
Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer

Abstract
The survival rates for women with ovarian cancer have shown scant improvement in recent years, with a 5-year survival rate of less than 40% for women diagnosed with advanced ovarian cancer. High-grade serous ovarian cancer (HGSOC) is the most lethal subtype where the majority of women develop recurrent disease and chemotherapy resistance, despite over 70%-80% of patients initially responding to platinum-based chemotherapy. The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway regulates many vital processes such as cell growth, survival and metabolism. However, this pathway is frequently dysregulated in cancers including different subtypes of ovarian cancer, through amplification or somatic mutations of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), amplification of AKT isoforms, or deletion or inactivation of PTEN. Further evidence indicates a role for the PI3K/AKT/mTOR pathway in the development of chemotherapy resistance in ovarian cancer. Thus, targeting key nodes of the PI3K/AKT/mTOR pathway is a potential therapeutic prospect. In this review, we outline dysregulation of PI3K signaling in ovarian cancer, with a particular emphasis on HGSOC and platinum-resistant disease. We review pre-clinical evidence for inhibitors of the main components of the PI3K pathway and highlight past, current and upcoming trials in ovarian cancers for different inhibitors of the pathway. Whilst no inhibitors of the PI3K/AKT/mTOR pathway have thus far advanced to the clinic for the treatment of ovarian cancer, several promising compounds which have the potential to restore platinum sensitivity and improve clinical outcomes for patients are under evaluation and in various phases of clinical trials.
Keywords
INTRODUCTION
Ovarian cancer is currently the sixth most common cause of cancer death in the UK, with approximately 4100 deaths recorded in 2017[1], and worldwide is the second leading cause of deaths from gynecological malignancies in westernized countries[2]. The overall survival rates for women with ovarian cancer have seen little improvement over the 30 years, despite advances in surgical techniques, imaging technologies, and introduction of new targeted therapies, e.g., PARP inhibitors and anti-angiogenics such as bevacizumab. Due to the lack of reliable biomarkers and the vague symptom profile of ovarian cancer, over 70% of patients will present with advanced stage disease (FIGO III or IV). Current standard-of-care for patients with ovarian cancer involves cytoreductive surgery and platinum-based chemotherapy usually in combination with a taxane. However, many patients will ultimately relapse and eventually develop chemotherapy-resistant disease, at which point treatment options are entirely palliative.
The most prevalent type of ovarian cancer, epithelial ovarian cancer (EOC), is categorized into different histological subtypes: serous (high or low grade), clear cell, endometrioid and mucinous. The classical dualistic classification of EOC into two types (I and II)[3] has been revised and expanded in recent years to take into account numerous molecular and histological studies, which have provided new important insights into EOC[4], and the updated WHO[5,6] and FIGO classifications[7,8]. Type I tumors, which account for approximately 25% of all EOC, encompass the subtypes low grade serous ovarian cancer (LGSC), endometrioid, mucinous, clear cell, and the rare Brenner tumor subtype. They are largely detected at an earlier stage (FIGO I-II), have high genomic stability, usually p53 wild-type, characterized by different mutations: LGSC (KRAS, BRAF), endometrioid (PTEN, PI3KCA), mucinous (KRAS, p53), and clear cell (PI3KCA, ARID1A), and are relatively resistant to platinum-based chemotherapy[9]. The more common Type II tumors are generally diagnosed at an advanced stage (FIGO III-IV), while they are initially more responsive to platinum-based chemotherapy, tend to recur and become resistant to chemotherapy, and are responsible for the majority of deaths from EOC. Subtypes of Type II include high-grade serous ovarian cancer (HGSOC), high-grade endometrioid, undifferentiated cancers and carcinosarcomas. Type II cancers are typified by high genomic instability, a near 100% p53 mutation rate, defects in homologous recombination repair, mutations in BRCA1 or 2, and extensive copy number aberrations[4].
HGSOC accounts for around 75% of EOC cases. The average 5-year survival rate for HGSOC is less than 40%, approximately 20% of patients do not respond to initial primary treatment efforts[10] and many of the remaining patients will relapse, acquiring resistance to platinum chemotherapy[11]. The complete set of mechanisms giving rise to platinum resistance in HGSOC and how they cooperate have not yet been fully elucidated. Genomic analyses of HGSOC revealed widespread clonal diversity exists before chemotherapy treatment, and more recent analyses of tumors collected at relapse have identified different mechanisms of acquired resistance to platinum chemotherapy[12-16]. Different models of evolution of resistance to platinum chemotherapy have been proposed (reviewed in Rottenberg et al.[17]). Broadly, one model proposes the presence of genomically heterogeneous sub-clones in the chemo-naive state, and subsequent chemotherapy treatments preferentially select these resistant clones leading to eventual recurrence of the disease[14,16,18]. Another model proposes that treatments with DNA-damaging platinum chemotherapy cause mutations that give rise to resistance. Proposed mechanisms contributing to acquired platinum resistance in HGSOC involve activation of AKT signaling[19], reversion of BRCA1/2 germline mutations, loss of BRCA1 methylation, extensive desmoplastic stroma, and overexpression of the ABCB1 multidrug transporter[15,20]. A further model hypothesizes the role of cancer stem cells (CSC) and epithelial-to-mesenchymal transition (EMT) in causing relapse following platinum chemotherapy[21]. CSCs and mesenchymal-like cells characteristically have a low cycling rate suggesting that they could be more resistant to the standard cytotoxic treatments such as platinum that target actively proliferating cells[22,23]. Studies have shown a direct correlation between CSC abundance and the onset of relapse, suggesting CSCs promote chemotherapy resistance[24-26]. For example, expression of different biomarkers for CSCs, CD44, ALDH, CD133 and MyD88, was observed to be associated with chemotherapy resistance and poor patient outcome in EOC[27-29]. Recently, the transcription factor NFATC4 (nuclear factor of activated T cells cytoplasmic 4) was identified as a regulator of quiescence in ovarian cancer, enriched in ovarian CSCs and associated with chemotherapy resistance and poor prognosis[30]. Using a model of ovarian malignant ascites in a heterospheroid assay of CSCs and carcinoma-associated mesenchymal stem/stromal cells, Raghavan et al.[28] demonstrated that PDGF (platelet-derived growth factor) signaling in these heterospheroids significantly increased stemness, metastatic potential and platinum resistance of CSCs. TWIST1 was suggested to be a regulator of EOC stemness through controlling stem cell differentiation via regulation of miR-199a and miR-214[31-33]. Furthermore, increased TWIST1 expression in CSCs has been shown to promote their differentiation into mesenchymal cells with CSC-like properties and capacity for migration[32,34]. Profiling of HGSOC tumors using the Oxford Classic classifier for molecular stratification of tumors demonstrated that EMT-high tumors were associated with poor survival and linked to immunosuppression[35]. However, the mechanisms of CSC and EMT-associated platinum resistance require further investigation to translate targeting of these CSC populations into improved treatment and outcomes of patients with platinum-resistant ovarian cancer.
This review aims to provide an overview of dysregulation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway in epithelial ovarian cancer, with a particular focus on the HGSOC subtype and chemotherapy resistant disease, and the potential targeting of the pathway as a therapeutic option for patients with ovarian cancer. We will present pre-clinical evidence and clinical trial outcomes for inhibitors targeting different nodes of the PI3K/AKT/mTOR pathway, and highlight past and upcoming clinical trials for ovarian cancers.
PI3K/AKT/mTOR signaling pathway overview
Extensive characterization of the PI3K/AKT/mTOR signaling pathway over the last two decades has led to a much greater understanding of the molecular mechanisms underlying this pathway’s regulation of essential cellular processes such as cell growth, survival, proliferation, angiogenesis, metabolism, transcription, and translation[36]. The classical mechanisms behind the canonical PI3K/AKT/mTOR pathway activation and its functions are described in Figure 1. Phosphoinositide 3-kinases (PI3Ks) are lipid kinases which are major downstream effectors of G protein coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs). There are three classes of PI3K (classes I, II, III), in which class I is comprised of class IA PI3K (α, β, δ) and class IB PI3K (γ). Each sub-class of PI3K class IA or IB is activated by receptors RTKs or GPCRs, respectively. Class IA PI3Ks are composed of a p85 regulatory subunit of which there are five variants and a p110 catalytic subunit[37,38]. There are three class IA p110 isoforms (α, β, δ) expressed by the genes phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), PIK3CB and PIK3CD, respectively, of which PIK3CA is the most frequently mutated in multiple cancer types, including EOC[37,39].
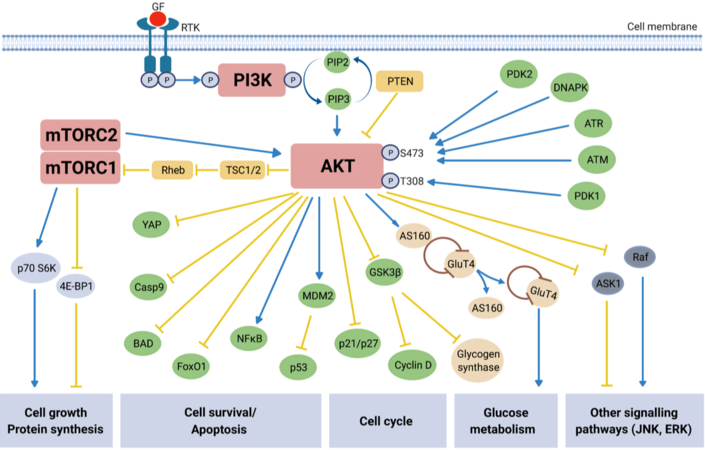
Figure 1. Overview of PI3K/AKT/mTOR signaling pathway. Activation (blue arrows) of growth factor (GF) receptor tyrosine kinase (RTK), resulting in autophosphorylation on tyrosine residues, recruits Phosphatidyl-inositol-3-kinase (PI3K) to the cell membrane. Direct binding of PI3K to the tyrosine residues causes activation via the PI3K catalytic subunit[42]. Activated PI3K in turn phosphorylates secondary messenger phosphatidylinositol-3,4,5-bisphospate (PIP2) which converts to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 is responsible for the recruitment of the protein kinase AKT to the cell surface where its subsequent activation/inhibition by multiple molecules leads to involvement in numerous downstream signaling pathways[43,44]. AKT is activated via phosphorylation at two key residues: S473 and T308[40]. Protein serine/threonine kinase-3’-PDK1 and PDK2, recruited to and activated at the cell surface are responsible for activating AKT, along with protein kinases ATM and ATR, and HSP90. DNA-PK, a nuclear serine/threonine kinase essential for non-homologous end-joining (NHEJ) repair, activates AKT via phosphorylation at S473 in response to cisplatin-induced DNA damage in platinum resistant EOC cells[19]. PTEN, tuberous sclerosis protein 1 (TSC1) and TSC2 are the main negative regulators (yellow arrows) of the pathway, with phosphorylation of TSC2 by AKT releasing the inhibitory effect on mTORC1 via the GTP-binding protein Rheb[42]. mTORC1 activates p70S6K and inhibits 4E-BP1, resulting in protein synthesis and cell growth, and mTORC2 activates AKT itself; overall mTOR activation leads to cell growth and survival. Inactivation of pro-apoptotic molecules YAP, Procaspase 9 (Casp9) and BAD, as well as inhibiting Forkhead transcription factors [e.g., Forkhead box protein O1 (FoxO1)] result in increased cell survival[45,46]. AKT is an essential part of the insulin signaling pathway. Activated in response to insulin stimulation, AKT causes Glucose Transporter 4 (GLUT4) to translocate to the cell surface, facilitating glucose uptake. Additionally, the inhibition of glycogen synthase kinase 3 beta (GSK-3β) by AKT increases glycogen production[47]. The role of AKT in glucose metabolism is again apparent through its inhibition of FoxO1, which suppresses hepatic glucose production[48]. The PI3K/AKT/mTOR signaling pathway includes points of cross-regulation and communication with other common signaling pathways, e.g., JNK and RAS-ERK. The AKT substrate c-RAF is activated by RAS and initiates a kinase cascade leading to ERK activation[49]. The phosphorylation of c-RAF by AKT inhibits its activity and subsequent downstream activity in the RAS/RAF/MEK/ERK signaling cascade known for its role in apoptosis and cell differentiation[50]. AKT involvement with the JNK pathway, a stress- and nutrient-response pathway, directly modifies activation of target genes, many of which are involved in adaptations to extra-cellular stresses[51].
In brief, the pathway becomes activated following phosphorylation of RTKs in response to extracellular signals, thus activating PI3K (p85, p110)[36]. PI3K converts phosphoinositide 4,5-biphosphate (PIP2) to phosphatidylinositol (3,4,5)-triphosphate (PIP3). PIP3 activates AKT (protein kinase B) via phosphoinositide-dependent kinase-1 (PDK1)-mediated phosphorylation at the Threonine 308 (T308) site of AKT and is recruited to the cell surface. PIP3 is later dephosphorylated by the tumor suppressor and negative regulator of the pathway phosphatases and tensin homolog (PTEN) back to PIP2. The full activation of AKT is achieved with participation of the mammalian target of rapamycin complex 2 (mTORC2) complex, which phosphorylates AKT at the Serine 473 (S473) site[40]. Protein kinases ATM (ataxia-telangiectasia mutant), ATR (ataxia-telangiectasia and Rad3 related) and DNA-PK (DNA-dependent protein kinase), and HSP90 (heat shock protein 90) can also activate AKT via phosphorylation at S473, demonstrating the diversity of cellular contexts in which AKT plays a role[19,41].
PI3K/AKT/mTOR pathway alterations in ovarian cancer
Dysregulation of the PI3K pathway is frequently observed in human cancers, comprising of amplification and somatic mutations of PIK3CA, deletion or inactivation of PTEN and amplification or mutation of AKT isoforms (reviewed in Janku et al.[52]). In EOC, early genomic investigations demonstrated frequencies of genetic alterations for PIK3CA of 4%-12% (somatic mutations) in epithelial ovarian tumors[39,53,54], 20%-46% of clear cell ovarian cancers[53,55], 20% of endometrioid ovarian cancers, and 2.3%-3% of HGSOC[53,56]. Frequent amplification of AKT2 was identified in different ovarian cancer cohorts, for example in 18.2% of patients with HGSOC[57] and 10% of patients with advanced ovarian cancer (cohort comprised of 89% HGSOC)[58]. Loss of PTEN was observed in different histological subtypes of ovarian cancer, via different mechanisms (deletion, loss-of-heterozygosity, epigenetic silencing or loss-of-function mutations)[59-62]. Significant advances in sequencing technologies in the last decade have further increased our understanding of the genomic dysregulation of the PI3K pathway. In 2011, the Cancer Genome Atlas (TCGA) study demonstrated that 45% of serous ovarian tumors had dysregulation of the PI3K pathway[13]. In particular, 7% had deletions of PTEN, < 1% had PTEN mutations, 18% had amplification or mutations of PIK3CA, and amplification of AKT1 and AKT2 was found in 3% and 6% of cases, respectively[13]. Dysregulated components of the PI3K pathway were also found to be linked to chemotherapy resistance in HGSOC. In the ICGC cohort of chemotherapy sensitive, refractory and resistant HGSOC tumors, inactivation of genes by disruption of transcriptional units (gene breakage) was observed for tumor suppressor genes including PTEN, and gene breakage was associated with acquired chemotherapy resistance[15]. Recently, seven copy number signatures were derived for HGSOC which may predict both overall survival and the probability of platinum-resistant relapse. Across these 7 signatures, signature exposures 4 and 6 were associated with aberrant PI3K signaling (mutation of PIK3CA, amplification of AKT2 and RICTOR)[63]. Table 1 outlines the frequencies of the main genomic alterations (amplification, deletion, and mutations) observed in HGSOC in the TCGA, ICGC and BriTROC-1 cohorts[13,15,63]. In HGSOC, genomic amplification of PIK3CA, AKT isoforms (AKT2, AKT3) or RICTOR or deletion of PTEN are relatively common, whereas mutations in these genes are rare.
PI3K/AKT/mTOR pathway genomic alterations for HGSOC in TCGA*, ICGC OV-AU§ and BriTROC-1 cohorts
| Gene | Alteration | Frequency of alteration % | ||
| TCGA* | ICGC(OV-AU)§ | BriTROC-1 | ||
| AKT1 | Amplification | 2.9-5.2 | 2.2 | |
| Deletion | 0-0.2 | |||
| Mutation | 0.3 | 3.3 | ||
| AKT2 | Amplification | 5.8-8 | 9.8 | 11.4 |
| Deletion | 0.3-1 | |||
| Mutation | 3.3 | |||
| AKT3 | Amplification | 4.1-9.5 | 4.3 | |
| Deletion | 0.2-0.4 | |||
| Mutation | 1 | 7.6 | ||
| PIK3CA | Amplification | 18-29 | 22.8 | |
| Mutation | 1 | 2.2 | 4.6 | |
| PIK3CB | Amplification | 3.9-11.4 | 13.0 | |
| Mutation | 0.3 | 1.1 | 2.3 | |
| PIK3R1 | Amplification | 0-0.2 | ||
| Deletion | 2-3.5 | |||
| Mutation | 2 | 2.2 | ||
| PTEN | Amplification | 0.7-1.2 | ||
| Deletion | 4.5-6.1 | 2.2 | ||
| Mutation | 1 | 6.5 | ||
| RICTOR | Amplification | 3.5-7.1 | 3.3 | 9.1 |
| Mutation | 1.1 | |||
Several studies investigating PI3K signaling in ovarian cancer have demonstrated enhanced signaling of different nodes of the pathway and how dysregulation of the PI3K/AKT/mTOR pathway contributes to cell proliferation, migration, and chemotherapy resistance. PI3K signaling outputs were determined by quantifying p-AKT, p-p70S6K and p-GSK3β in ascites samples from patients with advanced ovarian cancer, predominately the HGSOC subtype. Significantly higher levels of p-p70S6K levels were detected in patients who did not respond to chemotherapy[58]. Immunohistochemical staining of tissue microarrays (TMA) of different ovarian cancer subtypes showed significantly increased staining of p-4EBP1, p-p70S6K and p-S6, in particular p-4EBP1 expression correlated with high-grade tumors and poor prognosis[64]. A multi-center study from the Ovarian Tumor Tissue Analysis Consortium of over 5400 patient tumors investigated PTEN loss as a putative driver in different histological subtypes of ovarian cancer[65]. Downregulation of cytoplasmic PTEN expression was most frequent in endometrioid and clear cell ovarian cancers and associated with longer overall survival in HGSOC. PTEN loss was demonstrated to be a frequent driver in ovarian cancer, with strong associations with expression of the Androgen, Estrogen and Progesterone hormone receptors, and CD8+ TILs (tumor infiltrating lymphocytes) in HGSOC and clear cell ovarian cancers[65]. In a study by Huang et al.[66], p-AKT-S473 and p110α overexpression was demonstrated to be significantly associated with decreased survival in a cohort of 522 serous ovarian tumors. They also examined how loss of a number of PI3K-associated proteins affected vital cellular processes. Following small interfering RNA (siRNA) knockdown of PIK3CA in PIK3CA-mutant A2008 and PIK3CA copy-gain UPN251 cells, cell proliferation decreased by almost 50%[66]. The same study showed a more prominent increase of apoptosis (5.5-fold) and decrease of proliferation (51%) following knockdown of AKT2 in the OVCAR8 cell line, known to have a copy-gain of AKT2, compared to the other ovarian cancer lines tested. None of the PIK3CA, PIK3CB or AKT2 siRNAs induced apoptosis in normal ovarian surface epithelial cell lines[66]. Montero et al.[67] aimed to establish the functional relevance of mammalian target of rapamycin complex 1 (mTORC1) and mTORC2 in epithelial ovarian cancer cell lines (OVCAR8, SKOV3, A2780, and IGROV-1) as potential clinical targets. Using siRNAs against raptor or rictor to target mTORC1 and mTORC2, respectively, demonstrated that knockdown of either protein led to a significant decrease in cell proliferation. However, knockdown of raptor/mTORC1 displayed greater inhibitory effects than of rictor/mTORC2, suggesting a more important role for mTORC1 in ovarian cancer cell proliferation[67]. Raptor knockdown led to decreased levels of phosphorylated ribosomal S6 kinase-S240/244 and p4E-BP1, and also an increase of p-AKT-S473, indicating a negative feedback orchestrated by mTORC1 over mTORC2. Overall, data suggested targeting of both mTORC complexes as a clinical strategy. Furthermore, as the mTOR inhibitor BEZ-235 displayed a synergistic effect with cisplatin chemotherapy in vitro, combining mTOR inhibitors with standard-of-care chemotherapy was proposed as a viable option for ovarian cancer patients[67].
The role of AKT in chemotherapy resistance and targeting AKT as a strategy to re-sensitize chemotherapy resistant HGSOC cells to platinum-based chemotherapy has been explored. A study by Yang et al.[68] investigated the role of AKT in caspase-independent apoptosis in cisplatin-resistant OC cells. Cisplatin treatment induced the release of apoptosis-initiating factor (AIF), a mediator of caspase-independent apoptosis, in cisplatin-sensitive cells only, and overexpression of AIF in cisplatin-resistant cells re-sensitized cells to cisplatin. Furthermore, AKT negatively regulated AIF, and downregulation of AKT re-sensitized resistant cells to cisplatin-induced AIF-dependent apoptosis, suggesting that AKT plays a significant role in chemotherapy resistance and its inhibition can re-sensitize platinum-resistant cells to cisplatin[68]. In concordance, research by Stronach et al.[19] identified AKT as a potential mediator of platinum resistance in immortalized patient-acquired platinum-resistant HGSOC cell lines. Upon cisplatin treatment of platinum-resistant HGSOC cells, AKT relocated to the nucleus where it was phosphorylated at S473 by DNA-PK, inducing an AKT-dependent DNA damage response, and this activation of AKT led to an inhibition of platinum-mediated apoptosis in platinum-resistant cells. Furthermore, inhibition of AKT with the allosteric AKT inhibitor triciribine, led to a decrease in phosphorylated AKT-S473 in the presence or absence of cisplatin. Inhibition of AKT in combination with cisplatin treatment enhanced apoptotic induction in platinum-resistant HGSOC cells. Knockdown or inhibition of DNA-PK had a similar effect as triciribine treatment in platinum-resistant cells, enhancing apoptosis and decreasing accumulation of phosphorylated AKT-S473 in combination with cisplatin administration. Interestingly, targeting DNA-PK did not affect insulin-mediated activation of AKT, which is an alternative route for AKT activation upon its direct inhibition. Thus, it could be concluded that DNA-PK inhibition could target AKT without causing toxicity that is often observed when direct inhibitors of AKT are used, while still enhancing apoptosis[19].
Targeting of PI3K/AKT/mTOR pathway in ovarian cancer
Frequent activation of the PI3K/AKT/mTOR pathway in many cancers, including ovarian cancer, suggests the PI3K/AKT/mTOR pathway to be an attractive target for therapeutic intervention[52,69]. Following the approval of the mTORC1 inhibitors everolimus and temsirolimus over a decade ago for breast cancer and renal cell carcinoma[70,71], several inhibitors of the pathway have been developed as a potential monotherapy or in combination with other therapeutics (e.g., olaparib[72], and bevacizumab[73]) or chemotherapy drugs (e.g., carboplatin[74], and docetaxel[75]). Inhibitors targeting different nodes of the PI3K/AKT/mTOR pathway can be broadly categorized into four groups: PI3K inhibitors, mTOR inhibitors, dual PI3K/mTOR inhibitors, and AKT inhibitors. Clinical trials have been established for several compounds from each class of pathway inhibitor involving patients with ovarian cancer including platinum-refractory or resistant ovarian cancer cohorts. Tables 2 and 3 outline examples of PI3K/AKT/mTOR pathway inhibitors from each group in clinical trials that have been completed or recently completed and awaiting results [Table 2], or active and/or recruiting [Table 3] for ovarian cancer or solid malignancies including ovarian cancer patients. While a number of PI3K pathway inhibitors have been approved for use in patients by the Food and Drug Administration (FDA) for other cancer types, no compounds have yet progressed to clinical use for ovarian cancer. The majority of trials for PI3K/AKT/mTOR pathway inhibitors specifically targeting ovarian cancer cohorts are early-phase trials including several first-in-human, establishing pharmacokinetics (PK) and pharmacodynamic (PD) profiles, dose escalation, and combination studies. Enrolment based on a biomarker profile of different dysregulated nodes (e.g., PIK3CA mutation or amplification, loss of PTEN) of the pathway is a prerequisite in only a small number of the trials highlighted in Tables 2 and 3. However, most studies report including retrospective profiling of trial samples for potential biomarkers of clinical response. In the following sections, we will highlight the findings of some of the key pre-clinical studies and clinical trials for inhibitors of the PI3K/AKT/mTOR pathway, according to inhibitor groups.
Completed or ongoing clinical trials for inhibitors of the PI3K/AKT/mTOR pathway according to highest phase reached for trials including ovarian cancer patients, indication, monotherapy or combination therapy, clinical trials and publication references
| Compound/generic name | Mechanism of action | Status/highest phase | Indication (± biomarker enrolment criteria) | Monotherapy/combination therapy | Clinical trials reference | Clinical trial outcome/reference |
| PI3K | ||||||
| Buparlisib BKM120 | Class I pan-PI3K inhibitor | Phase II | Advanced solid malignancies incl. ovarian cancer patients with PI3K-activated tumors | Monotherapy | NCT01833169 | ORR 1.4% (n = 2); CBR 15.1% (n = 22)[94] |
| CH5132799 | Class I PI3K, in particular PI3Kα | Phase I | Advanced solid malignancies incl. ovarian cancer patients; dose escalation study | Monotherapy | NCT01222546 | MTD of 48mg BID; n = 1 PR by GCIG CA125[91] |
| Pictilisib GDC-0941 | Class I pan-PI3K inhibitor | Phase I | Advanced solid malignancies incl. ovarian cancer patients; first-in-human dose escalation study | Monotherapy | NCT00876122 | Well tolerated; 3% n = 2 PR by RECIST or GCIG CA125. RP2D of 330mg QD[95] |
| PX-866 | Class I pan-PI3K inhibitor | Phase I | Advanced solid malignancies incl. ovarian cancer patients; dose escalation study | Docetaxel | NCT01204099 | PR 6% n = 2; SD 63% n = 22; PD 31% n = 11. Median PFS 73.5 days (range: 1-569). RP2D of 8mg[75] |
| Alpelisib BYL-719 | PI3Kα | Phase Ib | Recurrent ovarian, fallopian tube, or primary peritoneal cancer of HGS histology or recurrent TNBC; ± known germline BRCA mutations; dose escalation and expansion trial | Olaparib | NCT01623349 | MTD and RP2D of 200mg alpelisib QD + olaparib 200mg BID; EOC pts PR 36% (n = 10), SD 50% (n = 14) by RECIST[93] |
| IPI-549 | PI3Kγ | Phase I/Ib | Advanced solid tumors; first in human study | Nivolumab | NCT02637531 | MTD not reached. PR n = 2 at 8wk assessment. RP2D IPI-549 40 mg QD + nivo 240 mg Q2W[96] |
| TAK117 Serabelisib | PI3Kα | Phase Ib (ongoing) | Advanced ovarian, endometrial, or breast cancer | Sapanisertib Paclitaxel | NCT03154294 | ORR 46% in 13 evaluable pts. CBR of 69%; PFS = 10 months. CR n = 2 pts[97] |
| PI3K/mTOR | ||||||
| Apitolisib GDC-0980 G-038390 | Class I pan-PI3K/mTOR inhibitor | Phase I | Advanced solid tumors incl. ovarian; first in human | Monotherapy | NCT00854152 | PR n = 10 pts by RECIST; RP2D of 40mg QD 28/28 schedule[98] |
| Bimiralisib PQR309 | Dual PI3K mTORC1/2 inhibitor | Phase I | Advanced solid tumors (SAKK 67/13), incl. ovarian patients; first-in human, dose escalation trial | Monotherapy | NCT01940133 | MTD and RP2D is 80 mg QD; PR n = 1, SD > 16 weeks n = 1 by RECIST[99] |
| SF1126 | Pan-PI3K/mTORC prodrug | Phase I | Advanced or metastatic tumors incl. ovarian cancer patients; first-in human | Monotherapy | NCT00907205 | SD in 58% (n = 19) of evaluable pts. MTD not reached[100] |
| BGT226 | Class I pan-PI3K/mTOR inhibitor | Phase I/II | Advanced solid malignancies incl. ovarian cancer patients; dose escalation study | Monotherapy | NCT00600275 | MTD 125mg/day TIW; SD 30% (n = 17); SD ≥ 16 weeks 16% (n = 9) by RECIST[101] |
| Gedatolisib PKI-587 PF-05212384 | Class I pan-PI3K/mTOR inhibitor | Phase I | Solid tumors, incl. ovarian cancer patients; first-in human | Monotherapy | NCT00940498 | MTD of 154mg QW; PR 2.5% (n = 2); SD > 6mths of 10% (n = 8) by RECIST[102] |
| LY3023414 Samotolisib | ATP competitive class I PI3K inhibitor, mTOR and DNA-PK | Phase Ib | Advanced or metastatic tumors incl. ovarian cancer patients | Prexasertib | NCT02124148 | No results posted to date for LY3023414 and Prexasertib combination |
| XL765 SAR245409 | Dual inhibitor of mTOR/PI3K | Phase II | Patients with previously treated unresectable low grade ovarian cancer | Pimasertib | NCT01936363 | ORR of 9.4% in combination arm; median PFS of 7.23 mths pimasertib alone and 9.99 mths pimasertib + SAR245409[103] |
| mTOR | ||||||
| Temsirolimus CCI-779 | mTORC1 inhibitor | Phase II | Ovarian Clear Cell Cystadenocarcinoma, Stage III Ovarian Cancer, Stage IV Ovarian Cancer | Carboplatin Docetaxel Paclitaxel | NCT01196429 | US/Korea cohort CR 31%, PR 23%; Japan cohort CR 6%, PR 65%[104] |
| Everolimus RAD-001 | mTORC1 inhibitor | Phase II | Recurrent ovarian, peritoneal, and Fallopian tube cancer (RADBEV trial) | Bevacizumab | NCT01031381 | 24% of pts progression-free at 6mths. CR n = 1; PR n = 6 pts; SD n = 35 pts[73] |
| Sirolimus | mTORC1 inhibitor | Phase II | Stage II-IV ovarian epithelial, Fallopian tube, or primary peritoneal cavity cancer | Vaccine Therapy | NCT01536054 | No results posted to date |
| BI-860585 | mTORC1/mTORC2 inhibitor | Phase I | Advanced and/or metastatic solid tumors incl. ovarian cancer patients | Exemestane Paclitaxel | NCT01938846 | BI 860585 + exemestane PR n = 4; BI 860585 + paclitaxel PR n = 4, CR n = 1[105] |
| CC-115 | mTOR/DNA-PK | Phase I | Advanced solid tumors and hematologic malignancies; first in human dose escalation and expansion study | Monotherapy | NCT01353625 | MTD reached in 5 tumour types; SD in ovarian pts (n = 2)[106] |
| AKT | ||||||
| Uprosertib GSK-2141795 | Pan-AKT isoform inhibitor | Phase I | Recurrent or persistent ovarian cancer | Monotherapy | NCT01266954 | CBR of 27% by RECIST; 30% by CA125 GCIG[107-109] |
| Afuresertib GSK-2110183 | Pan-AKT isoform inhibitor | Phase Ib/II | Recurrent platinum-resistant ovarian cancer | Carboplatin Paclitaxel | NCT01653912 | ORR of 32% by RECIST, 52% by GCIG CA125 Median PFS was 7.1 mths[74] |
| MK-2206 | Pan-AKT isoform inhibitor | Phase II | Recurrent platinum-resistant ovarian, fallopian tube, or peritoneal cancer | Monotherapy | NCT01283035 | SD in 4 of 5 evaluable pts; n = 1 SD for 19wks; n = 1 SD for 10.8 wks. Median PFS estimated 19wks[110] |
| Capivasertib AZD5363 | Pan-AKT isoform inhibitor | Phase I | Recurrent endometrial, TNBC, and ovarian, primary peritoneal, or fallopian tube cancer. Biomarker enrolment: germline BRCA1/2-mutant, or BRCA1/2 wild-type cancers harboring somatic DDR or PI3K–AKT pathway alterations | Olaparib | NCT02338622 | 44.6% (25/56) evaluable pts had CBR (RECIST CR/PR or SD ≥ 4 months)[72] |
| ARQ-092 Miransertib mesylate | Pan-AKT isoform inhibitor | Phase I (terminated) | Advanced, inoperable metastatic and/or recurrent solid tumors, ovarian or endometrial cancer. Enrolment criteria for endometrial cancer: AKT1, PIK3CA, or PIK3R1 mutations with or without PTEN deficiency | Carboplatin Paclitaxel Anastrozole (endometrial cancer) | NCT02476955 | Responses in EC 4/8 pts: CR n = 1 (confirmed), PR n = 3 (unconfirmed)[111] |
| PTX-200 Triciribine | AKT inhibitor | Phase I | Recurrent or persistent, platinum-resistant epithelial ovarian, fallopian tube or primary peritoneal carcinoma | Carboplatin | NCT01690468 | Study terminated due to change in strategic focus |
| Perifosine D-21266 | Pan-AKT isoform inhibitor | Phase I | Recurrent ovarian cancer; in combination with Doxetaxel | Doxetaxel | NCT00431054 | Median PFS of 1.9 mths; OS of 4.5 mths[112] |
Active or currently recruiting clinical trials for inhibitors of the PI3K/AKT/mTOR pathway according to the highest phase including ovarian cancer patients, indication (± biomarkers), monotherapy or combination therapy, clinical trials and year opened
| Compound/generic name | Mechanism of action | Status/highest phase | Indication (± biomarker enrolment criteria) | Monotherapy/combination therapy | Clinical trials reference | Year opened |
| PI3K | ||||||
| BGB-10188 | PI3Kδ inhibitor | Phase I/II (recruiting) | Mature B-Cell malignancies and combination with Tislelizumab in patients with solid tumors (including ovarian cancer) | Tislelizumab | NCT04282018 | 2020 |
| CYH-33 | PI3Kα inhibitor | Phase I (recruiting) | Patients with DDR gene mutations and/or PIK3CA mutations, in patients who progressed on prior PARP inhibitor, and patients with recurrent HGSOC, fallopian tube, or primary peritoneal cancer who are platinum resistant or refractory | Olaparib | NCT04586335 | 2020 |
| Linperlisib YY-20394 | PI3Kδ inhibitor | Phase I (recruitment to start) | Advanced solid tumors | Monotherapy | NCT04049929 | 2019 |
| Eganelisib IPI-549 | PI3Kγ inhibitor | Phase I/Ib (recruiting) | Advanced metastatic TNBC or ovarian cancer | Etrumadenant; PLD; Nab-paclitaxel | NCT03719326 | 2018 |
| Copanlisib BAY-806946 | Class I pan-PI3K inhibitor | Phase II (recruiting) | Recurrent endometrial and recurrent ovarian, primary peritoneal, or Fallopian tube cancer or BRCA mutant ovarian cancer | Niraparib | NCT03586661 | 2018 |
| TAK117 Serabelisib | PI3Kα | Phase Ib (recruiting) | Patients with advanced ovarian, endometrial, or breast cancer | Sapanisertib Paclitaxel | NCT03154294 | 2017 |
| Pictilisib or Taselisib | Class I pan-PI3K inhibitor | Phase Ib (active) | Advanced solid tumors and breast cancer. PI3KCA mutant tumors. PIPA trial | Palbociclib | NCT02389842 | 2015 |
| mTOR | ||||||
| Everolimus | mTORC1 inhibitor | Phase I (recruiting) | Advanced ovarian (platinum resistant or refractory) and breast cancer (triple negative or hormone receptor positive only) | Niraparib | NCT03154281 | 2017 |
| Sirolimus | mTORC1 inhibitor | Phase IV (recruiting) | Refractory Solid Tumors (PIK3CA mutation, PIK3CA amplification, PIK3CA-AKT pathway aberration) | Monotherapy | NCT02688881 | 2017 |
| AKT | ||||||
| Afuresertib | Pan-AKT isoform inhibitor | Phase II (recruiting) | Platinum-resistant ovarian cancer (PROFECTA-II) | Paclitaxel | NCT04374630 | 2020 |
| Ipatasertib GDC-0068 | Pan-AKT isoform inhibitor | Phase I/II (active) | Advanced breast cancer, ovarian cancer (HGSOC or endometrioid or clear cell epithelial ovarian, Fallopian tube, or primary peritoneal cancer), or prostate cancer | Rucaparib | NCT03840200 | 2019 |
| ARQ751 | Pan-AKT isoform inhibitor | Phase Ib (active) | Solid tumors with PIK3CA/AKT/PTEN mutations | Paclitaxel Fulvestrant | NCT02761694 | 2016 |
PI3K inhibitors
Commonly PI3K isoforms are categorized into three classes: class I, class II and class III PI3Ks[76]. Class I PI3Ks are the main isoforms targeted in cancer drug development[37]. PI3K inhibitors can be subdivided in to those that target one or two specific isoforms (α, β, γ, δ), and pan-PI3K inhibitors, which target the catalytic activity of all four PI3K class I isoforms[69,77]. While theoretically, targeting all four isoforms may appear to be a useful therapeutic strategy, this may lead to an increased risk of off-target effects and toxicity for patients. Consequently, the clinical development of the majority of pan-PI3K inhibitors has been severely limited due to poor outcomes and safety concerns such as increased drug-related toxicities, including hyperglycemia, gastrointestinal and neuropsychiatric side-effects[78]. Class I isoforms have different activation mechanisms which suggest that each isoform has distinct biological functions[76,79], therefore targeting individual isoforms may have a wider therapeutic benefit.
The first generation of pan-PI3K inhibitors developed, LY294002 and wortmannin, were shown to exert anti-cancer effects in a number of pre-clinical ovarian cancer cell models[36,80,81]. Hu et al.[82] demonstrated that LY294002 could inhibit tumor formation in vivo in a mouse model of ovarian cancer, and could inhibit proliferation of OVCAR-3 platinum-resistant ovarian cancer cells in vitro. Similarly Fekete et al.[83] showed that LY294002 is able to enhance the cytotoxic effects of the chemotherapeutic agents carboplatin and paclitaxel in the platinum-resistant ovarian cancer cell lines SKOV-3 and IGROV-1. In addition, the pan-PI3K inhibitor wortmannin has been shown both to enhance apoptosis in platinum-resistant ovarian cancer cells (A2780cis)[84], and to sensitize ovarian cancer cells to cisplatin in Caov3 mouse models[85].
Buparlisib (BKM120) is one of the most widely studied pan-PI3K inhibitors. Bendell et al.[86] conducted a Phase I study looking at BKM120 as single agent treatment in patients with advanced solid tumors including ovarian cancer; one patient out of 35 achieved partial response and 16 had a stable response for more than six weeks. Buparlisib has also been tested in combination with other agents in a number of ovarian cancer trials, for example a Phase II trial was performed administering buparlisib in combination with the MEK1/2 inhibitor trametinib where ovarian cancer patients were shown to have a 29% overall response rate and median progression-free survival of seven months[87]. In vitro buparlisib has been tested in combination with the PARP inhibitor olaparib and shown to effectively inhibit proliferation, survival and invasion in the PIK3CA mutant ovarian cancer cell lines SKOV-3, IGROV-1, and HEYA-8[88]. A dose escalation Phase I trial was recently conducted testing buparlisib in combination with olaparib in a cohort of 69 patients including 46 ovarian cancer patients, showing a 29% response rate irrespective of platinum sensitivity[89].
Other pan-PI3K inhibitors (XL147, GDC0941, CH5132799, and PX-866) have been under clinical investigation in Phase I-II trials [Table 2][90]. For example, the pan-PI3K inhibitor PX-866 has been tested in combination with docetaxel in a Phase I multi-center study in 43 patients with advanced solid tumors, including 5 patients with ovarian cancer. In the trial overall, 6% of patients achieved a partial response, 63% of patients had stable disease, and 31% of patients had disease progression[75]. A Phase I trial in patients with metastatic cancer by Blagden et al.[91] studying toxicity, PK and PD of CH5132799, a pan-PI3K inhibitor particularly inhibiting PI3Kα, revealed one patient with PIK3CA-mutant clear cell carcinoma of the ovary achieving a partial response (GCIC CA125 criteria) (NCT03767335).
Isoform-specific inhibitors have been developed to reduce off-target effects and the cumulative toxicity observed with pan-PI3K isoform inhibition[52,69]. Pre-clinical investigations of a considerable number of isoform-specific PI3K inhibitors in solid tumors and leukemia, e.g., alpelisib (PI3Kα), SAR260301 (PI3Kβ), IPI-549 (PI3Kγ), and idelalisib (PI3Kδ)[36,52,92], have led to Phase I trials, but limited progress has been achieved beyond Phase I with these inhibitors[52,69]. The efficacy of alpelisib was recently evaluated in a Phase I trial in combination with olaparib in patients with recurrent ovarian cancer or TNBC, based on pre-clinical data showing that treatment with PI3K inhibitors may sensitize homologous recombination repair-proficient EOC tumors to PARP inhibition. The EOC patients (n = 28) within the trial cohort achieved 36% partial response and 50% of patients had stable disease, with no unexpected toxic effects observed (grade 3-4 adverse events observed in low numbers of patients), showing that this synergistic combination deserves further clinical investigation[93]. There are a number of actively recruiting Phase I clinical trials looking at both pan- and isoform-specific PI3K inhibitors in advanced solid tumors and in particular recurrent ovarian cancer [Table 3], as monotherapy or in combination with PARP inhibitors (olaparib or niraparib), or immunotherapy agents (tislelizumab). While toxic side effects and low clinical efficacy have limited the advancement of many pan-PI3K inhibitors, PI3K isoform specific inhibitors may provide a better alternative with less off-target effects and adverse events for patients.
mTOR inhibitors
The multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) comprise the serine threonine protein kinase mTOR. Inhibitors of mTOR are either allosteric targeting either mTORC1 or mTORC2, or target both mTORC1/mTORC2 (non-allosteric/catalytic inhibitors)[52]. A number of in vitro studies have investigated allosteric and catalytic mTORC inhibitors as single agents or in combination with other agents in platinum-resistant ovarian cancer among other solid tumors, with promising findings. Pre-clinical in vitro studies with the allosteric mTORC1 inhibitor rapamycin (sirolimus), demonstrated that treatment of the platinum-resistant SKOV3 ovarian cancer cell line with sirolimus enhanced cisplatin-mediated apoptosis[113]. Likewise, Mabuchi et al.[114] showed that treatment with another allosteric inhibitor, everolimus (RAD001), enhanced cisplatin-induced apoptosis in platinum-resistant ovarian cancer cell lines (SKOV3 and OVCAR-10) with high AKT/mTOR activity, and inhibition of tumor growth and angiogenesis in mouse SKOV3 xenograft models.
The first PI3K/AKT/mTOR pathway inhibitors developed targeting mTORC1, everolimus and temsirolimus, are the most widely investigated mTOR inhibitors for ovarian cancer. Several studies have progressed to Phase I/II clinical trials, showing that mTOR inhibitors exhibit more promising results in combination with anti-angiogenics and/or chemotherapeutic agents than as a monotherapy[36]. A Phase II study conducted in 54 patients with persistent/recurrent epithelial ovarian cancer/primary peritoneal cancer, tested temsirolimus as a weekly intravenous (IV) single agent, showing modest activity and found 24% patients had progression-free survival (PFS) > 6 months (median 3.1 months)[115]. A Phase I study by Kollmannsberger et al.[116] administered temsirolimus in combination with carboplatin and paclitaxel in six ovarian cancer patients, resulting in disease stabilization (n = 3) or partial response (n = 3) in patients[117]. A further Phase Ib trial administered tesmirolimus in combination with pegylated liposomal doxorubicin in a cohort of patients with advanced breast, endometrial and ovarian cancers. Patients with ovarian cancer demonstrated durable partial responses of over 10 months (n = 2) or stable disease (median 6.4 months)
Clinical trials have been established to determine the efficacy of mTOR inhibitors in combination with anti-angiogenic agents. Following on from a dose-finding Phase I clinical trial in solid malignancies[119] and two Phase II trials in refractory metastatic colorectal cancer[120] and renal cell carcinomas[121], a recent study by Taylor et al.[73] investigated the efficacy of everolimus and bevacizumab in a Phase II trial in recurrent ovarian, peritoneal and fallopian tube cancers. The oral administration of everolimus in combination with the IV administration of bevacizumab every two weeks resulted in 24% of patients (95%CI: 16.67%-42.71%) being progression-free at six months[73].
Work to date on mTOR inhibitors has mainly focused on the allosteric mTORC1 or 2 inhibitors, but dual non-allosteric inhibition of mTORC1/mTORC2 has been demonstrated to overcome the feedback loop activation of PI3K and AKT in the pathway observed following inhibition with allosteric mTORC1 inhibitors, and therefore non-allosteric mTORC1/2 inhibitors may provide a greater degree of inhibition[36,122]. As a result, increasing numbers of non-allosteric mTORC1/2 inhibitors, e.g., AZD2014, AZD8055, OSI-027, and INK128/MLN128, have been investigated in vitro and in vivo. David-West et al.[122] demonstrated that platinum-resistant ovarian cancer cells (OVCAR-3) can be re-sensitized to carboplatin by inhibition of mTORC1/2 using INK128/MLN128. Similarly, results from the study using the non-allosteric mTORC1/2 inhibitor vistusertib (AZD2014) showed that the combination treatment of vistusertib and paclitaxel resulted in a significant reduction in tumor growth and increase in apoptosis in a cisplatin-resistant xenograft (A2780cis) model, which led to the initiation of a clinical trial to evaluate this drug combination (NCT02193633)[123]. A further study examined the combination of AZD8055 with the PI3K inhibitor GDC0941 and MEK1/2 inhibitor selumetinib in ovarian clear cell carcinoma cell lines and PDX models. Low-dose triple combination of inhibitors reduced kinase activity in both PI3K/AKT/mTOR and mitogen-activated protein kinase pathways, inhibited proliferation in vitro, and significantly reduced tumor growth in PDX models, suggesting this combination merits further clinical exploration[124].
Whether non-allosteric mTOR1/2 inhibitors will offer any clinical superiority over allosteric inhibitors remains unclear, and although overall success in ovarian cancer is currently limited, both drug classes continue to be tested in Phase I and II studies. Despite positive trial outcomes with temsirolimus, another Phase II trial investigating the temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma was terminated due to efficacy not meeting the pre-defined levels required as approximately 48% of OC and 40% of EC patients had progressive disease after eight weeks of treatment (NCT00926107)[125]. A Phase II trial evaluating the tolerability and activity of the single agent allosteric mTORC1 inhibitor ridaforolimus in women with recurrent endometrial cancer revealed a weak treatment response and significant toxicity (NCT00770185)[126]. However, a Phase I open-label trial of the allosteric mTORC1 inhibitor everolimus in combination with the PARP inhibitor niraparib in patients with advanced ovarian and breast cancer is currently recruiting (NCT03154281), as well as a Phase IV study in South Korea (NCT02688881) dispensing sirolimus as a monotherapy in patients with refractory solid tumors (PIK3CA mutation, PIK3CA amplification, PIK3CA-AKT pathway aberration). Results were recently reported from a first-in-human Phase I study (NCT01353625) in patients with advanced solid (including ovarian) or hematological malignancies for an inhibitor of mTORC1/2 which is also a potent inhibitor of DNA-PK (CC-115), concluding that CC-115 was well-tolerated with toxicities consistent with other mTOR inhibitor treatments, and suggesting that this novel dual mTOR/DNA-PK inhibitor could be a favorable treatment for cancer patients[106].
Dual PI3K/mTOR inhibitors
By inhibiting all four PI3K isoforms and both mTORC1 and mTORC2 complexes, the feedback activation of PI3K by mTORC can be overcome resulting in suppression of multiple significant nodes in the PI3K/AKT/mTOR pathway, theoretically leading to a more complete inhibition of the pathway[36,52,90]. As dual PI3K/mTOR inhibitors have a comparable poor toxicity profile to pan-PI3K inhibitors, with common adverse events including fatigue, nausea, vomiting and diarrhea, no dual PI3K/mTOR inhibitors have yet advanced for clinical use for any cancer type. Limited numbers of dual PI3K/mTOR inhibitors have progressed to clinical trials in ovarian cancer, despite numerous pre-clinical studies showing positive anti-tumor activity and outcomes. For example, Yuan et al.[127] tested the dual pan-PI3K/mTOR inhibitor PF-04691502 as a single agent in ovarian cancer SKOV3 xenograft models and observed dose-dependent reduction in tumor volume in PIK3CA mutant SKOV3 xenografts, which also correlated with plasma concentration, induced time- and dose-dependent target modulation and following oral administration, and was well tolerated without body weight loss. The PI3K/mTOR inhibitor CMG002 was demonstrated to re-sensitize chemo-resistant ovarian cancer cells (paclitaxel-resistant SKpac17 and A2780cis) to chemotherapies in vitro, and showed a marked decrease in tumor growth in xenograft mouse models of each cell line, either alone or in combination with paclitaxel or cisplatin[128]. Lezzi et al.[129] showed that the combined treatment of the dual PI3K/mTOR inhibitor gedatolisib and c-MET inhibitor crizotinib indicated that crizotinib was able to potentiate the activity of gedatolisib in ovarian cell lines (A2780 and SKOV3) and due to favorable tolerability, it could be a potential combination in settings in which either inhibitor alone already had some significant activity. A recent study profiling the anti-tumor effects of 16 different PI3K/AKT/mTOR pathway inhibitors in combination with paclitaxel in a panel of ovarian cancer cell lines and primary ovarian tumor cells covering the four main subtypes of EOC, found that the dual PI3K/mTOR inhibitor GSK458 in particular was a potent inhibitor of proliferation and cell migration in vitro, and was able to reduce tumor growth and metastasis in both SKOV3 xenograft and PDCX models in vivo[130]. Thus, GSK458 was proposed as an attractive candidate for further investigation as a treatment for chemotherapy-resistant ovarian cancer. Deng et al.[131] investigated the association of the PI3K/AKT/mTOR pathway with EMT and CSC in EOC platinum resistance, using the dual PI3K/mTOR inhibitor BEZ235. Treatment with BEZ235 and cisplatin inhibited the PI3K/AKT/mTOR pathway, reversed EMT and decreased the expression of CSC markers in platinum resistant A2780cis and IGROV1cis cell lines in vitro compared to inhibitor or cisplatin alone, suggesting BEZ235 as a good candidate for treating chemoresistant CSCs[131].
To date only early phase clinical trials have been established for dual PI3K/mTOR inhibitors in ovarian cancer cohorts [Table 2]. The PI3K/mTOR peptide pro-drug SF1126 was investigated in a Phase I first-in-human trial for patients with advanced solid tumors (n = 39) including ovarian (n = 5). Stable disease in 58% of patients was the best response reached and the pro-drug was found to be well tolerated[100]. XL765 (SAR245409) was tested in a first-in-human clinical trial to evaluate safety, maximum tolerated dose, PK, PD and efficacy; stable disease was achieved in 48% of patients with evaluable disease, supporting further development of XL765[132]. Other Phase I clinical trials have since been completed with XL765 in solid tumors, including ovarian cancer, and lymphoma, as a monotherapy (NCT01587040; NCT01596270) or in combination with other agents, e.g., erlotinib (NCT00777699), and letrozole (NCT01082068). A Phase II randomized double-blind placebo-controlled trial (NCT01936363) was completed testing the combination MEK1/2 inhibitor (pimasertib) with XL765 in patients with previously treated unresectable borderline or low grade ovarian cancer. Results reported show that while the combination was safe with manageable toxicities for patients, the ORR was 9.4% (80%CI: 3.5-19.7) for the combination treatment, with single agent pimasertib showing an ORR of 12%. The study was terminated due to low ORR and over 50% rate of patients discontinuing the trial[103].
There are a number of on-going Phase I/II trials currently investigating dual-PI3K/mTOR inhibitors in combination with chemotherapy or targeted therapies in breast cancer, renal cell and prostate cancers with promising results that may prove useful for ovarian cancer trials in the future. In particular, the dual PI3K/mTOR inhibitor, LY3023414 (samotolisib), also reported to inhibit DNA-PK, is under investigation in number of Phase I and II trials as a monotherapy in patients with endometrial cancer (NCT02549989), and in combination with chemotherapy or other agents in patients with other solid malignancies, e.g., TNBC trial with prexasertib (NCT04032080), metastatic breast cancer with combination therapies (NCT02057133), and prostate cancer with enzalutamide (NCT02407054)[52,133,134]. Due to pre-clinical data showing synergistic inhibition of proliferation in HGSOC cells in vitro and significantly enhancing efficacy (P < 0.001) in combination compared to single agent in HGSOC OV-90 and Cov504 xenografts[135], a Phase Ib trial (NCT02124148) was established (and recently completed, results pending) to assess the safety of prexasertib with LY3023414 in patients with solid tumors including ovarian. Although there have been multiple promising pre-clinical studies and early phase clinical trials, whether dual PI3K/mTOR inhibitors will advance through later phase clinical trials and into clinical use will depend on overcoming significant toxicities observed with dual PI3K/mTOR pathway inhibitor treatments. Despite being able to overcome unwanted pathway feedback activation, as inhibitors of multiple kinases, the considerable off-target and toxic effects observed may deter further clinical development.
AKT inhibitors
Targeting AKT is an attractive therapeutic prospect as inhibiting this key effector node of the PI3K/AKT/mTOR pathway leads to blocking mTORC1 activation and controlling the downstream effects of the pathway signaling cascade[36,90]. Several small molecule inhibitors of AKT have been developed to target the three mammalian isoforms of AKT: AKT1, AKT2, and AKT3. Depending on their mechanism of action, AKT inhibitors can be further classified as lipid-based phosphatidylinositol (PI) analogues, ATP-competitive, or allosteric inhibitors[90,136]. While most ATP-competitive AKT inhibitors target all isoforms of AKT (pan-AKT inhibitors), a number of allosteric inhibitors were developed in an attempt to selectively target AKT isoforms and have displayed some level of selectivity[136]. As increasing numbers of AKT inhibitors are advancing to clinical trial for treatment of advanced solid malignancies, including ovarian cancer, a number of trials in particular have focused on targeting recurrent chemotherapy-resistant ovarian cancer patients based on pre-clinical evidence of efficacy.
The allosteric AKT inhibitor MK-2206 has been investigated in many pre-clinical cancer studies, including ovarian. In particular, MK-2206 was tested in combination with a number of chemotherapy agents and targeted agents (iapatinib and erlotinib), and synergistic cell growth inhibition in combination with cytotoxic drugs in A2780 ovarian cancer cell lines and a reduction in tumor volume in SKOV3 xenografts in combination with iapatinib was observed[137]. A further study in multiple ovarian cell lines with differing PI3K or RAS/RAF pathway alterations, RB1 loss and wild-type for the alterations mentioned, demonstrated synergistic effects of MK-2206 and MEK (PD0325901) inhibitors in PI3K- and RAS-activated ovarian cancer cell lines and xenografts[138]. MK-2206 has progressed to clinical trials as a monotherapy or in combination with chemotherapy or other targeted agents. A phase II study tested MK-2206 as a monotherapy in a small cohort of patients with recurrent platinum-resistant ovarian, fallopian tube, or peritoneal cancer (NCT01283035). Poor accrual of patients led to early termination of the trial, but preliminary findings suggested that MK2206 was not clinically effective in the patient cohort (selected due to PI3K/AKT pathway alterations)[110]. The non-ATP competitive AKT inhibitor TAS-117 has been tested in vitro in combination with chemotherapeutic and targeted agents in A2780 ovarian cancer cells and xenografts, with results indicating that TAS-117 effectively enhances the cytotoxic effects of fluorouracil and cisplatin in vitro, and improves the anti-tumor effect of carboplatin in vivo[139]. One of the most studied AKT inhibitors, the PI-analogue perifosine, was tested in combination with paclitaxel in a study by Sun et al.[140] demonstrating an increase in ovarian cancer cell apoptosis in the CaOV-3 cell line. Perifosine has been investigated in over 40 clinical trials in multiple cancer types including ovarian (ClinicalTrials.gov). In a Phase I trial (NCT00431054), Fu et al.[112] examined the combined effects of perifosine and docetaxel in taxane and platinum-resistant or refractory epithelial ovarian cancer, and reported outcomes of median 1.9 months progression-free survival and 4.5 months overall survival, and no dose-limiting toxicity.
Numerous clinical trials for ATP-competitive pan-AKT inhibitors have been completed showing promising results with more in progress for ovarian cancer and other cancer types [Tables 2 and 3]. The pan-AKT inhibitor uprosertib (GSK2141795) has been studied in a number of dose-escalation trials in combination with other targeted therapies for example with BRAF or MEK inhibitors. A dose-escalation and expansion Phase I study in TNBC and BRSAF-wild type advanced melanoma was terminated early due to a low ORR (< 5%), minimal clinical activity, and the combination was not tolerated[141]. A Phase I dose-escalation trial (NCT01266954) in recurrent platinum-resistant ovarian cancer patients (n = 12) was established to investigate the PK and PD of repeat escalating doses of uprosertib by 18F FDG-PET. No relationship was observed for dose-response between uprosertib PK and fluoro-deoxyglucose F18 PET PD measures, but single-agent activity was observed with a clinical benefit rate of 27% and 30% CA125 response in the cohort[108,109]. A similar ATP-competitive AKT inhibitor afurosertib (GSK2110183) was investigated in a Phase Ib/II dose escalation study in combination with carboplatin and paclitaxel in a recurrent platinum-resistant ovarian cancer cohort, with positive findings of an ORR of 32% by Response evaluation criteria in solid tumors (RECIST) 1.1 and 52% by GCIC CA125 criteria, and a median progression-free survival of 7.1 months[74]. The PROFECTA-II (phase II) trial has recently opened and is recruiting patients with platinum-resistant ovarian cancer for combination treatments of afuresertib and paclitaxel (NCT04374630).
The pan-AKT inhibitor capivasertib (AZD5363) was first tested in patients with multiple solid tumor types including PIK3CA mutated breast and gynecologic cancers in a Phase I, open-label, first-in-human evaluation study (NCT01226316), and reported that capivasertib was well tolerated and had robust target modulation in tumors[142]. More recently capivasertib has been tested in a Phase I trial in combination with the PARP inhibitor olaparib for multiple solid tumors (NCT02338622) based on preclinical studies demonstrating synergy between PARP and PI3K/AKT pathway inhibitors in BRCA1/2 deficient and proficient tumors. In the 64 patients enrolled (n = 25 ovarian) capivasertib was well tolerated, overall 44.6% of patients achieved clinical benefit (RECIST1.1 complete response/partial response or stable disease > 4 months), 44% of ovarian patients had a median duration of response of 26.6 weeks (11.3-115), and 8/11 platinum resistant ovarian cancer patients showed clinical benefit[72]. In general, several AKT inhibitors are well tolerated and continue to be investigated both in vitro and in clinical trials in multiple advanced solid tumors. Data from early phase trials examining combinations of AKT inhibitors with chemotherapy agents and targeted therapies for platinum resistant ovarian cancer patients show positive response rates and clinical outcomes. In particular, findings suggest that the combination of AKT inhibition with platinum-based chemotherapy is effective and durable for patients with platinum resistance, and merits further investigation.
CONCLUSION AND FUTURE PERSPECTIVES
The last two decades have seen an exponential growth in the number of pre-clinical studies and early phase trials in ovarian cancers, targeting different nodes in the PI3K signaling cascade, greatly increasing our understanding of the dysregulation of PI3K/AKT/mTOR pathway. However, disappointingly to date no inhibitors directed towards the PI3K/AKT/mTOR pathway have progressed to late phase clinical trials for ovarian cancer patients. Significant advances are paramount to accelerate new pathway inhibitors to the clinic including characterizing new potential predictive biomarkers in the pathway, exploring new drug combinations, e.g., DDR inhibitors, cell cycle checkpoint inhibitors, and implementing innovative trial designs. Translational research studies including genomic-, proteomic-, and metabolomic-based analyses, should be incorporated into clinical trial designs to help uncover new clinical biomarkers and further investigations into potential mechanisms of resistance to PI3K pathway inhibitors and the effects of pathway inhibitors on ovarian CSC populations. A recent large phase II trial, the MATCH (Molecular Analysis for Therapy Choice) Screening trial (NCT02465060), was established for patients with advanced refractory solid tumors (including ovarian), lymphomas or multiple myeloma and is an ongoing collaborative effort between the NCI Precision Medicine Initiative and several pharmaceutical companies, aiming to match targeted therapy directed by genetic testing of the patient tumor. As mentioned previously, actionable mutations are rare and variable in ovarian cancer, e.g., PIK3CA mutations are present in 20%-46% of clear cell ovarian cancers[53,55,143], but this histological subtype is responsible for approximately 5%-10% of EOC. Therefore the study coordinators suggest that to evaluate whether personalized targeting of tumors is effective for cancers such as ovarian with multiple subtypes, as many patients as possible should be screened to determine whether molecular therapy is a beneficial strategy[144]. This multi-study trial aims to enroll up to 6452 patients. Inhibitors of the PI3K/AKT/mTOR pathway such as capivasertib or ipatasertib are indicated for patients with AKT mutations, or the PI3Kβ inhibitor GSK2636771 is indicated for patients with PTEN mutation, deletion and PTEN expression. On a smaller scale, the MyTACTIC trial (NCT04632992) aims to evaluate targeted therapies in patients with advanced solid tumors with genomic alterations or protein expression patterns predictive of response in an open-label Phase II study, and includes GDC-0077 indicated for patients with a positive biomarker result for PIK3CA or ipatasertib for patients with a positive biomarker result for either AKT1/2/3 activating mutation or loss/loss of function of PTEN.
As medicine moves more towards personalized care, the results of these types of basket trials will be important for future trial designs, identification and validation of actionable predictive biomarkers associated with clinical activity, and selection of the correct targeted therapies for patients with low-frequency molecular alterations such as the different PI3K pathway aberrations observed in ovarian and other gynecological cancers. Development of new compounds with fewer off-target effects is also necessary to limit toxicities and adverse events for patients, as well as explore new rational combination strategies and dosing schedules. Results from monotherapy trials for inhibitors of different nodes of the PI3K/AKT/mTOR pathway report limited responses to single agent inhibitors, but data from early phase combination trials indicate combination therapies to be more effective than single agent treatments. Targeting other nodes of the PI3K/AKT/mTOR pathway is another emerging field with inhibitors in pre-clinical development against eIF4E, S6K, MNK and PIM[37]. A greater understanding of the effects of PI3K pathway inhibitors on the different aspects of the tumor microenvironment is also required, which could open up new avenues for combination treatments with immunotherapies. Positive findings have been observed for PI3Kα or AKT inhibitors in combination with olaparib treatment for recurrent and platinum-resistant ovarian cancer patients[72,93], and further combinations with DDR therapeutics warrant investigation in these patient cohorts. It is becoming increasingly evident that PI3K/AKT/mTOR pathway inhibitors, particularly in combination with other chemotherapeutic and/or targeted therapies, may hold significant promise for the future treatment and outcomes of women with chemotherapy-resistant ovarian cancer.
DECLARATIONS
AcknowledgmentsFigure 1 was illustrated using BioRender.com.
Authors’ contributionsResearch, image illustration and contributed to writing of review manuscript: Rinne N, Ardasheva A
Performed data acquisition for tables: Christie E, Kwok CH, Demchenko N, Tralau-Stewart C
Substantial contributions to conception and planning of review and editing, as well as provided material support: Low C, Fotopoulou C
Conception, writing and editing of review manuscript: Fotopoulou C, Cunnea P
Availability of data and materialsNot applicable.
Financial support and sponsorshipWe thank the Myrovlytis Trust (Cunnea P, Fotopoulou C), Rosetrees Trust (Cunnea P), Imperial College Healthcare Charity [Ovarian fund (Cunnea P, Fotopoulou C)] and Imperial Private Healthcare (Rinne N) for funding.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. UK CR. Ovarian cancer statistics. 2020. Available from: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/ovarian-cancer [Last accessed on 10 Dec 2020].
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424.
3. Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 2010;34:433-43.
4. Kurman RJ, Shih Ie M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol 2016;186:733-47.
5. Meinhold-Heerlein I, Fotopoulou C, Harter P, et al. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch Gynecol Obstet 2016;293:1367.
6. Meinhold-Heerlein I, Fotopoulou C, Harter P, et al. Statement by the Kommission Ovar of the AGO: The new FIGO and WHO classifications of ovarian, fallopian tube and primary peritoneal cancer. Geburtshilfe Frauenheilkd 2015;75:1021-7.
7. J; FIGO Committee on Gynecologic Oncology. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet 2014;124:1-5.
8. Zeppernick F, Meinhold-Heerlein I. The new FIGO staging system for ovarian, fallopian tube, and primary peritoneal cancer. Arch Gynecol Obstet 2014;290:839-42.
9. Dion L, Carton I, Jaillard S, et al. The landscape and therapeutic implications of molecular profiles in epithelial ovarian cancer. J Clin Med 2020:9.
10. Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d'Investigateurs Nationaux Pour les Etudes des Cancers de l'Ovaire (GINECO). Cancer 2009;115:1234-44.
11. Bowtell DD, Böhm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015;15:668-79.
12. Bashashati A, Ha G, Tone A, et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol 2013;231:21-34.
13. Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609-15.
14. Cooke SL, Brenton JD. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol 2011;12:1169-74.
15. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489-94.
16. Schwarz RF, Ng CK, Cooke SL, et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic analysis. PLoS Med 2015;12:e1001789.
17. Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer 2021;21:37-50.
18. Lambrechts S, Smeets D, Moisse M, et al. Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur J Cancer 2016;53:51-64.
19. Stronach EA, Chen M, Maginn EN, et al. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011;13:1069-80.
20. Christie EL, Bowtell DDL. Acquired chemotherapy resistance in ovarian cancer. Ann Oncol 2017;28:viii13-5.
21. Zyl B, Tang D, Bowden NA. Biomarkers of platinum resistance in ovarian cancer: what can we use to improve treatment. Endocr Relat Cancer 2018;25:R303-R18.
22. Marchini S, Fruscio R, Clivio L, et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer 2013;49:520-30.
23. Nassar D, Blanpain C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol 2016;11:47-76.
24. Gao Y, Foster R, Yang X, et al. Up-regulation of CD44 in the development of metastasis, recurrence and drug resistance of ovarian cancer. Oncotarget 2015;6:9313-26.
25. Meng E, Long B, Sullivan P, et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis 2012;29:939-48.
26. Sabini C, Sorbi F, Cunnea P, Fotopoulou C. Ovarian cancer stem cells: ready for prime time? Arch Gynecol Obstet 2020;301:895-9.
27. Alvero AB, Chen R, Fu HH, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009;8:158-66.
28. Raghavan S, Snyder CS, Wang A, et al. Carcinoma-associated mesenchymal stem cells promote chemoresistance in ovarian cancer stem cells via PDGF signaling. Cancers :12.
29. Ruscito I, Darb-Esfahani S, Kulbe H, et al. The prognostic impact of cancer stem-like cell biomarker aldehyde dehydrogenase-1 (ALDH1) in ovarian cancer: A meta-analysis. Gynecol Oncol 2018;150:151-7.
30. Cole AJ, Iyengar M, Panesso-Gómez S, et al. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. JCI Insight 2020:5.
31. Chen R, Alvero AB, Silasi DA, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 2008;27:4712-23.
32. Nuti SV, Mor G, Li P, Yin G. TWIST and ovarian cancer stem cells: implications for chemoresistance and metastasis. Oncotarget 2014;5:7260-71.
33. Yin G, Chen R, Alvero AB, et al. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 2010;29:3545-53.
34. Yin G, Alvero AB, Craveiro V, et al. Constitutive proteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene 2013;32:39-49.
35. Hu Z, Cunnea P, Zhong Z, et al. The Oxford Classic Links Epithelial-to-Mesenchymal Transition to Immunosuppression in Poor Prognosis Ovarian Cancers. Clin Cancer Res 2021; doi: 10.1158/1078-0432.CCR-20-2782.
36. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol 2019;59:147-60.
37. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140-56.
38. Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol 2015;137:173-9.
39. Levine DA, Bogomolniy F, Yee CJ, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 2005;11:2875-8.
40. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489-501.
41. Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci 2005;118:5675-8.
42. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol 2014;4:64.
44. Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 2004;30:193-204.
45. Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev 2000;14:1027-47.
46. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 2001;98:10983-5.
47. Asano T, Fujishiro M, Kushiyama A, et al. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol Pharm Bull 2007;30:1610-6.
48. Avruch J, Hara K, Lin Y, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006;25:6361-72.
50. Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004;18:189-218.
51. Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014;39:159-69.
52. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 2018;15:273-91.
53. Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004;64:7678-81.
54. Wang Y, Helland A, Holm R, Kristensen GB, Børresen-Dale AL. PIK3CA mutations in advanced ovarian carcinomas. Hum Mutat 2005;25:322.
55. Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009;174:1597-601.
56. Matulonis UA, Hirsch M, Palescandolo E, et al. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS One 2011;6:e24433.
57. Nakayama K, Nakayama N, Kurman RJ, et al. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol Ther 2006;5:779-85.
58. Carden CP, Stewart A, Thavasu P, et al. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol Cancer Ther 2012;11:1609-17.
59. Kurose K, Zhou XP, Araki T, Cannistra SA, Maher ER, Eng C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am J Pathol 2001;158:2097-106.
60. McConechy MK, Ding J, Senz J, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol 2014;27:128-34.
61. Obata K, Morland SJ, Watson RH, et al. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res 1998;58:2095-7.
62. Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res 2000;60:7052-6.
63. Macintyre G, Goranova TE, De Silva D, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet 2018;50:1262-70.
64. Castellvi J, Garcia A, Rojo F, et al. Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer 2006;107:1801-11.
65. Martins FC, Couturier D-L, Paterson A, et al. Clinical and pathological associations of PTEN expression in ovarian cancer: a multicentre study from the Ovarian Tumour Tissue Analysis Consortium. Br J Cancer 2020;123:793-802.
66. Huang J, Zhang L, Greshock J, et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer 2011;50:606-18.
67. Montero JC, Chen X, Ocaña A, Pandiella A. Predominance of mTORC1 over mTORC2 in the regulation of proliferation of ovarian cancer cells: therapeutic implications. Mol Cancer Ther 2012;11:1342-52.
68. Yang X, Fraser M, Abedini MR, Bai T, Tsang BK. Regulation of apoptosis-inducing factor-mediated, cisplatin-induced apoptosis by Akt. Br J Cancer 2008;98:803-8.
69. Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 2014;13:1021-31.
70. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520-9.
71. Hudes G, Carducci M, Tomczak P, et al. Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271-81.
72. Yap TA, Kristeleit R, Michalarea V, et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2 and Non–BRCA1/2-Mutant Cancers. Cancer Discov 2020;10:1528-43.
73. Taylor SE, Chu T, Elvin JA, Edwards RP, Zorn KK. Phase II study of everolimus and bevacizumab in recurrent ovarian, peritoneal, and fallopian tube cancer. Gynecol Oncol 2020;156:32-7.
74. Blagden SP, Hamilton AL, Mileshkin L, et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-resistant Ovarian Cancer. Clin Cancer Res 2019;25:1472-8.
75. Bowles DW, Ma WW, Senzer N, et al. A multicenter phase 1 study of PX-866 in combination with docetaxel in patients with advanced solid tumours. Br J Cancer 2013;109:1085-92.
76. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010;11:329-41.
77. Hillmann P, Fabbro D. PI3K/mTOR Pathway Inhibition: opportunities in oncology and rare genetic diseases. Int J Mol Sci 2019:20.
78. Hanker AB, Kaklamani V, Arteaga CL. Challenges for the clinical development of PI3K inhibitors: strategies to improve their impact in solid tumors. Cancer Discov 2019;9:482-91.
79. Fruman DA. Towards an understanding of isoform specificity in phosphoinositide 3-kinase signalling in lymphocytes. Biochem Soc Trans 2004;32:315-9.
80. Sheridan C, Downward J. Inhibiting the RAS-PI3K pathway in cancer therapy. Inhibitors of the Ras superfamily G-proteins, Part B. Elsevier; 2013. pp. 107-36.
81. Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 1994;269:5241-8.
82. Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin Cancer Res 2000;6:880-6.
83. Fekete M, Santiskulvong C, Eng C, Dorigo O. Effect of PI3K/Akt pathway inhibition-mediated G1 arrest on chemosensitization in ovarian cancer cells. Anticancer Res 2012;32:445-52.
84. Zhao JX, Liu H, Lv J, Yang XJ. Wortmannin enhances cisplatin-induced apoptosis in human ovarian cancer cells in vitro. Eur Rev Med Pharmacol Sci 2014;18:2428-34.
85. Ohta T, Ohmichi M, Hayasaka T, et al. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer models. Endocrinology 2006;147:1761-9.
86. Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282-90.
87. Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res 2015;21:730-8.
88. Wang D, Wang M, Jiang N, et al. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016;7:13153-66.
89. Matulonis UA, Wulf GM, Barry WT, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol 2017;28:512-8.
90. Li H, Zeng J, Shen K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch Gynecol Obstet 2014;290:1067-78.
91. Blagden S, Omlin A, Josephs D, et al. First-in-human study of CH5132799, an oral class I PI3K inhibitor, studying toxicity, pharmacokinetics, and pharmacodynamics, in patients with metastatic cancer. Clin Cancer Res 2014;20:5908-17.
92. Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat Rev 2017;59:93-101.
93. Konstantinopoulos PA, Barry WT, Birrer M, et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 2019;20:570-80.
94. Piha-Paul SA, Taylor MH, Spitz D, et al. Efficacy and safety of buparlisib, a PI3K inhibitor, in patients with malignancies harboring a PI3K pathway activation: a phase 2, open-label, single-arm study. Oncotarget 2019;10:6526-35.
95. Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2015;21:77-86.
96. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Wolchok JD. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol 2018;36:3013.
97. Williams CB, Williams KA, Krie AK, et al. Results of a phase Ib trial evaluating the safety and clinical activity of sapanisertib (TAK 228) in combination with serabelisib (TAK 117) and paclitaxel in patients with advanced ovarian, endometrial, or breast cancer. J Clin Oncol 2020;38(15_suppl):3604.
98. Dolly SO, Wagner AJ, Bendell JC, et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res 2016;22:2874-84.
99. Wicki A, Brown N, Xyrafas A, et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur J Cancer 2018;96:6-16.
100. Mahadevan D, Chiorean EG, Harris WB, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer 2012;48:3319-27.
101. Markman B, Tabernero J, Krop I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol 2012;23:2399-408.
102. Shapiro GI, Bell-McGuinn KM, Molina JR, et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin Cancer Res 2015;21:1888-95.
103. Arend RC, Davis AM, Chimiczewski P, et al. EMR 20006-012: A phase II randomized double-blind placebo controlled trial comparing the combination of pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) to pimasertib alone in patients with previously treated unresectable borderline or low grade ovarian cancer. Gynecol Oncol 2020;156:301-7.
104. Farley JH, Brady WE, Fujiwara K, et al. A phase II evaluation of temsirolimus in combination with carboplatin and paclitaxel followed by temsirolimus consolidation as first-line therapy in the treatment of stage III-IV clear cell carcinoma of the ovary. J Clin Oncol 2016;34(15_suppl):5531.
105. de Braud F, Machiels JH, Boggiani D, et al. A Phase 1 Study of mTORC1/2 Inhibitor BI 860585 as a Single Agent or with Exemestane or Paclitaxel in Patients with Advanced Solid Tumors. Cancers ;12:1425.
106. Munster P, Mita M, Mahipal A, et al. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag Res 2019;11:10463-76.
107. Cheraghchi-Bashi A, Parker CA, Curry E, et al. A putative biomarker signature for clinically effective AKT inhibition: correlation of in vitro, in vivo and clinical data identifies the importance of modulation of the mTORC1 pathway. Oncotarget 2015;6:41736-49.
108. Gungor H, Saleem A, Babar S, et al. Dose-Finding Quantitative 18F-FDG PET Imaging Study with the Oral Pan-AKT Inhibitor GSK2141795 in Patients with Gynecologic Malignancies. J Nucl Med 2015;56:1828-35.
109. Gungor H SA, Agarwal R, Blagden S, Micheal A, Stronach EA. Pharmacokinetic (PK)/pharmacodynamic (PD) analysis of escalating repeat doses of the AKT inhibitor GSK2141795 (GSK795) in patients (pts) with ovarian cancer. J Clin Oncol 2011;29:5064.
110. Lee EK, Tan-Wasielewski Z, Aghajanian C, et al. Results of an abbreviated phase II study of AKT inhibitor MK-2206 in the treatment of recurrent platinum-resistant high grade serous ovarian, fallopian tube, or primary peritoneal carcinoma (NCT 01283035). Gynecol Oncol Rep 2020;32:100546.
111. Hyman DM, Bonafede M, O'Cearbhaill R, Grisham RN, Makker V. Abstract CT035: A phase Ib study of miransertib (ARQ 092) in combination with anastrozole in patients with PIK3CA or AKT1-mutant ER+ endometrial or ovarian cancer. Cancer Res 2018;78(13 Supplement):CT035-CT.
112. Fu S, Hennessy BT, Ng CS, et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol Oncol 2012;126:47-53.
113. Shi Y, Frankel A, Radvanyi LG, et al. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer Res 1995;55:1982-8.
114. Mabuchi S, Altomare DA, Cheung M, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res 2007;13:4261-70.
115. Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol 2011;123:19-26.
116. Kollmannsberger C, Hirte H, Siu LL, et al. Temsirolimus in combination with carboplatin and paclitaxel in patients with advanced solid tumors: a NCIC-CTG, phase I, open-label dose-escalation study (IND 179). Ann Oncol 2012;23:238-44.
117. Gasparri ML, Bardhi E, Ruscito I, et al. PI3K/AKT/mTOR pathway in ovarian cancer treatment: are we on the right track? Geburtshilfe Frauenheilkd 2017;77:1095-103.
118. Boers-Sonderen MJ, de Geus-Oei LF, Desar IM, et al. Temsirolimus and pegylated liposomal doxorubicin (PLD) combination therapy in breast, endometrial, and ovarian cancer: phase Ib results and prediction of clinical outcome with FDG-PET/CT. Target Oncol 2014;9:339-47.
119. Zafar Y, Bendell J, Lager J, Yu D, Hurwitz H. Preliminary results of a phase I study of bevacizumab (BV) in combination with everolimus (E) in patients with advanced solid tumors. J Clin Oncol 2006;24(18_suppl):3097.
120. Altomare I, Bendell JC, Bullock KE, et al. A phase II trial of bevacizumab plus everolimus for patients with refractory metastatic colorectal cancer. Oncologist 2011;16:1131-7.
121. Harshman LC, Barbeau S, McMillian A, Srinivas S. A phase II study of bevacizumab and everolimus as treatment for refractory metastatic renal cell carcinoma. Clin Genitourin Cancer 2013;11:100-6.
122. David-West G, Ernlund A, Gadi A, Schneider RJ. mTORC1/2 inhibition re-sensitizes platinum-resistant ovarian cancer by disrupting selective translation of DNA damage and survival mRNAs. Oncotarget 2018;9:33064-76.
123. Wong Te Fong AC, Thavasu P, Gagrica S, et al. Evaluation of the combination of the dual m-TORC1/2 inhibitor vistusertib (AZD2014) and paclitaxel in ovarian cancer models. Oncotarget 2017;8:113874-84.
124. Caumanns JJ, van Wijngaarden A, Kol A, et al. Low-dose triple drug combination targeting the PI3K/AKT/mTOR pathway and the MAPK pathway is an effective approach in ovarian clear cell carcinoma. Cancer Lett 2019;461:102-11.
125. Emons G, Kurzeder C, Schmalfeldt B, et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol Oncol 2016;140:450-6.
126. Tsoref D, Welch S, Lau S, et al. Phase II study of oral ridaforolimus in women with recurrent or metastatic endometrial cancer. Gynecol Oncol 2014;135:184-9.
127. Yuan J, Mehta PP, Yin MJ, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther 2011;10:2189-99.
128. Choi HJ, Heo JH, Park JY, et al. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol Oncol 2019;153:135-48.
129. Iezzi A, Caiola E, Broggini M. Acitivity of pan-class I isofrom PI3K/mTOR Inhibitor PF-05212384 in combination with crizotinib in ovarian cancer xenografts and PDX. Transl Oncol 2016;9:458-65.
130. Xiao Y, Yu Y, Jiang P, et al. The PI3K/mTOR dual inhibitor GSK458 potently impedes ovarian cancer tumorigenesis and metastasis. Cell Oncol ;43:669-80.
131. Deng J, Bai X, Feng X, et al. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019;19:618.
132. Papadopoulos KP, Tabernero J, Markman B, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res 2014;20:2445-56.
133. Rubinstein MM, Hyman DM, Caird I, et al. Phase 2 study of LY3023414 in patients with advanced endometrial cancer harboring activating mutations in the PI3K pathway. Cancer 2020;126:1274-82.
134. Bendell JC, Varghese AM, Hyman DM, et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res 2018;24:3253-62.
135. Dempsey J, Donoho G, Iversen P, et al. Abstract 1761: Inhibition of PI3K/AKT/mTOR signaling by a dual PI3K/mTOR inhibitor (LY3023414) potentiates the antitumor efficacy of the Chk1 inhibitor prexasertib (LY2606368) in models of human high-grade serous ovarian cancer (HGSOC). Cancer Res 2019;79(13 Supplement):1761.
136. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009;8:627-44.
137. Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010;9:1956-67.
138. Hanrahan AJ, Schultz N, Westfal ML, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov 2012;2:56-67.
139. Ichikawa K AT, Nagase H, Saito H, et al. TAS-117, a highly selective non-ATP competitive inhibitor of AKT demonstrated antitumour activity in combination with chemotherapeutic agents and molecular targeted drugs. Molecular Targets and Cancer Therapeutics 2013;12:19-23.
140. Sun H, Yu T, Li J. Co-administration of perifosine with paclitaxel synergistically induces apoptosis in ovarian cancer cells: more than just AKT inhibition. Cancer Lett 2011;310:118-28.
141. Tolcher AW, Kurzrock R, Valero V, et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother Pharmacol 2020;85:673-83.
142. Banerji U, Dean EJ, Perez-Fidalgo JA, et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin Cancer Res 2018;24:2050-9.
143. Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol 2010;347:21-41.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Rinne N, Christie EL, Ardasheva A, Kwok CH, Demchenko N, Low C, Tralau-Stewart C, Fotopoulou C, Cunnea P. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist 2021;4:573-95. http://dx.doi.org/10.20517/cdr.2021.05
AMA Style
Rinne N, Christie EL, Ardasheva A, Kwok CH, Demchenko N, Low C, Tralau-Stewart C, Fotopoulou C, Cunnea P. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resistance. 2021; 4(3): 573-95. http://dx.doi.org/10.20517/cdr.2021.05
Chicago/Turabian Style
Rinne, Natasha, Elizabeth L. Christie, Anastasia Ardasheva, Chun Hei Kwok, Nikita Demchenko, Caroline Low, Catherine Tralau-Stewart, Christina Fotopoulou, Paula Cunnea. 2021. "Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer" Cancer Drug Resistance. 4, no.3: 573-95. http://dx.doi.org/10.20517/cdr.2021.05
ACS Style
Rinne, N.; Christie EL.; Ardasheva A.; Kwok CH.; Demchenko N.; Low C.; Tralau-Stewart C.; Fotopoulou C.; Cunnea P. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021, 4, 573-95. http://dx.doi.org/10.20517/cdr.2021.05
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 111 clicks
Cite This Article 111 clicks

 Like This Article 2
likes
Like This Article 2
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.