
Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy

Abstract
More than half of cancer patients are treated with radiotherapy, which kills tumor cells by directly and indirectly inducing DNA damage, including cytotoxic DNA double-strand breaks (DSBs). Tumor cells respond to these threats by activating a complex signaling network termed the DNA damage response (DDR). The DDR arrests the cell cycle, upregulates DNA repair, and triggers apoptosis when damage is excessive. The DDR signaling and DNA repair pathways are fertile terrain for therapeutic intervention. This review highlights strategies to improve therapeutic gain by targeting DDR and DNA repair pathways to radiosensitize tumor cells, overcome intrinsic and acquired tumor radioresistance, and protect normal tissue. Many biological and environmental factors determine tumor and normal cell responses to ionizing radiation and genotoxic chemotherapeutics. These include cell type and cell cycle phase distribution; tissue/tumor microenvironment and oxygen levels; DNA damage load and quality; DNA repair capacity; and susceptibility to apoptosis or other active or passive cell death pathways. We provide an overview of radiobiological parameters associated with X-ray, proton, and carbon ion radiotherapy; DNA repair and DNA damage signaling pathways; and other factors that regulate tumor and normal cell responses to radiation. We then focus on recent studies exploiting DSB repair pathways to enhance radiotherapy therapeutic gain.
Keywords
Introduction
Ionizing radiation has been used to treat cancer for more than 120 years, and radiotherapy is widely used to treat many types of cancer. More than half of cancer patients receive radiation as monotherapy or in combination with surgery, genotoxic chemotherapy, and targeted therapy. Radiation is usually delivered with external beams, but radioactive implants (brachytherapy) are used to treat prostate, head and neck, breast, eye, and other cancers[1]. Regardless of the mode of delivery, ionizing radiation is effective because it causes cytotoxic DNA damage (i.e., it is genotoxic), and in this way it is similar to genotoxic chemotherapy. However, radiotherapy is only effective for local tumor control and isolated metastases, whereas genotoxic chemotherapy, delivered systemically, can also treat widespread metastatic disease. There is evidence that radiotherapy may be effective against distant disease, through immune-mediated, non-targeted abscopal effects, but this approach is currently limited to pre-clinical studies[2]. Radiotherapy has several benefits for patients: It is non-invasive, painless, and has low rates of severe side-effects, highlighting another difference from systemic, genotoxic chemotherapy which often causes side effects that compromise patient quality of life. Although metastatic disease is ultimately responsible for most cancer deaths, the importance of local tumor control should not be underestimated. As noted in a widely used radiation oncology textbook, “…for tumors with high metastatic potential, such as breast, prostate, and lung…improved locoregional control by radiotherapy with or without chemotherapy enhances overall [patient] survival”[3]. Among the ongoing challenges in the radiotherapy field are the adverse effects of radiation on sensitive, normal tissues adjacent to tumors, in particular brain, spinal cord, and heart. In contrast, systemic genotoxins cause widespread damage, in particular to proliferative normal tissues including gastrointestinal lining and bone marrow, causing nausea and anemia, as well as non-proliferating brain tissue, causing chemotherapy-induced cognitive impairment or “chemo-brain”[4]. For both genotoxic chemotherapeutics and radiation, there is great interest in understanding mechanisms of intrinsic and acquired tumor cell resistance to these agents[5-8].
The goal of radiotherapy is to completely eradicate tumor cells while sparing nearby normal tissue. The efficacy of radiotherapy has greatly improved with the development of advanced techniques for diagnostic imaging, beam-focusing, and beam-shaping[9,10], and treatment outcomes continue to improve as combination therapeutic strategies mature[11]. Two ways that combination therapies can improve therapeutic gain are to radiosensitize tumor cells, especially those with high intrinsic or acquired radioresistance, and protect normal tissue. There are many biological parameters that modulate tumor and normal cell responses to radiation, such as cell type, cell cycle phase, tissue/tumor microenvironment, oxygen levels, DNA repair capacity, and others. We begin with a synopsis of radiation damage to cellular components; cellular responses to radiation damage; environmental and cellular factors that determine normal and tumor cell radiosensitivity; and strategies used to counter tumor radioresistance or protect normal tissue from radiation damage. We then discuss how DNA repair and DNA damage response (DDR) pathways can be exploited to radiosensitize tumor cells and protect normal tissue during radiotherapy.
Ionizing radiation damege to cellular components and cell responses
Genotoxic chemotherapeutics and ionizing radiation kill cells by directly or indirectly damaging DNA or interfering with DNA metabolism (DNA polymerases, topoisomerases, or chromosome segregation machinery). Ionizing radiation, whether delivered by X-rays, protons, or carbon ions, causes damage to cellular components through direct energy absorption or indirectly by ionizing water to generate reactive oxygen species (ROS), including hydroxyl radicals, superoxide, and hydrogen peroxide[12]. ROS are highly reactive and interact almost immediately with cellular components, causing oxidative and other damage to proteins, nucleic acids, and membrane components. ROS are also generated during normal cell metabolism, primarily from mitochondrial function[13,14]. Cells survive and thrive despite > 100,000 spontaneous DNA lesions/cell/day, including ~10,000 single-strand breaks and ~50 DNA double-strand breaks (DSBs)[15-17]. Nearly all DNA lesions block DNA replication, although some can be bypassed by error-prone translesion DNA polymerases[18]. The ability of cells to manage this remarkable daily lesion load is a reflection of the high efficiency of DNA repair systems. That said, DNA damage can cause mutations, chromosome structural alterations, cell cycle arrest, senescence, and cell death. Among the hundreds of types of DNA lesions, DSBs are among the most cytotoxic, and the cytotoxicity of genotoxic chemicals and ionizing radiation is largely due to DSBs[19,20]. Other double-strand lesions, such as inter-strand crosslinks, are also highly cytotoxic[21].
Cells respond to DNA damage by activating checkpoint signaling and DNA repair pathways, collectively termed the DDR. DDR promotes cell survival and suppresses cancer by promoting genome stability, but it also triggers programmed cell death when damage is excessive. Altered expression or mutation of DDR proteins predispose to cancer, determine tumor response to chemo- and radiotherapy, and underlie several congenital conditions including multiple types of Seckel syndrome, primordial dwarfism, and premature aging syndromes[22-24]. The DDR is a major determinant of cancer cell responses to chemo- and radiotherapy, and is thus an enticing target to augment cancer therapy[25-30]. DDR components are often defective in cancer, but because the DDR is a complex network of interacting/cross-talking pathways, cells can respond to alterations in one pathway with compensatory changes in other pathways. Compensatory pathways within the DDR network represent formidable obstacles to successful cancer treatment. A better understanding of DDR pathways can reveal synthetic lethal relationships that can be exploited to augment cancer therapy in general, and to develop personalized therapies[31-35].
The DDR includes two checkpoint signaling pathways, one centered on ataxia telangiectasia mutated (ATM), a kinase that responds to DSBs and one centered on ataxia telangiectasia and Rad3 related (ATR) kinase that is triggered by single-stranded DNA (ssDNA) generated by 5’-3’ resection of DSB ends and by decoupling of the replication machinery from MCM helicase at stalled replication forks[36-39]. ATM and ATR, along with DNA-PKcs, are PI3 kinase-like kinases (PIKKs) that are “early responders” to DSBs and replication stress. PIKKs phosphorylate large networks of proteins[40-42] including the downstream effector kinases Chk1 and Chk2 that phosphorylate p53 and other targets to arrest the cell cycle in response to damage, promote DNA repair, and promote programmed cell death pathways when damage exceeds a threshold[43-46][Figure 1]. The DDR thus presents two broad targets to manipulate for therapeutic gain: inhibiting DNA repair sensitizes cells to damage and inhibiting checkpoint signaling prevents cell cycle arrest in response to damage, increasing replication stress, fork collapse to DSBs, genome instability, and cell death[20,47-50].
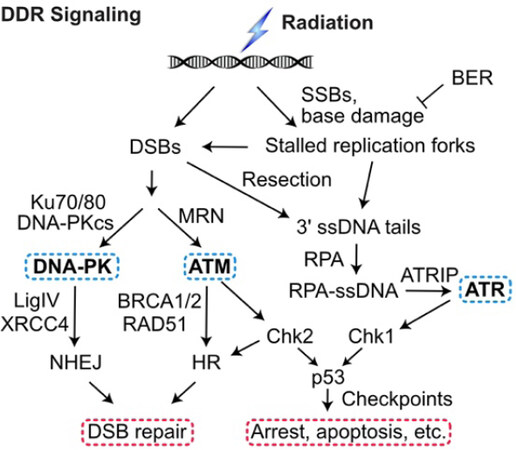
Figure 1. DDR signaling. Ionizing radiation and genotoxic chemotherapy create single- and double-strand DNA damage including DSBs that activate three PIKKs: DNA-PK, ATM, and ATR. Single-strand breaks and base damage, if not repaired by base excision repair (BER), block replication, which produces ssDNA when the replisome decouples from the MCM helicase or stalled forks are cleaved to produce DSBs, which, along with frank DSBs, are resected to 3’ single-stranded tails that are coated by RPA. This activates ATR to signal checkpoint responses through Chk1 and p53. Non-resected DSB ends are bound by the Ku70/Ku80 heterodimer, which recruits and activates DNA-PKcs in the DNA-PK holoenzyme, LigIV/XRCC4 ligates DNA ends to effect NHEJ. The competing HR pathway initiates with limited DSB end resection by MRE11/RAD50/NBS1 (MRN), more extensive resection by Exo1 and Dna2, and RAD51 binding to ssDNA (mediated by BRCA1, BRCA2, and other proteins) to yield the RAD51-ssDNA nucleoprotein filament that effects HR. DDR: DNA damage response; DSBs: DNA double-strand breaks
DSBs are repaired by error-prone non-homologous end-joining (NHEJ) or by homologous recombination (HR) repair [Figure 2][51,52], templated from sister chromatids (restricted to S/G2 phases), homologous chromosomes, or short sequence repeats if the double-strand damage occurs within or nearby repeated sequences - not uncommon given the human genome comprises > 50% repetitive elements (Alu, MIRs, SINEs, LINEs, etc.)[53]. HR is generally accurate, but it does pose risks of genome rearrangements including large-scale loss of heterozygosity and translocations that can initiate tumorigenesis and drive tumor progression[27,54,55]. When the primary NHEJ or HR pathways fail, even more error-prone DSB repair pathways serve as back-up, including alternative (microhomology-mediated) NHEJ, single-strand annealing, and break-induced replication[56-62].
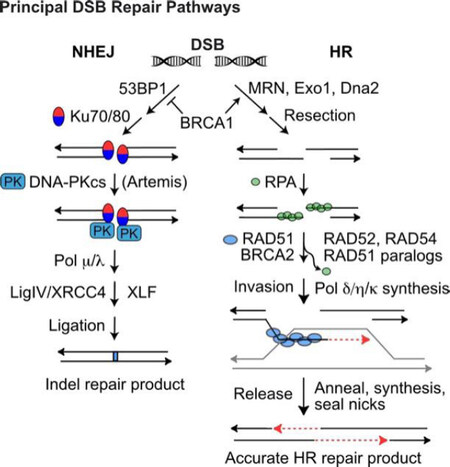
Figure 2. DSB repair by NHEJ and HR. Pathway choice is determined by end-resection, prevented by 53BP1, or promoted by BRCA1/MRN and Exo1/Dna2 exonucleases. (Left) NHEJ of unresected DSB ends initiates with Ku70/80 binding to ends and recruitment of DNA-PKcs to form the activated DNA-PK holoenzyme. Artemis is required to trim certain types of end-structures, and small gaps may be filled with polymerases m and l prior to LigIV/XRCC4/XLF-mediated ligation. NHEJ repair usually produces small indels (1-20-bp deletions, few-bp insertions). (Right) Resected 3’ single-strand ends are coated with RPA, which is then exchanged with RAD51, mediated by BRCA2, RAD52, RAD54, and RAD51 paralogs. The RAD51 nucleoprotein filament seeks and invades a homologous donor duplex (grey). RAD51 dissociates before repair synthesis; the newly synthesized strand (red dash) is released from the donor duplex and anneals with the complementary strand on the opposite side of the DSB. A second round of repair synthesis and nick sealing completes repair. DDR: DNA damage response; DSB: DNA double-strand break; NHEJ: non-homologous end-joining; HR: homologous recombination
Radiobiological properties of therapeutic ionizing radiation
Three types of external beam radiation are used to treat cancer. X-rays and protons are low linear energy transfer (LET) radiation, although proton LET varies (see below). LET is a measure of ionization density, thus low LET X-rays (and protons for the most part) are sparsely ionizing. This means that most X-ray lesions, including DSBs, are widely dispersed. X-rays are massless photons that interact weakly with tissue, thus the highest X-ray doses are near the skin at the entrance point. To concentrate X-ray doses within tumors, beams are intensity modulated and delivered to patients from several angles, spreading low doses to a large volume of normal tissue[3]. Protons have a small mass and a single positive charge. Proton interactions with tissue slow and eventually stop these particles at a defined depth (within a tumor), termed the Bragg peak[63]. This feature provides a clear benefit as normal tissue beyond the tumor receives essentially no dose. Carbon ions with high mass and six positive charges are high LET radiation. Because of their mass, carbon ions also stop at depth and eliminate exit dose, similar to protons. However, the high mass and high charge of carbon ions produces dense ionization tracks, especially at the end of the track as particles slow and stop[64,65].
X-rays, protons, and carbon ions induce the same number of DSBs per unit dose (~40 DSBs/Gy). Exposures to 1 Gy of X-rays or protons kills ~10%-20% of cells[66-68]. In contrast, the same dose of carbon ions kills 2-3-fold more cells, hence the relative biological effect (RBE) of carbon ions is ~2.5. Proton LET increases somewhat in the distal region of the Bragg peak, and RBE correspondingly increases to perhaps as high as 1.7[65,69]. The high RBE of carbon ions reflects the fact that these ions efficiently induce clustered DSBs, defined as two or more DSBs separated by < 200 bp[64,68,70]. Clustered DSBs are repaired inefficiently and are hence more cytotoxic than isolated DSBs. Low LET X-rays and protons induce occasional clustered DSBs - it is thought that these lesions primarily determine low LET radiation cytotoxicity, not the more prevalent isolated DSBs[64,68,71-73]. The greater cytotoxicity (RBE) of carbon ions reflects their greater efficiency at inducing clustered DSBs. NHEJ, the dominant DSB repair pathway, initiates with Ku70/Ku80 (Ku) binding to DNA ends and recruitment of DNA-PKcs [Figure 2][51]. Ku appears to efficiently bind both large and small DNA fragments, generated by isolated and clustered DSBs, respectively. However, short fragments do not activate DNA-PKcs kinase[74], which has critical roles in NHEJ, HR, DDR signaling, and checkpoint activation[75]. Thus, short DNA fragments appear to be refractory to repair by NHEJ, and this may account for both the greater cytotoxicity of clustered vs. isolated DSBs, and the shift from NHEJ toward HR in cells exposed to high LET radiation[64,76-79]. A greater dependence on HR was also observed with protons than X-rays[80], perhaps reflecting the higher proton LET in the Bragg peak. However, a more recent study showed minimal differences when cells were treated with X-rays vs. protons, and inhibitors of NHEJ or HR[81], suggesting additional factors determine repair pathway choices among cell types. That cells struggle to repair clustered DSBs may reflect their rarity in nature and the lack of selective pressure to evolve repair systems for this class of complex DNA lesion.
Low and high LET radiation are distinguished in two other ways. Low LET X-rays and protons induce ROS most efficiently in well-oxygenated tissue. At low oxygen levels, the cytotoxic effects of X-rays and protons is reduced ~3-fold, the so-called oxygen enhancement ratio (OER)[82]. Importantly, high LET carbon ions show far less reliance on oxygen (lower OER), owing to the greater ionization potential of these high mass/high charge ions[82,83]. Radiosensitivity varies during the cell cycle. Low LET X-rays and protons show highest cytotoxicity during G1 and M phases, and ~2-fold less cytotoxicity during S-phase, termed S-phase radioresistance[84]. Interestingly, high LET carbon ions show the opposite effect: ~2-3-fold S-phase radiosensitivity relative to G1 cells (Kato, unpublished results). This suggests one mechanism by which mixed high and low LET exposures might yield synergistic cell killing[85-87].
The highly damaging effects of high LET radiation initially raised concerns about the safety of carbon ions in radiotherapy[88], but serious side effects occur no more often than with X-rays or protons[89-91]. This safety profile probably reflects the fact that high LET ions behave similarly to low LET X-rays and protons while traveling through (normal) tissue at high speed, gaining their high LET properties only when slowing and stopping at the end of their tracks (in tumors)[63,92]. Thus, carbon ion LET and RBE are relatively low in the entrance region and increase dramatically in the Bragg peak, and the most damaging effects are confined to the tumor volume[63,93,94].
Cellular radiosensitivity and radioresistance
Many physical, biological, and environmental factors influence cell responses to ionizing radiation, including those that determine the level and types of damage to cell components; cell state (proliferating or quiescent, cell cycle phase); DDR signaling and DNA repair capacity; propensity for programmed cell death; cellular “memory” of past adaptive exposure; and tissue macro- and microenvironments. For example, RB status influences intrinsic radiosensitivity among individuals[95,96], and such biomarkers can be exploited to personalize radiotherapy treatment planning[97,98]. The physical natures of ionizing radiation (photon vs. particle and large vs. small mass/charge) determine lesion spatial distributions, reparability, and cytotoxicity. Nonetheless, as noted by Willers, Xia, and colleagues[99], “there is no absolute resistance to radiation”. If enough radiation can be delivered, all tumor cells will be eradicated regardless of environmental, genetic, or metabolic factors. The practical limitation, of course, is collateral damage to normal tissue. Hence, any strategy that increases radiation dose to tumors, decreases doses to normal tissues, increases tumor-specific cytotoxic effects of radiation, or protects normal tissue from unavoidable exposure can improve therapeutic gain and/or reduce side effects.
Hypoxia
An important environmental factor that regulates DNA damage induction is oxygen level, which varies among tumor types, within different regions of a tumor, and between tumor and normal tissue. Normal tissue is well-oxygenated, but tumors are often hypoxic as they struggle to supply oxygen during their rapid growth. To a degree, tumors adapt to the hypoxic state, for example, by stabilizing HIF1α, which regulates oxygen metabolism and angiogenesis via vascular endothelial growth factor, among other effects[100]. Although certain solid tumors are frequently characterized as “hypoxic”, e.g., head and neck and pancreatic cancers, it is now clear that most solid tumors have hypoxic regions. The degree of hypoxia is regulated by passive oxygen diffusion, creating somewhat stable oxygen gradients across tumor masses, and by transient effects such as altered perfusion by tumor vasculature[100]. Given the importance of oxygen for ROS production during irradiation (OER), hypoxic regions within tumors are naturally radioresistant; this is a particularly vexing problem given that normal (well-oxygenated) tissue may suffer greater ROS damage than adjacent tumors, reducing therapeutic gain. Several strategies have been proposed to mitigate hypoxia-related radioresistance, including modulation of dose fractionation, inflammatory responses, and hypoxia itself[101,102]. For example, investigators have explored hyperbaric oxygen to radiosensitize tumors, and tourniquets to promote normal tissue radioresistance, but these approaches have fallen out of favor[3]. Another idea is to mimic oxygen with agents such as nitroimidazoles, which radiosensitize hypoxic tumors. Although these are effective, clinical use has been restricted because of associated neurotoxicity[103,104].
Cell proliferation rates
Solid tumors comprise rapidly growing (“bulk”) tumor cells and small numbers of so-called cancer stem cells (CSCs). Much of tumor sensitivity to genotoxic chemo- and radiotherapeutics reflects the fact that rapidly dividing, bulk tumor cells are more sensitive to DNA damage than most (non-dividing) normal cells. CSCs, similar to normal stem cells, divide more slowly than bulk tumor cells, hence CSCs are naturally radioresistant. Because CSCs are tumor-initiating cells that support both local tumor growth and seed distant metastases, CSC radioresistance is a significant barrier to durable chemo- and radiotherapy treatment responses[105-107]. Similar to CSCs, some tumor cells may be quiescent; tumor dormancy is seen locally and at metastatic sites, it can be induced by therapy, and it confers radioresistance[108]. Changing fractions of bulk, CSC, and quiescent tumor cells may cause regional variations in tumor radioresistance, complicating radiotherapy treatment planning.
Hyperthermia
The sensitizing effects of hyperthermia have long been investigated in vitro and in pre-clinical models, but it has not yet advanced to clinical practice[109]. Hyperthermia alters tissue perfusion to mitigate hypoxia, and it has been debated whether it directly induces DNA damage, but there is clear evidence that it triggers DDR signaling and suppresses DNA repair[110,111]. Ultrasound waves generate heat, and this technology is being explored to induce local hyperthermia for tumor radiosensitization[112].
Radioprotectors and radiosensitizers
Because most radiation damage is induced indirectly through ROS, intrinsic and extrinsic modulation of cellular re-dox status strongly affects radioresistance. Re-dox mechanisms have been investigated to sensitize tumors and/or protect normal tissue during radiotherapy, including modulating NAD+, glucose, and other re-dox metabolic pathways; use of antioxidants (e.g., vitamins C and E) and isoflavones; use of Mn-porphyrin compounds such as manganese-dependent superoxide dismutase (Mn-SOD) mimetics; modulating superoxide dismutase; and modifying patient exercise routines[113-119]. Metformin, which reduces hypoxia by reducing oxygen consumption, and melatonin, a natural hormone with antioxidant and anti-inflammatory effects, are also under investigation for radiosensitization or protection[120-123]. Radiation countermeasures are designed to protect individuals from adverse effects of accidental or intentional (i.e., dirty bomb) total-body irradiation; these strategies may be useful for normal tissue protection during radiotherapy[124-126].
Adaptive responses
Cells exposed to a low dose of radiation and then subsequently challenged by a high, cytotoxic dose show enhanced survival compared to cells that did not receive a “priming” dose. This effect, termed the adaptive response, typically refers to enhanced cell survival, but radioadaptive responses have been observed with other endpoints, including chromosome aberrations, mutation, micronuclei formation, sister chromatid exchange, delayed genome instability, and cellular transformation[127-133]. These radioadaptive responses are transient, usually subsiding within 24 h of the priming dose. Several regulatory proteins are known to positively or negatively influence cell survival adaptive responses to radiation, including Mn-SOD, NFkB, p53, and NOX4, several of which are mediated by the anti-apoptosis factor survivin[134-139]. Adaptive responses may be problematic, for example, if CT scans used to locate tumors induce tumor radioresistance[134], but other radioadaptive effects, such as immunomodulatory responses, may prove beneficial[140,141]. These transient radioadaptive responses are distinct from two other types of tumor adaptative responses to therapy: adaptive (upregulated) mutagenesis, which accelerates tumor evolution, and modulation of tumor microenvironments, both of which can drive tumor resistance to radio- and chemotherapy[142,143].
Targeting DSB repair to enhance radiotherapy
DSB repair is a major determinant of cellular radioresistance, and key NHEJ and HR proteins are attractive tumor radiosensitization targets. Because DNA repair and DDR systems are tightly integrated, radiosensitization can be achieved by interfering with these networks in a multitude of ways. In addition, “omics” analyses hold promise for personalizing radiotherapy doses based on radiation response profiles[144]. For additional perspectives on these topics, readers are referred to these recent reviews[31,35,97,145-149]. Current experimental and therapeutic options that target DSB repair and DDR factors are listed in Table 1 and discussed in the following sections.
DSB repair and DDR targets to enhance radiotherapy
| Pathway(s) | Target proteins | Inhibitors* | Ref. |
|---|---|---|---|
| NHEJ | DNA-PKcs | LY294002, LY3023414, NU7026, NU7441, NU5455, VX-984, CC-115, BCCA621C, IC86621, IC87102, IC87361, OK-1035, SU11752, KU-00600648 | [150-157] |
| EGFR | Cetuximab | [148,158-160] | |
| HR | RAD51 | RI-1, B02, CYT-0851, 3E10 (antibody), Fab-F2-iPTD (antibody fragment) | [161-166] |
| BRCA2 | BRCA2 siRNA | [167] | |
| PARP1 | AZD2281, olaparib, veliparib, rucaparib, PJ34, E7016, AG14361, GPI-15427, 4-amino-1,8-naphthalimide | [168-170] | |
| DDR | ATM, ATR, Chk1/2, Wee1 | KU-55933, KU-60019, KU-59403, CP466772, AZD0156, VX-970, VE-821, AZD-6738, (–)-Schisandrin B, NVP-BEZ235, ETP-46464, AZ-20, AZD-7762, PF-00477736, XL-844, SCH-900776, prexasertib (LY2606368), LY2880070, SRA737, GDC-0575, AZD-1775, CJM061 | [34] |
| PI3K/AKT/mTOR | CC-115, BEZ235, PI103, BKM120, rapamycin, NVP-BEZ235 | [156,171-173] | |
| Multiple | Hsp90 (DNA-PKcs + RAD51) | 17AAG, PU-871, TAS-116 | [174-180] |
| DNA-PKcs + PARP1 | Rucaparib + NU7441, AZD7648 + olaparib, | [181,182] | |
| ATM/ATR/Chk1 + PD-L1 | shRNA-ATM + PD-L1 antibody | [183] |
Targeting NHEJ
DNA-PKcs is activated when complexed with Ku-bound DNA ends at DSBs, leading to phosphorylation of itself and other targets including Ku, RPA, and H2AX. DNA-PKcs autophosphorylation at two clusters (ABCDE and PQR, including T2609 and T2056 residues) is critical for subsequent NHEJ steps[51,75,184], and DNA-PKcs inhibitors are strong radiosensitizers. However, because NHEJ is active in all nucleated cells, and cells need to repair spontaneous DSBs, inhibiting NHEJ non-specifically may adversely affect normal tissues, especially those within the radiation field. In certain solid tumors, such as ovarian and liver cancers, DNA-PK activity is elevated and this correlates with poor prognoses[185,186]. In these cases, DNA-PKcs inhibition may improve therapeutic gain. Several small molecule DNA-PKcs inhibitors, and other targeted approaches, have shown promising results in vitro and in pre-clinical models to enhance radio- and/or chemotherapy, but few have advanced to human clinical trials, due at least in part to challenges associated with cross-inhibitory effects against PIKKs (ATM, ATR, and mTOR) or bioavailability.
NU7441 is a fairly specific DNA-PKcs inhibitor that showed promising results as a radiosensitizer against nasopharyngeal and liver cancer[150,151], and low concentrations of NU7441 enhance radiosensitivity of lung cancer cells to both X-rays and carbon ions[152]. Targeting DNA-PKcs with NU7441 in combination with the PARP1 inhibitor rucaparib radiosensitized Ewing sarcoma cells[181]. The DNA-PKcs inhibitor VX-984 radiosensitizes glioblastoma cells in vitro and in orthotopic tumors[153]. Two recently developed small molecule DNA-PKcs inhibitors are NU5455 and AZD7648. NU5455 is a highly selective DNA-PKcs inhibitor that increases the efficacy of radiotherapy and genotoxic chemotherapy treatment of lung cancer xenografts[154]. AZD7648 is a highly selective and potent DNA-PKcs inhibitor that enhances radiotherapy of lung tumor xenografts alone and when combined with the PARP1 inhibitor olaparib; this drug is advancing to clinical trials[182]. Precise selectivity is not necessarily required: the DNA-PKcs inhibitors, LY3023414 and CC-115, cross-inhibit mTOR (another PIKK) and show promising pre-clinical results. LY3023414 has advanced to clinical trials[155,156]. In preclinical studies, selective radiosensitization of hypoxic tumors was achieved using the hypoxia-activated pro-drug BCCA621C to inhibit DNA-PKcs[157].
Many tumors overexpress wild-type or mutant versions of the epidermal growth factor receptor (EGFR). The EGFR pathway feeds into the PI3K/AKT/mTOR pathway that drives cell cycle progression. Interestingly, EGFR pathway activation stimulates DSB repair, and this was traced, at least in part, to an interaction between AKT1 and DNA-PKcs[187]. In a parallel EGFR pathway, radioresistance of tumor cells that overexpress Rab5C, Ku70, and Ku80 was traced to Rab5C regulation of EGFR internalization and its translocation to the nucleus, where EGFR stimulates Ku70/Ku80 expression[188]. Cetuximab, a clinically useful monoclonal antibody that targets EGFR, inhibits DNA-PKcs[158] and enhanced radiotherapy in early clinical trials to treat cutaneous squamous cell carcinoma[159]. EGFR nuclear translocation is stimulated by radiation mediated by Cavelolin-1 (CAV-1), and CAV-1 knockdown radiosensitizes triple-negative breast cancer, a tumor type for which there are no current targeted therapies and poor prognoses[189]. Mutant forms of EGFR (D746-750, L858R, and the targeted-therapy resistant T790M mutant) confer radiosensitivity to hypoxic lung cancer cells, at least in part due to downregulation of RAD50, a member of the MRE11/RAD50/NBS1 complex that plays early end-processing and signaling roles in NHEJ and HR[190]. These results suggest that tumor EGFR status can be used to personalize radiotherapy treatment plans and augmentation with NHEJ inhibitors. The link between EGFR and DSB repair suggests strategies to modulate tumor radiosensitivity by inhibiting NHEJ indirectly with available drugs that target EGFR and AKT1/3 pathways[148,160].
Targeting HR
A key step in HR is formation of RAD51 nucleoprotein filaments that seek and invade homologous duplex DNA repair template [Figure 2]. RAD51 sub-nuclear foci are observed ~1 h after irradiation and are often interpreted as evidence of “HR activity”. However, RAD51 nucleoprotein filament formation marks only the initial phase of HR; once the filament invades a donor duplex, RAD51 must dissociate to allow extension of the invading strand by repair-associated DNA polymerases[191]. Thus, RAD51 foci are markers of HR initiation, but persistent RAD51 foci may reflect failure to complete HR due to downstream HR defects[192]. Functional HR, therefore, is best assayed by directly detecting HR products. There are several types of HR assay systems, including plasmid transfection systems, integrated HR repeat substrates, and HR-mediated gene editing[193,194]. When assaying RAD51-dependent HR using linked (direct or inverted) repeats, it is important that the design detects RAD51-dependent gene conversion but not RAD51-independent single-strand annealing[62]. Plasmid transfection assays are convenient, but substrates may not be chromatinized before or during HR, and therefore may not accurately reflect the full constellation of HR functions in chromatin[195]. Similarly, gene editing involves transfection of a non-chromatinized, homologous donor DNA sequence. Plasmid and gene editing assays are useful in rapid HR screens that can be complemented by analysis of HR products in a chromosomal context.
HR is important for repair of frank DSBs, but its other critical role is repairing single-ended DSBs that arise when replication stress causes fork collapse [Figure 3][196]. A 1-Gy dose of ionizing radiation induces ~40 frank DSBs, but hundreds of single-strand lesions that can cause “secondary DSBs” due to fork collapse[197,198]. HR is critical for repair of these one-ended DSBs because mis-repair by NHEJ necessarily involves a distant DSB end (from a different broken replication fork or a frank DSB), causing large-scale genome rearrangements including deletions, translocations, and dicentric chromosomes that can trigger cell death or genome instability from persistent bridge-breakage-fusion cycles[199]. Thus, care must be taken when interfering with HR to enhance radiotherapy, as HR is critical for maintaining genome stability in normal tissues to prevent induction of secondary cancers.
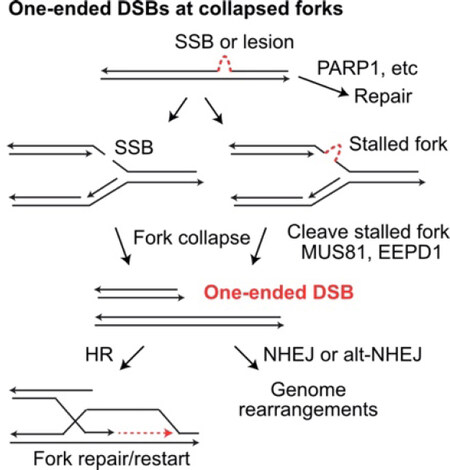
Figure 3. Repair and mis-repair of one-ended DSBs at collapsed replication forks. Single-strand breaks (SSB) and single-strand DNA lesions may be repaired prior to replication. If encountered by a replication fork, SSBs and single-strand lesions cause fork collapse and fork stalling, respectively. Stalled forks may collapse when cleaved by MUS81 or EEPD1. Collapsed forks create one-ended DSBs that can initiate fork restart by HR, or they may be joined to other DSB ends (at collapsed forks or frank DSBs) by NHEJ or alt-NHEJ, creating large-scale genome rearrangements. DSBs: DNA double-strand breaks; HR: homologous recombination; NHEJ: non-homologous end-joining
Because RAD51 plays a central role in HR, it is an attractive target for radiosensitization. The Bishop and Connell labs developed a small molecule RAD51 inhibitor, RI-1, that blocks RAD51 binding to ssDNA[200] and radiosensitizes glioma and glioblastoma cells[161,162]. New RAD51 inhibitors have been developed, including one that blocks D-loop formation (strand invasion) and HR but does not affect RAD51 binding to ssDNA or formation of radiation-induced RAD51 foci[201,202]. A recently developed antibody fragment linked to a cell-penetrating peptide blocks RAD51 DNA binding, sensitizes cells to radiation, and is synthetically lethal with PTEN defects in glioma and melanoma cells[163-165]. Another small molecule RAD51 inhibitor, CYT-0851, is currently in a clinical trial as monotherapy against several types of cancer[166].
HR defects pre-dispose to cancer, including breast, ovarian, and other cancers with defects in BRCA1, BRCA2, PALB2, MRE11, and RAD51, as well as DDR factors that regulate HR, such as ATM[203-206]. HR proteins function as tumor suppressors by maintaining genome stability by promoting accurate DSB repair, stabilizing stressed replication forks, and repairing and restarting collapsed replication forks[207]. PARP1 inhibitors cause replication stress by inhibiting PARP1-dependent repair of single-strand damage and from PARP1-trapping on damaged DNA, accounting for the synthetic lethality of PARP1 inhibitors in HR-deficient tumor cells[35,208]. PARP1 inhibitors are widely used in clinical management of HR-defective breast and ovarian cancers[209,210] and are being explored as adjuncts to radiotherapy[168-170]. To inhibit proteins such as BRCA2 for which there are no small molecule inhibitors, genetic approaches such as siRNA knockdown offer another means to transiently induce HR defects to enhance radiosensitivity[167]. HR defects, whether intrinsic to the tumor or induced by drugs or other means, may be particularly useful when paired with high LET carbon ions given the greater importance of HR in repair of clustered DSBs[64,76-78,167]. Just as HR defects sensitize cells to radiation and genotoxic chemotherapy, therapeutic resistance to these agents, and to PARP1 inhibitors, correlates with restoration or upregulation of HR[211-215]. Radiosensitization of tumors with HR inhibitors may thus be most effective against cancers that upregulate HR.
Targeting DDR signaling factors
The DDR is important for tumor suppression, and it also comprises important targets that mediate therapeutic resistance to radiation and chemotherapy[216]. ATM and ATR are key regulators of critical HR factors, including MRE11, NBS1, CtIP, p53, RPA, BRCA1, PALB2, H2AX, and RAD51[37,192,217,218]. ATM, ATR, and DNA-PKcs collaborate to regulate HR, NHEJ, and DNA damage checkpoint responses[30]. Targeting these PIKKs and other DDR factors, including Chk1, Chk2, and Wee1, are very active research topics[35,97,146,148,149,219]. Some DDR inhibitors show significant toxicity, hence delivery during protracted, fractionated radiotherapy raises safety concerns; these might be mitigated by using localized drug delivery. Nonetheless, several DDR inhibitors have advanced to clinical trials, including two phase 1 trials to augment radiotherapy with the ATR inhibitors VX-970 and AZD6738[34,220]. ATM inhibitors, including AZD1390 and AZD0156, have shown promise for radiosensitizing various solid tumors in preclinical studies, including glioblastoma, head and neck cancer, and lung cancer[34,221-223]. ATM and ATR inhibitors are also being tested for synthetic lethal effects with PARP1 inhibitors[220-222]; such combinations may also augment radiotherapy. The PI3K/AKT/mTOR pathway has well-defined roles in suppressing apoptosis and promoting cell proliferation, but it also interfaces with the DDR, promoting both HR and NHEJ[171]. PI3K/AKT/mTOR inhibitors sensitize tumor cells to PARP1 inhibitors[224,225] and to radiotherapy[172]. HPV, the causative agent for most cervical cancers, modulates the DDR to confer therapeutic resistance, and DDR inhibitors are being explored to improve cervical cancer outcomes[226]. HPV is not alone: many viruses hijack different parts of the DDR to complete their life cycles[227]. ATM, ATR, and Chk1 signaling modulates PD-L1 expression in response to DSBs induced by radiation or chemotherapeutics[141]. In preclinical studies, inhibition of ATM during radiotherapy enhanced tumor immunogenicity and tumor sensitivity to PD-L1 immune checkpoint blockade[183]. These findings highlight the pleiotropic effects of PIKK signaling networks and suggest new opportunities for combination therapy to radiosensitize tumors and exploit anti-tumor activity of the immune system.
Simultaneous targeting of NHEJ and HR with Hsp90 inhibitors
Given the importance of DSB repair for cell survival, and the central roles of NHEJ and HR in DSB repair, simultaneously blocking these pathways can exquisitely sensitize tumors to radio- and chemotherapy. Hsp90 inhibitors have emerged as important tools for simultaneous downregulation of NHEJ and HR. Hsp90 is a protein chaperone that regulates stress responses and tumor growth proteins, and Hsp90 inhibitors are being used to treat cancer in monotherapy and to augment traditional therapies[228-230]. Although Hsp90 is not mutated in tumor cells, it has an altered conformation and higher ATPase activity than in normal cells. Hsp90 inhibitors exploit this difference to selectively affect tumor cells[174,229,231,232]. The radiosensitizing effects of Hsp90 inhibitors to low and high LET radiation have been studied for more than a decade[174-179]. The Hsp90 inhibitor 17-AAG, suppresses HR[176], radiosensitizes tumor cells, and suppresses tumor growth after radiotherapy[174]. Interestingly, the greatest radiosensitization was observed with carbon ions[174], another example of how HR inhibition potentiates radiosensitization with high LET radiation. Because protein chaperones affect many cellular processes, Hsp90 inhibitors can have pleiotropic effects, and early Hsp90 inhibitors caused serious side effects including ocular degeneration[233,234]. Second and third generation Hsp90 inhibitors (PU-H71 and TAS-116) proved to be safer alternatives. These drugs are tumor-specific radiosensitizers that suppress both NHEJ and HR by downregulating RAD51, RAD51 foci, and DNA-PKcs Ser2056/Thr2609 phosphorylation[175,178]. TAS-116 showed promising results in a phase 1 trial as monotherapy against advanced, heavily pre-treated gastrointestinal and lung cancers, with an acceptable safety profile (e.g., no greater than grade 1 ocular disorders and nausea) and anti-tumor activity[180]. It will be interesting to test TAS-116 as an adjunct to radiotherapy, and to carbon ion radiotherapy in particular.
Summary and future perspectives
DDR signaling, DNA repair, and DNA replication systems are tightly integrated, and they are key regulators of genome integrity, genome replication, and cell viability/cell proliferative capacity. This means that agents that target DDR and DNA repair factors can be highly effective against tumors, especially when exploiting a tumor-specific synthetic lethal weakness. Unfortunately, these systems are also critical in normal cells, and DDR and DNA repair inhibitors can cause unacceptable normal tissue damage, especially if delivered systemically, reducing patient quality of life, both short- and long-term, and potentially reducing lifespan due to organ failure, accelerated tumor progression, or secondary cancers. This delicate balance is exemplified by a recent study showing that ATM counters toxic NHEJ at collapsed replication forks - an important finding because it points to new synthetic lethal approaches to treat ATM-defective tumors[235]. However, it also raises the possibility of ATM inhibition enhancing NHEJ-mediated mis-repair of single-ended DSBs during (therapy-induced) replication stress. This would destabilize the genome and may accelerate progression of surviving tumor cells or induce secondary cancers.
Once radio-modulators are proven effective in pre-clinical studies, it is important to determine safe and effective ways to administer to patients. These will vary depending the type of radio- or chemotherapy being augmented, the types of agents administered, tumor location, and the organs at risk. Therapeutic efficacy can be increased, and side effects decreased, by employing multi-targeted approaches[236]. For example, the Li lab combined physical (radiation) targeting with two other targeting approaches. The first was an oncolytic adenovirus to deliver hTERT promoter-driven E1a gene for conditional replication in hTERT-positive (tumor) cells, and the second was a replication-defective adenovirus expressing shRNA to repress DNA-PKcs[237]. This downregulated NHEJ specifically in tumor cells within the (physically-targeted) radiation beam. Another tumor-specific targeting approach is illustrated by recent studies targeting triple-negative breast cancer. Here, CRISPR/Cas9 designed to knock out the Lcn2 oncogene was delivered to breast cancer cells using a tumor-tropic, ICAM1 antibody-linked nanomaterial[238,239]. These and other targeting strategies can be combined to enhance a wide variety of therapeutic interventions.
The adaptive response raised concerns about improved tumor cell survival when tumors are “primed” with 5-10 mGy diagnostic CT scans to localize tumors before treatment with a 2-10 Gy “challenge” (therapeutic) dose[134]. It may be possible to invert this paradigm and exploit the adaptive response to protect normal tissue and increase therapeutic gain. This might be done, for example, by using a transverse photon (X-ray) beam to expose normal tissue above the tumor to low (mGy) doses. This could induce a transient adaptive response in at-risk normal tissue [specifically, organs at risk (OAR)], protecting this tissue from high dose radiotherapy delivered with a perpendicular beam [Figure 4A]. Such a strategy might be optimized with particle radiation, as priming doses can be delivered to just the normal tissue region that will be subsequently exposed to therapeutic doses in the entrance region, and particles also spare distal tissue [Figure 4B].
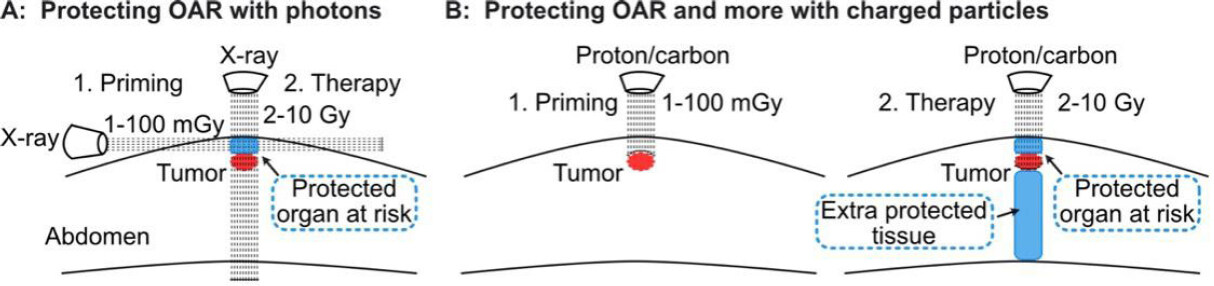
Figure 4. Proposed approach to protect normal tissue by stimulating radioadaptive responses. Horizontal X-ray beam delivers a priming dose to protect OAR (blue), but not the tumor (red) from subsequent, high doses delivered with a vertical beam(A); low priming dose of charged particles (left) protect OAR (blue) from subsequent high doses (right) (B). With charged particles, priming and therapeutic doses can be delivered along the same beamline since particles stop at predetermined depths. Charged particles also protect normal tissue distal to the tumor (larger blue section). OAR: organs at risk
In conclusion, multi-targeted strategies that combine DNA repair and DDR-modulated tumor-specific radiosensitization, advanced photon and particle beam focusing, and radioprotection of normal tissues are a rational path to tumor cures with minimal side effects.
Declarations
AcknowledgmentsWe thank Ryuichi Okayasu, Akira Fujimori, Tom Borak, Susan Bailey, Michael Weil, Claudia Wiese, and members of the Nickoloff and Kato labs for many helpful discussions. We thank the anonymous Reviewers for their helpful suggestions.
Authors’ contributionsconception and preparation of this manuscript: Nickoloff JA, Taylor L, Sharma N, Kato TA
Availability of data and materialsNot applicable.
Financial support and sponsorshipResearch in the Nickoloff lab was supported by NIH (R01 GM084020); American Lung Association grant (LCD-686552); CSU Office of the Vice President for Research; and the Japan National Institute of Radiological Sciences Open Laboratory program. Research in the Kato lab was supported by the Dr. Akiko Ueno Radiobiology Research fund.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Otter SJ, Stewart AJ, Devlin PM. Modern brachytherapy. Hematol Oncol Clin North Am 2019;33:1011-25.
2. Daguenet E, Louati S, Wozny AS, et al. Radiation-induced bystander and abscopal effects: important lessons from preclinical models. Br J Cancer 2020;123:339-48.
3. Halperin EC, Wazer DE, Perez CA, Brady LW. Perez & Brady’s Principles and Practice of Radiation Oncology. Lippincott Williams & Wilkins; 2018. p. 24.
4. Mounier NM, Abdel-Maged AE, Wahdan SA, Gad AM, Azab SS. Chemotherapy-induced cognitive impairment (CICI): An overview of etiology and pathogenesis. Life Sci 2020;258:118071.
5. Delou JMA, Souza ASO, Souza LCM, Borges HL. Highlights in resistance mechanism pathways for combination therapy. Cells 2019;8:1013.
6. Sato K, Shimokawa T, Imai T. Difference in acquired radioresistance induction between repeated photon and particle irradiation. Front Oncol 2019;9:1213.
7. Ali MY, Oliva CR, Noman ASM, et al. Radioresistance in glioblastoma and the development of radiosensitizers. Cancers 2020;12:2511.
8. Zhan Y, Fan S. Multiple mechanisms involving in radioresistance of nasopharyngeal carcinoma. J Cancer 2020;11:4193-204.
9. Thariat J, Hannoun-Levi JM, Sun Myint A, Vuong T, Gerard JP. Past, present, and future of radiotherapy for the benefit of patients. Nat Rev Clin Oncol 2013;10:52-60.
10. Karpuz M, Silindir-Gunay M, Ozer AY. Current and future approaches for effective cancer imaging and treatment. Cancer Biother Radiopharm 2018;33:39-51.
11. Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene 2015;34:3617-26.
12. Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM. Effects of ionizing radiation on biological molecules--mechanisms of damage and emerging methods of detection. Antioxid Redox Signal 2014;21:260-92.
13. Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis 2017;22:1321-35.
14. Mailloux RJ. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020;9:472.
15. Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017;168:644-56.
16. Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA 2003;100:12871-6.
17. Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014;6:a016428.
18. Yang W, Gao Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem 2018;87:239-61.
19. Allen C, Ashley AK, Hromas R, Nickoloff JA. More forks on the road to replication stress recovery. J Mol Cell Biol 2011;3:4-12.
20. Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015;15:276-89.
21. Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014;19:135-42.
22. Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res 2014;24:291-9.
23. Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 2014;83:519-52.
24. Casper AM, Durkin SG, Arlt MF, Glover TW. Chromosomal instability at common fragile sites in Seckel syndrome. Am J Hum Genet 2004;75:654-60.
25. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther 2016;160:65-83.
26. Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015;15:166-80.
27. Nickoloff JA. Paths from DNA damage and signaling to genome rearrangements via homologous recombination. Mutat Res 2017;806:64-74.
28. Nickoloff JA, Boss MK, Allen CP, LaRue SM. Translational research in radiation-induced DNA damage signaling and repair. Transl Cancer Res 2017;6:S875-S91.
29. Desai A, Yan Y, Gerson SL. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018;66-67:24-9.
30. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 2017;66:801-17.
31. Hengel SR, Spies MA, Spies M. Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol 2017;24:1101-19.
32. Killock D. Targeted therapies: DNA polymerase theta-a new target for synthetic lethality? Nat Rev Clin Oncol 2015;12:125.
33. Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 2019;16:81-104.
34. Carrassa L, Damia G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat Rev 2017;60:139-51.
35. Nickoloff JA, Jones D, Lee S-H, Williamson EA, Hromas R. Drugging the cancers addicted to DNA repair. J Natl Cancer Inst 2017;109:djx059.
36. Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet 2016;50:155-73.
37. Serrano MA, Li Z, Dangeti M, et al. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene 2013;32:2452-62.
39. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 2015;149:124-38.
40. Williams RM, Yates LA, Zhang X. Structures and regulations of ATM and ATR, master kinases in genome integrity. Curr Opin Struct Biol 2020;61:98-105.
42. Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007;316:1160-6.
44. Merry C, Fu K, Wang J, Yeh IJ, Zhang Y. Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle 2010;9:279-83.
45. Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010;108:73-112.
46. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin Cancer Biol 2016;37-38:51-64.
47. Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol 2016;23:103-9.
49. Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res 2014;329:85-93.
50. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014;16:2-9.
51. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 2017;18:495-506.
52. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 2018;293:10524-35.
53. de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 2011;7:e1002384.
54. Piazza A, Heyer WD. Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol 2019;29:135-49.
55. Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. Is homologous recombination really an error-free process? Front Genet 2014;5:175.
56. Sfeir A, Symington LS. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci 2015;40:701-14.
57. Costantino L, Sotiriou SK, Rantala JK, et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014;343:88-91.
58. Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 2015;793:166-75.
59. Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev 2013;23:271-9.
60. Sotiriou SK, Kamileri I, Lugli N, et al. Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol Cell 2016;64:1127-34.
61. Tutt A, Bertwistle D, Valentine J, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J 2001;20:4704-16.
62. Bhargava R, Onyango DO, Stark JM. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 2016;32:566-75.
63. Karger CP, Peschke P. RBE and related modeling in carbon-ion therapy. Phys Med Biol 2017;63:01TR02.
64. Nickoloff JA, Sharma N, Taylor L. Clustered DNA double-strand breaks: biological effects and relevance to cancer radiotherapy. Genes 2020;11:99-116.
65. Paganetti H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys Med Biol 2014;59:R419-72.
66. Howard M, Beltran C, Sarkaria J, Herman MG. Characterization of relative biological effectiveness for conventional radiation therapy: a comparison of clinical 6 MV X-rays and 137Cs. J Radiat Res 2017;58:608-13.
67. Asaithamby A, Chen DJ. Cellular responses to DNA double-strand breaks after low-dose γ-irradiation. Nucleic Acids Res 2009;37:3912-23.
68. Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol) 2013;25:578-85.
69. Vitti ET, Parsons JL. The radiobiological effects of proton beam therapy: impact on DNA damage and repair. Cancers 2019;11:946.
70. Nickoloff JA. Photon, light ion, and heavy ion cancer radiotherapy: paths from physics and biology to clinical practice. Ann Transl Med 2015;3:336.
71. Bukowska B, Karwowski BT. The clustered DNA lesions - types, pathways of repair and relevance to human health. Curr Med Chem 2018;25:2722-35.
72. Mladenova V, Mladenov E, Iliakis G. Novel biological approaches for testing the contributions of single DSBs and DSB clusters to the biological effects of high LET radiation. Front Oncol 2016;6:163.
73. Sage E, Shikazono N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic Biol Med 2017;107:125-35.
74. Pang D, Winters TA, Jung M, et al. Radiation-generated short DNA fragments may perturb non-homologous end-joining and induce genomic instability. J Radiat Res 2011;52:309-19.
75. Chan DW, Chen BP-C, Prithivirasingh S, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002;16:2333-8.
76. Okayasu R, Okada M, Okabe A, et al. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non-homologous end-joining pathway. Radiat Res 2006;165:59-67.
77. Wang H, Zhang X, Wang P, et al. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res 2010;38:3245-51.
78. Hada M, Sutherland BM. Spectrum of complex DNA damages depends on the incident radiation. Radiat Res 2006;165:223-30.
79. Gerelchuluun A, Manabe E, Ishikawa T, et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat Res 2015;183:345-56.
80. Fontana AO, Augsburger MA, Grosse N, et al. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother Oncol 2015;116:374-80.
81. Vitti ET, Kacperek A, Parsons JL. Targeting DNA double-strand break repair enhances radiosensitivity of HPV-positive and HPV-negative head and neck squamous cell carcinoma to photons and protons. Cancers 2020;12:1490.
82. Cartwright IM, Su C, Haskins JS, et al. DNA repair deficient chinese hamster ovary cells exhibiting differential sensitivity to charged particle radiation under aerobic and hypoxic conditions. Int J Mol Sci 2018;19:2228.
83. Antonovic L, Lindblom E, Dasu A, et al. Clinical oxygen enhancement ratio of tumors in carbon ion radiotherapy: the influence of local oxygenation changes. J Radiat Res 2014;55:902-11.
84. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 2004;59:928-42.
85. Higgins PD, DeLuca PM Jr., Gould MN. Effect of pulsed dose in simultaneous and sequential irradiation of V-79 cells by 14.8-MeV neutrons and 60Co photons. Radiat Res 1984;99:591-5.
86. Cheng L, Brzozowska B, Sollazzo A, et al. Simultaneous induction of dispersed and clustered DNA lesions compromises DNA damage response in human peripheral blood lymphocytes. PLoS One 2018;13:e0204068.
87. Staaf E, Brehwens K, Haghdoost S, Czub J, Wojcik A. Gamma-H2AX foci in cells exposed to a mixed beam of X-rays and alpha particles. Genome Integr 2012;3:8.
88. Newhauser WD, Durante M. Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer 2011;11:438-48.
89. Tsujii H, Kamada T, Shirai T, et al. Carbon-Ion Radiotherapy Principals, Practices, and Treatment Planning. Tokyo: Springer; 2014. p. 312.
90. Tsujii H, Kamada T. A review of update clinical results of carbon ion radiotherapy. Jpn J Clin Oncol 2012;42:670-85.
91. Kamada T, Tsujii H, Blakely EA, et al. Carbon ion radiotherapy in Japan: an assessment of 20 years of clinical experience. Lancet Oncol 2015;16:e93-e100.
92. Allen CP, Borak TB, Tsujii H, Nickoloff JA. Heavy charged particle radiobiology: using enhanced biological effectiveness and improved beam focusing to advance cancer therapy. Mutat Res 2011;711:150-7.
93. Sunada S, Cartwright IM, Hirakawa H, et al. Investigation of the relative biological effectiveness and uniform isobiological killing effects of irradiation with a clinical carbon SOBP beam on DNA repair deficient CHO cells. Oncol Lett 2017;13:4911-6.
94. Buglewicz DJ, Banks AB, Hirakawa H, Fujimori A, Kato TA. Monoenergetic 290 MeV/n carbon-ion beam biological lethal dose distribution surrounding the Bragg peak. Sci Rep 2019;9:6157.
95. Kato TA, Wilson PF, Nagasaw H, et al. Variations in radiosensitivity among individuals: a potential impact on risk assessment? Health Phys 2009;97:470-80.
96. Wilson PF, Nagasawa H, Fitzek MM, Little JB, Bedford JS. G2-phase chromosomal radiosensitivity of primary fibroblasts from hereditary retinoblastoma family members and some apparently normal controls. Radiat Res 2010;173:62-70.
97. Kirsch DG. Current opportunities and future vision of precision medicine in radiation oncology. Int J Radiat Oncol Biol Phys 2018;101:267-70.
98. Speers C, Zhao S, Liu M, et al. Development and validation of a novel radiosensitivity signature in human breast cancer. Clin Cancer Res 2015;21:3667-77.
99. Willers H, Azzoli CG, Santivasi WL, Xia F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J 2013;19:200-7.
100. Hill RP, Bristow RG, Fyles A, et al. Hypoxia and predicting radiation response. Semin Radiat Oncol 2015;25:260-72.
101. Kim BM, Hong Y, Lee S, et al. Therapeutic implications for overcoming radiation resistance in cancer therapy. Int J Mol Sci 2015;16:26880-913.
102. Hu J, Li H, Luo X, et al. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int J Cancer 2017;141:1722-9.
103. Wardman P. Nitroimidazoles as hypoxic cell radiosensitizers and hypoxia probes: misonidazole, myths and mistakes. Br J Radiol 2019;92:20170915.
104. Bonnet M, Hong CR, Wong WW, et al. Next-generation hypoxic cell radiosensitizers: nitroimidazole alkylsulfonamides. J Med Chem 2018;61:1241-54.
105. Olivares-Urbano MA, Grinan-Lison C, Marchal JA, Nunez MI. CSC radioresistance: a therapeutic challenge to improve radiotherapy effectiveness in cancer. Cells 2020;9:1651.
106. Liu Y, Yang M, Luo J, Zhou H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. Int J Oral Sci 2020;12:19.
107. Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers 2019;11:862.
108. Talukdar S, Bhoopathi P, Emdad L, et al. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv Cancer Res 2019;141:43-84.
109. Peeken JC, Vaupel P, Combs SE. Integrating hyperthermia into modern radiation oncology: What evidence Is necessary? Front Oncol 2017;7:132.
110. Oei AL, Vriend LE, Crezee J, Franken NA, Krawczyk PM. Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol 2015;10:165.
111. Genet SC, Fujii Y, Maeda J, et al. Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol 2013;228:1473-81.
112. Zhu L, Altman MB, Laszlo A, et al. Ultrasound hyperthermia technology for radiosensitization. Ultrasound Med Biol 2019;45:1025-43.
113. Lewis JE, Singh N, Holmila RJ, et al. Targeting NAD+ Metabolism to Enhance Radiation Therapy Responses. Semin Radiat Oncol 2019;29:6-15.
114. Ashcraft KA, Warner AB, Jones LW, Dewhirst MW. Exercise as adjunct therapy in cancer. Semin Radiat Oncol 2019;29:16-24.
115. Schoenfeld JD, Alexander MS, Waldron TJ, et al. Pharmacological ascorbate as a means of sensitizing cancer cells to radio-chemotherapy while protecting normal tissue. Semin Radiat Oncol 2019;29:25-32.
116. Floberg JM, Schwarz JK. Manipulation of glucose and hydroperoxide metabolism to improve radiation response. Semin Radiat Oncol 2019;29:33-41.
117. Aykin-Burns N, Pathak R, Boerma M, Kim T, Hauer-Jensen M. Utilization of vitamin E analogs to protect normal tissues while enhancing antitumor effects. Semin Radiat Oncol 2019;29:55-61.
118. Hillman GG. Soy isoflavones protect normal tissues while enhancing radiation responses. Semin Radiat Oncol 2019;29:62-71.
119. Boss MK, Deegan R, Batinic-Haberle I, et al. Manganese porphyrin and radiotherapy improves local tumor response and overall survival in orthotopic murine mammary carcinoma models. Radiat Res 2020. (in press)
120. Mortezaee K, Shabeeb D, Musa AE, Najafi M, Farhood B. Metformin as a radiation modifier; implications to normal tissue protection and tumor sensitization. Curr Clin Pharmacol 2019;14:41-53.
121. Lin A, Maity A. Molecular pathways: A novel approach to targeting hypoxia and improving radiotherapy efficacy via reduction in oxygen demand. Clin Cancer Res 2015;21:1995-2000.
122. Fernandes JM, Jandrey EHF, Koyama FC, et al. Metformin as an alternative radiosensitizing agent to 5FU during neoadjuvant treatment for rectal cancer. Dis Colon Rectum 2020;63:918-26.
123. Farhood B, Goradel NH, Mortezaee K, et al. Melatonin as an adjuvant in radiotherapy for radioprotection and radiosensitization. Clin Transl Oncol 2019;21:268-79.
125. Livingston K, Schlaak RA, Puckett LL, Bergom C. The role of mitochondrial dysfunction in radiation-induced heart disease: from bench to bedside. Front Cardiovasc Med 2020;7:20.
126. Thermozier S, Hou W, Zhang X, et al. Anti-ferroptosis drug enhances total-body irradiation mitigation by drugs that block apoptosis and necroptosis. Radiat Res 2020;193:435-50.
127. Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984;223:594-7.
128. Huang L, Kim PM, Nickoloff JA, Morgan WF. Targeted and non-targeted effects of low-dose ionizing radiation on delayed genomic instability in human cells. Cancer Res 2007;67:1099-104.
129. Ikushima T. Radio-adaptive response: characterization of a cytogenetic repair induced by low-level ionizing radiation in cultured Chinese hamster cells. Mutat Res 1989;227:241-6.
130. Rigaud O, Moustacchi E. Radioadaptation for gene mutation and the possible molecular mechanisms of the adaptive response. Mutat Res 1996;358:127-34.
131. Redpath JL, Antoniono RJ. Induction of an adaptive response against spontaneous neoplastic transformation in vitro by low-dose gamma radiation. Radiat Res 1998;149:517-20.
132. Barquinero JF, Barrios L, Caballin MR, et al. Occupational exposure to radiation induces an adaptive response in human lymphocytes. Int J Radiat Biol 1995;67:187-91.
133. Monsieurs MA, Thierens HM, Vral AM, et al. Adaptive response in patients treated with 131I. J Nucl Med 2000;41:17-22.
134. Grdina DJ, Murley JS, Miller RC, et al. A survivin-associated adaptive response in radiation therapy. Cancer Res 2013;73:4418-28.
135. Grdina DJ, Murley JS, Miller RC, et al. A manganese superoxide dismutase (SOD2)-mediated adaptive response. Radiat Res 2013;179:115-24.
136. Grdina DJ, Murley JS, Miller RC, Woloschak GE, Li JJ. NFkappaB and survivin-mediated radio-adaptive response. Radiat Res 2015;183:391-7.
137. Murley JS, Miller RC, Weichselbaum RR, Grdina DJ. TP53 mutational status and ROS effect the expression of the survivin-associated radio-adaptive response. Radiat Res 2017;188:579-90.
138. Murley JS, Arbiser JL, Weichselbaum RR, Grdina DJ. ROS modifiers and NOX4 affect the expression of the survivin-associated radio-adaptive response. Free Radic Biol Med 2018;123:39-52.
139. Unruhe B, Schroder E, Wunsch D, Knauer SK. An old flame never dies: Survivin in cancer and cellular senescence. Gerontology 2016;62:173-81.
140. Coleman CN, Eke I, Makinde AY, et al. Radiation-induced adaptive response: new potential for cancer treatment. Clin Cancer Res 2020;26:5781-90.
141. Sato H, Niimi A, Yasuhara T, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8:1751.
143. Russo M, Crisafulli G, Sogari A, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019;366:1473-80.
144. Scott JG, Berglund A, Schell MJ, et al. A genome-based model for adjusting radiotherapy dose (GARD): a retrospective, cohort-based study. Lancet Oncol 2017;18:202-11.
145. Trenner A, Sartori AA. Harnessing DNA double-strand break repair for cancer treatment. Front Oncol 2019;9:1388.
146. Kirsch DG, Diehn M, Kesarwala AH, et al. The future of radiobiology. J Natl Cancer Inst 2018;110:329-40.
147. Kelley MR, Logsdon D, Fishel ML. Targeting DNA repair pathways for cancer treatment: what’s new? Future Oncol 2014;10:1215-37.
148. Toulany M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes 2019;10:25.
149. Mohiuddin IS, Kang MH. DNA-PK as an emerging therapeutic target in cancer. Front Oncol 2019;9:635.
150. Dong J, Ren Y, Zhang T, et al. Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair. Oncol Rep 2018;39:912-20.
151. Yang C, Wang Q, Liu X, et al. NU7441 enhances the radiosensitivity of liver cancer cells. Cell Physiol Biochem 2016;38:1897-905.
152. Sunada S, Kanai H, Lee Y, et al. Nontoxic concentration of DNA-PK inhibitor NU7441 radio-sensitizes lung tumor cells with little effect on double strand break repair. Cancer Sci 2016;107:1250-5.
153. Timme CR, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther 2018;17:1207-16.
154. Willoughby CE, Jiang Y, Thomas HD, et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J Clin Invest 2020;130:258-71.
155. Smith MC, Mader MM, Cook JA, et al. Characterization of LY3023414, a vovel PI3K/mTOR dual inhibitor eliciting transient target modulation to impede tumor growth. Mol Cancer Ther 2016;15:2344-56.
156. Tsuji T, Sapinoso LM, Tran T, et al. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017;8:74688-702.
157. Lindquist KE, Cran JD, Kordic K, et al. Selective radiosensitization of hypoxic cells using BCCA621C: a novel hypoxia activated prodrug targeting DNA-dependent protein kinase. Tumour Microenv Ther 2013;1:46-55.
158. Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol 2005;76:157-61.
159. Joseph K, Alkaabi K, Warkentin H, et al. Cetuximab-radiotherapy combination in the management of locally advanced cutaneous squamous cell carcinoma. J Med Imaging Radiat Oncol 2019;63:257-63.
160. Qi Y, Lang J, Zhu X, et al. Down-regulation of the radiation-induced pEGFRThr654 mediated activation of DNA-PK by Cetuximab in cervical cancer cells. RSC Adv 2020;10:1132-41.
161. Balbous A, Cortes U, Guilloteau K, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016;16:604.
162. King HO, Brend T, Payne HL, et al. RAD51 Is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Reports 2017;8:125-39.
163. Pastushok L, Fu Y, Lin L, et al. A novel cell-penetrating antibody fragment inhibits the DNA repair protein RAD51. Sci Rep 2019;9:11227.
164. Turchick A, Liu Y, Zhao W, Cohen I, Glazer PM. Synthetic lethality of a cell-penetrating anti-RAD51 antibody in PTEN-deficient melanoma and glioma cells. Oncotarget 2019;10:1272-83.
165. Turchick A, Hegan DC, Jensen RB, Glazer PM. A cell-penetrating antibody inhibits human RAD51 via direct binding. Nucleic Acids Res 2017;45:11782-99.
166. Cyteir Therapeutics I. A phase 1/2 study of CYT-0851, an oral RAD51 inhibitor, in B-cell malignancies and advanced solid tumors. In; 2019.
167. Yu D, Sekine E, Fujimori A, Ochiya T, Okayasu R. Down regulation of BRCA2 causes radio-sensitization of human tumor cells in vitro and in vivo. Cancer Sci 2008;99:810-5.
168. Hirai T, Shirai H, Fujimori H, et al. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high linear energy transfer radiation. Cancer Sci 2012;103:1045-50.
169. Hirai T, Saito S, Fujimori H, et al. Radiosensitization by PARP inhibition to proton beam irradiation in cancer cells. Biochem Biophys Res Commun 2016;478:234-40.
170. Jannetti SA, Zeglis BM, Zalutsky MR, Reiner T. Poly(ADP-ribose)polymerase (PARP) inhibitors and radiation therapy. Front Pharmacol 2020;11:170.
171. Chang L, Graham PH, Hao J, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis 2014;5:e1437.
172. Schotz U, Balzer V, Brandt FW, et al. Dual PI3K/mTOR inhibitor NVP-BEZ235 enhances radiosensitivity of head and neck squamous cell carcinoma (HNSCC) cell lines due to suppressed double-strand break (DSB) repair by non-homologous end joining. Cancers 2020;12:467.
173. Gil del Alcazar CR, Hardebeck MC, Mukherjee B, et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin Cancer Res 2014;20:1235-48.
174. Hirakawa H, Fujisawa H, Masaoka A, et al. The combination of Hsp90 inhibitor 17AAG and heavy-ion irradiation provides effective tumor control in human lung cancer cells. Cancer Med 2015;4:426-36.
175. Lee Y, Li HK, Masaoka A, et al. The purine scaffold Hsp90 inhibitor PU-H71 sensitizes cancer cells to heavy ion radiation by inhibiting DNA repair by homologous recombination and non-homologous end joining. Radiother Oncol 2016;121:162-8.
176. Noguchi M, Yu D, Hirayama R, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun 2006;351:658-63.
177. Segawa T, Fujii Y, Tanaka A, et al. Radiosensitization of human lung cancer cells by the novel purine-scaffold Hsp90 inhibitor, PU-H71. Int J Mol Med 2014;33:559-64.
178. Lee Y, Sunada S, Hirakawa H, et al. TAS-116, a novel Hsp90 inhibitor, selectively enhances radiosensitivity of human cancer cells to X-rays and carbon ion radiation. Mol Cancer Ther 2017;16:16-24.
179. Fujii Y, Kato T, Kubota N, et al. p53 independent radio-sensitization of human lymphoblastoid cell lines by Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Oncol Rep 2010;23:199-203.
180. Shimomura A, Yamamoto N, Kondo S, et al. First-in-human Phase I study of an oral HSP90 inhibitor, TAS-116, in patients with advanced solid tumors. Mol Cancer Ther 2019;18:531-40.
181. Vormoor B, Schlosser YT, Blair H, et al. Sensitizing Ewing sarcoma to chemo- and radiotherapy by inhibition of the DNA-repair enzymes DNA protein kinase (DNA-PK) and poly-ADP-ribose polymerase (PARP) 1/2. Oncotarget 2017;8:113418-30.
182. Fok JHL, Ramos-Montoya A, Vazquez-Chantada M, et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun 2019;10:5065.
183. Zhang Q, Green MD, Lang X, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res 2019;79:3940-51.
184. Chen BP, Uematsu N, Kobayashi J, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem 2007;282:6582-7.
185. Cornell L, Munck JM, Alsinet C, et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin Cancer Res 2015;21:925-33.
186. Abdel-Fatah TM, Arora A, Moseley P, et al. ATM, ATR and DNA-PKcs expressions correlate to adverse clinical outcomes in epithelial ovarian cancers. BBA Clin 2014;2:10-7.
187. Toulany M, Maier J, Iida M, et al. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov 2017;3:17072.
188. Baptistella AR, Landemberger MC, Dias MVS, et al. Rab5C enhances resistance to ionizing radiation in rectal cancer. J Mol Med 2019;97:855-69.
189. Zou M, Li Y, Xia S, et al. Knockdown of CAVEOLIN-1 sensitizes human basal-like triple-negative breast cancer cells to radiation. Cell Physiol Biochem 2017;44:778-91.
190. Saki M, Makino H, Javvadi P, et al. EGFR mutations compromise hypoxia-associated radiation resistance through impaired replication fork-associated DNA damage repair. Mol Cancer Res 2017;15:1503-16.
191. Amunugama R, Fishel R. Homologous recombination in eukaryotes. Prog Mol Biol Transl Sci 2012;110:155-206.
192. Shrivastav M, Miller CA, De Haro LP, et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair 2009;8:920-9.
193. Nickoloff JA, Brenneman MA. Analysis of recombinational repair of DNA double-strand breaks in mammalian cells with I-SceI nuclease. Methods Mol Biol 2004;262:35-52.
194. Yoshino Y, Endo S, Chen Z, et al. Evaluation of site-specific homologous recombination activity of BRCA1 by direct quantitation of gene editing efficiency. Sci Rep 2019;9:1644.
195. Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell 2013;152:1344-54.
196. Kaushal S, Freudenreich CH. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 2019;58:270-83.
197. Wray J, Liu J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res 2008;68:2699-707.
198. Groth P, Orta ML, Elvers I, et al. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res 2012;40:6585-94.
200. Budke B, Logan HL, Kalin JH, et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 2012;40:7347-57.
201. Budke B, Lv W, Kozikowski AP, Connell PP. Recent developments using small molecules to target RAD51: How to best modulate RAD51 for anticancer therapy? ChemMedChem 2016;11:2468-73.
202. Lv W, Budke B, Pawlowski M, Connell PP, Kozikowski AP. Development of small molecules that specifically inhibit the D-loop activity of RAD51. J Med Chem 2016;59:4511-25.
203. Mersch J, Jackson MA, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015;121:269-75.
204. Sekhar D, Pooja S, Kumar S, Rajender S. RAD51 135G > C substitution increases breast cancer risk in an ethnic-specific manner: a meta-analysis on 21,236 cases and 19,407 controls. Sci Rep 2015;5:11588.
206. Jette NR, Kumar M, Radhamani S, et al. ATM-deficient cancers provide new opportunities for precision oncology. Cancers 2020;12:687.
207. Byrum AK, Vindigni A, Mosammaparast N. Defining and modulating ‘BRCAness’. Trends Cell Biol 2019;29:740-51.
208. Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17.
209. Yi M, Dong B, Qin S, et al. Advances and perspectives of PARP inhibitors. Exp Hematol Oncol 2019;8:29.
210. del Rivero J, Kohn EC. PARP inhibitors: the cornerstone of DNA repair-targeted therapies. Oncology 2017;31:265-73.
211. Gil Del Alcazar CR, Todorova PK, Habib AA, Mukherjee B, Burma S. Augmented HR repair mediates acquired temozolomide resistance in glioblastoma. Mol Cancer Res 2016;14:928-40.
212. Zhang X, Ma N, Yao W, Li S, Ren Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int 2019;19:356.
213. Liu X, Han EK, Anderson M, et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol Cancer Res 2009;7:1686-92.
214. Noordermeer SM, van Attikum H. PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol 2019;29:820-34.
215. D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018;71:172-6.
216. Tian H, Gao Z, Li H, et al. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett 2015;358:8-16.
217. Bakr A, Oing C, Kocher S, et al. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res 2015;43:3154-66.
218. Ahlskog JK, Larsen BD, Achanta K, Sorensen CS. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep 2016;17:671-81.
220. Glorieux M, Dok R, Nuyts S. Novel DNA targeted therapies for head and neck cancers: clinical potential and biomarkers. Oncotarget 2017;8:81662-78.
221. Riches LC, Trinidad AG, Hughes G, et al. Pharmacology of the ATM inhibitor AZD0156: potentiation of irradiation and olaparib responses preclinically. Mol Cancer Ther 2020;19:13-25.
222. Zhou C, Parsons JL. The radiobiology of HPV-positive and HPV-negative head and neck squamous cell carcinoma. Expert Rev Mol Med 2020;22:e3.
223. Ferri A, Stagni V, Barila D. Targeting the DNA damage response to overcome cancer drug resistance in glioblastoma. Int J Mol Sci 2020;21:4910.
224. Philip CA, Laskov I, Beauchamp MC, et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017;17:638.
225. Wang D, Li C, Zhang Y, et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol 2016;142:548-56.
226. Cossar LH, Schache AG, Risk JM, et al. Modulating the DNA damage response to improve treatment response in cervical cancer. Clin Oncol 2017;29:626-34.
227. Weitzman MD, Fradet-Turcotte A. Virus DNA replication and the host DNA damage response. Annu Rev Virol 2018;5:141-64.
228. Jafari A, Rezaei-Tavirani M, Farhadihosseinabadi B, Taranejoo S, Zali H. HSP90 and co-chaperones: impact on tumor progression and prospects for molecular-targeted cancer therapy. Cancer Invest 2020;38:310-28.
229. Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 2013;14:e358-69.
230. Jhaveri K, Modi S. HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv Pharmacol 2012;65:471-517.
231. Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003;425:407-10.
232. Moulick K, Ahn JH, Zong H, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol 2011;7:818-26.
233. Sessa C, Shapiro GI, Bhalla KN, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res 2013;19:3671-80.
234. Renouf DJ, Velazquez-Martin JP, Simpson R, Siu LL, Bedard PL. Ocular toxicity of targeted therapies. J Clin Oncol 2012;30:3277-86.
235. Balmus G, Pilger D, Coates J, et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun 2019;10:87.
236. Nickoloff JA. Improving cancer therapy by combining cell biological, physical, and molecular targeting strategies. Chin J Cancer Res 2013;25:7-9.
237. Kon T, Zhang X, Huang Q, et al. Oncolytic virus-mediated tumor radiosensitization in mice through DNA-PKcs-specific shRNA. Transl Cancer Res 2012;1:4-14.
238. Guo P, Yang J, Jia D, Moses MA, Auguste DT. ICAM-1-targeted, Lcn2 siRNA-encapsulating liposomes are potent anti-angiogenic agents for triple negative breast cancer. Theranostics 2016;6:1-13.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Nickoloff JA, Taylor L, Sharma N, Kato TA. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist 2021;4:244-63. http://dx.doi.org/10.20517/cdr.2020.89
AMA Style
Nickoloff JA, Taylor L, Sharma N, Kato TA. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resistance. 2021; 4(2): 244-63. http://dx.doi.org/10.20517/cdr.2020.89
Chicago/Turabian Style
Nickoloff, Jac A., Lynn Taylor, Neelam Sharma, Takamitsu A. Kato. 2021. "Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy" Cancer Drug Resistance. 4, no.2: 244-63. http://dx.doi.org/10.20517/cdr.2020.89
ACS Style
Nickoloff, JA.; Taylor L.; Sharma N.; Kato TA. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist. 2021, 4, 244-63. http://dx.doi.org/10.20517/cdr.2020.89
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 2
likes
Like This Article 2
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.