
Improving TRAIL-induced apoptosis in cancers by interfering with histone modifications

Abstract
Epigenetic regulation refers to alterations to the chromatin template that collectively establish differential patterns of gene transcription. Post-translational modifications of the histones play a key role in epigenetic regulation of gene transcription. In this review, we provide an overview of recent studies on the role of histone modifications in carcinogenesis. Since tumour-selective ligands such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) are well-considered as promising anti-tumour therapies, we summarise strategies for improving TRAIL sensitivity by inhibiting aberrant histone modifications in cancers. In this perspective we also discuss new epigenetic drug targets for enhancing TRAIL-mediated apoptosis.
Keywords
Introduction
In humans, the genetic information (DNA) is contained in 23 chromosome pairs. These chromosomes are composed of DNA and histone proteins that form highly condensed chromatin. In parallel to genetics, the term “epigenetics” was originally defined to describe heritable changes that are not encoded in the DNA. Currently, epigenetics is used as a common term to describe chromatin modifications that regulate DNA-based processes including heritable and non-heritable changes[1]. The main players in epigenetic regulations are DNA modifications, histone modifications and non-coding RNAs. Histone modifications regulate, among other things, chromatin remodelling, which is closely related to regulation of gene transcription. For example, heterochromatin is usually tightly packed and prohibits gene transcription, while euchromatin is usually loosely packed and enables gene transcription[2]. Since epigenetics plays a crucial role in DNA-based processes, histone modifications are very important in cell growth in normal and disease states such as carcinogenesis.
Among various strategies to treat cancers, the selective induction of cellular apoptosis in cancer cells is considered as a promising therapeutic strategy. A well-known ligand to induce apoptosis is tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). Dulanermin is a TRAIL-based therapeutic containing amino acids 114-281 of human TRAIL, which has been developed as a clinical anti-cancer drug. An early phase I clinical study showed that dulanermin was well-tolerated by patients with advanced cancer. However, only 3% of the patients in this study responded to dulanermin treatment for a period longer than 6 months[3]. This may be due to TRAIL-resistance, which occurs in various type of cancer cells. TRAIL-resistance can be attributed to impaired TRAIL binding to death receptors, modified levels of apoptosis-related proteins, and reduced caspase functions[4]. In spite of this, TRAIL-based therapeutics are currently under clinical investigation. For instance, a phase I clinical study is recruiting participants to study the application of the novel TRAIL trimer SCB-313 for the treatment of malignant pleural effusions and peritoneal malignancies (NCT03869697 and NCT03443674). Moreover, phase I/II clinical studies with lung cancer patients using TRAIL expressed by mesenchymal stem cells are ongoing (NCT03298763). This demonstrates an active interest in the clinical application of TRAIL-based therapeutics.
In this review, we provide an overview of post-translational modifications of histones and the enzymes involved in the addition or removal of these modifications. We discuss small molecules targeting these enzymes and their anti-tumour effects. We connect this to targets involved in apoptosis as potential approach in cancer therapy. Finally, we summarize the current understanding of epigenetic mechanisms involved in sensitivity to TRAIL-induced apoptosis.
Histone modifications
Histones are the central components of nucleosomes, in which a DNA string wraps around an octamer containing two copies of four core histones (H3, H4, H2A and H2B). These nucleosomes are organized like “beads” on DNA strings and are connected by histone protein H1 and further compacted to 30 nm-chromatin fibres, which are eventually condensed to form a chromosome. Therefore, histones provide structural support for chromosomes to provide organized packing of the DNA inside the nucleus. Unstructured histone tails are excluded from nucleosome cores and these tails are rich in lysine and arginine residues. Lysine residues are positively charged and provide charge-charge interactions with the negatively charged DNA, thus compacting the chromatin structure. Post-translational modifications occur mostly on the N-terminal tails of histones. These modifications play versatile roles in regulation of the structure and accessibility of the chromatin for transcription factors [Table 1].
Histone modifications
| Amino acids | Modifications | Positions | Nomenclature | Ref. |
|---|---|---|---|---|
| Arginine | Methylation | *H3R2/R8/R17/R26, H4R3, H2AR3 | R-me1, R-me2s, R-me2a | [5,6] |
| Citullination | *H3R2/R8/R17/R26/R42, H4R3, H2A, and H1 | R-citrulline | [7] | |
| Lysine | Methylation | *H3K9/K4/K36/K79/K27, H4K5/K20 | K-me1, K-me2, K-me3 | [8] |
| Acetylation | *H3K9/K14/K56, H4K5/K12/K16 | K-acetyl | [9,10] | |
| Propionylation | *H3K14 | K-propionyl | [11] | |
| Butyrylation | *H3K14, H4K5/K8 | K-butyryl | [11,12] | |
| 2-hydroxyisobutyrylation | H2AK5/9/36/74/75/95/118, H2BK5/12/20/23/24/34/43/46/57/85/108/116/120,
H3K4/9/14/18/23/27/36/56/64/79/122 H4K5/8/12/16/31/44/59/77/79/91 | K-2-hydroxyisobutyryl | [13] | |
| Malonylation | *H2AK119 | K-malonyl | [14] | |
| Succinylation | *H3K79 | K-succinyl | [15] | |
| Crotonylation | H2AK36/118/119/125, H2BK5/11/12/15/16/20/23/34
H3K4/9/18/23/27/56, H4K5/8/12/16 | K-crotonyl | [16] |
Modifications of arginine
Arginine methylation
Biologically, arginine methylation refers to a reaction in which a methyl group is transferred from S-adenosyl-L-methionine (SAM) to one or both omega nitrogens of an arginine amino acid residue. This transfer leads to formation of monomethylarginine (MMA), asymmetric dimethylarginine (ADMA) and/or symmetric dimethylarginine (SDMA). This methylation reaction is catalysed by N-arginine methyltransferases (PRMTs). All of the PRMTs can catalyse monomethylation of arginine to provide MMA. Type I PRMTs, including PRMT1, 2, 3, 4, 6 and 8, methylate MMA further to provide ADMA. Type II PRMTs, including PRMT5 and 9, methylate MMA further to provide SDMA. PRMT7 is classified as a type III enzyme that catalyses methylation of various substrates. Histone arginine methylation is directly associated with gene transcription. For instance, methylation at H3R2 blocks the ability to methylate H3K4, which is responsible for recruiting chromatin-remodelling enzymes to maintain a transcriptionally active state[17]. H4R3 has been identified as a binder of DNA methyltransferase DNMT3A[18].
In contrast to arginine methylation, it is less clear which enzymes catalyse arginine demethylation. JMJD6 was initially reported to demethylate H3R2 and H4R3[19], however this was disputed in later studies[20-22]. Recently, a new study reported that JMJD1B, a lysine demethylase, also demethylates arginine at H4R3[23].
Arginine citrullination
A recently identified arginine post-translational modification is citrullination. This post-translational modification was already found in dozens of proteins, such as proteases, metabolic enzymes, and histones. The citrullination of histones is well-known to be involved in the formation of neutrophil extracellular traps (NETs), which is connected to innate immunity. In the process of clearing bacteria, the neutrophils secrete DNA, histones, and intracellular proteins to the extracellular space where they form NETs[24]. In comparison to the involvement of histone citrullination in immune response, the exact biological significance of histone citrullination in carcinogenesis is largely unclear[7].
Modifications of lysine
Lysine methylation
Lysine methylation is tightly regulated by “writers” (KMTs, methyltransferases) and “erasers” (KDMs, demethylases). Similar to PRMTs, KMTs also employ SAM as co-factor to transfer one, two, or three methyl groups to specific histone lysine residues. More than 50 human KMTs and 30 KDMs have been identified[25]. Instead of global regulation of gene expression across different types of cells, KMTs may be involved in the regulation of genes with specific roles in normal or cancer cells. For instance, there are 6 homologues of H3K4 methyltransferases, denoted KMT2A to KMT2E, that are involved in methylation at this position. Moreover, one recent study has shown that KMT2A and KMT2B control different genomic regions in brain cells to regulate memory function[25]. Therefore, KMTs may serve as potential biomarkers in patients for individualized treatment. Depending on the lysine position, methylation state, and amino acids environment, histone lysine methylation can activate or repress gene transcription. Generally, methylation on H3K4, H3K36, and H3K79 are considered to activate gene transcription, while methylation on H3K9, H3K27 and H4K20 are thought to repress gene transcription[8]. In contrast to KMTs, one KDM can catalyse demethylation on several lysine residues. For instance, LSD1 (also called KDM1A) is specific to H3K4 and H3K9 residues[26].
Short-chain lysine acylation
A classically studied lysine modification is acetylation of histone lysine residues. In a lysine acetylation reaction, an acetyl group from acetylated coenzyme A is transferred to the e-amino from a lysine residue, which results in neutralization of the positive charge and thus weakening of the electrostatic interaction with the DNA. This change leads to a more open chromatin structure, which allows access of DNA binding proteins. In general, acetylation is related to increased gene transcription, while deacetylation is connected to repression of gene transcription [Figure 1]. This dynamic process is catalysed by three groups of enzymes: (1) histone acetyltransferases (HATs), also known as “writers”, are responsible for transferring acetyl groups to targeted lysine residues; (2) histone deacetylases (HDACs), known as “erasers”, are found to remove acetyl groups; and (3) bromodomain proteins, known as “readers”, specifically recognize acetylated lysine residues.
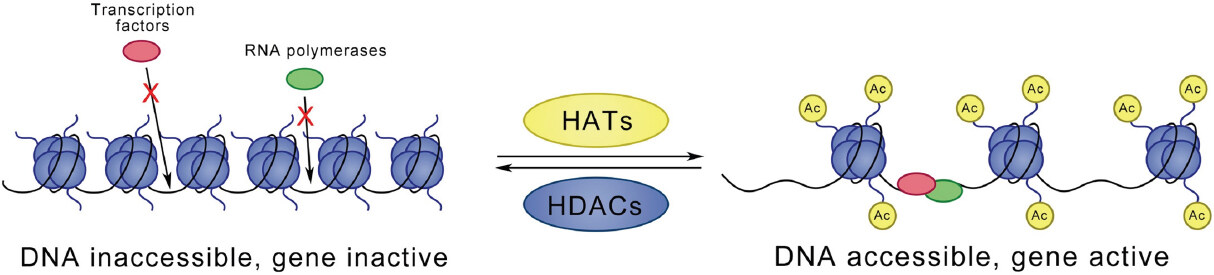
Figure 1. Acetylation or deacetylation of histone lysine residues is catalysed by HATs and HDACs, respectively. Lysine acetylation is connected to loosening of the chromatin structure. This change enables DNA binding and eventually leads to activation of gene transcription. In contrast, deacetylation closes the chromatin structure and represses gene transcription. HATs: histone acetyltransferases; HDACs: histone deacetylases
Besides histone lysine acetylation, recent studies show that other short-chain CoAs, such as propionyl-CoA, butyryl-CoA[27], 2-hydroxyisobutyryl-CoA[13], crotonyl-CoA[16], malonyl-CoA and succinyl-CoA[28], can be used as substrates to acylate histone lysine residues.
Others
Besides the aforementioned methylation and acetylation, other types of post-translational modifications are identified on histones, such as lysine ubiquitinoylation, sumoylation and ADP-ribosylation. These modifications are mostly reported to relate to DNA damage and repair. Moreover, phosphorylation of histone serine and threonine residues is a globally found modification, which plays important roles in diverse nuclear processes. Details for these modifications are discussed in recent reviews[29-31].
Aberrant histone modifications in cancers and the development of small-molecule inhibitors
Inhibitors to target arginine modifications
Overexpression of PRMTs has been observed in various types of human cancers[32]. For instance, the overexpression of PRMT5 has been observed in non-Hodgkin lymphoma[33,34]. Additionally, recent studies show that PRMT5 promotes survival of lymphoma cells via WNT and AKT-mediated proliferation signalling[35,36]. Interfering with PRMT5 activity prevents the maintenance of malignant phenotypes[37]. Therefore, PRMT5 is a rational target for treating lymphoma. Small-molecule inhibitors specifically targeting PRMT5 have been developed and two inhibitors, JNJ-64619178 and GSK3326595, were patented and are now under clinical investigation. Besides the development of PRMT5 inhibitors, type I PRMT inhibitors have also gained interest due to the high expression of type I PRMTs in various types of cancers[38-41]. Moreover, PRMT1 is identified as an essential component of mixed lineage leukaemia (MLL) and specific knockdown of PRMT1 suppresses MLL-mediated transformation[42]. Interestingly, a recent study shows that GSK3368715, a type I PRMT inhibitor, synergizes with the anti-tumour effect of PRMT5 inhibition[43][Table 2].
Inhibitors of histone methylation in clinical studies
| Name | Type of histone modification | Target | Clinical phase | Condition or disease in clinic | Disease in preclinical studies |
|---|---|---|---|---|---|
| Pinometostat
(EPZ-5676) | Lysine methylation | DOT1L | 1 | Advanced acute leukemia, particularly MLL-r[44] | Rearranged mixed lineage leukemia (MLL-r)[45-47] |
| CPI-1205 | EZH2 | 1 | B-cell lymphomas[48] | B-cell lymphomas[49] | |
| Tazemetostat
(EPZ-6438) | 2 | Elapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours[50] | Non-Hodgkin lymphoma[51,52] Rhabdoid tumour models[53] | ||
| GSK2879552 | LSD1 | 1 | Relapsed or refractory SCLC[54] | Small cell lung carcinoma[55] | |
| JNJ-64619178 | Arginine methylation | PRMT5 | 1 | Relapsed/refractory B cell non-Hodgkin lymphoma (NHL) or advanced solid tumours | Human NSCLC and SCLC cancer mouse xenograft models[49] |
| GSK3326595
(EPZ015938) | PRMT5 | 1 | Advanced or metastatic solid tumours and non-Hodgkin’s lymphoma[56,57] | Hematologic and solid tumour cells lines[58] | |
| GSK3368715
(EPZ019997) | Type I PRMTs | 1 | Solid tumours and diffuse large B-cell lymphoma | Lymphoma and AML cell lines[43] |
Inhibitors to target lysine modifications
Numerous studies have shown that mutation, dysregulation, or overexpression of lysine modifying enzymes such as KMTs, KDMs, HATs, or HDACs are associated with cancers and other diseases. Therefore, these enzymes were recognized as potential drug targets for cancer treatment[59,60].
As listed in Table 2, several inhibitors targeting lysine methylation have been described. EZH2 (enhancer of zeste) homolog is becoming a potential target for treating lymphoma. EZH2 is a catalytic components of polycomb repressive complexes 2 (PRC2), which methylate H3K27[61]. Gain-of-function mutations of EZH2 are mainly detected in diffuse large B cell lymphoma and follicular lymphoma among all categories of lymphomas and lymphoid leukaemia[62]. Moreover, a mutation at Y641 within the catalytic domain of EZH2 proved to increase methylation of H3K27, because the mutant EZH2 shows higher catalytic efficiency compared to wild type EZH2. This increased methylation contributes to the pathogenesis of germinal centre B-cell lymphomas[63]. Another EZH2 mutation, A677G, also increases methylation of H3K27me3[64]. These insights triggered the development of EZH2 inhibitors for therapeutic use. For instance, tazemetostat is a promising inhibitor that is under investigation in phase II clinical trials.
Previously, the FDA approved several pan-HDAC inhibitors for the treatment of cancers. For instance, vorinostat (SAHA) is approved for the treatment of cutaneous manifestations of cutaneous T-cell lymphoma[65]. Belinostat (Beleodaq) is approved for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL)[66], and panobinostat (Farydak) is approved for patients with relapsed multiple myeloma (MM)[67]. Besides these pan-HDAC inhibitors, a class I specific HDAC inhibitor romidepsin (Isodax) is approved for the treatment of PTCL. Further developments are aimed at the application of more isoenzyme-selective HDAC inhibitors. Moreover, several specific inhibitors are shown that were developed for cancer treatment over the last decade. Among these inhibitors, HDAC6-selective inhibitors show a promising anti-tumour effect to various cancers. For instance, ricolinostat (ACY-1215) shows strong potential at treating MM alone or with other drugs[68-70]. Several clinical trials using ricolinostat for patients with MM are currently ongoing (NCT01323751, NCT02189343, NCT01997840, and NCT01583283) [see Table 3].
Specific HAT and HDAC inhibitors developed between 2009 and 2019, and their applications in cancer in vitro
| Name | Target | Links to cancer |
|---|---|---|
| BG45 | Class I HDAC | multiple myeloma[71-73] |
| TMP-195 | Class IIa HDAC | Breast tumour[74] |
| LMK235 | HDAC4,5 | Chemoresistant cancer cells[75] multiple myeloma[76] pancreatic neuroendocrine tumours[77] |
| Tubastatin A | HDAC6 | cholangiocarcinoma[78] melanoma[79] |
| Ricolinostat (ACY-1215) | multiple myeloma[68-70] | |
| SKLB-23bb | solid and hematologic tumour[80] | |
| Cay 10603 | Burkitt’s lymphoma[81] lung carcinoma[82] | |
| Nexturastat A | myeloma[83-85] | |
| PCI-34051 | HDAC8 | neuroblastoma[86] T-cell lymphomas[87] malignant peripheral nerve sheath tumours[88] |
| A485 | P300/CBP | myeloma[89,90] |
In comparison to HDAC inhibitors, the development of potent and specific HAT inhibitors is lagging. C646 was firstly considered as a p300 and CBP selective inhibitor[91]. However, a recent study shows that C646 binds off-target to other kinases[92]. A novel p300 and CBP specific inhibitor A485 was synthesized and shows inhibition of proliferation in myeloma cells[90,93]. This new inhibitor holds promise for further exploration in myeloma.
Bromodomains are protein modules that are present in 46 different human proteins[94]. An important bromodomain family is the bromodomain and extra-terminal domain-containing (BET) protein family, which consists of two bromodomains (BD1 and BD2) and one extra-terminal domain. BET proteins recognize and bind specific peptide sequence in the chromatin, which plays an enabling role in the assembly of protein-protein complexes on the chromatin[95]. Currently, BET protein modules are considered as a promising group of targets for treatment of cancer, which triggered the development of BET inhibitors[96-98]. Whereas early studies provided BET inhibitors with limited selectivity, more recent studies promise to provide therapeutically relevant BET inhibition with improved selectivity profiles. For instance, ABBV-744 and BY27 are BD2-specific inhibitors that are proven to inhibit tumour cell growth in vitro[99,100]. A phase I study is ongoing in which the safety and pharmacokinetics of BET inhibitor ABBV-744 is evaluated for treatment of patients with acute myeloid leukaemia (NCT03360006).
Improved trail-induced apoptosis by targeting enzymes involved in histone modifications
TRAIL-induced apoptosis pathways
TRAIL is a member of the TNF superfamily and it binds to five receptors, including death receptor 4 (DR4), death receptor 5 (DR5), decoy receptor 1 (DcR1), decoy receptor 2 (DcR2), and osteoprotegerin. DR4 and DR5 both contain an intracellular death domain (DD), which initiates apoptotic signalling transduction. In contrast, DcR1 and DcR2 do not induce apoptosis due to the truncated DD in DcR1 and the absent DD in DcR2. The mechanisms of TRAIL-induced apoptosis have been intensively investigated and pathways identified are shown in Figure 2[101-103]. Extrinsic apoptotic signalling is initiated upon binding of a TRAIL trimer to DR4 or DR5, which initiates formation of a death-inducing signalling complex (DISC). In this DISC, FAS-associated protein with death domain (FADD) is connected with DR4 or DR5 via DDs. Initiator caspases, like pro-caspase-8 or 10, are recruited to FADD via the interaction between death effector domains. This recruitment also actives self-dimerization of pro-caspase-8 or 10, leading to auto-proteolytic processing at consensus cleavage sites. Executioner caspases, like caspase-3 or 7, are cleaved by initiator caspases to create a mature functional protease, which coordinates to the degradation phase of apoptosis, including DNA fragmentation, membrane blebbing and cell shrinkage. Single active executioner caspase can cleave and activate other caspases, resulting in activation of the caspase cascade. In addition, caspase-8 or 10 engages the intrinsic apoptosis pathway through cleavage of the BH3-interacting domain death agonist (Bid) to facilitate the release of cytochrome C from mitochondria. In fact, the truncated Bid translocates from the cytoplasm to mitochondria and stimulates oligomerization of Bax or Bak. At the same time, Bax and Bak permeabilize the membrane of the mitochondrion, also called mitochondrial outer membrane permeabilization (MOMP). Following MOMP, the mitochondrial inner membrane releases cytochrome C or second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO) into the cytosol. With the binding of cytochrome C to adaptor protein apoptotic protease-activating factor-1, dATP and the initiator caspase caspase-9 are recruited to form the apoptosome. Finally, active caspase-9 directly cleaves executioner caspases caspase-3 or 7.
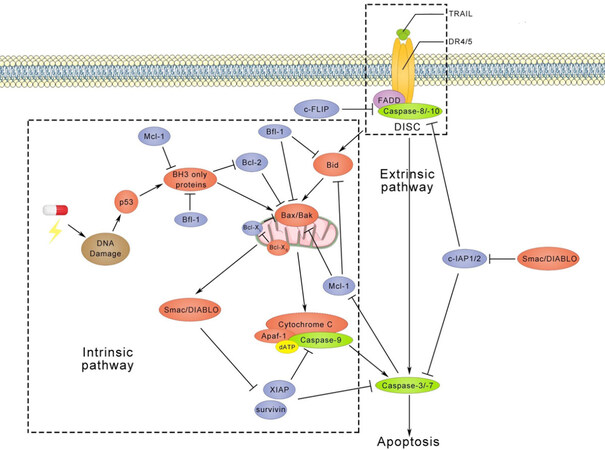
Figure 2. TRAIL-induced apoptotic pathways. After trimerization, TRAIL binds to death receptors, which triggers the formation of the DISC and activates caspase-8/10. Subsequently, activated caspase-8/10 induces cleavage of caspase-3/7, which leads to apoptosis. On the other hand, cleaved caspase-8/10 can also recruit Bid to trigger apoptosis via the intrinsic pathways. The intrinsic pathway is usually activated by DNA damage followed by p53 activation, whereas TRAIL-induced intrinsic apoptotic pathway is independent of p53. Interestingly, p53 has also been found to regulate TRAIL receptors DR4, DR5, DcR1, and DcR2[104-107]. Anti-apoptotic proteins, including c-FLIP, c-IAP1/2, Bcl-2, Bfl-1, Mcl-1, Bcl-XL, XIAP, and survivin, are shown in blue circles. DISC: death-inducing signalling complex; DR4: death receptor 4; DR4: death receptor 5; DcR1: decoy receptor 1; DcR2: decoy receptor 2
Anti-apoptotic proteins are also involved in these apoptotic signalling pathways. For instance, cellular-FLIP (c-FLIP) and cellular inhibitors of apoptotic proteins (cIAP1 and cIAP2) disturb the formation of DISC. X-linked IAP (XIAP) and survivin, on the other hand, block executioner caspases and the apoptosome. Moreover, anti-apoptotic Bcl-2 family members, like Bcl-2, Bcl-XL, Mcl-1, and Bfl-1 are able to prevent MOMP.
Improving TRAIL-induced apoptosis
Although, TRAIL has promising tumour-cell selective apoptosis-inducing properties, various tumour cells are resistant to TRAIL treatment. Therefore, it is important to improve TRAIL-sensitivity. Here, we discuss the strategies of improving TRAIL-sensitivity by targeting histone modifying enzymes that are involved in methylation and acetylation. Examples of the use of selective inhibitors as TRAIL sensitizers to overcome TRAIL-resistance are shown in Table 4.
Improved TRAIL-induced apoptosis pathway using inhibitors targeting enzymes in histone modifications
| Target | Small molecule | Regulation mechanisms | Cancer type | Ref. |
|---|---|---|---|---|
| Euchromatic histone-lysine N-methyltransferase 2 (EHMT2, G9a) | BIX-01294 | Downregulation of Survivin and Upregulation of DR5 | Renal carcinoma | [108] |
| Upregulation of DR5 | Breast cancer | [109] | ||
| PRC2 | Retinoic acid (RA) or 3-deazaneplanocin A (DZNep) | Increased DR5 transcript level | Colon cancer | [110] |
| Class I HDAC | Entinostat
(MS-275) | Restore expression of Coxsackie Adenovirus receptor | Prostate cancer | [111] |
| Upregulation of DR4, DR5, Bax, Bak | Breast cancer | [112] | ||
| Decrease degradation of endogenous TRAIL | Anaplastic thyroid carcinoma | [113] | ||
| Expression of endogenous TRAIL | Acute myeloid leukemia | [114] | ||
| HDAC3 | RGFP966 | Upregulation of DR4 | Colon cancer | [115] |
| HDAC8 | PCI34051 |
Histone methylation
The enzyme EHMT2 catalyses the dimethylation of H3K9me2, which is associated with silencing of tumour suppressor genes. The PRC2 complex plays an important role in H3K27me3, which is also related to transcriptional repression of tumour suppressor genes. When combined with TRAIL, inhibitors of either EHMT2 or PRC2 increase the number of apoptotic cells through upregulation of DR5[108,109]. These results indicate that the expression of DR5 may be be related to the reduced methylation of histones.
Additionally, a recent study shows that silencing KDM2B, a H3K36-specific histone demethylase, can cause a de-repression of a pro-apoptotic gene Harakiri (HRK) in glioblastoma multiforme cells. This study also shows that the silencing of KDM2B cooperates with TRAIL to reduce cell viability[116].
As discussed above, EZH2 is a promising therapeutic target for lymphoma. Therefore, EZH2-specific inhibitors may enhance the sensitivity of lymphoma cells to TRAIL. Additionally, another methyltransferase PRMT5 has been identified as a novel TRAIL receptor binding protein at the plasma membrane, which is involved in the early stage of signal initiation for induction of the NF-κB signalling pathways[117]. Moreover, a study shows that the overexpression of PRMT5 increased expression of c-FLIPL by decreasing the ubiquitination via inhibition of the interaction between c-FLIPL and ITCH, leading to decreased apoptotic cells induced by doxorubicin in human lung cancer cells[118]. Therefore, targeting PRMT5 by specific inhibitors may improve sensitivity to TRAIL.
Histone acetylation
Previously, studies have shown that the combination of pan-HDAC inhibitors, such as panobinostat, with TRAIL downregulates anti-apoptotic proteins, c-FLIP and XIAP, thereby improving sensitivity to TRAIL[119,120]. This study indicates a close relationship of histone acetylation and the TRAIL signalling pathways.
Moreover, highly acetylated Ku70, a DNA repair protein, disrupts the formation of the Ku70-FLIP complex and triggers the degradation of FLIP by polyubiquitination. Therefore, using the HDAC inhibitor vorinostat increases apoptosis through the stabilization of the Ku70-FLIP complex in colon cancer models in vivo. Interestingly, this study also shows that the HDAC6-specific inhibitor tubacin increases apoptosis[121]. With the increasing development of HDAC-specific inhibitors, the combination of HDAC specific inhibitors with TRAIL may be an interesting choice [Table 4].
Interestingly, the BET inhibitor JQ1 was reported to reduce the expression of c-FLIP and XIAP at mRNA and protein level in KRAS-mutated NSCLC cells. Combined JQ1 with TRAIL significantly enhanced apoptosis[122]. Furthermore, JO1 combined with the HDAC inhibitor vorinostat increases apoptosis via the extrinsic pathway in CTLC cells[123]. These results indicate that BET inhibitors play an important role in regulating proteins in the apoptotic signalling pathway axis. Therefore, the combination of BET inhibitor with TRAIL may be a promising strategy for the development of cancer therapeutics.
Conclusion
Due to intensive research efforts over the past decades, the knowledge of epigenetic regulation in carcinogenesis is expanding rapidly. This knowledge provides new insights into the role of histone modifications in oncogenic gene transcription. Consequently, histone modifying enzymes have been recognized as drug targets. In this review, we summarize recent discoveries involving histone modifications and the enzymes involved. We focus on small molecules targeting these enzymes involved, and we highlight their effects on TRAIL-induced apoptosis. Finally, we indicate new targets in Table 4 for enhancing TRAIL sensitivity.
Declarations
Authors’ contributionsConceptualization: Zhang BJ
Writing original draft, review and editing: Zhang BJ, Chen D, Dekker FJ, Quax WJ
Figure creating: Chen D
Resources: Dekker FJ, Quax WJ
Funding acquisition, Supervision: Quax WJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis research was partly funded by The Dutch Technology Foundation (STW) (No. 11056) and European Fund for Regional Development (KOP/EFRO) (No. 068 and No. 073). Zhang BJ and Chen D have received a PhD scholarship from China Scholarship Council.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 2012;13:436-47.
3. Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol 2010;28:2839-46.
4. Wong SHM, Kong WY, Fang CM, Loh HS, Chuah LH, et al. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit Rev Oncol Hematol 2019;143:81-94.
5. Fulton MD, Brown T, Zheng YG. Mechanisms and Inhibitors of Histone Arginine Methylation. Chem Rec 2018;18:1792-807.
7. Song S, Yu Y. Progression on citrullination of proteins in gastrointestinal cancers. Front Oncol 2019;9:1-6.
8. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med 2017;49.
9. Sanaei M, Kavoosi F. Histone deacetylases and histone deacetylase inhibitors: molecular mechanisms of action in various cancers. Adv Biomed Res 2019;8:63.
10. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521.
11. Kebede AF, Nieborak A, Shahidian LZ, Le Gras S, Richter F, et al. Histone propionylation is a mark of active chromatin. Nat Struct Mol Biol 2017;24:1048-56.
12. Goudarzi A, Zhang D, Huang H, Barral S, Kwon OK, et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol Cell 2016;62:169-80.
13. Dai L, Peng C, Montellier E, Lu Z, Chen Y, et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol 2014;10:365-70.
14. Ishiguro T, Tanabe K, Kobayashi Y, Mizumoto S, Kanai M, et al. Malonylation of histone H2A at lysine 119 inhibits Bub1-dependent H2A phosphorylation and chromosomal localization of shugoshin proteins. Sci Rep 2018;8:1-10.
15. Wang Y, Guo YR, Liu K, Yin Z, Liu R, et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017;552:273-7.
16. Tan M, Luo H, Lee S, Jin F, Yang JS, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011;146:1016-28.
17. Iberg AN, Espejo A, Cheng D, Kim D, Michaud-Levesque J, et al. Arginine methylation of the histone H3 tail impedes effector binding. J Biol Chem 2008;283:3006-10.
18. Zhao Q, Rank G, Tan YT, Li H, Moritz RL, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 2009;16:304-11.
19. Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science 2007;318:444-7.
20. Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, et al. JMJD6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009;325:90-3.
21. Hong X, Zang J, White J, Wang C, Pan CH, et al. Interaction of JMJD6 with single-stranded RNA. Proc Natl Acad Sci U S A 2010;107:14568-72.
22. Mantri M, Krojer T, Bagg EA, Webby CJ, Butler DS, et al. Crystal structure of the 2-Oxoglutarate- and Fe(II)-dependent lysyl hydroxylase JMJD6. J Mol Biol 2010;401:211-22.
23. Li S, Ali S, Duan X, Liu S, Du J, et al. JMJD1B demethylates H4R3me2s and H3K9me2 to facilitate gene expression for development of hematopoietic stem and progenitor cells. Cell Rep 2018;23:389-403.
24. Hamam HJ, Palaniyar N. Post-translational modifications in netosis and nets-mediated diseases. Biomolecules 2019;9:1-25.
25. Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep 2017;20:538-48.
26. Shi YG, Tsukada Y. The discovery of histone demethylases. Cold Spring Harb Perspect Biol 2013;5:2-4.
27. Chen Y, Sprung R, Tang Y, Ball H, Sangras B, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics 2007;6:812-9.
28. Xie Z, Dai J, Dai L, Tan M, Cheng Z, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 2012;11:100-7.
29. Jeusset L, McManus K. Developing targeted therapies that exploit aberrant histone ubiquitination in cancer. Cells 2019;8:165.
30. Cubeñas-Potts C, Matunis MJ. SUMO: a multifaceted modifier of chromatin structure and function. Dev Cell 2013;24:1-12.
32. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2012;13:37-50.
33. Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, et al. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 2007;26:3558-69.
34. Chung J, Karkhanis V, Tae S, Yan F, Smith P, et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) Silencing. J Biol Chem 2013;288:35534-47.
35. Zhu F, Guo H, Bates PD, Zhang S, Zhang H, et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019;33:2898-911.
36. Chung J, Karkhanis V, Baiocchi RA, Sif S. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/β-catenin and AKT/GSK3β proliferative signaling. J Biol Chem 2019;294:7692-710.
37. Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015;125:2530-43.
38. Kim YR, Lee BK, Park RY, Nguyen NTX, Bae JA, et al. Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 2010;10:1-13.
39. Takahashi Y, Iwai M, Kawai T, Arakawa A, Ito T, et al. Aberrant expression of tumor suppressors CADM1 and 4.1B in invasive lesions of primary breast cancer. Breast Cancer 2012;19:242-52.
40. Zhong J, Cao RX, Zu XY, Hong T, Yang J, et al. Identification and characterization of novel spliced variants of PRMT2 in breast carcinoma. FEBS J 2012;279:316-35.
41. Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 2011;128:562-73.
42. Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol 2007;9:1208-15.
43. Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, et al. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019;36:100-14.e25.
44. Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018;131:2662-9.
45. Waters NJ, Daigle SR, Rehlaender BN, Basavapathruni A, Campbell CT, et al. Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J Control Release 2015;220:758-65.
46. Waters NJ. Preclinical pharmacokinetics and pharmacodynamics of pinometostat (EPZ-5676), a first-in-class, small molecule S-adenosyl methionine competitive inhibitor of DOT1L. Eur J Drug Metab Pharmacokinet 2017;42:891-901.
47. Campbell CT, Haladyna JN, Drubin DA, Thomson TM, Maria MJ, et al. Mechanisms of pinometostat (EPZ-5676) treatment-emergent resistance in MLL-rearranged leukemia. Mol. Cancer Ther 2017;16:1669-79.
48. Blum KA, Abramson J, Maris M, Flinn I, Goy A, et al. 41OA phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann Oncol 2018;29:2018.
49. Wu T, Millar H, Gaffney D, Beke L, Mannens G, et al. JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Available from: https://cancerres.aacrjournals.org/content/78/13_Supplement/4859. [Last accessed on 17 Sep 2020].
50. Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018;19:649-59.
51. Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 2014;13:842-54.
52. Brach D, Johnston-Blackwell D, Drew A, Lingaraj T, Motwani V, et al. EZH2 inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL. Mol Cancer Ther 2017;16:2586-97.
53. Kurmasheva RT, Sammons M, Favours E, Wu J, Kurmashev D, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2017;64.
54. Bauer TM, Besse B, Martinez-Marti A, Trigo JM, Moreno V, et al. Phase I, open-label, dose-escalation study of the safety, pharmacokinetics, pharmacodynamics, and efficacy of GSK2879552 in relapsed/refractory SCLC. J Thorac Oncol 2019;14:1828-38.
55. Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015;28:57-69.
56. Rasco D, Tolcher A, Siu LL, Heinhuis K, Postel-Vinay S, et al. A phase I, open-label, dose-escalation study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of GSK3326595 in subjects with solid tumors and non-Hodgkin’s lymphoma. Available from: https://cancerres.aacrjournals.org/content/77/13_Supplement/CT038. [Last accessed on 17 Sep 2020].
57. Siu LL, Rasco DW, Vinay SP, Romano PM, J Menis FLO, et al. A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumors. nn Oncol 2019;30:159.
58. Gerhart S V., Kellner WA, Thompson C, Pappalardi MB, Zhang XP, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 2018;8:1-15.
59. Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struc. Mol Biol 2019;26:880-9.
60. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012;11:384-400.
61. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity of Zeste protein complex containing the Enhancer associated with a human multiprotein. Genes Dev 2002;16:2893-905.
62. Li B, Chng WJ. EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications. J Hematol Oncol 2019;12:118.
63. Lue JK, Amengual JE. Emerging EZH2 Inhibitors and Their Application in Lymphoma. Curr Hematol Malig Rep 2018;13:369-82.
64. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012;109:2989-94.
65. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007;12:1247-52.
66. Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res 2015;21:2666-70.
67. Raedler LA. Farydak (Panobinostat): first HDAC inhibitor approved for patients with relapsed multiple myeloma. Am Heal drug benefits 2016;9:84-7.
68. Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012;119:2579-89.
69. Cosenza M, Civallero M, Marcheselli L, Sacchi S, Pozzi S. Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 2017;22:827-40.
70. Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res 2017;23:3307-15.
71. Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014;28:680-9.
72. Tang S, Cheng B, Zhe N, Ma D, Xu J, et al. Histone deacetylase inhibitor BG45-mediated HO-1 expression induces apoptosis of multiple myeloma cells by the JAK2/STAT3 pathway. Anticancer Drugs 2018;29:61-74.
73. Harada T, Ohguchi H, Grondin Y, Kikuchi S, Sagawa M, et al. HDAC3 regulates DNMT1 expression in multiple myeloma: therapeutic implications. Leukemia 2017;31:2670-7.
74. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017;543:428-32.
75. Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, et al. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem 2013;56:427-36.
76. Li X, Guo Y, Kuang X, Zhao L, Li H, et al. Histone deacetylase inhibitor LMK-235-mediated HO-1 expression induces apoptosis in multiple myeloma cells via the JNK/AP-1 signaling pathway. Life Sci 2019;223:146-57.
77. Wanek J, Gaisberger M, Beyreis M, Mayr C, Helm K, et al. Pharmacological inhibition of class IIA HDACs by LMK-235 in pancreatic neuroendocrine tumor cells. Int J Mol Sci 2018;19:3128.
78. Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, et al. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res 2013;73:2259-70.
79. Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol 2015;9:1447-57.
80. Wang F, Zheng L, Yi Y, Yang Z, Qiu Q, et al. SKLB-23bb, A HDAC6-Selective inhibitor, exhibits superior and broad-spectrum antitumor activity via additionally targeting microtubules. Mol Cancer Ther 2018;17:763-75.
81. Ma XJ, Xu G, Li ZJ, Chen F, Wu D, et al. HDAC-selective inhibitor cay10603 has single anti-tumour effect in Burkitt’s lymphoma cells by impeding the cell cycle. Curr Med Sci 2019;39:228-36.
82. Wang Z, Tang F, Hu P, Wang Y, Gong J, et al. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncol Rep 2016;36:589-97.
83. Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, et al. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem 2012;55:9891-9.
84. Wu H, Yang K, Zhang Z, Leisten ED, Li Z, et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem 2019;62:7042-57.
85. Sun X, Xie Y, Sun X, Yao Y, Li H, et al. The selective HDAC6 inhibitor nexturastat A induces apoptosis, overcomes drug resistance and inhibits tumor growth in multiple myeloma. Biosci Rep 2019;39:1-11.
86. Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis 2015;6:1-14.
87. Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, et al. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008;22:1026-34.
88. Lopez G, Bill KLJ, Bid HK, Braggio D, Constantino D, et al. HDAC8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (MPNST). PLoS One 2015;10:1-12.
89. Wang R, He Y, Robinson V, Yang Z, Hessler P, et al. Targeting lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol Cancer Ther 2018;17:2543-50.
90. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128-32.
91. Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol 2010;17:471-82.
92. Dahlin JL, Nelson KM, Strasser JM, Barsyte-Lovejoy D, Szewczyk MM, et al. Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat Commun 2017;8:1-14.
93. Wang R, He Y, Robinson V, Yang Z, Hessler P, et al. Targeting Lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol Cancer Ther 2018;17:2543-50.
94. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012;149:214-31.
95. Wai DCC, Szyszka TN, Campbell AE, Kwong C, Lorna EWW, et al. The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators. J Biol Chem 2018;293:7160-75.
96. Cochran AG, Conery AR, Sims RJ. Bromodomains: a new target class for drug development. Nat Rev Drug Discov 2019;18:609-28.
97. Sahni JM, Keri RA. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol Res 2018;129:156-76.
98. Bechter O, Schöffski P. Make your best BET: the emerging role of BET inhibitor treatment in malignant tumors. Pharmacol Ther 2020;208:107479.
99. Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306-10.
100. Chen D, Lu T, Yan Z, Lu W, Zhou F, et al. Discovery, structural insight, and bioactivities of BY27 as a selective inhibitor of the second bromodomains of BET proteins. Eur J Med Chem 2019;182:111633.
101. Rathore R, McCallum JE, Varghese E, Florea AM, Büsselberg D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017;22:898-919.
102. Von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer 2017;17:352-66.
103. Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol 2017;13:217-33.
104. Liu X, Yue P, Khuri FR, Sun SY. Decoy receptor 2 (<em>DcR2</em>) is a p53 target gene and regulates chemosensitivity. Cancer Res 2005;65:9169-75.
105. Mansour NM, Bernal GM, Wu L, Crawley CD, Cahill KE, et al. Decoy receptor DcR1 is induced in a p50/Bcl3-dependent manner and attenuates the efficacy of temozolomide. Cancer Res 2015;75:2039-48.
106. Liu X, Yue P, Khuri FR, Sun SY. p53 upregulates death receptor 4 expression through an intronic p53 Binding site. Cancer Res 2004;64:5078-83.
107. Shetty S, Graham BA, Brown JG, Hu X, Vegh-Yarema N, et al. Transcription factor NF-κB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol Cell Biol 2005;25:5404-16.
108. Woo SM, Seo SU, Min KJ, Kwon TK. BIX-01294 sensitizes renal cancer Caki cells to TRAIL-induced apoptosis through downregulation of survivin expression and upregulation of DR5 expression. Cell Death Discov 2018;4:29.
109. Kim SY, Hong MN, Heo SH, Park S, Kwon TK, et al. Inhibition of euchromatin histone-lysine N-methyltransferase 2 sensitizes breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through reactive oxygen species-mediated activating transcription factor 4-C/EBP homologous protein-de. Mol Carcinog 2018;57:1492-506.
110. Benoit YD, Laursen KB, Witherspoon MS, Lipkin SM, Gudas LJ. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J Cell Physiol 2013;228:764-72.
111. Kasman L, Onicescu G, Voelkel-Johnson C. Histone deacetylase inhibitors restore cell surface expression of the coxsackie adenovirus receptor and enhance CMV promoter activity in castration-resistant prostate cancer cells. Prostate Cancer 2012;2012:137163.
112. Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther 2010;9:3254-66.
113. Borbone E, Berlingieri MT, De Bellis F, Nebbioso A, Chiappetta G, et al. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene 2010;29:105-16.
114. Nebbioso A, Carafa V, Conte M, Tambaro FP, Ciro A, et al. C-Myc modulation and acetylation is a key HDAC inhibitor target in cancer. Clin Cancer Res 2017;23:2542-55.
115. Zhang B, Liu B, Chen D, Setroikromo R, Haisma HJ, et al. Histone deacetylase inhibitors sensitize TRAIL-induced apoptosis in colon cancer cells. Cancers (Basel) 2019;11:645.
116. Kurt IC, Sur I, Kaya E, Cingoz A, Kazancioglu S, et al. KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis 2017;8:e2897.
117. Tanaka H, Hoshikawa Y, Oh-hara T, Koike S, Naito M, et al. PRMT5, a novel TRAIL receptor-binding protein, inhibits TRAIL-induced apoptosis via nuclear factor-κB activation. Mol Cancer Res 2009;7:557-69.
118. Li M, An W, Xu L, Lin Y, Su L, et al. The arginine methyltransferase PRMT5 and PRMT1 distinctly regulate the degradation of anti-apoptotic protein CFLAR L in human lung cancer cells. J Exp Clin Cancer Res 2019;38:1-13.
119. Kauh J, Fan S, Xia M, Yue P, Yang L, et al. C-FLIP degradation mediates sensitization of pancreatic cancer cells to trail-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One 2010;5:e10376.
120. Symanowski J, Vogelzang N, Zawel L, Atadja P, Pass H, et al. A histone deacetylase inhibitor LBH589 downregulates XIAP in mesothelioma cell lines which is likely responsible for increased apoptosis with TRAIL. J Thorac Oncol 2009;4:149-60.
121. Kerr E, Holohan C, McLaughlin KM, Majkut J, Dolan S, et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ 2012;19:1317-27.
122. Klingbeil O, Lesche R, Gelato KA, Haendler B, Lejeune P. Inhibition of bet bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis 2016;7:1-13.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zhang BJ, Chen D, Dekker FJ, Quax WJ. Improving TRAIL-induced apoptosis in cancers by interfering with histone modifications. Cancer Drug Resist 2020;3:791-803. http://dx.doi.org/10.20517/cdr.2020.58
AMA Style
Zhang BJ, Chen D, Dekker FJ, Quax WJ. Improving TRAIL-induced apoptosis in cancers by interfering with histone modifications. Cancer Drug Resistance. 2020; 3(4): 791-803. http://dx.doi.org/10.20517/cdr.2020.58
Chicago/Turabian Style
Zhang, Bao-Jie, Deng Chen, Frank J. Dekker, Wim J. Quax. 2020. "Improving TRAIL-induced apoptosis in cancers by interfering with histone modifications" Cancer Drug Resistance. 3, no.4: 791-803. http://dx.doi.org/10.20517/cdr.2020.58
ACS Style
Zhang, B.J.; Chen D.; Dekker FJ.; Quax WJ. Improving TRAIL-induced apoptosis in cancers by interfering with histone modifications. Cancer Drug Resist. 2020, 3, 791-803. http://dx.doi.org/10.20517/cdr.2020.58
About This Article
Copyright

Data & Comments
Data


 Cite This Article 109 clicks
Cite This Article 109 clicks

 Like This Article 19
likes
Like This Article 19
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.