
Molecular mechanisms of enzalutamide resistance in prostate cancer

Abstract
An estimated 30,000 men in the United States will die of metastatic prostate cancer (PCa) each year due to the development of therapy resistance, most notably resistance to second-generation antiandrogen enzalutamide. The vast majority of PCa is driven by the androgen receptor (AR). Enzalutamide is an AR antagonist, which extends patient survival and is widely used in the clinic for the treatment of castration-resistant prostate cancer (CRPC); however, many patients will have primary or develop acquired resistance and continue to progress. Characterization of the molecular mechanisms of enzalutamide resistance provides insight into potentially efficacious therapies for enzalutamide-resistant CRPC (ER-CRPC). Understanding these mechanisms is critical for the identification of biomarkers predictive of therapy resistance and the development of therapeutic strategies to target ER-CRPC.
Keywords
Introduction
Nearly 30,000 men in the United States will die of metastatic prostate cancer (PCa) in 2019[1,2]. Despite the introduction of seven new Food and Drug Administration-approved therapeutic agents since 2007, metastatic PCa is incurable. The mainstay of treatment for metastatic PCa remains androgen deprivation therapy (ADT), via pharmacological or surgical castration. The majority of patients will initially respond to ADT; however, a significant proportion becomes therapy-resistant and develops castration-resistant prostate cancer (CRPC)[3,4].
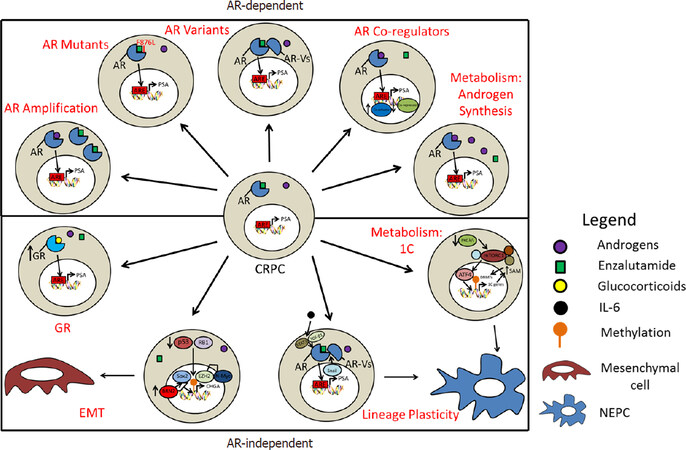
Figure 1. Model of androgen receptor (AR)-dependent and AR-independent mechanisms that enable a castration-resistant prostate cancer (CRPC) to become an enzalutamide resistant-CRPC. EMT: epithelial-mesenchymal transition; GR: glucocorticoid receptor; 1C: one-carbon; NEPC: neuroendocrine prostate cancer
The androgen receptor (AR) is a steroid hormone nuclear receptor that drives the vast majority of PCa. CRPC remains dependent on AR signaling and second-generation antiandrogens, such as the AR antagonist enzalutamide or the androgen synthesis inhibitor abiraterone, extend the overall survival of metastatic CRPC patients by a median of 4-5 months[5,6]. However, primary or acquired resistance to these agents is common with cancer recurrence and progression. Multiple mechanisms of enzalutamide resistance have been defined and offer insights into further therapeutic strategies against enzalutamide-resistant CRPC (ER-CRPC)[7-9]. In this review, we present a critical evaluation of the defined molecular mechanisms, potential biomarkers, and therapeutic options for ER-CRPC.
AR amplification
For receptor-ligand interactions, the relative concentrations of receptors, agonists, and antagonists in a cell can dictate whether antagonists will be efficacious in inhibiting their target. Stoichiometric ratios of AR, enzalutamide, and androgens determine whether AR is bound to agonist or antagonist, and whether AR signaling is active. Sequencing of CRPC and ER-CRPC patient tumors demonstrates that 70% of patients have significant AR pathway alterations, with the vast majority containing AR gene amplifications[10,11]. Thus, AR antagonists, like enzalutamide, apalutamide, or darolutamide, may be effectively unable to antagonize all AR proteins in this context. Furthermore, since CRPC and ER-CRPC tumors may synthesize their own androgens, sustained or enhanced AR signaling is noted, even under castrate-level serum androgen conditions[12]. Thus, AR amplification and increased AR signaling are robust mechanisms of resistance to first- and second-generation antiandrogens[10,11,13-16].
AR mutants
AR point mutations that convert first-generation antiandrogens, such as flutamide and bicalutamide, from antagonists to agonists have been well-characterized. These mutations are in the ligand binding domain (LBD), and include T877A and W741C point mutations, which mediate resistance to flutamide and bicalutamide, respectively[13-16]. Similarly, resistance to enzalutamide has been modeled to be driven by an AR F876L point mutation. This mutation has been infrequently identified in ER-CRPC samples and confers agonistic properties to both enzalutamide and apalutamide[17-19]. Importantly, since these mutations are specific for each antiandrogen, other antiandrogens can have activity against AR. For example, CRPC cells with T877A and W741C mutations are resistant to flutamide and bicalutamide, respectively, but are sensitive to enzalutamide and apalutamide[17]. While AR mutations can confer resistance to enzalutamide, the low observed prevalence of this mutation in ER-CRPC patients does not support a significant clinical role for this mutation in enzalutamide resistance.
AR variants
There are a large number of known AR splice variants (AR-Vs)[20-24]. AR variants primarily appear after the selection pressure of castration and more frequently after enzalutamide and abiraterone treatment[20-22,24-31]. The most common AR-V is AR-V7 (AR3), which contains exons 1, 2, 3, and a cryptic exon 3b but lacks the LBD[21,26,27]. AR-Vs can be constitutively active, and detection of AR-Vs in CRPC correlate with poor survival, progression, and therapy resistance[20-22,24-31]. Assays to detect AR-Vs in circulating tumor cells are commercially available and predict therapy response to enzalutamide and abiraterone[25]. AR-Vs can substitute for the full-length AR (AR-FL) and can bind to canonical androgen responsive elements (AREs) on the chromatin, heterodimerize with AR-FL, and drive transcription from AREs[31-34]. AR-Vs have been shown to be capable of independently driving transcription, cell proliferation, and DNA repair in CRPC[35]. However, AR-Vs are rarely seen without significant amplification of AR-FL, and its activity may be dependent on full-length AR[31,32,36].
Importantly, the ability of AR-Vs to independently drive ER-CRPC has not been proven. In vitro studies with knockdown of variants in CRPC cell lines that express both AR-FL and AR-Vs indicate that AR-V expression confers a distinct growth advantage, when treated with antiandrogens[31-34,37]. Treatment with niclosamide inhibits AR-V7 recruitment to AR target genes, reduces AR-V7 protein levels in a proteasome-dependent manner, and re-sensitizes ER-CRPC cells to enzalutamide[38]. Induction of AR-Vs through NF-κB2/p52 can also enhance enzalutamide resistance[39-41]. While these data suggest that AR-Vs can contribute to ER-CRPC growth and can mediate resistance to enzalutamide, they do not definitively establish that AR-Vs drive resistance to enzalutamide.
The true contribution of AR-Vs in driving resistance can only be clearly established from drugs that specifically target AR-Vs and not AR-FL; however, no such agent has been developed. Since AR-FL and AR-Vs share a common N-terminal domain (NTD), compounds designed to target the NTD will target both AR-FL and AR-Vs. For example, ESSA Pharma’s EPI-506, which was designed to target the AR NTD, could target both AR-Vs and AR-FL, but demonstrated no activity against enzalutamide- and abiraterone-resistant CRPC patients in Phase I clinical trials[42]. Ongoing clinical trials with AR degraders will also likely target AR-FL and AR-Vs, and are unlikely to establish AR-Vs as independent molecular drivers of ER-CRPC.
AR co-regulators
Upon binding androgens and activation, AR alters its protein interactome, translocates to the nucleus, and regulates a pro-proliferative transcriptional program. AR interacts with a number of co-activators to enhance transcription, like members of the p160 steroid receptor coactivator (SRC) family, histone acetyltransferase CBP/p300, and pioneer factor forkhead box A1 (FOXA1), and co-repressors to inhibit transcription, such as zinc finger and BTB domain containing 16 (ZBTB16)[43-48]. Modulating the expression of these cofactors can enhance or attenuate AR transcriptional activity. For example, alterations in expression of coactivators SRC-1, SRC-2, SRC-3, CBP/p300, and FOXA1 and co-repressor ZBTB16 are frequently seen CRPC and ER-CRPC patients and are associated with worse prognosis[10,11,49-53]. Since enzalutamide can modulate the recruitment of AR cofactors, altering the expression of co-activators or co-repressors in CPRC can potentially bypass the inhibitory effects of enzalutamide and confer resistance[48,54,55].
Glucocorticoid receptor
Like AR, the glucocorticoid receptor (GR) is a steroid hormone nuclear receptor, which binds DNA as a homodimer in an inverted repeat fashion[56,57]. Upon binding its ligand, GR transcriptionally activates a stress response program, which enhances the expression of anti-inflammatory genes and suppresses pro-inflammatory genes[58-60]. Overexpression of GR was noted in ER-CRPC and mediates enzalutamide resistance, where it may bypass the need for AR signaling[61]. Primary GR-dependent enzalutamide resistance may be observed in a subset of CRPC tumors that have increased basal expression of GR. Acquired GR-dependent enzalutamide resistance entails a de-repression mechanism, whereby AR normally inhibits GR expression, and enzalutamide enables enhanced GR expression by blocking AR signaling[61]. Interestingly, chromatin immunoprecipitation-sequencing (ChIP-seq) studies in enzalutamide-resistant cells identified GR binding over 50% of AR binding sites on the chromatin, with the strongest AR-regulated genes also being regulated by GR[61]. Thus, overexpressed GR functionally substitutes for AR. Therapeutic targeting of GR in ER-CRPC has been proposed, with at least two companies, ORIC Pharmaceuticals and Corcept Therapeutics, developing GR antagonists.
Epithelial-mesenchymal transition
The epithelial-mesenchymal transition (EMT) is a process by which epithelial cells become more mesenchymal, a state characterized by increased invasive capacity, apoptotic resistance, and enhanced motility and metastatic potential[62-65]. Acute enzalutamide treatment induces EMT through a number of mechanisms, including increasing TGF-β1 expression and STAT3 activation, as well as Snail induction[66-68]. Metformin blocks enzalutamide-induced EMT and improves PCa sensitivity to enzalutamide[66]. In addition, autocrine IL-6 can facilitate CRPC growth and confers enzalutamide resistance mediated by STAT3 activation[38]. Furthermore, AR directly represses Snail transcription, and acute enzalutamide treatment enhances Snail expression and EMT[67]. Importantly, in models of chronic enzalutamide treatment, enzalutamide resistance can be mediated by Snail induction of both AR and AR-V7 expression, leading to increased AR signaling[69,70]. Whether in an AR-dependent or -independent manner, programs that enable cell transitions in response to selective pressures are important mechanisms of resistance to enzalutamide.
Metabolic alterations
Most molecular mechanisms identified in ER-CRPC have focused on AR transcriptional regulation and maintenance of AR signaling, despite AR inhibition. Evaluations of the downstream programs that confer enzalutamide resistance often culminate in metabolic alterations, as the metabolic state governs whether cells will resist stress and proliferate.
Intracrine androgen synthesis
Increased androgen synthesis can overwhelm the ability of enzalutamide to block AR signaling[12]. Intracrine androgen synthesis has recently been shown to confer enzalutamide resistance through upregulation of steroid synthesis genes, such as aldo-keto reductase family 1 member C3 (AKR1C3)[12,30,71-75]. AKR1C3 catalyzes the conversion of androstenedione and 5 α-androstanedione to testosterone and DHT, respectively, and is enriched in acquired and de novo ER-CRPC[12,76,77]. Increased AKR1C3 levels results in upregulated intracrine androgen synthesis and confers resistance to enzalutamide[12].
Hypoxia
An AR-independent mechanism of enzalutamide resistance involves hypoxia and the metabolic consequences of hypoxia-induced programs driven by hypoxia-inducible factor (HIF)[78]. PCa cells that are capable of stimulating hypoxia-induced survival programs through the upregulation of hypoxia response genes, such as glucose-6-phosphate isomerase (GPI), are clonally selected to become AR-independent and resistant to enzalutamide[78]. Under normoxic and normal androgenic conditions, AR enhances glycolysis and the pentose phosphate pathway (PPP)[79,80]. Under hypoxic and normal androgenic conditions, the PPP is slightly upregulated, AR inhibits GPI, and glycolysis is inhibited. However, under hypoxic and castrate conditions, the PPP is inhibited, GPI is upregulated, and glycolysis is stimulated[78]. PCa cells that are thus able to redirect glucose away from the PPP and toward glycolysis are able to evade stress and proliferate normally[78]. Glycolytic inhibitors, such as 2-deoxyglucose, may be useful in this context; however, toxicity is a concern due to lack of selectivity. Selective glycolytic inhibitors in development may be efficacious against some forms of ER-CRPC. Of note, GPI is preferentially overexpressed in neuroendocrine prostate cancer (NEPC) tumors, attesting to the importance of metabolic rewiring in driving neuroendocrine disease[78].
One-carbon metabolism
Increased reliance on serine and one-carbon (1C) metabolism promotes enzalutamide resistance in NEPC[81]. In CRPC cells, loss of protein kinase C (PKC) λ/ι allows cells to transition from a luminal, AR-dependent phenotype to a basal, AR-independent phenotype through enhanced one-carbon metabolism and resulting epigenetic changes[81]. This upregulation in one-carbon metabolism is dependent on mammalian target of rapamycin 1 (mTORC1) and cyclic AMP-dependent transcription factor 4 (ATF4) and culminates in an increase in S-adenosylmethionine (SAM), which supports epigenetic reprogramming (DNA methylation)[81]. Enhancer of zeste homolog 2 (EZH2) inhibition has been shown to reverse NEPC to a more AR-dependent state sensitive to antiandrogens, which demonstrates the potential efficacy of this strategy and further indicates the importance of epigenetic changes for the development of NEPC[11,82]. Additionally, DNA methylation inhibitors, such as decitabine, may be efficacious in targeting neuroendocrine disease. mTORC1 inhibitors, such as everolimus, may also show benefits in NEPC patients with a PKCλ/ι deficiency.
Lineage plasticity
Lineage plasticity is a mechanism through which cells can acquire characteristics of a lineage that no longer requires a certain drug target[83]. With enzalutamide treatment, cells become AR-independent and therefore enzalutamide-resistant. In PCa, lineage plasticity is a state characterized by significant epigenetic changes, decreased AR signaling, and an increased expression of neuroendocrine and stem cell markers[82-84].
p53 and retinoblastoma 1 loss
Enzalutamide resistance can develop from loss of tumor suppressors tumor protein p53 (TP53) and retinoblastoma 1 (Rb1) and a downstream SRY-box 2 (SOX2)-driven shift[83] .The proposed mechanism involves increasing cell plasticity, which confers resistance through lineage switching to an AR-independent, basal-like cell[83]. Similarly, loss of p53 and Rb1 creates a stem cell-like epigenetic environment due to derepression of EZH2 (and SOX2), which allows for adaptation to selective pressures, such as enzalutamide[82].
BRN2
Regulators of Sox2, like POU-domain transcription factor BRN2 (POU3F2), drive the emergence of NEPC and enzalutamide resistance[84]. BRN2 expression is inhibited by AR, is required for the expression of neuroendocrine markers, and expressed in NEPC. Enzalutamide derepresses AR inhibition of BRN2 in CRPC and enables BRN2-driven transdifferentiation into enzalutamide-resistant NEPC[84]. Furthermore, BRN2 regulates SOX2, and these proteins directly interact at the enhancers of neuronal genes and cooperate to drive a neuroendocrine phenotype[84]. Targeting BRN2 remains an attractive option for preventing lineage plasticity and the development of AR-independent PCa.
N-Myc and EZH2
N-Myc overexpression is found in 5% of primary PCa patients, 20% of CRPC patients, and roughly 40% of NEPC patients[85-87]. N-Myc and EZH2 cooperate to drive transdifferentiation into NEPC and enzalutamide resistance[86]. N-Myc differentially regulates the DNA damage response in a context-dependent manner. Upregulation of N-Myc inhibits ataxia-telangiesctasia mutated (ATM), which allows PCa to become CRPC. In CRPC, overexpression of N-Myc with EZH2 blocks ATM inhibition, leading to ATM upregulation[88] and the development of enzalutamide-resistant NEPC. Given the dependence of NEPC on epigenetic reprogramming and EZH2 in particular, targeting EZH2 may be an effective therapeutic option. While some EZH2 inhibitors have failed clinical trials [NCT01897571], other agents, such as Constellation Pharmaceuticals’ CPI-1205 and Daiichi-Sankyo’s DS-3201b, may offer hope for selected patients with NEPC [NCT03480646, NCT03110354].
Conclusion
Discovering the molecular underpinnings of enzalutamide resistance has led to a greater understanding of the factors that drive progression and the heterogeneity that belies ER-CRPC. Ongoing studies will enable the identification of biomarkers predictive of therapy resistance and the development of targeted therapies to overcome therapeutic resistance.
Declarations
Authors’ contributionsMade substantial contributions to both the writing and editing of this review: Blatt EB, Raj GV
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Department of Defense (W81XWH017-1-0674) and the Prostate Cancer Foundation (18CHAL16), as well as support from the Cole Foundation and the Wilson Foundation.
Conflicts of interestDr. Raj GV is a named inventor in several patents in drugs that may be used in enzalutamide-resistant prostate cancers.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. American Cancer Society. Cancer Facts & Figures 2018. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2018.html. [Last accessed on 14 May 2019].
3. Antonarakis ES, Blackford AL, Garrett-Mayer E, Eisenberger MA. Survival in men with nonmetastatic prostate cancer treated with hormone therapy: a quantitative systematic review. J Clin Oncol 2007;25:4998-5008.
4. Armstrong AJ, Garrett-Mayer E, de Wit R, Tannock I, Eisenberger M. Prediction of survival following first-line chemotherapy in men with castration-resistant metastatic prostate cancer. Clin Cancer Res 2010;16:203-11.
5. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33-9.
6. Loriot Y, Fizazi K, de Bono JS, Forer D, Hirmand M, et al. Enzalutamide in castration-resistant prostate cancer patients with visceral disease in the liver and/or lung: outcomes from the randomized controlled phase 3 AFFIRM trial. Cancer 2017;123:253-62.
7. Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res 2015;21:2315-24.
8. Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov 2011;1:466-8.
9. Alanee S, Moore A, Nutt M, Holland B, Dynda D, et al. Contemporary incidence and mortality rates of neuroendocrine prostate cancer. Anticancer Res 2015;35:4145-50.
10. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215-28.
11. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nature medicine 2016;22:298-305.
12. Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res 2015;75:1413-22.
13. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239-43.
14. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 2013;63:920-6.
15. Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res 2003;63:149-53.
16. Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, et al. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol 1992;41:665-9.
17. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 2013;3:1020-9.
18. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013;2:e00499.
19. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol 2013;31:3525-30.
20. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009;69:16-22.
21. Guo Z, Yang X, Sun F, Jiang R, Linn DE, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009;69:2305-13.
22. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clinical Invest 2010;120:2715-30.
23. Ahrens-Fath I, Politz O, Geserick C, Haendler B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 2005;272:74-84.
24. Kohli M, Ho Y, Hillman DW, Van Etten JL, Henzler C, et al. Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin Cancer Res 2017;23:4704-15.
25. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028-38.
26. Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 2011;6:e27970.
27. Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 2011;6:e19059.
28. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008;68:5469-77.
29. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, et al. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res 2011;71:2108-17.
30. Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 2011;17:5913-25.
31. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013;73:483-9.
32. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A 2010;107:16759-65.
33. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 2012;72:3457-62.
34. Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011;71:1656-67.
35. Cronauer MV, Hittmair A, Eder IE, Hobisch A, Culig Z, et al. Basic fibroblast growth factor levels in cancer cells and in sera of patients suffering from proliferative disorders of the prostate. Prostate 1997;31:223-33.
36. Luo J, Attard G, Balk SP, Bevan C, Burnstein K, et al. Role of androgen receptor variants in prostate cancer: report from the 2017 Mission Androgen Receptor Variants Meeting. Eur Urol 2018;73:715-23.
37. Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem 2012;287:19736-49.
38. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res 2014;20:3198-210.
39. Nadiminty N, Chun JY, Lou W, Lin X, Gao AC. NF-kappaB2/p52 enhances androgen-independent growth of human LNCaP cells via protection from apoptotic cell death and cell cycle arrest induced by androgen-deprivation. Prostate 2008;68:1725-33.
40. Nadiminty N, Dutt S, Tepper C, Gao AC. Microarray analysis reveals potential target genes of NF-kappaB2/p52 in LNCaP prostate cancer cells. Prostate 2010;70:276-87.
41. Nadiminty N, Tummala R, Liu C, Yang J, Lou W, et al. NF-kappaB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther 2013;12:1629-37.
42. Moigne RL, Zhou HJ, Obst JK, Banuelos CA, Jian K, et al. Lessons learned from the metastatic castration-resistant prostate cancer phase I trial of EPI-506, a first-generation androgen receptor N-terminal domain inhibitor. J Clin Oncol 2019;37:257.
43. Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J Biol Chem 1998;273:31853-9.
44. Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol 1999;19:8383-92.
45. Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem 2002;277:38087-94.
46. Reid J, Murray I, Watt K, Betney R, McEwan IJ. The androgen receptor interacts with multiple regions of the large subunit of general transcription factor TFIIF. J Biol Chem 2002;277:41247-53.
47. Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol 2003;17:1484-507.
48. Hsieh CL, Botta G, Gao S, Li T, Van Allen EM, et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res 2015;75:1944-8.
49. Agoulnik IU, Vaid A, Bingman WE 3rd, Erdeme H, Frolov A, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res 2005;65:7959-67.
50. Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE 3rd, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res 2006;66:10594-602.
51. Comuzzi B, Nemes C, Schmidt S, Jasarevic Z, Lodde M, et al. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J Pathol 2004;204:159-66.
52. Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, et al. p300 in prostate cancer proliferation and progression. Cancer Res 2003;63:7638-40.
53. Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res 2005;65:7976-83.
54. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787-90.
55. Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res 2016;22:4466-77.
56. Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, et al. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991;352:497-505.
57. Savory JG, Prefontaine GG, Lamprecht C, Liao M, Walther RF, et al. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol Cell Biol 2001;21:781-93.
58. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 2002;16:61-71.
59. Lu NZ, Collins JB, Grissom SF, Cidlowski JA. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol 2007;27:7143-60.
60. Ren R, Oakley RH, Cruz-Topete D, Cidlowski JA. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 2012;153:5346-60.
61. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013;155:1309-22.
62. Kiemer AK, Takeuchi K, Quinlan MP. Identification of genes involved in epithelial-mesenchymal transition and tumor progression. Oncogene 2001;20:6679-88.
63. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341-50.
64. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002;156:299-313.
65. Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res 2003;63:3386-94.
66. Liu Q, Tong D, Liu G, Xu J, Do K, et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-beta1/STAT3 axis-regulated EMT. Cell Death Dis 2017;8:e3007.
67. Miao L, Yang L, Li R, Rodrigues DN, Crespo M, et al. Disrupting androgen receptor signaling induces Snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res 2017;77:3101-12.
68. Martin SK, Pu H, Penticuff JC, Cao Z, Horbinski C, et al. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res 2016;76:912-26.
69. Ware KE, Somarelli JA, Schaeffer D, Li J, Zhang T, et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget 2016;7:50507-21.
70. Kong D, Sethi S, Li Y, Chen W, Sakr WA, et al. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate 2015;75:161-74.
71. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011;71:6503-13.
72. Ishizaki F, Nishiyama T, Kawasaki T, Miyashiro Y, Hara N, et al. Androgen deprivation promotes intratumoral synthesis of dihydrotestosterone from androgen metabolites in prostate cancer. Sci Rep 2013;3:1528.
73. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 2008;68:6407-15.
74. Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res 2011;71:1486-96.
75. Fankhauser M, Tan Y, Macintyre G, Haviv I, Hong MK, et al. Canonical androstenedione reduction is the predominant source of signaling androgens in hormone-refractory prostate cancer. Clin Cancer Res 2014;20:5547-57.
76. Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, et al. The key role of 17 beta-hydroxysteroid dehydrogenases in sex steroid biology. Steroids 1997;62:148-58.
77. Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alpha-androstane-3alpha, 17beta-diol to 5alpha-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol endocrinol 2006;20:444-58.
78. Geng H, Xue C, Mendonca J, Sun XX, Liu Q, et al. Interplay between hypoxia and androgen controls a metabolic switch conferring resistance to androgen/AR-targeted therapy. Nat commun 2018;9:4972.
79. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 2011;30:2719-33.
80. Tsouko E, Khan AS, White MA, Han JJ, Shi Y, et al. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 2014;3:e103.
81. Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, et al. Increased serine and one-carbon pathway metabolism by PKClambda/iota deficiency promotes neuroendocrine prostate cancer. Cancer Cell 2019;35:385-400.e9.
82. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78-83.
83. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84-8.
84. Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov 2017;7:54-71.
85. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 2011;1:487-95.
86. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016;30:563-77.
87. Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013;15:1-10.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Blatt EB, Raj GV. Molecular mechanisms of enzalutamide resistance in prostate cancer. Cancer Drug Resist 2019;2:189-97. http://dx.doi.org/10.20517/cdr.2019.25
AMA Style
Blatt EB, Raj GV. Molecular mechanisms of enzalutamide resistance in prostate cancer. Cancer Drug Resistance. 2019; 2(2): 189-97. http://dx.doi.org/10.20517/cdr.2019.25
Chicago/Turabian Style
Blatt, Eliot B., Ganesh V. Raj. 2019. "Molecular mechanisms of enzalutamide resistance in prostate cancer" Cancer Drug Resistance. 2, no.2: 189-97. http://dx.doi.org/10.20517/cdr.2019.25
ACS Style
Blatt, EB.; Raj GV. Molecular mechanisms of enzalutamide resistance in prostate cancer. Cancer Drug Resist. 2019, 2, 189-97. http://dx.doi.org/10.20517/cdr.2019.25
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 35 clicks
Cite This Article 35 clicks

 Like This Article 0
likes
Like This Article 0
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.