivity to Venetoclax[24,25].
In preclinical models, IDH mutation in AML conferred high BCL-2 dependence and sensitivity to BCL-2 inhibition. The mechanism involved oncometabolite (R)-2 hydroxyglutarate (2-HG), which inhibits mitochondrial cytochrome c oxidase, causing a reduction of mitochondrial threshold for apoptosis induction[26]. A recent study by Stuani et al.[27] described additional mechanisms behind IDH mutant sensitivity to BCL-2 inhibitor through inhibition of mitochondrial respiration. Mutant IDH cells and patient-derived xenografts (PDX) were found to have a larger capacity for mitochondrial oxidative phosphorylation (OXPHOS), as evident by increased electron transport chain (ETC) complex I activity, NADH production by tricarboxylic acid cycle enzymes, mitochondrial ATP content and oxygen consumption rate[27]. Gene set enrichment analysis on IDH1 mutant cells also showed enhanced fatty acid oxidation (FAO) gene signature, especially CPT1a, an acyl-carnitine transporter during FAO, and its transcriptional regulator, CEBPα[27]. While 2-HG was shown to drive CPT1a and CEBPα-dependent FAO and OXPHOS, abrogation of 2-HG production by IDH inhibitor did not impact FAO rate or OXPHOS in the treated AML cell lines, suggesting maintenance of OXPHOS phenotype independent of 2-HG[27]. Inhibition of OXPHOS and IDH1 mutation subsequently showed synergistic cytotoxic activity and improved cell differentiation in vitro and in vivo[27].
Venetoclax, a BCL-2 inhibitor, was reported to target leukemia stem cells (LSCs) metabolism through reduction of ETC complex II activity and OXPHOS[28]. Consistent with preclinical data, IDH mutant cells showed heightened sensitivity upon OXPHOS inhibition by Venetoclax therapy[27]. In the phase 2 trial of a single agent Venetoclax, IDH1/2 mutated R/R AML subset reached a higher objective response rate of 33% as compared to 10% ORR in the IDH-wildtype AML subset[4]. Mirroring synergy seen in preclinical study[27], a preliminary analysis from an ongoing phase 1/2 clinical trial demonstrated encouraging composite complete remission of 75% with median overall survival (OS) of 9.7 months in the R/R cohort upon treatment with a combination of Ivosidenib (an IDH1 inhibitor), Venetoclax and Azacitidine “triplet”[29]. Future updates are anticipated to evaluate clinical characteristics and molecular predictors of response. In parallel, a phase 1b/2 study of Enasidenib (an IDH2 inhibitor) and Venetoclax combination is underway (NCT04092179) after demonstrated efficacy in preclinical studies[30,31].
Utilizing highly sensitive quantitative reverse transcription polymerase chain reaction (RT-qPCR), a clinical study by DiNardo et al.[32] found a strikingly high rate of mutation clearance in NPM1 mutated patients upon treatment with a combination of Venetoclax and HMA or LDAC (4/4 cases). As expected, a high molecular remission rate translated into an excellent survival with relapse-free survival not reached after a median follow-up of 20 months in patients who initially had persistent mutation despite intensive chemotherapy[33]. Unlike IDH mutated AML, the mechanistic basis of Venetoclax sensitivity in NPM1 mutated AML is not well understood[32]. However, clinical findings indicate NPM1 (without FLT3 co-mutation) as an important predictor of Venetoclax sensitivity.
ASXL1 mutation was recently found to drive response to Venetoclax in vitro. ASXL1 acts as an epigenetic regulator via PRC2-mediated chromatin modification to keep various genes in a repressed state[34]. Thus, ASXL1 mutation leads to aberrant activation of its target genes, including BCL2[34]. In preclinical study of Rahmani et al.[34], ATAC-sequencing analysis suggested higher chromatin accessibility on the BCL-2 locus of ASXL1 mutant KBM5 cells due to failure of PRC2 complex recruitment, resulting in BCL-2 overexpression[34]. BH3 profiling further showed higher BCL2 anti-apoptotic protein dependency in ASXL1 mutant cells, which explained ASXL1 sensitivity to Venetoclax in vitro[34]. Rahmani et al.[34] also demonstrated enhanced global cytosine methylation, which led to sensitivity to Azacitidine, a DNMT inhibitor. Thus, treatment of ASXL1 mutant cells with single agent Venetoclax or Azacitidine resulted in increased cell differentiation, decreased cell growth and viability[34]. Analysis of data from clinical trials is needed to prove this finding in the clinical setting.
VENETOCLAX RESISTANCE: PRECLINICAL STUDIES
Cellular mechanism of resistance to Venetoclax: other BCL-2 family proteins
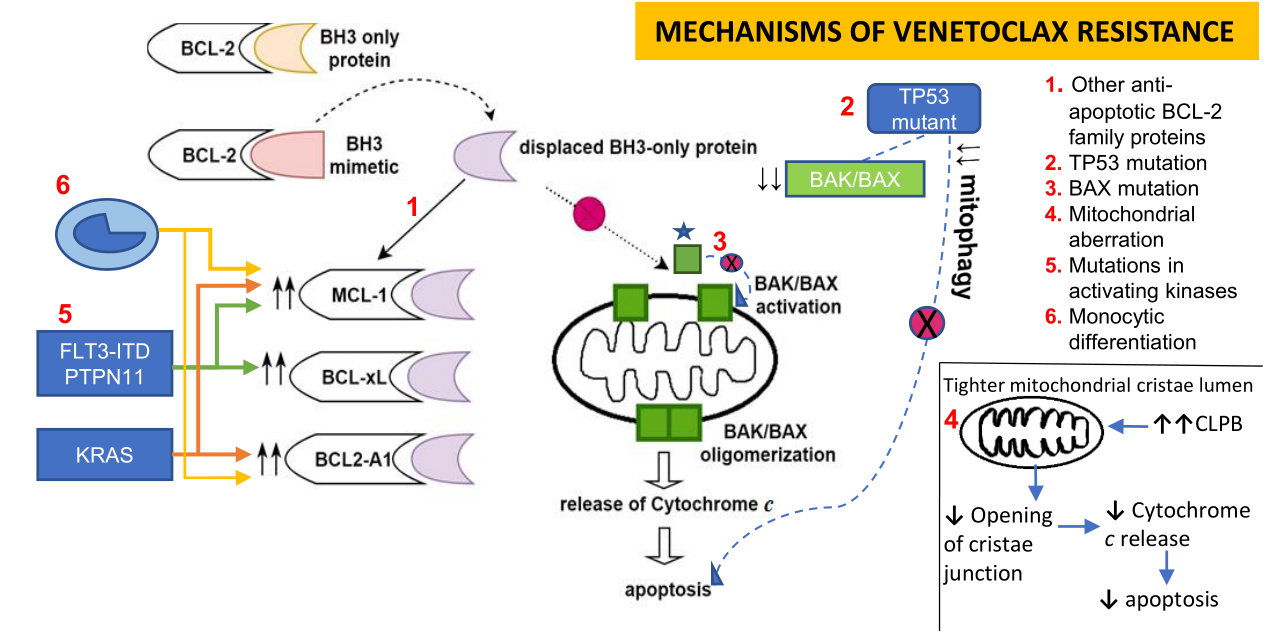
Dependencies on other anti-apoptotic BCL-2 family members, including BCL2-A1, MCL-1, and BCL-xL, have been demonstrated as the key contributors to primary or adaptive Venetoclax resistance[4,7,13,35-37]. Venetoclax resistance is associated with sequestration of BIM freed from Venetoclax binding to BCL-2 by other Bcl-2 family members, which consequently prevents apoptosis [Figure 1][38,39]. Preclinical work demonstrated lower expression of MCL-1 and BCL-xL in Venetoclax sensitive AML cell lines and lower BCL-2 protein levels in resistant cells[13]. When resistance was induced in AML cell lines through exposure to Venetoclax over several months, a shift was seen toward upregulation of MCL-1 and or BCL-xL with less dependency on BCL-2[35,40]. To investigate this potential mechanism, BCL-xL and MCL-1 were inactivated with BCL-xL inhibitor (WEHI-539) and short hairpin RNA (shRNA), respectively, which resulted in the restoration of Venetoclax sensitivity in resistant AML cell lines[35]. Utilizing BH3 profiling, a phase 2 study of Venetoclax monotherapy for R/R AML showed shorter durability of Venetoclax responses in patients with MCL-1 or BCL-xL dependence, suggesting that these mechanisms are operational in the clinical setting[4].

Figure 1. Binding of BH3 mimetic, Venetoclax to BCL-2 anti-apoptotic protein releases bound BH3-only protein, subsequently allowing interaction between BH3-only protein and BAK/BAX. Upregulation of MCL-1, BCL-xL, and BCL2-A1 confers Venetoclax resistance by sequestration of BH3 only proteins, preventing them from interacting with BAK/BAX and avoidance of apoptosis[35-40].
The knowledge around common co-dependencies in leukemia and cancer led to the exploration of selective BCL-xL and MCL-1 inhibitors. Navitoclax (ABT263), a predecessor of Venetoclax, possessed a high affinity for BCL-2, BCL-xL and BCL-w[41]. However, its clinical use has been limited by on-target toxicity of thrombocytopenia due to reliance on BCL-xl for platelet survival[42]. Recently there is a growing interest in the exploration of MCL-1 inhibitors, which have shown striking synergy with BCL-2 inhibitors in resistant AML cell lines and xenograft models[40,43,44]. Multiple MCL-1 inhibitors are under development, and several are being evaluated in clinical trials[45-47].
In recent studies, transcriptomic analysis of AML patients’ samples showed differential expression of BCL2A1 in resistant cells[36,37]. Upregulation of BCL2A1 expression correlated with higher Venetoclax’s area under the curve (AUC) and less apoptosis after treatment with Venetoclax with or without Azacitidine and Cytarabine. Knockdown of BCL2A1 restored apoptosis and reduced cell growth in the resistant AML cells without substantial effect on the CD34+ hematopoietic stem and progenitor cell (HSPC)[37]. Therefore, there is a potential for synergy between Venetoclax and BCL2A1 inhibitor in selective AML subsets, similar to Venetoclax and MCL-1 inhibitor[37,44].
Mutations in activating kinases and Venetoclax resistance
Activation of intracellular signaling pathways by KRAS/PTPN1 or FMS-like tyrosine kinase 3 (FLT3) mutant proteins is postulated to induce Venetoclax resistance. Genomic biomarkers were analyzed before and after treatment with Venetoclax monotherapy[24]. Three out of 14 patients with pre-treatment FLT3-internal tandem duplication (ITD) and 4 out of 14 patients with PTPN11 mutation failed to achieve bone marrow blast reduction, suggesting intrinsic resistance to Venetoclax. In addition, a subset of patients at the time of relapse were found to harbor FLT3-ITD and/or PTPN11 mutations not identified prior to therapy, strongly indicating the emergence or selection of these mutations as secondary or acquired resistance[24].
FLT3-ITD mutation occurs in about 25% of adult AML cases and confers an adverse prognosis[27]. FLT3-ITD mutation promotes survival via activation of PI3K-protein kinase B (Akt), RAS-MAPK, and STAT5 pathway[14,38]. While the precise downstream molecular pathways are yet to be defined, FLT3-ITD mutation is known to induce higher expression of BCL-xL and MCL-1, which may contribute to Venetoclax resistance [Figure 2][48-50]. Several studies demonstrated the involvement of the molecular pathway downstream of FLT3 in modulating BCL-xL and MCL-1 expression. STAT5, which was activated by FLT3-ITD, but not by their wild-type counterpart[50], was found to regulate transcription of the BCL-xL gene[51]. Akt, a downstream substance of PI3K pathway, was shown to influence MCL-1 stabilization by inactivation of glycogen synthase kinase 3 (GSK3), leading to sequestration of BIM and prevention of MOMP[52]. Yoshimoto et al.[50] further reported the role of STAT5 and Akt in MCL-1 upregulation. Suppression of STAT5 by small interfering RNA (siRNA) reduced the level of MCL-1 protein and mRNA in FLT3-ITD+ MV4-11 cell lines. In addition to direct stimulation of MCL-1 transcription, STAT5 also enhances phosphorylation of Akt, which indirectly increases MCL-1 expression in FLT3-ITD cells[50].

Figure 2. FLT3-ITD mutation causes an increased level of BCL-xL and MCL-1 via activation of downstream PI3K-AKT, RAS-MAPK, and STAT5 pathways. AKT and ERK promoted inhibitory phosphorylation of GSK3, leading to a reduction of MCL1 ubiquitination and degradation. In addition to upregulation of MCL-1 and BCL-xL, STAT5 also increases MCL-1 indirectly through AKT activation. In summary, FLT3-ITD mutation confers Venetoclax resistance by upregulation of BCL-xL and MCL-1[48-53].
Several preclinical studies evaluated the efficacy of combining FLT3 inhibitor and Venetoclax in FLT3-ITD+ cell lines, patient samples, and xenograft models. Ma et al.[38] assessed the efficacy of Midostaurin (1st generation type 1 FLT3 inhibitor)[3] and Gilteritinib (2nd generation type 1 FLT3 inhibitor)[3] in combination with Venetoclax. Midostaurin or Gilteritinib in combination with Venetoclax synergistically induced apoptosis in FLT3-ITD+ cell lines and patient samples[38]. The combination of Gilteritinib and Venetoclax also resulted in higher survival of the MV4-11 xenograft model compared to either drug alone, with four out of five treated mice remaining disease-free on day 190[38]. A similar finding was reported by Zhu et al.[14] The combination of Gilteritinib with Venetoclax outperformed their respective monotherapy in halting the proliferation of FLT3-ITD+ cells and reducing tumor burden in the FLT3-ITD+ patient-derived xenograft (PDX) and resistant MOLM-14 model[14]. Another set of experiments with a combination of 2nd generation type 2 FLT-3 inhibitor (Quizartinib)[3] and Venetoclax in vitro and in vivo, resonated with prior studies[49]. Co-treatment with Quizartinib and Venetoclax in the xenograft model produced a longer response with a delay in tumor resurgence for up to three months post-treatment[24].
In these studies, mechanisms driving the synergy between FLT3 inhibitor and Venetoclax were interrogated. Ma et al.[38] and Zhu et al.[14] demonstrated that treatment with FLT3 inhibitor (Midostaurin or Gilteritinib) alone or in combination with Venetoclax reduced the expression of MCL-1 in vitro[14,38]. Utilizing western blot, reduced phosphorylation of ERK, AKT, STAT5 was seen after 24 hours of treatment with Gilteritinib or Quizartinib. This finding suggests FLT3 inhibition modulates MCL-1 by simultaneous suppression of multiple kinase pathways including RAS-MAPK, PI3K-AKT, and STAT5[14,38]. Co-immunoprecipitation assay in FLT-ITD+ cell lines further revealed decreased binding of BIM to MCL-1 and increased binding of BIM to BCL-2 after Gilteritinib treatment, while the opposite was seen with Venetoclax treatment[14,38]. Interestingly, co-treatment with Gilteritinib and Venetoclax also increased the binding of BIM to BAX without increasing BIM binding to other BCL-2 anti-apoptotic proteins, specifically BCL-xL[14]. Thus, combination therapy with FLT3 inhibitor and Venetoclax in vitro reduced BIM binding to both BCL-2 and MCL-1, liberating BIM to interact with BAX and induce apoptosis[14].
In addition to FLT3, mutations in other activating kinases can confer resistance to Venetoclax (so-called “signaling” mutations). Genomic data from primary patient samples in the BEAT AML database was analyzed along with Venetoclax AUC established in vitro[37]. Samples with KRAS or PTPN11 mutations were found to have higher Venetoclax AUC. Venetoclax resistance was reproduced when AML cell lines were transduced to overexpress G12D KRAS and A72D PTPN11[37]. Analysis with RT-PCR and immunoblot showed decreased BCL-2 and BAX with increased MCL-1 and BCL2A1 levels in G12D KRAS cells, as well as increased MCL-1 and BCL-xL levels in A72D PTPN11 cells [Figure 3][37]. G12D KRAS cells showed a reduction in cellular viability after treatment with MCL-1 inhibitors (AZD5991)[37]. However, neither BCL-2 blockade nor simultaneous blockade of BCL-2, BCL-w, and BCL-xL by ABT263 and ABT737 produced a similar response[37]. This finding implied KRAS mutation dependency on MCL-1 to drive Venetoclax resistance. In A72D PTPN11, AZD5991 was also capable of suppressing PTPN mutant cells, while partial response only was seen with BCL-2/Bcl-XL dual inhibitors ABT-263 and ABT 737. This finding suggests partial dependence of PTPN-induced Venetoclax resistance on MCL-1 and BCL-xL[37]. As expected, the combination of AZD5991 and Venetoclax showed synergy and fully rescued mutant cell lines from KRAS- and PTPN11-mediated resistance. Hence, combinatorial therapy with Venetoclax and MCL-1 inhibitor is expected to demonstrate high efficacy in AML patients harboring these mutations[37].

Figure 3. Both KRAS and PTPN11 mutations confer Venetoclax resistance. KRAS mutation causes upregulation of MCL-1 and BCL2A1, while PTPN11 mutation causes upregulation of MCL-1 and BCL-xL. KRAS mutation also downregulates BCL-2 and BAX[37].
Roles of TP53, BAX, and mitochondria in Venetoclax resistance
In a recent report, BAX variants were found by deep sequencing performed on samples derived from AML patients who relapsed after initially achieving remission with Venetoclax-based regimens, signifying acquired BAX mutation as adaptive Venetoclax resistance[54]. Reduced survival was also seen when BAX deficient OCI-AML3 cells were transplanted into the AML xenograft model[54]. BAX deficient cells and xenograft model were resistant to cell death induced by Venetoclax, MCL-1 inhibitor (S63845), or a combination of both[54]. This is contrary to a prior study which showed sensitivity of BAX knockout (KO) cells to a different MCL-1 inhibitor (AZD-5991) with similar resistance to Venetoclax and BCL-2/BCL-xL inhibitor (AZD-4320)[55]. Hence, particular attention to BAX mutant subsets is warranted in future studies to evaluate its impact on response to BH3 mimetics as single agents or combinations.
Genome-scale CRISPR/Cas9 screening identified BAX along with TP53 and PMA1P1 (NOXA) as genes whose inactivation confers Venetoclax resistance[40,55,56]. Gene enrichment and protein-protein interaction analysis identified these genes as an essential part of the mitochondrial apoptosis pathway[55,56]. TP53 is activated by cellular stress and functions as a transcription factor for genes controlling various cellular processes, including apoptosis and cell cycle arrest[55]. Several BH3-only proteins, including BAK, BAX, PUMA, and NOXA, are also TP53 target genes[55,57,58]. As expected, a lower level of BAK1, PUMA, and NOXA was observed in TP53 KO cells[55]. Interestingly, transcriptional changes were observed outside TP53 target genes with an increased ratio of BCL-xL/BCL-2, which may further confer Venetoclax resistance in TP53 KO cells[55].
Several preclinical studies demonstrated that TP53 mutated cells and xenograft models are resistant to single-agent therapy with Venetoclax[55,59] or MCL-1 inhibitor[55,59,60]. Contesting these findings, Thijssen et al.[60] reported observation that the lack of TP53 function did not preclude cell killing by sub-lethal BCL-2 or MCL-1 inhibitor monotherapy. However, this response was not durable as surviving TP53 deficient cells outgrew TP53 wild-type cells over a longer period of exposure, indicating a competitive survival advantage[60]. Delayed activation of BAX/BAK and subsequent apoptosis upon BH3 mimetics treatment was thought to cause reduced BH3 mimetics efficacy by lifting the early apoptosis threshold in the absence of TP53. Notably, simultaneous inhibition of BCL-2 and MCL-1 was able to overcome the apoptotic delay with improved durability of response[60], resonating with synergistic action described in past studies[55,59].
The impact of TP53 and BAX on mitochondrial function and morphology was evaluated. TP53 and BAX KO cells demonstrated less mitophagy when exposed to stress by a mitochondrial uncoupler and increased cellular respiration, reflected by higher production of cellular reactive oxygen species (ROS)[55]. Furthermore, TP53 and BAX KO cells also showed aberrant metabolic profiles with increased nucleotide synthesis and parallel decreases in glucose, pyruvate, amino acids, and urea cycle intermediates levels, suggesting priority shifting on carbon usage to support cancer cell proliferation[55]. These findings highlight the crucial role of mitochondria in mediating sensitivity or resistance to Venetoclax.
A deeper dive into mitochondrial biology found that aberrant mitochondrial architecture and bioenergetics were implicated in the apoptotic response to Venetoclax[56]. CRISPR/Cas9 screening further identified a negative association between Venetoclax resistance and mitochondrial chaperonin CLPB, which regulates mitochondrial cristae and metabolism[56]. Chen et al.[56] reported overexpression of CLPB in AML which led to tighter mitochondrial cristae lumen. On the other hand, loss of CLPB resulted in wider crista and stimulation of mitochondrial stress response, which in turn triggered cell cycle arrest and lowered the mitochondrial threshold for apoptosis[56]. A combination of CLPB deletion and Venetoclax was found to rescue Venetoclax resistant AML cells even in the presence of TP53 mutation[56]. Another genomic CRISPR-Cas9 knockout screen demonstrated a negative selection of DAP3, MRPL54, MRPL17, RBFA genes, which are part of the mitochondrial translation machinery[61]. When mitochondrial protein synthesis was blocked by tedizolid or doxycycline, reduction of AML cell viability was only observed upon co-treatment with Venetoclax, but not with Venetoclax or tedizolid or doxycycline alone[61]. A subsequent investigation by Sharon et al.[61] demonstrated that the combination of Venetoclax and tedizolid led to augmentation of the integrated stress response with associated reduction of OXPHOS, and decreased glycolytic capacity, resulting in ATP consumption and cell death.
Monocytic AML in Venetoclax resistance
Several preclinical studies suggested that AML may respond differently to Venetoclax-based therapies, depending on the blast differentiation stage. In the studies discussed below, AML was classified based on cell morphology according to the French, American, and British (FAB) classification system[62]. Guided by flow cytometry, ex vivo drug sensitivity testing on patient samples with primary AML showed a progressive increase in Venetoclax resistance through the cell maturation phase from the most primitive AML (M0) to monocytic AML (M5)[63,64]. Aligned with this observation, analysis of the BEAT AML dataset also noticed higher Venetoclax AUC in the leukemic blasts with high expression of CD14 and CLEA7A (gene encoding CD369) that are usually present in M4/M5 AML, suggesting Venetoclax resistance in AML with myelomonocytic or monocytic differentiation[37].
Further analysis also indicated different expression levels of alternative BCL-2 anti-apoptotic protein at the selected stages of cell maturation, which correlates with Venetoclax resistance. For instance, a gradual decline in BCL-2 expression with a concurrent increase in MCL-1 expression from M0 to M5 was observed, suggesting a lineage-associate switch to MCL-1 in monocytic AML[63,64]. In addition, the link between high expression of CD14 and CLEA7A, presence of KRAS mutation and increased BCL2A1 were seen in M4/5 AML[37,63,64]. Hence, Venetoclax resistance in AML with myelomonocytic differentiation may additionally be governed by BCL2A1 through upstream mutant KRAS[37,63].
Consistent with these laboratory predictions, analysis of matched patient samples at diagnosis and relapse post-Venetoclax revealed the coexistence of primitive and monocytic features at diagnosis, suggesting developmental heterogenicity within the leukemic blast[64]. Monocytic clone subsequently expanded under the selective pressure of Venetoclax and Azacitidine, with the contemporaneous vanishing of primitive population at the time of relapse[64]. Interestingly, a portion of relapsed monocytic subclone also showed increased HOXA9 expression (similar to that observed in MLL-rearranged leukemia)[65], which was not present in their parental clone[64]. However, both diagnosis and relapsed monocytic clone retained their MCL-1 dependency[64]. These findings suggest the potential role of menin inhibitor or MCL-1 inhibitor to overcome Venetoclax resistance in monocytic disease[64,65].
VENETOCLAX-BASED COMBINATION THERAPIES: LESSONS FROM CLINICAL STUDIES
Venetoclax-based combination therapies
Given low response rates to Venetoclax as a single agent in R/R AML Phase II study, further studies focused on combination regimens. Several lines of clinical trials investigated the combination of Venetoclax with HMA or low-dose cytarabine in both frontline and R/R settings[66-69]. Monotherapy with HMA was commonly used in unfit or older patients with AML, but its benefit is limited by low response rates of ~30% and modest median survival at 8-10 months[70-73]. The synergy between Venetoclax and HMA therapy was reported in preclinical studies. Bogenberger et al.[74,75] demonstrated increased sensitivity of HMA after inhibition of BCL-2 family proteins in AML samples. Tsao et al.[76] suggested Azacitidine has activity against MCL-1, and it induces synergism with Venetoclax. Subsequent clinical trials validated the clinical efficacy of these regimens.
In a phase 1b study, Venetoclax was combined with either Decitabine or Azacitidine to treat 145 older, newly diagnosed patients unfit for induction chemotherapy. Sixty-seven percent of patients achieved CR/CRi, including 60% of CR/CRi rates in patients with poor cytogenetics risk. The median OS in the study was 17.5 months, and the median duration of response was 11.3 months, with a tolerable safety profile[66]. Following phase 2 study of 10-day decitabine and Venetoclax for intensive chemotherapy ineligible patients, including 70 newly diagnosed AML and 55 R/R AML, confirmed acceptable safety profile and high efficacy with CR/CRi rate of 74%. The study showed a median OS of 18.1 months in newly diagnosed de-novo AML and six months in R/R AML[68]. A randomized phase 3 clinical trial (VIALE-A) comparing Azacitidine monotherapy to combination therapy with Azacitidine and Venetoclax as a frontline treatment confirmed improved response rate and survival in Azacitidine/Venetoclax treated patients compared with Azacitidine alone. CR/CRi rate was 66% in the combination therapy arm compared to 28% in patients who received single Azacitidine[77]. OS was longer in the combination group at 14.7 months with a median event-free survival (EFS) of 9.8 months compared to 9.6 months of OS and seven months of EFS in the single-agent Azacitidine group.
Cytarabine, a commonly used cytotoxic chemotherapy agent in AML, was likewise shown to have synergism with Venetoclax through inhibition of MCL-1, increased BH3 activity, and upregulation of pro-apoptotic protein, Bim[78]. A phase 1b/2 study of low-dose Cytarabine (LDAC) combined with Venetoclax in newly diagnosed older AML patients demonstrated excellent safety data and a high response rate with CR/CRi of 54% and median OS of 10.1 months[69]. This is encouraging compared to results from prior clinical studies of low-intensity cytotoxic therapy, which showed response rates of less than 20% and median OS of less than six months. Thus, the combination of LDAC and Venetoclax showed significantly improved efficacy compared to conventional chemotherapy[72,79]. In a randomized phase 3 trial in 211 newly diagnosed AML patients ineligible for intensive treatment, the combination of Venetoclax and LDAC was compared to LDAC with placebo. The combination group had a higher CR/CRi rate of 48% compared to 13% of the control group, with an EFS of 4.7 months in the combination group compared to 2 months in the control group[80]. The combination group also had longer OS at 7.2 months compared to 4.1 months in the control group, with a 25% reduction in risk of death[80].
In a phase 1b/2 study of Venetoclax with high-intensity chemotherapy FLAG-IDA (Fludarabine, Cytarabine, granulocyte colony-stimulating factor and Idarubicin), 68 younger AML patients with a median age of 46 years old were enrolled, with 29 de novo and 39 R/R patients[81]. The study demonstrated high clinical efficacy of the combined regimen with median survival not reached at the 12 months median follow-up, and a high minimal residual disease (MRD) negative CR rate of 96% in ND-AML patients and 69% in R/R AML patients. The majority of patients treated with this regimen proceeded towards allogeneic stem-cell transplant (69% of de novo AML and 46% of R/R AML patients)[81].
In another phase 1b study, Venetoclax was given in a dose-escalated fashion up to 600 mg prior to Cytarabine and Idarubicin infusion (5 + 2) in fit older patients with AML age 60 years and above (CAVEAT study)[82]. The combination regimen was well tolerable and efficacious, with ORR of 97% and 43% in de novo AML and secondary AML, respectively. Median OS was 11.2 months, with longer median OS seen in de novo AML of 31.3 months and 29.5 months in patients who achieved CR[82]. This study proved the acceptable tolerability and high efficacy of Venetoclax when given in combination with standard intensive chemotherapy in fit older adults with AML[82].
An ongoing phase 2 trial of the combination of Venetoclax with Cladribine plus LDAC alternating with hypomethylating agents in 48 newly diagnosed older AML patients demonstrated a high CR/CRi rate of 94% with MRD-negativity of 92%. Median OS was not reached over the 11-month median follow-up, and 24% of patients received an allogeneic stem-cell transplant[83].
Molecular landscape associated with response to Venetoclax combination therapies
Analysis of pre-treatment patients’ characteristics and paired AML samples pre-treatment and at the time of progression was performed to delineate the molecular patterns associated with treatment response to the combination of Venetoclax with HMA or with low dose Cytarabine (LDAC). In the frontline setting, AML patients with NPM1 or IDH1/2 mutations achieved high rates of CR/CRi and had sustained response for > 12 months[32,66]. Median duration of CR/CRi and median OS was not reached in IDH1/2 or NPM1 mutated patients[66]. Real-world experience supported the observations of IDH1/2 and NPM1 mutations as molecular predictors of response, even in the R/R setting[84,85].
It is noteworthy that deep molecular remissions were seen in patients with NPM1 mutation[32,33]. Highly sensitive RT-qPCR demonstrated MRD clearance in all recruited patients with NPM1 mutation upon treatment with Venetoclax and HMA or LDAC (4/4 cases)[32]. A subsequent study by Tiong et al.[33] also reproduced this finding in NPM1 mutated patients with molecular persistence or relapse after treatment with standard intensive chemotherapy. In addition, there was no molecular or hematological relapse observed in NPM1 mutated patients who achieved MRD negative remission after a median follow-up of 11.8 months[33]. Notably, two of the older adult patients unfit for stem cell transplantation (SCT) in this study were alive at 6 and 12 months of follow-up[33], validating Venetoclax-based therapy as a safe and well-tolerated therapy in the frail older adult population. This finding further highlights Venetoclax as a highly efficacious agent to treat NPM1 mutated AML even after intensive chemotherapy failure. A longer follow-up would be needed to evaluate the duration of MRD clearance with Venetoclax based therapy. If indeed durable, a Venetoclax-based regimen could be a well-tolerated and effective option in lieu of SCT in the unfit older population with NPM1 mutated AML. As deep remission was produced by Venetoclax-based therapy after failure of intensive chemotherapy treatment, further studies would be needed to assess Venetoclax-based therapy as a non-inferior or even a superior alternative to intensive chemotherapy in NPM1 mutated younger AML patients prior to transplantation.
Despite the general association with improved response to Venetoclax-based regimens, several points need to be taken into consideration with respect to NPM1 and IDH2 mutations. Despite responding well to combination therapy with Venetoclax and HMA or LDAC, persistent IDH2 mutation is commonly detectable in remission[32]. Although infrequent, new IDH2 mutation has been found at the time of relapse[84], raising questions if adding IDH inhibitor as part of maintenance or consolidative therapy would result in deeper remissions and longer survival. Future studies are needed to address these questions. While NPM1 mutation is generally eliminated by Venetoclax-based therapies, concurrent mutation with FLT3-ITD may confer resistance[32,65]. As NPM1 mutation is associated with heightened HOX/MEIS1 signature[32], the possible role of menin inhibitor merits further investigation and has been shown to enhance anti-AML efficacy of Venetoclax in NPM1-mutant AML models[86]. A combination of menin inhibitor (DS-1594) with Azacitidine and Venetoclax will be evaluated for NPM1c mutated R/R AML in an ongoing phase 1/2 clinical trial (NCT04752163)[65].
In keeping with the response seen with Venetoclax monotherapy, patients with spliceosomal mutation (SRSF2, U2AF1, SF3B1, ZRSR2) obtained a higher response rate to Venetoclax and HMA therapy compared to wild-type patients (CRi/CRh of 28% vs. 11%)[87]. Interestingly, the presence of co-mutation seems to influence differential responses seen among genes encoding spliceosome complex. SRSF2 appeared to be enriched with IDH mutations, especially IDH2, compared to the rest of spliceosomal genes cohort[87]. The impressive outcome was seen among SRSF2/IDH1/2 mutated patients with 1-year OS of 100% and 2-year OS of 88%[87]. Patients with SRSF2/IDH1/2 co-mutation also had statistically significant higher survival compared to SRSF2 mutated patients without concurrent IDH mutation, further substantiating the important presence of IDH mutation in driving Venetoclax sensitivity in the SRSF2 group[87]. On the other hand, U2AF1 mutation was enriched with RAS mutation with lower CRc and MRD negative CR rates[87].
Molecular determinants of resistance to Venetoclax combination therapies
In multiple clinical trials of Venetoclax and HMA or LDAC, TP53 mutation was found to be the predominant mutation in the AML patients that did not respond to Venetoclax-based therapy (7/20 cases)[32]. While most of the cases harbored a mutation in the DNA binding domain[88,89], different forms of TP53 abnormalities were observed, including single mutation without TP53 deletion (deletion 17p) and multiple mutations with or without chromosomal abnormality in the TP53 or non-TP53 domain[88]. Complex cytogenetics was seen in the majority of TP53 mutated cases[32,88,89], aligned with a known high prevalence of TP53 mutation in AML with complex karyotype[90]. These findings underscore TP53 mutation and complex cytogenetics as the most common cause of primary resistance to Venetoclax combination therapy.
Validating preclinical findings[55,59,60], TP53 mutation was associated with significantly lower response rates to Venetoclax and Decitabine combination therapy compared to wild-type group (ORR of 66% vs. 89%, CR/CRi of 57% vs. 77%, and MRD negative rate of 29% vs. 59%)[88]. There was a trend toward improved survival in patients who responded or initially responded prior to relapse compared to primary refractory cases. In responder cases, TP53 mutated AML cohort had a shorter duration of response of 3.5 months vs. not reached in the TP53 wild-type AML cohort[88]. These lower responses translated into poor survival with 60-day mortality and median OS of 26% and 5.2 months in the TP53 mutated patients compared to 4% and 19.4 months in the wild-type patients. Interestingly, TP53 mutation burden [variant allele frequency (VAF)] was not proven to be a predictor of response in this mutation cohort, although gain in VAF may be seen at relapse[88]. In line with this finding, single-cell DNA sequencing on AML patient samples upon relapse to Venetoclax-based therapy showed expansion of clones containing TP53 mutation under the selective pressure of therapy with a Venetoclax-based regimen[32]. While TP53 mutation may be detected at diagnosis, expansion of new TP53 variants is frequently detected at the time of relapse, with or without concurrent structural loss of 17p locus, suggesting a selection of clones with biallelic TP53 perturbations[32]. Given lower response rates, shorter duration of response, and poor survival, TP53 mutated patients do not derive long-term benefits from Venetoclax-based therapies and should be offered clinical trials whenever feasible. Research to develop novel agents and treatment strategies is urgently needed given the poor prognosis with limited treatment options in this population.
A focused analysis of relapse patterns after Venetoclax-based combination therapy reported the most common emerging mutations in genes involved in signaling pathways (NF1, FLT3-ITD, NRAS, JAK1), in line with the above-mentioned preclinical findings; RNA splicing (U2AF1, U2AF2, SRSF2, ZRSR2), and transcription factors (IKZF1, SETBP1, RUNX1, STAT5A), followed by tumor suppressors (TP53, WT1), and epigenetic modifiers (BCOR, CREBBP)[7]. At relapse, concurrent expansion of clones with different types of activating kinase mutations was shown, including FLT3-ITD, FLT3-TKD, FLT3 N676, RAS, CBL[32]. The emergence of multiple new mutations was also observed in a study by Stahl et al.[84], corroborating previous findings. These observations indicate that adaptive resistance may be governed by the complex interaction between various clones rather than single driver mutation. The polyclonal nature of the clonal expansion adds a tremendous challenge to salvage management of AML patients relapsing post Venetoclax-based induction, which is currently associated with extremely poor outcomes and median survival of less than three months[7,32].
Future directions and strategies to mitigate resistance
Current understanding of the mechanisms behind Venetoclax resistance identified in preclinical studies led to the development of several combination strategies that have entered clinical trials, as summarized in Table 1. Amongst these regimens, the combination of Venetoclax with FLT-3 tyrosine kinase inhibitors (TKIs) is under rigorous clinical investigation[91,92]. In a phase 1b trial, the combination of Gilteritinib and Venetoclax reached an ORR of 90% in FLT-3 mutated R/R AML, with similarly high responses in patients who failed prior TKIs[91]. Preliminary result from the “triplet” with Quizartinib, Decitabine and Venetoclax is promising, with a composite response rate (CRc) of 69% and median OS of 7.1 months in the R/R setting, while median OS was not reached in the frontline setting[92]. Recruitment is ongoing for Quizartinib as Venetoclax “doublet” (NCT03735875) or “triplet” (NCT03661307) therapy, and updated results are eagerly anticipated[92,93].
Clinical trials evaluating venetoclax-based combination regimens
| Drug Regimen | Treatment Category | Mutation (if required for eligibility) | Clinicaltrials.gov identifier | Target Number of patient enrollment++ | Phase | Year of study initiation |
| Azacitidine + Venetoclax | Frontline | NCT03466294 | 42 | II | 2018 | |
| Venetoclax + Decitabine | Both+ | NCT03404193 | 400 | II | 2018 | |
| ASTX727 (Decitabine and Cedazuridine) + Venetoclax | Both+ | NCT04657081 | 124 | I/II | 2021 | |
| ASTX727 (Decitabine and Cedazuridine) + Venetoclax | Both+ | NCT04746235 | 40 | II | 2021 | |
| Venetoclax + Cladribine + LDAC induction followed by Cladribine+ LDAC + Azacitidine | Frontline | NCT03586609 | 85 | II | 2018 | |
| LDAC + Venetoclax^^ | Frontline | NCT02287233 | 94 | I/II | 2014 | |
| LDAC + Venetoclax vs LDAC + placebo | Frontline | NCT03069352 | 211 | III | 2017 | |
| CPX-351 (Liposome-encapsulated Daunorubicin-Cytarabine) + Venetoclax | Both+ | In RR subset, (+) RAS pathway activating mutation: KIT, HRAS/NRAS/KRAS, BRAF, CBL or PTPN11 or loss of function mutation of NF1 | NCT03629171 | 52 | II | 2018 |
| Ivosidenib (IDH1 inhibitor) + Venetoclax +/- Azacitidine | Both+ | IDH1+ | NCT03471260 | 30 | I/II | 2018 |
| Enasidenib (IDH2 inhibitor) + Venetoclax | Both+ | IDH2 (+) | NCT04092179 | 48 | I/II | 2020 |
| Gilteritinib (FLT3 inhibitor) + Venetoclax^^ | Salvage | NCT03625505 | 61 | I | 2018 | |
| Gilteritinib (FLT3 inhibitor) + Azacitidine + Venetoclax | Salvage | FLT3 | NCT04140487 | 42 | I/II | 2019 |
| Gilteritinib (FIT3 inhibitor) + ASTX727 (Decitabine and Cedazuridine) + Venetoclax | Salvage (phase I), Both (phase II)+ | FLT3 | NCT05010122 | 42 | I/II | 2021 |
| Quizartinib (FLT3 inhibitor) + Venetoclax | Salvage | FLT3 | NCT03735875 | 32 | I/II | 2019 |
| Quizartinib (FLT3 inhibitor) + Decitabine + Venetoclax | Both+ | FLT3 | NCT03661307 | 52 | I/II | 2018 |
| S64315 (MCL-1 inhibitor) + Venetoclax | Salvage | NCT03672695 | 40 | I | 2018 | |
| AZD5991 (MCL-1 inhibitor) + Venetoclax** | Salvage | NCT03218683 | 144 | I/II | 2017 | |
| Pevonedistat (NAE inhibitor) +/- Venetoclax + Azacitidine | Frontline | NCT03862157 | 40 | I/II | 2019 | |
| Cobimetinib (MEK inhibitor) + Venetoclax; Idasanutlin (MDM2 inhibitor) + Venetoclax^^ | Salvage | NCT02670044 | 88 | I | 2016 | |
| Trametinib (MEK inhibitor) + Azacitidine + Venetoclax | Both+ | (+) RAS pathway activating mutation in R/R subset | NCT04487106 | 40 | II | 2020 |
| Dinaciclib (CDK inhibitor) + Venetoclax | Salvage | NCT03484520 | 48 | I | 2018 | |
| Alvocidib (CDK inhibitor) + Venetoclax^^ | Salvage | NCT03441555 | 36 | I | 2018 | |
| CYC065 (CDK inhibitor) + Venetoclax | Salvage | NCT04017546 | 25 | I | 2019 | |
| APR-246 + Venetoclax +/- Azacitidine | Frontline | TP53 + | NCT04214860 | 51 | I | 2019 |
| Magrolimab + Venetoclax + Azacitidine or MEC or CC-486 (oral Azacitidine) | Both+ | NCT04778410 | 164 | II | 2021 | |
| Magrolimab + Azacitidine + Venetoclax | Frontline (phase I), Salvage (phase II) | NCT04435691 | 98 | I/II | 2021 | |
| ALX148 (Evorpacept) + Venetoclax + Azacitidine | Both+ | NCT04755244 | 97 | I/II | 2021 | |
| TTI-622 (SIRPα-IgG4 Fc) + AZA +/-VEN | Frontline | TP53 +/- | NCT03530683 | 150 | I | 2018 |
| Ph I/IIb DS-1594 (Menin inhibitor) +/- Azacitidine + Venetoclax or miniHCVD | Salvage | Presence of MLL rearrangement, NPM1 (+) | NCT03735875 | 32 | I/II | 2019 |
MCL-1 undoubtfully plays an important role in mediating Venetoclax resistance through numerous upstream mediators, both genomic and epigenetic. Hence, targeting MCL-1 directly or indirectly is rational to improve sensitivity to Venetoclax[38-40,46]. The progress in replicating the success of direct MCL-1 inhibitor from bench to bedside has been hindered by observed troponin leak, possibly related to the regulation of mitochondrial homeostasis by MCL-1[47,94,95]. S64315, a direct MCL-1 inhibitor, is being evaluated in the early phase of clinical trial in combination with Venetoclax (NCT03672695)[46,47,93], and additional data on safety profile and efficacy is anticipated.
Several approaches have been pursued to target MCL-1 indirectly, including inhibition of MCL-1 transcription by CKD9 inhibitors, upregulation of NOXA to increase MCL-1 neutralization by NEDD8-activating enzyme (NAE) inhibitor, and targeting RAS-RAF-MEK-ERK (MAPK) pathway to promote MCL-1 degradation by MEK inhibitor and MDM2 inhibitor (a P53 activator)[53,96-98]. Based on synergy seen in the preclinical studies[53,96,98], multiple clinical trials evaluating the combination of Venetoclax and these novel small molecule inhibitors are being conducted, as summarized in Table 1. Several clinical trials have published their preliminary finding. In the newly diagnosed secondary AML population, the combination of Pevonidostat (NAE inhibitor), Azacitidine and Venetoclax (NCT03862157) was safe and efficacious with a CR/CRi rate of 70% and 6-months OS of 82%[99]. A phase 1 clinical trial (NCT02670044) of Venetoclax and Cobimetinib (MEK inhibitor) or Idasanutlin (MDM2 inhibitor) is completed. In R/R/ AML, a preliminary result showed an ORR of 18% in Venetoclax and Cobimetinib cohort[100]. However, Venetoclax 600 mg and Idasanutlin 200 mg arm achieved an ORR of 38%[100]. This finding is encouraging as the achieved response rate was higher compared to Venetoclax monotherapy or combination with LDAC or HMA in R/R population[4,84]. In terms of toxicity, gastrointestinal side effects were predominant with diarrhea, nausea, and vomiting, more with the Cobimetinib combination than idasanutlin[100].
TP53 mutation confers Venetoclax resistance and is associated with worse outcomes[55,84]. MDM2 inhibitor activates wild-type p53 by disrupting p53-MDM2 interaction, which consequently prevents proteosomal p53 degradation, increases p53-regulated, and reduces p53 nuclear export[53,101]. However, MDM2 inhibitor depends on intact p53 function to exert apoptotic activity[102]. In the setting of mutant TP53, APR-246 (eprenetapopt) was developed[103]. Through covalent binding of its reactive electrophile form (methylene quinuclidinone) to mutant p53, APR-246 (methylated PRIMA-1) restores its function and induces p53-dependent apoptosis[103]. A combination of APR-246, Venetoclax and Azacitidine is being evaluated in phase 1 clinical trial (NCT04214860)[93].
Another strategy is harnessing the innate immune system. Leukemic cells evade macrophage-mediated phagocytosis by overexpressing CD47 which binds to the signal regulatory protein alpha (SIRPα) receptor on the macrophage to produce a “do not eat me” signal[3,104]. CD47 becomes a potential therapeutic target as it is highly expressed on LSCs[105]. Several compounds have been developed to block CD47 and SIRPα interaction[104]. Magrolimab (Hu5F9-G4), an anti-CD47 antibody, was the first one to enter the clinical phase[104]. In a phase 1 study, Magrolimab was well tolerated and reduced LSC fraction in patients who achieved a response[106]. Anemia was a notable on-target effect managed by priming and maintenance dose[106]. Importantly, Magrolimab was not observed to augment Azacitidine’s toxicity when used in combination[104,106]. This suggests that adding another cytotoxic agent like Venetoclax in combination with Magrolimab is likely safe and synergistic due to the enhanced pro-phagocytic signal[104,106,107]. Clinical trials on the combination of Venetoclax and anti-CD47 antibody (Magrolimab, ALX148), or SIRPαFc fusion protein (TTI-622) are ongoing to validate the safety and efficacy of this novel immunotherapy regimen [Table 1].
Being protected by the bone marrow microenvironment (BMM), eliminating leukemia stem cells (LSCs) is challenging to any cytotoxic regimen, including Venetoclax[108]. In addition to protection, BMM also provides a survival signal and promotes stemness features in resistant AML cells[109,110]. Yu et al.[110] recently discovered the role of CD44, an adhesion molecule expressed by LSCs, in governing Venetoclax resistance induced by stimulation of the CXCR4-CXCL12 pathway. Higher MCL-1 expression was also observed after CXCL12 stimulation, which was abrogated by CD44 knockout[110]. Loss of CD44 function also reduced the viability of Venetoclax resistant cells[110]. With several small molecule inhibitors in development[111], a combination of CD44 or CXCR4-CXC12 pathway blockade with Venetoclax-based therapies may be worth pursuing.
CONCLUSIONS
The advent of Venetoclax as a BCL-2 inhibitor has transformed AML management. Multiple Venetoclax-based combination therapies are developed based on current knowledge about mechanisms of responsiveness and resistance to Venetoclax. The heterogenous nature of the disease with rapid and divergent clonal evolution poses a unique challenge when it comes to designing an optimal maintenance or salvage therapy. Molecular profiling would be helpful in personalizing treatment plans, especially in selecting a frontline regimen and prior to changing therapy upon MRD persistence, refractoriness or disease relapse.
Several treatment approaches are being evaluated in clinical trials, with remaining questions to be addressed in the future, all to tackle resistance and improve the durability of response. Current strategies are directed toward maximizing induction therapy by the introduction of “triplets” therapy which incorporates several novel targeted therapies to produce a deeper response upfront with the hope to prevent the emergence of mutation from the surviving clone. Sequential therapy is also of great interest, which can be done empirically [such as Cladribine + LDAC + Venetoclax alternating with Azacitidine+Venetoclax (NCT03586609)] or adaptively tailored to the molecular signature of the resistance clones. Each approach, however, comes with its own challenge. Triplets therapy may only be suitable for selected genomic subsets and may require Venetoclax’s dose adjustment to avoid prolonged myelosuppression. Adaptive sequential therapy is an attractive option. However, this strategy requires sensitive molecular techniques for early detection of rising clone(s), which are not readily available at the moment. Reliance on the limited targetable options further hinders its applicability. Hence, the most feasible option currently is a “shot-gun” combo approach aiming at avoiding resistance through the rotating nature of chemo- or immune-therapy agents with different mechanisms of action, aided by Venetoclax as a universal sensitizer.
Considering these limitations, intensive multi-agent chemotherapy and Venetoclax combination would be the best approach for fit younger AML patients currently as a bridge to stem cell transplantation. Immune-based therapy is another promising approach, given its theoretical efficacy across genomic subsets. Current research is focusing on the benefit of immune-based therapy as “MRD erasers” in maintenance or consolidation therapy in patients with a low disease burden[2,3]. Several agents are currently in development, such as bispecific T-cell engagers (BiTEs), NK engagers, cancer antigen vaccines, and cellular therapies[2,3]. With multiple novel agents added to the AML armamentarium, additional questions remained unanswered, including the efficacy and duration of response of new combination regimens, the molecular pattern of drug response and resistance, duration of therapy after achieving response to treatment, and response to prior regimen after therapy interruption or discontinuation. Future research would be crucial in addressing these questions to evaluate how to optimally use these different types of therapeutic strategies and to identify patients that benefit the most.
DECLARATIONS
Authors’ contributionsConceptualized, wrote and edited manuscript: Ong F, Kim K, Konopleva M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestMarina Konopleva: Consultant for AbbVie, Genentech, F. Hoffman La-Roche, Stemline Therapeutics, Amgen, Forty-Seven, KisoJi, Janssen; serves as advisory board member for Stemline Therapeutics, F. Hoffman La-Roche, Janssen; holds shares from Reata Pharmaceuticals; honoraria from Forty-Seven and F. Hoffman La-Roche; research funding from AbbVie, Genentech, F. Hoffman La-Roche, Eli Lilly, Cellectis, Calithera, Stemline Therapeutics, Ablynx, Agios, Ascentage, Astra Zeneca, Rafael Pharmaceutical, Sanofi, and Forty-Seven.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Howlader N NA, Krapcho M, Miller D, (eds), et al. SEER Cancer Statistics Review, 1975-2017. National Cancer Institute. Available from: https://seer.cancer.gov/csr/1975_2017/ [Last accessed on 6 Apr 2022].
2. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424-47.
3. Short NJ, Konopleva M, Kadia TM, et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov 2020;10:506-25.
4. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 2016;6:1106-17.
5. Food and Drug Administration. FDA grants regular approval to venetoclax in combination for untreated acute myeloid leukemia. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-venetoclax-combination-untreated-acute-myeloid-leukemia [Last accessed on 6 Apr 2022].
6. DiNardo CD, Rausch CR, Benton C, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol 2018;93:401-7.
7. Maiti A, Rausch CR, Cortes JE, et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica 2021;106:894-8.
8. Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19:202-8.
9. Roberts AW. Therapeutic development and current uses of BCL-2 inhibition. Hematology Am Soc Hematol Educ Program 2020;2020:1-9.
10. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma 2017;58:1-17.
11. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 2018;25:65-80.
12. Kapoor I, Bodo J, Hill BT, Hsi ED, Almasan A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis 2020;11:941.
13. Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014;4:362-75.
14. Zhu R, Li L, Nguyen B, et al. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct Target Ther 2021;6:186.
15. Campos L, Rouault JP, Sabido O, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81(11):3091-3096. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7684624 [Last accessed on 6 Apr 2022].
16. Delia D, Aiello A, Soligo D, et al. bcl-2 proto-oncogene expression in normal and neoplastic human myeloid cells. Blood. 1992;79(5):1291-1298. Available from: https://www.ncbi.nlm.nih.gov/pubmed/1536952 [Last accessed on 6 Apr 2022].
17. Lauria F, Raspadori D, Rondelli D, et al. High bcl-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia 1997;11:2075-8.
18. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329-41.
19. Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 2015;7:279ra40.
20. Bensi L, Longo R, Vecchi A, et al. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica. 1995;80(2):98-102. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7628759 [Last accessed on 6 Apr 2022].
21. Ishizawa J, Kojima K, McQueen T, et al. Mitochondrial profiling of acute myeloid leukemia in the assessment of response to apoptosis modulating drugs. PLoS One 2015;10:e0138377.
22. Niu X, Wang G, Wang Y, et al. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014;28:1557-60.
23. Salah HT, DiNardo CD, Konopleva M, Khoury JD. Potential biomarkers for treatment response to the bcl-2 inhibitor venetoclax: state of the art and future directions. Cancers (Basel) 2021;13:2974.
24. Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol ;2018:E202-5.
25. Crews LA, Balaian L, Delos Santos NP, et al. RNA Splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell 2016;19:599-612.
26. Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 2015;21:178-84.
27. Stuani L, Sabatier M, Saland E, et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J Exp Med 2021;218:e20200924.
28. Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018;24:1859-66.
29. Lachowiez CA, Borthakur G, Loghavi S, et al. Phase Ib/II study of the IDH1-mutant inhibitor ivosidenib with the BCL2 inhibitor venetoclax +/- azacitidine in IDH1-mutated hematologic malignancies. JCO 2020;38:7500-7500.
30. Cathelin S, Sharon D, Subedi A, et al. Combination of enasidenib and venetoclax shows superior anti-leukemic activity against IDH2 mutated AML in patient-derived xenograft models. Blood 2018;132:562-562.
31. Daver N, Wei AH, Pollyea DA, Fathi AT, Vyas P, DiNardo CD. New directions for emerging therapies in acute myeloid leukemia: the next chapter. Blood Cancer J 2020;10:107.
32. DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020;135:791-803.
33. Tiong IS, Dillon R, Ivey A, et al. Venetoclax induces rapid elimination of NPM1 mutant measurable residual disease in combination with low-intensity chemotherapy in acute myeloid leukaemia. Br J Haematol 2021;192:1026-30.
34. Rahmani NE, Ramachandra N, Sahu S, et al. ASXL1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J 2021;11:157.
35. Lin KH, Winter PS, Xie A, et al. Targeting MCL-1/BCL-XL Forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep 2016;6:27696.
36. Bisaillon R, Moison C, Thiollier C, et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2020;34:63-74.
37. Zhang H, Nakauchi Y, Köhnke T, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer 2020;1:826-39.
38. Ma J, Zhao S, Qiao X, et al. Inhibition of Bcl-2 Synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer Res 2019;25:6815-26.
39. Niu X, Zhao J, Ma J, et al. Binding of released bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res 2016;22:4440-51.
40. Zhang Q, Riley-Gillis B, Han L, et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct Target Ther 2022;7:51.
41. Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008;68:3421-8.
42. Schoenwaelder SM, Jarman KE, Gardiner EE, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011;118:1663-74.
43. Moujalled DM, Pomilio G, Ghiurau C, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2019;33:905-17.
44. Ramsey HE, Fischer MA, Lee T, et al. A Novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov 2018;8:1566-81.
45. Wang H, Guo M, Wei H, Chen Y. Targeting MCL-1 in cancer: current status and perspectives. J Hematol Oncol 2021;14:67.
46. Roberts AW, Wei AH, Huang DCS. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 2021;138:1120-36.
47. Wei AH, Roberts AW, Spencer A, et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev 2020;44:100672.
48. Kasper S, Breitenbuecher F, Heidel F, et al. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J 2012;2:e60.
49. Singh Mali R, Zhang Q, DeFilippis RA, et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021;106:1034-46.
50. Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009;114:5034-43.
51. Dumon S, Santos SC, Debierre-Grockiego F, et al. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene 1999;18:4191-9.
52. Young AI, Timpson P, Gallego-Ortega D, Ormandy CJ, Oakes SR. Myeloid cell leukemia 1 (MCL-1), an unexpected modulator of protein kinase signaling during invasion. Cell Adh Migr 2018;12:513-23.
53. Pan R, Ruvolo V, Mu H, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell 2017;32:748-760.e6.
54. Moujalled DM, Brown FC, Pomilio G, et al. Acquired mutations in BAX confer resistance to BH3 mimetics in acute myeloid leukemia. Blood 2020;136:7-8.
55. Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov 2019;9:910-25.
56. Chen X, Glytsou C, Zhou H, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov 2019;9:890-909.
57. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 2018;25:104-13.
59. Carter BZ, Mak PY, Tao W, et al. Co-Targeting MCL-1 and BCL-2 is highly synergistic in BH3 mimetic- and venetoclax/hypomethylating agent-resistant and TP53 mutated AML. Blood 2020;136:7-7.
60. Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021;137:2721-35.
61. Sharon D, Cathelin S, Mirali S, et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med 2019;11:eaax2863.
62. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976;33:451-8.
63. Kuusanmäki H, Leppä AM, Pölönen P, et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 2020;105:708-20.
64. Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov 2020;10:536-51.
65. Issa GC, Ravandi F, DiNardo CD, Jabbour E, Kantarjian HM, Andreeff M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021;35:2482-95.
66. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019;133:7-17.
67. Jozal W Moore, Aryeh Pelcovits, John L Reagan. Azacitidine and venetoclax in AML. N Engl J Med 2020;383:2087-9.
68. Dinardo CD, Maiti A, Rausch CR, et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial. The Lancet Haematology 2020;7:e724-36.
69. Wei AH, Strickland SA Jr, Hou JZ, et al. Venetoclax Combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol 2019;37:1277-84.
70. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291-9.
71. Thépot S, Itzykson R, Seegers V, et al. Groupe Francophone des Myélodysplasies (GFM). Azacitidine in untreated acute myeloid leukemia: a report on 149 patients. Am J Hematol 2014;89:410-6.
72. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670-7.
73. He PF, Zhou JD, Yao DM, et al. Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: A systematic review and meta-analysis. Oncotarget 2017;8:41498-507.
74. Bogenberger JM, Kornblau SM, Pierceall WE, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014;28:1657-65.
75. Bogenberger JM, Delman D, Hansen N, et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma 2015;56:226-9.
76. Tsao T, Shi Y, Kornblau S, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol 2012;91:1861-70.
77. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020;383:617-29.
78. Samra B, Konopleva M, Isidori A, Daver N, DiNardo C. Venetoclax-based combinations in acute myeloid leukemia: current evidence and future directions. Front Oncol 2020;10:562558.
79. Dennis M, Hills RK, Russell NH, et al. An evaluation of 17 years of low dose cytarabine as therapy for AML patients not fit for intensive treatment, including patients with adverse cytogenetics, shows improving survival, potential underutilisation and highlights the need for new therapy. Blood ;130(Supplement 1):3874-3874.
80. Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood 2020;135:2137-45.
81. DiNardo CD, Lachowiez CA, Takahashi K, et al. Venetoclax combined with flag-ida induction and consolidation in newly diagnosed and relapsed or refractory acute myeloid leukemia. J Clin Oncol 2021;39:2768-78.
82. Chua CC, Roberts AW, Reynolds J, et al. Chemotherapy and venetoclax in elderly acute myeloid leukemia trial (CAVEAT): a phase ib dose-escalation study of venetoclax combined with modified intensive chemotherapy. J Clin Oncol 2020;38:3506-17.
83. Kadia TM, Borthakur G, Pemmaraju N, et al. Phase II study of venetoclax added to cladribine + low dose AraC (LDAC) alternating with 5-azacytidine demonstrates high rates of minimal residual disease (MRD) negative complete remissions (CR) and excellent tolerability in older patients with newly diagnosed acute myeloid leukemia (AML). Blood 2020;136:17-9.
84. Stahl M, Menghrajani K, Derkach A, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv 2021;5:1552-64.
85. Wang YW, Tsai CH, Lin CC, et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann Hematol 2020;99:501-11.
86. Carter BZ, Tao W, Mak PY, et al. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood 2021;138:1637-41.
87. Lachowiez CA, Loghavi S, Furudate K, et al. Impact of splicing mutations in acute myeloid leukemia treated with hypomethylating agents combined with venetoclax. Blood Adv 2021;5:2173-83.
88. Kim K, Maiti A, Loghavi S, et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021;127:3772-81.
89. Aldoss I, Zhang J, Pillai R, et al. Venetoclax and hypomethylating agents in TP53-mutated acute myeloid leukaemia. Br J Haematol 2019;187:e45-8.
90. Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer 2005;43:227-38.
91. Perl AE, Daver NG, Pratz KW, et al. Venetoclax in combination with gilteritinib in patients with relapsed/refractory acute myeloid leukemia: a phase 1b study. Blood 2019;134:3910-3910.
92. Yilmaz M, Kantarjian HM, Muftuoglu M, et al. Quizartinib with decitabine and venetoclax (triplet) is highly active in patients with FLT3-ITD mutated acute myeloid leukemia (AML). JCO 2021;39:e19019-e19019.
93. Yue X, Chen Q, He J. Combination strategies to overcome resistance to the BCL2 inhibitor venetoclax in hematologic malignancies. Cancer Cell Int 2020;20:524.
94. Bolomsky A, Vogler M, Köse MC, et al. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J Hematol Oncol 2020;13:173.
95. Rasmussen ML, Taneja N, Neininger AC, et al. MCL-1 Inhibition by selective BH3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. iScience 2020;23:101015.
96. Bogenberger J, Whatcott C, Hansen N, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017;8:107206-22.
97. Knorr KL, Schneider PA, Meng XW, et al. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ 2015;22:2133-42.
98. Han L, Zhang Q, Dail M, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica 2020;105:697-707.
99. Short N BP, Dinardo C, et al. Preliminary Results Of A Phase I/II Study of azacitidine, venetoclax and pevonedistat in patients with secondary acute myeloid leukemia who are unfit for intensive chemotherapy. HemaSphere. 2020;4:232. Available from: https://library.ehaweb.org/eha/2020/eha25th/294475/nicholas.short.preliminary.results.of.a.phase.i.ii.study.of.azacitidine.html [Last accessed on 6 Apr 2022].
100. Daver N, Pollyea DA, Yee KW, et al. Preliminary results from a phase ib study evaluating BCL-2 inhibitor venetoclax in combination with MEK inhibitor cobimetinib or MDM2 inhibitor idasanutlin in patients with relapsed or refractory (R/R) AML. Blood 2017;130:813-813.
101. Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther 2019;12:2903-10.
102. Lu J, Guan S, Zhao Y, et al. Novel MDM2 inhibitor SAR405838 (MI-773) induces p53-mediated apoptosis in neuroblastoma. Oncotarget 2016;7:82757-69.
103. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis 2018;9:439.
104. Chao MP, Takimoto CH, Feng DD, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol 2019;9:1380.
105. Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009;138:286-99.
106. Sallman DA, Donnellan WB, Asch AS, et al. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: Initial phase 1b results. JCO 2019;37:7009-7009.
107. Donio MJ, Wilson WC, Darwech I, et al. Pre-clinical combination of AO-176, a highly differentiated clinical stage CD47 antibody, with either azacitidine or venetoclax significantly enhances DAMP induction and phagocytosis of acute myeloid leukemia. Blood 2020;136:9-10.
109. Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat 2009;12:103-13.
110. Yu X, Munoz-Sagredo L, Streule K, et al. CD44 loss of function sensitizes AML cells to the BCL-2 inhibitor venetoclax by decreasing CXCL12-driven survival cues. Blood 2021;138:1067-80.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ong F, Kim K, Konopleva MY. Venetoclax resistance: mechanistic insights and future strategies. Cancer Drug Resist 2022;5:380-400. http://dx.doi.org/10.20517/cdr.2021.125
AMA Style
Ong F, Kim K, Konopleva MY. Venetoclax resistance: mechanistic insights and future strategies. Cancer Drug Resistance. 2022; 5(2): 380-400. http://dx.doi.org/10.20517/cdr.2021.125
Chicago/Turabian Style
Ong, Faustine, Kunhwa Kim, Marina Y. Konopleva. 2022. "Venetoclax resistance: mechanistic insights and future strategies" Cancer Drug Resistance. 5, no.2: 380-400. http://dx.doi.org/10.20517/cdr.2021.125
ACS Style
Ong, F.; Kim K.; Konopleva MY. Venetoclax resistance: mechanistic insights and future strategies. Cancer Drug Resist. 2022, 5, 380-400. http://dx.doi.org/10.20517/cdr.2021.125
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 134 clicks
Cite This Article 134 clicks

 Like This Article 30
likes
Like This Article 30
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.