
Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment
 0
0Abstract
Myeloid and lymphoid neoplasms with eosinophilia and FGFR1 rearrangements (MLN-eo FGFR1) disease is derived from a pluripotent hematopoietic stem cell and has a complex presentation with a myeloproliferative disorder with or without eosinophilia and frequently presents with mixed lineage T- or B-lymphomas. The myeloproliferative disease frequently progresses to AML and lymphoid neoplasms can develop into acute lymphomas. No matter the cell type involved, or clinical presentation, chromosome translocations involving the FGFR1 kinase and various partner genes, which leads to constitutive activation of downstream oncogenic signaling cascades. These patients are not responsive to treatment regimens developed for other acute leukemias and survival is poor. Recent development of specific FGFR1 inhibitors has suggested an alternative therapeutic approach but resistance is likely to evolve over time. Mouse models of this disease syndrome have been developed and are being used for preclinical evaluation of FGFR1 inhibitors. Cell lines from these models have now been developed and have been used to investigate the mechanisms of resistance that might be expected in clinical cases. So far, a V561M mutation in the kinases domain and deletion of PTEN have been recognized as leading to resistance and both operate through the PI3K/AKT signaling axis. One of the important consequences is the suppression of PUMA, a potent enforcer of apoptosis, which operates through BCL2. Targeting BCL2 in the resistant cells leads to suppression of leukemia development in mouse models, which potentially provides an opportunity to treat patients that become resistant to FGFR1 inhibitors. In addition, elucidation of molecular mechanisms underlying FGFR1-driven leukemias and lymphomas also provides new targets for combined treatment as another option to bypass the FGFR1 inhibitor resistance and improve patient outcome.
Keywords
INTRODUCTION
The involvement of chromosome region 8p11 in an atypical myeloproliferative disease was first described in 1992[1] and subsequently referred to[2,3] as 8p11 myeloproliferative syndrome (EMS). This syndrome is characterized cytogenetically by the presence of reciprocal chromosome translocations that invariably involve 8p11, often as the only cytogenetic abnormality[4], and is the only example of a malignancy that always involves abnormalities of FGFR1. At least 15 different chromosome translocations have been associated with this syndrome[5-7] but the invariant features are the translocation breakpoints occurring in intron 8 of FGFR1, just upstream of the kinase domain, and the presence of a dimerization/oligomerization domain in the partner translocated gene [Figure 1]. The most common rearrangements are t(8;13)(p11;q12), t(8;22)(p11;q11) and t(8;9)(p12;q33) giving rise to ZMYM2-FGFR1, BCR-FGFR1 and CNTRL-FGFR1 fusion kinases respectively. As a result of the rearrangement, an in-frame fusion event occurs in all cases, facilitating the activation of FGFR1 kinase as a result of dimerization and, since the partner genes are typically constitutionally active so too is the fusion kinase. The loss of the trans-membrane domain from FGFR1 in the fusion genes leads to constitutive and ligand-independent activation, which is responsible for the transformation of hematopoietic stem cells [Figure 2]. These rearrangements give rise to mixed lineage disease[8] suggesting a pluripotent stem cell origin, which led to a nomenclature of the Stem Cell Leukemia/Lymphoma (MLN-EO FGFR1) syndrome[9]. This syndrome has since been classified by the WHO[10] as myeloid and lymphoid malignancies associated with eosinophilia and FGFR1 rearrangement (MLN-eo FGFR1). Because of phenotypic overlap in clinical presentation, rearrangements involving PDGFRA and PDGFRB were added to the sub-classification[10,11], although there are still important clinical differences between each subgroup[6].
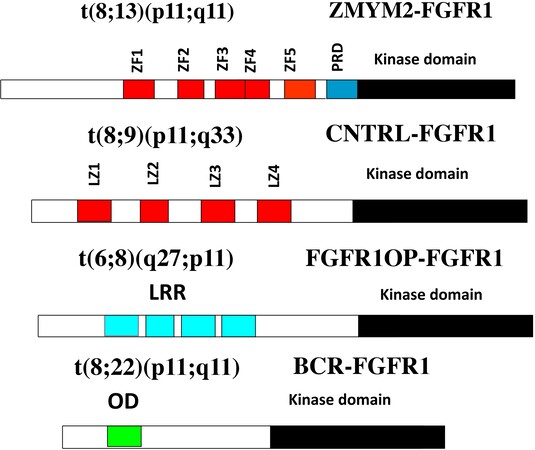
Figure 1. Examples of chimeric kinase motifs provided by partner genes include Zinc Fingers (ZF), Leucine zippers (LZ), Leucein rich repeats (LRR) and oligomerization domains (OD) all of which facilitate dimerization.
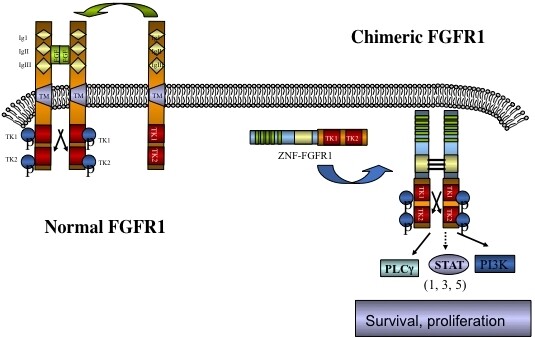
Figure 2. Schematic of the activation of chimeric kinases. Normal monomeric, membrane bound, FGFR1, on binding ligand, dimerizes and is phosphoactivated but remains tethered to the membrane. In the absence of ligand FGFR1 remains an inactive monomer. Chimeric kinases are dimerized through the partner gene motif leading to ligand independent activation and constitutive downstream signaling.
CLINICAL ANALYSIS
Presentation characteristics
The diagnosis of MLN-eo FGFR1 is complicated by the mixed lineage T-, B- and myeloid cell immune-phenotypes on the malignant cells suggesting a pluripotential stem cell origin. Patients show peripheral blood leukocytosis and splenomegaly due to extra medullary hematopoiesis. Typically presentation is of a chronic myeloproliferative neoplasm (MPN) with variable presence of eosinophilia[10]. These patients may also transform to acute lymphoblastic leukemia and/or AML within 1-2 years while some patients present with AML without the antecedent MPN. Depending on the specific chromosome translocation involved, these patients may also demonstrate T-lymphoblastic lymphoma, with aggressive development of blast phase secondary acute leukemia with a myeloid phenotype[9] or, as in the case of patients with the BCR-FGFR1 t(8;22) rearrangement, B-ALL and peripheral basophilia[12-15]. Cytogenetic analysis shows that the same fusion gene is present in the myeloid or lymphoid malignancies of the patient, regardless of their cellular immune phenotype, suggesting a common underlying cause[16,17]. A detailed summary of the variable clinical characteristics associated with MLN-eo FGFR1 patients have been described in detail elsewhere[4,9,14,18].
The t(8;13p)(11q12) rearrangement is the most common structural chromosome translocation which was shown to generate the ZNF198 (ZMYM2)-FGFR1 chimeric gene[19] that typically leads to T-lymphomas[9]. Other, less common, rearrangements that also lead to T-lymphomas include FGFR1OP2-FGFR1[20] and CNTRL-FGFR1[21,22]. Rearrangements involving the BCR gene, however, are more frequently associated with B-lymphomas[12,14,23]. While there has been extensive characterization of the function of the chimeric kinases in vitro[24-26], the common observation is the involvement of constitutive activation of FGFR1 leading to downstream activation of a variety of signaling pathways, although it has been proposed that the fusion partner can possibly influence the phenotypic outcome and survival characteristics from studies in animal models[27].
While the activation of FGFR1 through chromosome translocations is the hallmark of MLN-eo FGFR1, and considered the driver event in transfomation of HSCs, it has also been reported that additional cytogenetic abnormalities can occur in the leukemic cells, particularly associated with progression to blast phase[4-5,14]. In particular, trisomy 21 was seen as the most common secondary abnormality associated with progression[5,28]. There have also been reports of mutations involving RUNX1 frequently accompanying FGFR1 rearrangements[28-30] during progression. Thus, it is possible that these additional genetic changes may influence not only progression but also the phenotype of the leukemic cells and may possibly modify the response to FGFR1 inhibitors.
Incidence of MLN-eo FGFR1
MLN-Eo FGFR1 is generally described as an exceeding rare syndrome, where the incidence has been hard to quantify. The complex diversity in its presentation characteristics may contribute to under diagnosis and, unless definitive cytogenetic or molecular analysis is performed, may go undiagnosed. In a study from the Mayo Clinic involving over 24,000 sequential cases of leukemia analysed cytogenetically from this institution, only 4 cases (0.000164%) showed an FGFR1 rearrangement[4]. This, on a background of leukemias that represent only ~10% of all cancers confirms the rare nature of the disease in the population. As a result, our understanding of its pathobiology has been limited and conducting clinical trials has been challenging. Consequently, the majority of case reports describe only a single patient with limited molecular analysis and, where drug treatments were reported only single cases were involved[7,22,31-33]. To overcome these limitations, several groups have developed murine models of the disease to study the genetic changes responsible for transformation of the hematopoietic stem cell and to investigate the use of molecularly targeted therapies in suppressing the disease without the limitations of incidence and variable diagnosis imposed in human populations.
EXPERIMENTAL STUDIES OF DRUG RESISTANCE
Mouse models of MLN-eo FGFR1
Early models of MLN-eo FGFR1[34,35] involved transduction of hematopoietic stem cells with either the ZMYM2-FGFR1 or BCR-FGFR1 chimeric kinases. When transplanted into syngeneic hosts, the transformed bone marrow cells give rise to MLN-eo FGFR1 disease in the recipient mice, which faithfully recapitulated the splenomegaly and peripheral leukocytosis seen in the human disease. These models also developed MPN and associated lymphomas typical of the human disease. Similar models were developed independently and showed comparable phenotypes and disease progression[36-38]. In the ZMYM2-FGFR1 model reported by Ren et al.[39], the development of T-lymphomas highlighted their close genetic relationship with T-lymphomas from other origins, notably the upregulation of the Notch signaling pathway and the role of BCL2[39]. Subsequently other mouse models were developed for the BCR-FGFR1[37], CNTRL-FGFR1[38] and FGFR1OP2-FGFR1[40] chimeric kinases where, for the most part, the disease developed with a similar presentation to the human disease, although transformation of the myeloproliferative disorder to AML only occurred in the CNTRL-FGFR1 and FGFR1OP2-FGFR1 models. We have argued in the other cases the rapid onset of lymphomas may have overwhelmed the mice, which died before transformation to AML could occur. To extend these studies to be more representative of the human disease, in vivo models have also been developed for human cells derived from CD34+ cord blood cells transformed with the chimeric kinases. Using the same transduction and transplantation approaches developed for the mouse models, transplantable BCR-FGFR1[41], CNTRL-FGFR1[38] and ZMYM2-FGFR1 transformed human cells[42] were xenografted into immune-compromised mice where disease progression and presentation closely mimicked development of MPN and AML in the human disease after relatively long latency periods.
Cell lines were developed from the mouse models which, when xenografted into mice also produced representative MLN-eo FGFR1 disease. BBC2 cells express BCR-FGFR1 and lead to rapid (< 20 days) onset of B-cell leukemia/lymphoma. Both the ZMYM2-FGFR1 expressing ZNF112 and CNTRL-FGFR1 expressing CEP2A cells predominantly show a T-cell ALL immunophenotype. The FGFR1OP2-FGFR1 chimeric kinase was also detected[43] in a human cell line, KG1, which had unwittingly been developed 30 years earlier in Mel Greaves’s laboratory[44]. These cell lines have been used to develop concepts for drug resistance.
Treatment of MLN-eo FGFR1.
During the early years following recognition of MLN-eo FGFR1, treatments used were largely adapted from standard regimens in place for ALL or AML[9,18], where prognosis was typically poor. Clinical course of the disease is very aggressive with rapid progression to secondary acute leukemia within 1-2 years. In one early study of 65 cases, clinical remission rate was 27% and overall survival was 15 months after intensive chemotherapy[9]. Hematopoietic stem cell transplants (HSCT) were clearly indicated to improve these statistics but in the same study, mean survival time for transplant patients was only increased to 24 months. In a more recent study, although prognosis remained poor, allogenic HSCT provided some clinical benefit in selected patients[15,18] and provides the only possibility of curative treatment. Using multi-targeted kinase inhibitors such as PKC412 (midostaurin) and sorafinib only provided a short term hematological response[34,45]. The next generation of more specific drugs, largely only targeting the FGFR family, included ponatinib, which was originally developed to target the mutant BCR-ABL rearrangement in CML[46] but showed a reasonable specificity for FGFR1 at lower concentrations. In one case, after showing chemotherapy resistance, partial remission was achieved using ponatinib[32], although other studies suggested it was less effective[6]. Recently more successful remission was achieved using pemigatinib on a single patient with a t(8;9) rearrangement, although the patient died of unrelated complications[7].
Development of FGFR1 inhibitor regimens in mouse models of MLN-eo FGFR1
There have been suggestions about potential molecular targets in MLN-eo FGFR1 based on genetic studies in the mouse models of MLN-eo FGFR1 (see below). However, since the most consistent observation is the constitutive activation of FGFR1, this seemed like the rational target that might treat all manifestations of the disease whether T-, B-, stem or myeloid cells are involved, since they all carry the same FGFR1 kinase component. The mouse models described above have been used to assess the efficacy of using FGFR1 inhibitors that may translate to the human disease. In an extensive survey of the effects of ponatinib in a variety of cells expressing different fusion kinases, for example, we reported a remarkable efficacy in suppressing leukemogenesis in cell line xenografts in mice[47]. Since these early observations, however, several more specific inhibitors have been developed including the BGJ398[48], JNJ 42756493[49] and AZT4548[50]. In a side-by-side comparison of the effectiveness of these drugs in suppressing leukemogenesis in vitro, we demonstrated efficient suppression of cell growth, with BGJ398 being the most effective and at micro molar concentrations as well as improving survival in mouse models in vivo[51]. The exciting use of mouse models to investigate the potential utility of specific drugs to treat MLN-eo FGFR1 led to our studies that investigated the mechanisms of resistance to FGFR1 inhibitors.
Genetics of resistance to FGFR1 inhibitors
In anticipation of the more widespread use of FGFR1 targeted therapies for MLN-eo FGFR1 in the future, we investigated possible mechanisms of resistance to several FGFR1 inhibitors in the murine models. For the most part, the mode of action of FGFR1 inhibitors is to target the ATP binding domain of the FGFR1 kinases and so share a common mechanism with greater or lesser effectiveness. Various T- and B-lymphoma murine cell lines derived from primary tumors expressing ZMYM2-FGFR1[36], CNTRL-FGFR1[38] and BCR-FGFR1[37], as well as the human KG1 cell line, which expresses the FGFR1OP2-FGFR1 kinase, were treated with progressively increasing concentrations of ponatinib over many months leading to populations that became 1000-3000-fold resistant[52]. As anticipated from the underlying targeting mechanisms, cells that were resistant to ponatinib were also resistant to other inhibitors such as BGJ398, AZT4548 and JNJ42756493.
Consistent with targeting of the ATP binding domain, most mechanisms of resistance to tyrosine kinase inhibitors result from mutations in the ATP binding site[53]. Sequencing the FGFR1 kinase domain in the chimeric genes in the four resistant cell lines showed that two of them carried the same V561M mutation in the ATP binding domain. Chemical biology studies using bacterial synthesized proteins in vitro demonstrated that the mutant site was a significant activating event and that E3810[54] and AZT4547 showed a reduced binding affinity to the mutant protein[55] and induced strong resistance to PD173074 and BGJ398. In model cell systems, the V561M mutation increased FGFR1 auto-phosphorylation leading to hyper-activation. Together these studies provide confirmatory evidence that the V561M mutation is responsible for resistance to FGFR1 inhibitors.
To assess the mechanism in the other 50% of the cell lines, we investigated changes in cancer cell related signaling pathways using the representative proportional protein analysis. This technology uses antibodies not only to common cancer dysregulated genes but also their target phosphorylation sites in an array format to report changes in the resistant vs. sensitive cell pairs. Resistant MLN-EO FGFR1 cells that did not show the V561 mutation, instead showed deletion of exon 6 in the PTEN gene in both cases leading to a premature stop codon that abolishes PTEN function. Introduction of a wild type PTEN gene into these resistant cells re-conferred sensitivity to the FGFR1 inhibitors[52]. PTEN suppresses PI3K signaling[56] and, as a result of its deletion, the PI3K/AKT pathway was upregulated in the resistant cells[52]. The significance of PTEN upregulation in the resistant cells was re-enforced following pharmacological inhibition of PI3K activity using BEZ235, which led to reduced proliferation in vitro and increased survival in vivo[52].
Mechanisms underlying resistance to FGFR1 inhibitors
To extend our understanding of the downstream consequences of the mutations leading to resistance, we used RNA-Seq to compare gene expression changes in sensitive and resistant murine and human cell lines and demonstrated a consistent dysregulation of genes involved in various cell death pathways[57]. Particularly notable was the up regulation of the Bbc3 gene, which encodes the p53 upregulated modulator of apoptosis (PUMA) protein, a potent killer from the BCL2 family of proteins[58], which exerts its influence both through p53-dependent and independent mechanisms[59]. The proapoptotic PUMA protein regulates apoptosis by interacting with antiapoptotic members of the BCL2 family of proteins. As a result, the proapoptotic proteins BAX and BAK are released from the mitochondrial membrane resulting in activation of their proapoptotic action. In the absence of PUMA, BCL2 retains its strong suppressive influence over apoptosis[58].
In MLN-Eo FGFR1 cells, constitutive FGFR1 activation leads to activation of the PI3 Kinase, which in turn activates AKT [Figure 3] leading to downstream signaling cascades that promote oncogenesis. The V561M mutation in resistant cells is not affected by FGFR1 inhibitors and so AKT signaling is sustained. PUMA is regulated mostly at the transcription level and a number of different transcription factors have been implicated, including the 3A member of the forkhead (FOXO3A) transcription factor family[60,61]. AKT phosphorylation of FOXO3A sequesters it in the cytoplasm and so low levels of PUMA are maintained. Pharmacological suppression of FGFR1 activation, however, suppresses AKT activation by PI3K, allowing unphosphorylated FOXO3A to relocate into the nucleus and activate PUMA, which orchestrates apoptosis through BCL2. Since both FGFR1 mutation and loss of PTEN impact the activation of AKT, FOXO3A is phosphoactivated in resistant MLN-Eo FGFR1 cells, thus suppressing PUMA activation.
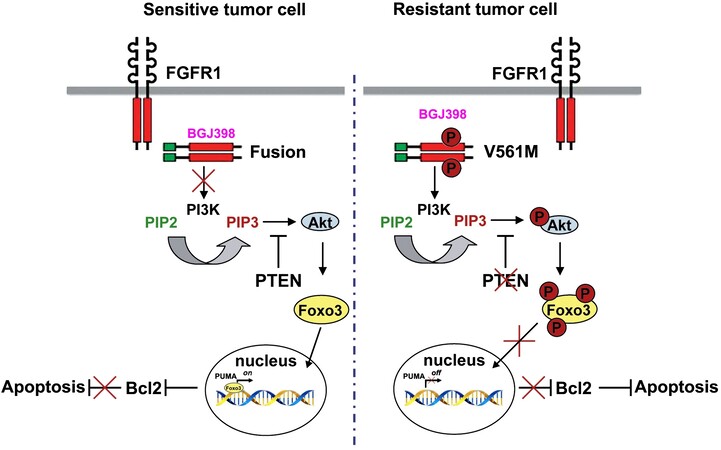
Figure 3. Mechanisms of resistance to FGFR1 inhibitors. In sensitive cells, treatment with an inhibitor (e.g., BGJ398) prevents activation of PI3K. As a result AKT is not activated and FOXO3A remains unphosphorylated and can move into the nucleus where it activates PUMA. PUMA then sequesters the antiapoptotic BCL2 gene, removing its restraint on apoptosis. In the resistant cells, BGJ398 cannot bind FGFR1 and so PI3K is activated leading to activation of AKT and phosphorylation of FOXO3A which is sequestered in the cytoplasm. As a result, PUMA is not activated and so cannot interact with and suppress BCL2 and so apoptosis is suppressed.
Since BCL2 is a major response indicator of FGFR1 inhibition, when MLN-eo FGFR1 cells with either FGFR1 mutations or PTEN deletions were treated with the potent ABT199 (ventocalx) BCL2 inhibitor[62,63], there was a dose-dependent relative decrease in cell survival and reduced leukemic cell expansion in vivo[57]. Thus, targeting the PI3K pathway downstream of PUMA increases cell apoptosis and suppresses cell viability and may suggest an alternative therapeutic approach to target cells resistant to FGFR1 inhibitors.
BIM is another protein in the BCL2 family that has overlapping function with PUMA in promoting apoptosis by binding with BAK and BAX to destabilize the mitochondrial membrane[64-67]. In MLN-Eo FGFR1 cells resistant to FGFR1 inhibitors, BIM was also inactivated[52]. While BIM can be regulated by posttranslational modification, it can also be regulated at the transcription level where FOXO3A is the key transcriptional regulator[68]. Thus, deregulation of AKT activation in cells resistant to FGFR1 inhibitors affects two key regulators of apoptosis.
An unsuspected mechanism to avoid sensitivity to FGFR1 inhibitors
Dimerization of the chimeric kinases in MLN-eo FGFR1 results in phospho-activation of FGFR1 and targeting the ATP binding site in the kinase has become the prime mechanism of suppressing FGFR1 activation, which is essential for transformation[27]. Activation of FGFR1 is presumed to phosphoactivate oncogenic downstream protein targets. Recently, however, it was shown that the chimeric kinases can be cleaved by granzyme B, which generates a truncated FGFR1 (nFGFR1) derivative which localizes exclusively to the nucleus[69]. These nFGFR1 derivatives lack the dimerization domains provided by the translocated partner genes and, as such, are not dimerized and not phosphoactivated. Through a study of MYC activation in MLN-eo FGFR1 cells, nFGFR1 has been shown to function as a cofactor for transcription regulation[69] and likely promotes expression of many other genes, some of which may be involved in transformation of HSCs. Since nFGFR1 does not have inherent DNA binding motifs, its function as a transcription regulator most likely involves associated with as yet unidentified partners. The critical role of granzyme B in generating the truncated nFGFR1 was shown through mutating the binding site and using pharmacological inhibitors. Treatment of BaF3 cells that express the fusion kinases with FGFR1 inhibitors in vitro shows suppression of cell growth but the cells transformed with nFGFR1 are insensitive to FGFR1 inhibitors. In serial transplant studies in mice, the nFGFR1 derivative became a dominant molecular species in advanced stages of disease evolution and indeed, in unpublished studies, we have shown that nFGFR1 can independently transform murine HSCs in in vivo transformation studies, albeit with a relatively long latency period. These observations have profound implications for the application of FGFR1 inhibitors as a means for treating MLN-eo FGFR1 since, although the full length kinases can be suppressed, the truncated derivative is still promoting mechanisms of oncogenesis that are not sensitive to these inhibitors. A better understanding of the molecular consequences of the generation of nFGFR1, therefore, will be essential to overcome this alternative mechanism of transformation.
Alternative regimens to overcome FGFR1 resistance in MLN-eo FGFR1
Resistance to FGFR1 inhibitors in FGFR1 overexpressing malignancies will undoubtedly be a consequence of monotherapies, requiring alternative and/or combinatorial regimens to treat MLN-eo FGFR1. Of note, FGFR1 overexpression is not restricted to MLN-eo FGFR1 and has been shown in 10%-20% of de novo AML[51], which are also sensitive to FGFR1 inhibition in vivo, thus broadening the relevance of overcoming resistance to FGFR1 inhibitors. Although new FGFR1 inhibitors will undoubtedly be developed, if targeting the ATP binding domain remains the main strategy, they will likely fall short of avoiding resistance too. As such, alternative approaches to overcome B- and T- lymphomas have been suggested from model systems.
Genomic analysis of T-lymphomas expressing chimeric FGFR1 kinases, for example, demonstrated deletion of the T-cell receptor alpha (TCRA) gene, upregulation of BCL2[39] and increased expression of Notch[39]. Activation of notch requires cleavage by gamma secretase and, as an early proof-of-principle, treatment of these cells with gamma secretase inhibitors (GSI) led to prolonged survival in mouse models in vivo[39]. Since GSIs have toxic side effects in normal cells that express Notch, other ways of targeting Notch function[70] could be considered as a useful strategy either alone or in combination with other drugs for FGFR1-driven T-lymphomas.
Activation of chimeric kinases also leads to activation of SRC[71] and treatment of MLN-eo FGFR1 cells with the SRC inhibitor dasatinib led to reduced proliferation in vitro and prolonged survival in vivo. In a CNTRL-FGFR1 model of MLN-eo FGFR1, upregulation of MYC was detected and when the JQ1 MYC inhibitor was combined with ponatinib, there was a synergistic effect on suppression of leukemogenesis in human cell models propagated in immunocompromised mice[38]. Up regulation of MYC is one of the most consistent observations in MLN-eo FGFR1 cells, regardless of the chimeric kinase involved, since it is directly activated by FGFR1[72]. Using the 10054-F8 MYC inhibitor, cell proliferation was suppressed in vitro and this effect synergized with BJG398. One of the consequences of MYC activation is the upregulation of the MYB oncogene and treatment of MLN-eo FGFR1 cells with mebendazole leding to increased apoptosis and reduced cell viability as well as suppression of leukemogenesis in vivo[73]. Thus, a detailed analysis of genetic mechanisms of MLN-eo FGFR1 development have suggested other targets that might be effective in treatment of FGFR1-driven malignancies resistant to FGFR1 inhibitors.
BCR-FGFR1 is unique amongst the various chimeric kinases in that it has dual kinase activity, with a serine-threonine kinase (STK) provided by the BCR component of the chimeric kinase, which activates SHP2 kinase amongst other targets[27]. Targeting SHP2 with the SHP099 pharmacological agent[74,75], which stabilizes SHP2 in an inactive conformation, reduced cell proliferation in vitro and leukemogenesis in vivo and, when combined with the BGJ398 FGFR1 inhibitor, showed synergistic effects in vivo[27]. When other MLN-eo FGFR1 cells expressing other chimeric kinases were treated with SHP099, the effects were less dramatic since they do not have STK activity. FGFR1 activation in MLN-eo FGFR1 is also accompanied by Rac activation[76] and when MLN-eo FGFR1 T- and B- lymphomas were treated with the Ehop016 pan-RAC inhibitor[77], cell viability in vitro and leukemogenesis of the BCR-FGFR-driven B-lymphomas in vivo, was also suppressed[27]. These studies begin to address the issue of identifying molecular targets that are specific to individual rearrangement-defined MLN-eo FGFR1 as part of a customized therapy approach.
Thus, while not directly overcoming the mechanisms of resistance to FGFR1 inhibitors, the targets identified through a fine-detail dissection of genetic events associated with lymphoma development in MLN-eo FGFR1 has provided alternative strategies that might be used in combination with FGFR1 inhibitors to prevent/overcome resistance to mono-therapies as well as provide alternative approaches to treat resistant cells. Importantly, a more detailed molecular analysis of tumors in individual MLN-eo FGFR1 patients at the time of diagnosis is required to be able to customize an effective treatment regimen for each patient. This will be particularly challenging given the rare nature of the disease, which precludes large clinical trials to evaluate the efficacy of individual approaches but, given the poor overall outcome, may require bold decisions to be made in the clinical management of these patients.
CONCLUSION
Developing methods of overcoming resistance to therapies targeting the driver event in rare forms of cancer can be challenging, since there are insufficient patients to conduct extensive clinical trials and in a reasonable time to benefit the patients that develop resistance. The development of representative mouse models for leukemia and lymphomas, however, can be a valuable addition to the understanding of the basis of drug resistance and also provide insight into therapeutic strategies that might be alternatives when resistance arises when used in concert with a detailed molecular profiling of the primary disease. This is especially the case where the human disease is rare and opportunities to develop approaches to overcome resistance are limited. Pre knowledge of the tumor specific genetic events that may be responsible for resistance, therefore, can streamline decision making. In the case of FGFR1 driven neoplasms, mechanisms of resistance that have been suggested in animal studies can be monitored during the course of standard ant-FGFR1 treatment to possibly detect emergence of resistant clones, although it cannot be excluded that novel mechanisms may also arise. In either event, the availability of preclinical data suggesting alternative therapeutic strategies may streamline the decision process in selecting alternative approaches.
DECLARATIONS
Author’s contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Cowell JK, Hu T
Availability of data and materialsRequests for data and materials should be directed to the corresponding author.
Financial supportThis work was supported by a grant from the National Institutes of Health, CA076167.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll animal experiments were conducted under protocols approved by the IACUC committee of the University of Augusta.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Abruzzo LV, Jaffe ES, Cotelingam JD, Whang-Peng J, Del Duca V Jr, Medeiros LJ. T-cell lymphoblastic lymphoma with eosinophilia associated with subsequent myeloid malignancy. Am J Surg Pathol 1992;16:236-45.
2. Macdonald D, Aguiar RC, Mason PJ, Goldman JM, Cross NC. A new myeloproliferative disorder associated with chromosomal translocations involving 8p11: a review. Leukemia 1995;9:1628-30.
3. Inhorn RC, Aster JC, Roach SA, et al. A syndrome of lymphoblastic lymphoma, eosinophilia, and myeloid hyperplasia/malignancy associated with t(8;13)(p11;q11): description of a distinctive clinicopathologic entity. Blood 1995;85:1881-7.
4. Patnaik MM, Gangat N, Knudson RA, et al. Chromosome 8p11.2 translocations: prevalence, FISH analysis for FGFR1 and MYST3, and clinicopathologic correlates in a consecutive cohort of 13 cases from a single institution. Am J Hematol 2010;85:238-42.
5. Savage N, George TI, Gotlib J. Myeloid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, and FGFR1: a review. Int J Lab Hematol 2013;35:491-500.
7. Kasbekar M, Nardi V, Dal Cin P, et al. Targeted FGFR inhibition results in a durable remission in an FGFR1-driven myeloid neoplasm with eosinophilia. Blood Adv 2020;4:3136-40.
8. Vega F, Medeiros LJ, Davuluri R, Cromwell CC, Alkan S, Abruzzo LV. t(8;13)-positive bilineal lymphomas: report of 6 cases. Am J Surg Pathol 2008;32:14-20.
9. Jackson CC, Medeiros LJ, Miranda RN. 8p11 myeloproliferative syndrome: a review. Hum Pathol 2010;41:461-76.
10. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937-51.
11. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405.
12. Demiroglu A, Steer EJ, Heath C, et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins. Blood 2001;98:3778-83.
13. Haslam K, Langabeer SE, Kelly J, Coen N, O'Connell NM, Conneally E. Allogeneic hematopoietic stem cell transplantation for a BCR-FGFR1 myeloproliferative neoplasm presenting as acute lymphoblastic leukemia. Case Rep Hematol 2012;2012:620967.
14. Montenegro-Garreaud X, Miranda RN, Reynolds A, et al. Myeloproliferative neoplasms with t(8;22)(p11.2;q11.2)/BCR-FGFR1: a meta-analysis of 20 cases shows cytogenetic progression with B-lymphoid blast phase. Hum Pathol 2017;65:147-56.
15. Konishi Y, Kondo T, Nakao K, et al. Allogeneic hematopoietic stem cell transplantation for 8p11 myeloproliferative syndrome with BCR-FGFR1 gene rearrangement: a case report and literature review. Bone Marrow Transplant 2019;54:326-9.
16. Kim SY, Oh B, She CJ, et al. 8p11 Myeloproliferative syndrome with BCR-FGFR1 rearrangement presenting with T-lymphoblastic lymphoma and bone marrow stromal cell proliferation: a case report and review of the literature. Leuk Res 2011;35:e30-4.
17. Murati A, Arnoulet C, Lafage-Pochitaloff M, et al. Dual lympho-myeloproliferative disorder in a patient with t(8;22) with BCR-FGFR1 gene fusion. Int J Oncol 2005;26:1485-92.
18. Umino K, Fujiwara SI, Ikeda T, et al. Clinical outcomes of myeloid/lymphoid neoplasms with fibroblast growth factor receptor-1 (FGFR1) rearrangement. Hematology 2018;23:470-7.
19. IH Cowell JK. The t(8;13) atypical myeloproliferative disorder: further analysis of the ZNF198 gene and lack of evidence for multiple genes disrupted on chromosome 13. Blood 1998;92:1456-8.
20. Grand EK, Grand FH, Chase AJ, et al. Identification of a novel gene, FGFR1OP2, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 2004;40:78-83.
21. Guasch G, Mack GJ, Popovici C, et al. FGFR1 is fused to the centrosome-associated proteinCEP110 in the 8p12 stem cell myeloproliferative disorder with t(8;9)(p12;q33). Blood 2000;95:1788-96.
22. Chen M, Wang K, Cai X, et al. Myeloid/lymphoid neoplasm with CEP110-FGFR1 fusion: An analysis of 16 cases show common features and poor prognosis. Hematology 2021;26:153-9.
23. Fioretos T, Panagopoulos I, Lassen C, et al. Fusion of the BCR and the fibroblast growth factor receptor-1 (FGFR1) genes as a result of t(8;22)(p11;q11) in a myeloproliferative disorder: the first fusion gene involving BCR but not ABL. Genes Chromosomes Cancer 2001;32:302-10.
24. Smedley D, Demiroglu A, Abdul-Rauf M, et al. ZNF198-FGFR1 transforms Ba/F3 cells to growth factor independence and results in high level tyrosine phosphorylation of STATS 1 and 5. Neoplasia 1999;1:349-55.
25. Baumann H, Kunapuli P, Tracy E, Cowell JK. The oncogenic fusion protein-tyrosine kinase ZNF198/fibroblast growth factor receptor-1 has signaling function comparable with interleukin-6 cytokine receptors. J Biol Chem 2003;278:16198-208.
26. Heath C, Cross NC. Critical role of STAT5 activation in transformation mediated by ZNF198-FGFR1. J Biol Chem 2004;279:6666-73.
27. Chong Y, Liu Y, Lu S, et al. Critical individual roles of the BCR and FGFR1 kinase domains in BCR-FGFR1-driven stem cell leukemia/lymphoma syndrome. Int J Cancer 2020;146:2243-54.
28. Goradia A, Bayerl M, Cornfield D. The 8p11 myeloproliferative syndrome: review of literature and an illustrative case report. Int J Clin Exp Pathol 2008;1:448-56.
29. Agerstam H, Lilljebjörn H, Lassen C, et al. Fusion gene-mediated truncation of RUNX1 as a potential mechanism underlying disease progression in the 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 2007;46:635-43.
30. Strati P, Tang G, Duose DY, et al. Myeloid/lymphoid neoplasms with FGFR1 rearrangement. Leuk Lymphoma 2018;59:1672-6.
31. Verstovsek S, Subbiah V, Masarova L, et al. Treatment of the myeloid/lymphoid neoplasm with FGFR1 rearrangement with FGFR1 inhibitor. Ann Oncol 2018;29:1880-2.
32. Khodadoust MS, Luo B, Medeiros BC, et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia 2016;30:947-50.
33. Chase A, Grand FH, Cross NC. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood 2007;110:3729-34.
34. Chen J, Deangelo DJ, Kutok JL, et al. PKC412 inhibits the zinc finger 198-fibroblast growth factor receptor 1 fusion tyrosine kinase and is active in treatment of stem cell myeloproliferative disorder. Proc Natl Acad Sci U S A 2004;101:14479-84.
35. Roumiantsev S, Krause DS, Neumann CA, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 2004;5:287-98.
36. Ren M, Li X, Cowell JK. Genetic fingerprinting of the development and progression of T-cell lymphoma in a murine model of atypical myeloproliferative disorder initiated by the ZNF198-fibroblast growth factor receptor-1 chimeric tyrosine kinase. Blood 2009;114:1576-84.
37. Ren M, Tidwell JA, Sharma S, Cowell JK. Acute progression of BCR-FGFR1 induced murine B-lympho/myeloproliferative disorder suggests involvement of lineages at the pro-B cell stage. PLoS One 2012;7:e38265.
38. Ren M, Qin H, Kitamura E, Cowell JK. Dysregulated signaling pathways in the development of CNTRL-FGFR1-induced myeloid and lymphoid malignancies associated with FGFR1 in human and mouse models. Blood 2013;122:1007-16.
39. Ren M, Cowell JK. Constitutive Notch pathway activation in murine ZMYM2-FGFR1-induced T-cell lymphomas associated with atypical myeloproliferative disease. Blood 2011;117:6837-47.
40. Qin H, Wu Q, Cowell JK, Ren M. FGFR1OP2-FGFR1 induced myeloid leukemia and T-cell lymphoma in a mouse model. Haematologica 2016;101:e91-4.
41. Cowell JK, Qin H, Chang CS, Kitamura E, Ren M. A model of BCR-FGFR1 driven human AML in immunocompromised mice. Br J Haematol 2016;175:542-5.
42. Ren M, Qin H, Wu Q, Savage NM, George TI, Cowell JK. Development of ZMYM2-FGFR1 driven AML in human CD34+ cells in immunocompromised mice. Int J Cancer 2016;139:836-40.
43. Gu TL, Goss VL, Reeves C, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 2006;108:4202-4.
44. Furley AJ, Reeves BR, Mizutani S, et al. Divergent molecular phenotypes of KG1 and KG1a myeloid cell lines. Blood 1986;68:1101-7.
45. Wakim JJ, Tirado CA, Chen W, Collins R. t(8;22)/BCR-FGFR1 myeloproliferative disorder presenting as B-acute lymphoblastic leukemia: report of a case treated with sorafenib and review of the literature. Leuk Res 2011;35:e151-3.
46. Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 2012;11:690-9.
47. Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia 2013;27:32-40.
48. Guagnano V, Furet P, Spanka C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011;54:7066-83.
49. Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther 2017;16:1010-20.
50. Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 2012;72:2045-56.
51. Wu Q, Bhole A, Qin H, et al. SCLLTargeting FGFR1 to suppress leukemogenesis in syndromic and de novo AML in murine models. Oncotarget 2016;7:49733-42.
52. Cowell JK, Qin H, Hu T, Wu Q, Bhole A, Ren M. Mutation in the FGFR1 tyrosine kinase domain or inactivation of PTEN is associated with acquired resistance to FGFR inhibitors in FGFR1-driven leukemia/lymphomas. Int J Cancer 2017;141:1822-9.
53. Bhullar KS, Lagarón NO, McGowan EM, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17:48.
54. Bello E, Colella G, Scarlato V, et al. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res 2011;71:1396-405.
55. Sohl CD, Ryan MR, Luo B, Frey KM, Anderson KS. Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles' heel of targeted therapy. ACS Chem Biol 2015;10:1319-29.
56. Georgescu MM. PTEN Tumor suppressor network in PI3K-Akt pathway control. Genes Cancer 2010;1:1170-7.
57. Liu Y, Cai B, Chong Y, et al. Downregulation of PUMA underlies resistance to FGFR1 inhibitors in the stem cell leukemia/lymphoma syndrome. Cell Death Dis 2020;11:884.
59. Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003;4:321-8.
60. You H, Pellegrini M, Tsuchihara K, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med 2006;203:1657-63.
61. Dudgeon C, Wang P, Sun X, et al. PUMA induction by FoxO3a mediates the anticancer activities of the broad-range kinase inhibitor UCN-01. Mol Cancer Ther 2010;9:2893-902.
62. Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19:202-8.
63. Peirs S, Matthijssens F, Goossens S, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014;124:3738-47.
64. Bean GR, Ganesan YT, Dong Y, et al. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal 2013;6:ra20.
65. Garrison SP, Phillips DC, Jeffers JR, et al. Genetically defining the mechanism of Puma- and Bim-induced apoptosis. Cell Death Differ 2012;19:642-9.
66. Erlacher M, Labi V, Manzl C, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 2006;203:2939-51.
67. Zhang LN, Li JY, Xu W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther 2013;20:1-7.
68. Dijkers PF, Medema RH, Lammers JJ, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Current Biology 2000;10:1201-4.
69. Hu T, Wu Q, Chong Y, et al. FGFR1 fusion kinase regulation of MYC expression drives development of stem cell leukemia/lymphoma syndrome. Leukemia 2018;32:2363-73.
70. Moore G, Annett S, McClements L, Robson T. Top notch targeting strategies in cancer: a detailed overview of recent insights and current perspectives. Cells 2020;9:1503.
71. Ren M, Qin H, Ren R, Tidwell J, Cowell JK. Src activation plays an important key role in lymphomagenesis induced by FGFR1 fusion kinases. Cancer Res 2011;71:7312-22.
72. Hu T, Chong Y, Lu S, et al. Loss of the BCR-FGFR1 GEF domain suppresses RHOA activation and enhances B-lymphomagenesis in mice. Cancer Res 2019;79:114-24.
73. Hu T, Chong Y, Cai B, Liu Y, Lu S, Cowell JK. DNA methyltransferase 1-mediated CpG methylation of the miR-150-5p promoter contributes to fibroblast growth factor receptor 1-driven leukemogenesis. J Biol Chem 2019;294:18122-30.
74. Sun X, Ren Y, Gunawan S, et al. Selective inhibition of leukemia-associated SHP2E69K mutant by the allosteric SHP2 inhibitor SHP099. Leukemia 2018;32:1246-9.
75. Chen YN, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148-52.
76. Hu T, Chong Y, Lu S, McGuinness M, Williams DA, Cowell JK. Rac1/2 activation promotes FGFR1 driven leukemogenesis in stem cell leukemia/lymphoma syndrome. Haematologica 2020;105:e68-71.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cowell JK, Hu T. Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment. Cancer Drug Resist 2021;4:607-19. http://dx.doi.org/10.20517/cdr.2021.30
AMA Style
Cowell JK, Hu T. Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment. Cancer Drug Resistance. 2021; 4(3): 607-19. http://dx.doi.org/10.20517/cdr.2021.30
Chicago/Turabian Style
Cowell, John K., Tianxiang Hu. 2021. "Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment" Cancer Drug Resistance. 4, no.3: 607-19. http://dx.doi.org/10.20517/cdr.2021.30
ACS Style
Cowell, JK.; Hu T. Mechanisms of resistance to FGFR1 inhibitors in FGFR1-driven leukemias and lymphomas: implications for optimized treatment. Cancer Drug Resist. 2021, 4, 607-19. http://dx.doi.org/10.20517/cdr.2021.30
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 1
likes
Like This Article 1
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.