
Drug resistance and combating drug resistance in cancer


Abstract
Cancer is the second leading cause of death in the US. Current major treatments for cancer management include surgery, cytotoxic chemotherapy, targeted therapy, radiation therapy, endocrine therapy and immunotherapy. Despite the endeavors and achievements made in treating cancers during the past decades, resistance to classical chemotherapeutic agents and/or novel targeted drugs continues to be a major problem in cancer therapies. Drug resistance, either existing before treatment (intrinsic) or generated after therapy (acquired), is responsible for most relapses of cancer, one of the major causes of death of the disease. Heterogeneity among patients and tumors, and the versatility of cancer to circumvent therapies make drug resistance more challenging to deal with. Better understanding the mechanisms of drug resistance is required to provide guidance to future cancer treatment and achieve better outcomes. In this review, intrinsic and acquired resistance will be discussed. In addition, new discoveries in mechanisms of drug resistance will be reviewed. Particularly, we will highlight roles of ATP in drug resistance by discussing recent findings of exceptionally high levels of intratumoral extracellular ATP as well as intracellular ATP internalized from extracellular environment. The complexity of drug resistance development suggests that combinational and personalized therapies, which should take ATP into consideration, might provide better strategies and improved efficacy for fighting drug resistance in cancer.
Keywords
Introduction
Cancer is the second leading cause of death in the US[1]. In 2017, about 1.7 million people were diagnosed with cancer and 0.6 million people died from the disease[2]. Drug resistance and the resulting ineffectiveness of the drug treatment are responsible for up to 90% of the cancer related deaths[3-7].
Drug resistance in cancer is a well-known phenomenon that results when cancer becomes tolerant to pharmaceutical treatment. Resistance to anticancer drugs arises from a wide variety of factors, such as genetic mutations and/or epigenetic changes, conserved but upregulated drug efflux, and various other cellular and molecular mechanisms.
Current major treatments for cancer management include surgery, cytotoxic chemotherapy, targeted therapy, radiation therapy, endocrine therapy and immunotherapy[8-12]. Despite the achievements made in treating cancers during the past decades, resistance to classical chemotherapeutic agents and/or novel targeted drugs continue to be a major problem in cancer therapies and responsible for most relapses, one of the major causes of death in cancer[3-7]. Many classical chemotherapeutic anticancer agents kill cancer cells by directly damaging their DNA, which has the problem of non-specificity and relatively high toxicity. In recent decades, more and more targeted drugs have been developed to precisely target/block changes that drive cancer growth and proliferation. Although these drugs show remarkable effects during initial treatment, a large majority of patients develop resistance as treatment proceeds. For example, 30%-55% of patients with non-small cell lung cancer (NSCLC) relapse and die from the disease afterwards[13]. The 50%-70% of ovarian adenocarcinomas reoccur within 1 year after surgery and associated chemotherapy[14]. About 20% of pediatric acute lymphoblastic leukemia patients develop recurrence[15].
Better understanding of mechanisms underlying the acquisition of drug resistance is urgently needed and will facilitate developing novel therapeutic strategies and lead to better clinical outcomes. In this review, we will describe intrinsic and acquired drug resistance, present new discoveries in specific mechanisms of drug resistance and discuss new strategies for fighting against drug resistance and improving anticancer efficacy.
Intrinsic and acquired drug resistance
Drug resistance can be categorized as intrinsic or acquired resistance based on the time when it is developed. Intrinsic resistance exists before drug treatment while the acquired resistance is induced after therapy, each occurring in about 50% of cancer patients with drug resistance[16,17].
Intrinsic resistance
Intrinsic resistance is usually defined as the innate resistance that exists before the patient is administered with (exposed to) drugs, which usually causes reduced efficacy of the drug treatment. Intrinsic resistance can be caused by: (1) pre-existing (inherent) genetic mutations in a majority of tumors that result in decreased responsiveness of cancer cells, such as triple negative breast cancer cells, to both chemo and target drugs; (2) heterogeneity of tumors in which pre-existed insensitive subpopulations, including cancer stem cells, will be selected upon drug treatment thus leading to relapse in later stages of therapeutic treatment; (3) activation of intrinsic pathways used as defense against environmental toxins (such as anticancer drugs).
Intrinsic drug resistance may exist in cancer cells prior to therapies due to the presence of genetic mutation(s) of genes involved in cancer cell growth and/or apoptosis. For example, poorer outcome of cisplatin treatment in gastric cancer patients was found to be associated with HER2 overexpression[18]. The high expression of HER2 gene promoted the upregulation of transcription factor Snail, inducing a morphological change similar to epithelial-mesenchymal transition (EMT), which renders cancer cells more resistant. Furthermore, the HER2/Snail double positive patients were found to have lower survival rate than patients with only single positive or double negative genetic makeup.
It was shown that transcriptional repressors Snail and Slug not only mediate EMT, but also resistance to p53 induced apoptosis, and a self-renewal program[19]. The latter two activities make cancer stem cells (CSCs) more resistant to radiation and chemotherapies. It was also shown that resistant cells are more mesenchymal-like[20,21]. These concomitant changes link intrinsic drug resistance with EMT and CSCs.
Pre-existing resistant subpopulations in tumors may also lead to relapse after chemotherapeutic treatment. Increasing evidence shows that existence of intratumoral genetic heterogeneity in primary tumors predates clinical intervention[22-24]. Patients would respond to therapy initially because the majority of the tumor cells are sensitive to the drug. However, the resistant subclones would proliferate after the drug treatment and cause recurrence[25-27]. This intrinsic drug resistance is sometimes mistaken for acquired resistance since the tumor would shrink first upon treatment and the resistance seems to be acquired due to therapy. CSCs are a subpopulation in tumors with the capacity of self-renewal and differentiation, participating in tumor initiation and progression[28]. They have been reported to be involved in resistance to chemotherapeutic drugs in multiple cancer types, including leukemia[29], glioblastoma[30] and pancreatic cancer[31]. Combined therapy targeting both CSCs and most tumor cells might be required to reduce drug resistance.
Therapeutic effects of a drug can be reduced by activation of intrinsic pathways that are used as a defense against environmental toxins, including anticancer drugs. Examples of these protective mechanisms include ATP binding cassette (ABC) transporter mediated drug efflux[32] and glutathione (GSH)/glutathione S-transferase system[33], working to reduce cellular drug accumulation or detoxify drug treated cancer cells respectively.
Acquired resistance
Acquired resistance can be identified by gradual reduction of anticancer efficacy of a drug after the drug treatment. Acquired resistance can be a result of: (1) activation of second proto-oncogene that becomes the newly emerged driver gene; (2) mutations or altered expression levels of the drug targets; (3) changes in tumor microenvironment (TME) after treatment.
Shrunk tumors can acquire resistance and regrow due to new mutations. In one study, genomic profiles before and after relapse of eight acute myeloid leukemia patients were analyzed using whole-genome sequencing[34]. The comparison of mutations between primary and relapse tumors revealed novel gene mutations. In addition, the results showed increased transversion mutations in relapsed tumors, suggesting that cytotoxic chemotherapeutic drugs caused DNA damage in cancer cells and might increase probability of the emergence of new mutations.
Cancer cells can acquire resistance to targeted drugs when the genes encoding target proteins develop new mutations or change expression levels. One example of secondary mutations within the target kinase is the threonine 315 to isoleucine (T315I) mutation in the BCR-ABL kinase domain. Tyrosine kinase inhibitor (TKI) imatinib targeting BCR-ABL is commonly used in chronic myelogenous leukemia treatment, but approximately 20%~30% of patients will experience resistance or relapse after treatment[35]. One mechanism of the resistance is due to a point mutation of T315I of the fusion tyrosine kinase protein[35-37]. The change of threonine 315 to isoleucine results in a loss of a hydrogen bond, which is necessary for the binding of imatinib to the ATP-binding site of BCR-ABL, leading to significantly reduced efficacy of the drug.
Drug resistance can also be acquired due to the dynamic changes of TME in the course of treatment. During malignancy progression and development of resistance, cross-talk exists between tumor cells and their microenvironment. Exosomes released by cancer and stromal cells are involved in the crosstalk. Researchers found that exosomes, which are released by cancer cells and carry certain miRNAs were used by cancer cells and tumor-associated macrophages (TAMs) in the TME to communicate with each other[38]. In cisplatin treated neuroblastoma (NBL) tumor, NBL cells secret exosomic miR-21 to induce TAMs to produce exosomic miR-155 to in turn silence the TERF1 gene in NBL cells. TERF1 protein is an inhibitor of telomerase, and decreased expression of TERF1 would result in increased telomerase activity and resistance to chemotherapy. Therefore, the exchange of exosomic miRNAs between tumor cells and stromal cells in TME can promote drug resistance.
Mechanisms of intrinsic and acquired resistance described above can co-exist during tumor progression and treatment. Mechanisms of acquired drug resistance can be totally different from the pre-existing intrinsic drug resistance. Alternatively, it can be a selective expansion of the intrinsic drug resistance.
The degree of intrinsic drug resistance pre-determines the sensitivity of given cancer cells to a specific drug. Genomic and other biochemical analyses should be performed before the drug treatment plan is designed to avoid potential pre-existing drug resistance. After acquired drug resistance is developed, therapeutic schemes need to be adjusted accordingly.
One of the objectives of drug treatment should be to effectively slow down or stop tumor growth without inducing acquired, or at least uncontrollable acquired drug resistance. The best drug treatment plan should take prevention or delay of acquired drug resistance into consideration.
Mechanisms of drug resistance
Although it is scientifically important to distinguish intrinsic and acquired resistances, the specific mechanisms of resistance are more clinically significant.
Increased efflux of drugs
Elevated efflux of anticancer agents, which leads to decreased intracellular drug accumulation, has been considered to be the major reason for chemotherapy resistance[7,39,40]. The resistance caused by the abnormally high rates of drug efflux could be either intrinsic or acquired, depending on if it exists prior to or develops after drug administration.
Transmembrane transporters responsible for the drug efflux are primarily from the ABC transporter superfamily. The human genome contains 48 ABC genes and they are classified into seven subfamilies (ABCA-ABCG)[41,42]. Among them, ABCB1, ABCC1 and ABCG2 are highly involved in the acquisition of multidrug resistance (MDR) to cancer chemotherapeutics.
ABCB1 (MDR1 or P-gp) is one of the most well-characterized ABC transporters. It is composed of two transmembrane domains that form a passage for substrates and two nucleotide-binding domains that bind and hydrolyze ATP. The binding and the subsequent hydrolysis of ATP is coupled with conformational changes in the transporter, leading to the pumping out of the transport substrates[43]. ABCB1 has multiple drug binding sites that can bind and pump a wide variety of substrates from the cell, such as etoposide, doxorubicin, paclitaxel and vinblastine[44-48]. High expression level of ABCB1 has been observed before chemotherapy in many different tumor types, including kidney, lung, liver, colon and rectum[49]. In contrast, initially low expression and then dramatic increased expression of ABCB1 post chemotherapy were observed in many hematological malignancies, such as AML and ALL[50-52].
ABCC1, or multidrug resistance-associated protein 1 (MRP1), similar to ABCB1, is also responsible for pumping out a wide variety of anticancer agents, such as vinca alkaloids, anthracyclines, epipodophyllotoxins, camptothecins, and methotrexate[53]. While ABCB1 transports amphipathic and lipid-soluble compounds, ABCC1 pumps organic anionic substrates such as compounds conjugated to glutathione, glucuronide, or sulfate[54-56]. Overexpression of ABCC1 has been shown to be associated with resistance in many cancer types including lung, breast and prostate cancers[53,57,58].
ABCG2, or breast cancer resistance protein, is the major drug efflux transporter in breast cancer associated resistance, as indicated by the name. ABCG2 is considered a marker of CSCs in some cancers, and responsible for the side-population effect. It can transport both positively- or negatively-charged drugs, ranging from chemotherapeutic drugs (mitoxantrone, bisantrene, epipodophyllotoxin, camptothecins, flavopiridol and anthracyclines) to several TKIs (imatinib and gefitinib)[48,59,60]. Besides breast cancer, ABCG2 overexpression was also found in many other cancer types including lung cancer and leukemia[60,61].
Other ABC transporters have also been studied for their substrates and functions in tumor resistance to anticancer drugs, providing additional explanations on the mechanisms of drug resistance[62]. For example, ABCC2 and ABCC3 can transport many chemotherapeutic drugs, including cisplatin, doxorubicin, and etoposide, and their overexpression results in multidrug resistance[62-65]. Mutations and overexpression of ABC transporters directly influence tumor sensitivity and drugs’ anticancer efficacy. An accurate and complete expression profile of ABC transporters in tumors is important for proper drug selection and better treatment outcomes.
Alteration of drug target
Compared to traditional chemotherapies which kill cancer cells by disrupting rapid cell proliferation and may affect normal dividing cells, targeted therapies can block the growth of cancer cells by inhibiting the activity of specific target proteins involved in tumor development, thus being more selective and effective to cancer cells and less harmful to normal cells. However, targeted therapy may also develop the problem of resistance, resulting from alteration of drug targets. The alteration of drug targets may be either a secondary mutation in the target protein or changes in expression levels due to epigenetic alterations.
For example, TKIs of the epidermal growth factor receptor (EGFR), such as erlotinib and gefitinib targeting NSCLC, have been reported to show high response rate at initial treatment[66,67]. However, almost 50% of the responsive patients would develop a T790M mutation on EGFR within one year, resulting in resistance to the first and second generations of TKIs[68-70]. The mutation from threonine to methionine led to a configuration change in EGFR and consequently enhanced ATP binding affinity and impaired binding of gefitinib/erlotinib for the kinase[70,71]. To overcome the resistance caused by T790M, third generation of TKIs, like osimertinib and rociletinib, has been developed and reported to show clinical efficacy with patients harboring T790M mutation[72,73]. However, resistance to third-generation inhibitors develops not long after their use, raising the need of developing fourth generation TKIs. One reported major mechanism of the new resistance is due to a mutation in EGFR known as C797S[74]. The loss of the cysteine residue, which is important for TKIs to target the ATP site, impairs the binding of the third generation TKIs to EGFR. Therefore, EAI045, a fourth generation TKI targeting both T790M and C797S, has been designed to bind an allosteric site located on EGFR, attempting to circumvent the mechanism patterns of resistance to the early generations of TKIs which all bind to the ATP sites[75,76]. The battle between generation of new genetic mutations and generation of new TKIs that restore drug sensitivity may become a new trend in the everlasting war against drug resistance.
The development and use of estrogen receptor inhibitors in breast cancer treatment provide another example for the resistance induced by alteration of the drug target. Tamoxifen (TAM) is commonly used for patients with ER-positive breast cancer, relying on its ability to compete with estrogen for the ligand binding site of ER. However, extended exposure of TAM often leads to drug resistance. Mechanisms of resistance vary in different cases, and mutations in the ER gene and decrease in ER expression level are among them[77,78]. Given the problems with TAM and the quest for alternative drugs, aromatase inhibitors (AIs) were developed, working by interfering with the last step of estrogen synthesis. Third-generation AIs are now being used as first-line therapy in postmenopausal women with hormone receptor positive breast cancer[79].
Enhanced DNA damage repair
Many chemotherapy drugs, like cisplatin and 5-fluorouracil (5-FU), kill cancer cells by inducing DNA damage. The DNA damage response (DDR) of affected cells to the anti-cancer drugs may result in reduced efficacy of the drugs by DNA lesion repairs, leading to drug resistance[80]. For example, genes involved in DNA repair, like FEN1, FANCG, RAD23B, were found to be upregulated in 5-FU resistant human colon cancer cell lines[81,82]. 5-FU treatment induced upregulation of p53-target genes on DNA damage response and repair. Success to repair the damages led to reduced cell cycle arrest and apoptosis in the resistant cell lines compared to parental cell lines[82].
Although deregulation of DDR may remit the resistance induced by DNA repair, it may also increase the risk of developing new mutations due to genomic instability, the accumulation of which may initiate a new round of carcinogenesis. Therefore, DNA damage response is a complex mechanism in cancer treatment and recurrence, and it requires thorough consideration when used as an anticancer therapeutic target.
Senescence escape
Cellular senescence refers to irreversible arrest of cell proliferation, largely leading to activation of tumor suppressive pathways mediated by p53 and/or p16INK4a[83]. Cellular senescence can be triggered by endogenous and exogenous stimuli, among which the three major stimuli include excessive mitogenic signaling produced by activated oncogenes, telomere shortening[84], and non-telomeric DNA damage caused by chemotherapeutic drugs. For example, Doxorubicin and Cisplatin used in chemotherapy by inducing cell death can also initiate senescence[85,86].
Escape from therapy-induced senescence (TIS) has been recognized as a mechanism for drug resistance and tumor recurrence/progression[87]. Cancer cells with TIS can gain stem-cell properties, which accounts for the escape from senescence and cancer relapse[88,89].
Epigenetic alterations
An emerging mechanism contributing to drug resistance is epigenetic alterations. Increasing evidence brought people’s attention to epigenetic modifications that also participate in the development of other mechanisms of resistance, including increased drug efflux, enhanced DNA repair, and impaired apoptosis.
Epigenetic modifications include DNA methylation, histone modification, chromatin remodeling, and non-coding RNA related alterations. For example, demethylation of DNA at the promoter region of an oncogene would upregulate the expression of the gene, resulting in drug resistance. A recent study demonstrated that a G-actin monomer binding protein thymosin β4 (Tβ4) was enriched through demethylation of DNA and active modification of histone H3 at the promoter region in a resistant hepatocellular carcinoma (HCC) cell line[95]. Overexpression of Tβ4 led to the acquisition of stem cell-like capacity in the HCC cell line and induced resistance to VEGFR inhibitor sorafenib in vivo[95].
Besides chromosomal modification, non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), also play an important role in drug resistance[96,97]. MiRNAs contain about 21-25 nucleotides (nt) while lncRNAs can range from 200 to more than 10,000 nt in length. MiRNAs are known to be important regulators of post-transcriptional gene expression by binding to their complementary mRNAs and mediating mRNA degradation and repression of protein synthesis. LncRNAs participate in gene expression regulation in different ways such as blocking the binding of transcription activators to key DNA sequences in genes or recruiting chromatin remodeling proteins. Both miRNA and lncRNA regulate expression of proteins related to cancer drug resistance. For example, lncRNA urothelial cancer-associated 1 (UCA1) was shown to be upregulated in cisplatin-resistant bladder cancer cells compared to sensitive cells[98]. Upregulation of UCA1 expression resulted in significantly increased mRNA and protein levels of wingless-type MMTV integration site family member 6 (Wnt6), promoting Wnt signaling and cell survival[98].
Tumor heterogeneity
Four levels of heterogeneity are present in tumors: genetic heterogeneity, cell type heterogeneity (cancer cells, stromal cells, immune cells, etc.), metabolic heterogeneity in oxygen/nutrient distribution, and temporal heterogeneity in dynamic tumor progression[99]. Tumor heterogeneity increases the complexity and difficulty of cancer treatment, making it almost impossible to kill all cancer cells using one single therapeutic. This problem led to the development of combinational therapies used in many cancer treatments such as FEC: 5-FU, epirubicin, cyclophosphamide for breast cancer. In this section, genetic heterogeneity will be the focus of discussion.
Ample evidence has shown that subpopulations of cancer cells with various genetic makeups co-exist in primary tumors like ovarian cancer[100], renal cell carcinoma[101], breast cancer[102], and chronic lymphocytic leukemia[103]. These clonal variants have different sensitivity to chemo or targeted drugs, so that initial treatment can kill only a portion of the tumor and those less sensitive cancer cells would survive. Once the resistant clones proliferate and grow, the tumor would come back with different cell composition portions that are insensitive to the initial chemotherapy. This genomic heterogeneity of the subpopulations evolves under drug treatment in a Darwinian selection manner, supported by evidence that subclonal compositions differ significantly at different stages of treatment[23,24,101,104]. Also, in heterogeneous populations of tumor cells, drug-resistant tumor cells can transfer microRNAs by exosome to drug-sensitive tumor cells and induce resistance to the latter[105].
The contribution of tumor heterogeneity to drug resistance is supported by studies reporting the loss of drug sensitivity to targeted drugs. The high specificity of targeted therapies, which is their advantage in increasing efficacy and reducing side effects, may become a limitation when dealing with tumor heterogeneity. Therefore, combined/cocktail therapies with more than one drug are required to overcome or delay the relapse of tumor.
Furthermore, heterogeneity among patients would result in differences in the patients’ response to the same treatment, urgently calling for the development of individualized therapies.
TME
Tumors are not bags of homogeneous cancer cells, but contain various types of cells and extracellular matrix (ECM) that work together to contribute to all aspects of the hallmarks of cancer[106,107]. Microenvironment of solid tumors includes ECM, immune and inflammatory cells, blood vessels, fibroblasts, and various nutrients and signaling molecules. They work in a coordinated manner to play vital roles in tumor growth and survival.
TME may contribute to intrinsic resistance to anticancer therapies. One of the TME factors is pH. In normal tissues and cells, extracellular pH is usually slightly higher than intracellular pH (pHe7.3-7.5 vs. pHi6.8-7.2)[108]. In contrast, cancer cells develop a so-called “reversed pH gradient”, with increased intracellular pH and decreased extracellular pH, through the proton pumping of proton transporters and the modulation of pH sensors[109,110]. The acidic (pH6.5-7.1) extracellular environment of cancer cells has been reported to be a contributor to the resistance to chemotherapeutics[111]. The reversed pH gradient impairs the distribution of weak base anticancer drugs, which is described as “ion trapping”, and enables cancer cells to evade apoptosis[112,113]. As an emerging hallmark of solid tumors, the low extracellular pH would be a potential target for cancer therapy. Therapeutic approaches attempting to reduce microenvironment acidity, such as proton pump inhibitors (PPIs), have been developed and shown good efficacy in shrinking tumor and sensitizing cancer cells to chemotherapy drugs. Lansoprazole, an example of PPI, has been reported to demonstrate synergistic effects when used in combination with paclitaxel in melanoma cells both in vitro and in vivo[114].
Changes in the composition of TME after treatment also contribute to the adaptation of cancer cells to chemo or targeted therapeutics, thereby reducing drug efficacy and inducing resistance. For example, TAMs play a role in the acquisition of resistance in response to anticancer therapies in glioblastoma multiforme (GBM), a severe type of brain tumor[115]. Macrophages secrete high levels of colony stimulating factor-1 (CSF-1) in GBM tumors, supporting cancer cell proliferation and survival[115,116]. Therefore, CSF-1 receptor (CSF-1R) has been targeted by small molecule inhibitors or antibodies in cancer treatment with promising in vivo effects[117-119]. However, more than 50% GBM patients suffer from recurrence, which is derived from elevated secretion of insulin-like growth factor-1 (IGF-1) from TAMs and IGF-1 induced elevation of phosphatidylinositol 3-kinase (PI3K) pathway signaling in GBM tumor cells[115]. Combining inhibition of CSF-1R with inhibition of IGF-1 receptor or PI3K signaling has been reported to extend overall survival in mouse models[115]. Thereby, combined therapies simultaneously targeting cancer cells and TME may produce much improved anticancer efficacy by reducing drug resistance.
Besides TME heterogeneity per se being one aspect of tumor heterogeneity, it also contributes to the enrichment of genetic heterogeneity. For example, due to variation and dynamic nature of vasculature inside tumors, fluctuating hypoxia is one characteristic of TME[120]. The frequent cycles of hypoxia and reoxygenation produce oxidative stress that could induce DNA damages in tumor cells, thus contributing to genetic instability that leads to accumulation of additional mutations and emergence of genetically divergent clonal subpopulations[121]. Moreover, as mentioned previously, cells in TME, like TAMs, interfere with the expression profiles of cancer cells by releasing miRNA-containing exosomes[38], thus contributing to tumor heterogeneity.
Therefore, TME plays very significant role in tumor progression and therapeutic resistance. Better understanding and targeting/manipulating TME and its interaction with tumor cells could substantially enhance therapy response and achieve better clinical outcomes.
EMT
EMT is a process during which epithelial cells lose their attachment to each other and gain the characteristics of mesenchymal stem cells. EMT has been known to be essential for the initiation of metastasis in tumors of epithelial origin, but it is not so clear about its role in other tumors such as sarcomas. Increasing evidence shows that EMT plays a critical role in chemotherapy resistance. Fischer et al.[122] reported that EMT promotes resistance to apoptosis induction activated by drug cyclophosphamide in an EMT lineage-tracing mouse system. However, the mechanisms of EMT-induced drug resistance are not fully understood although most recent studies suggest that EMT and CSC share some similarities and their involvements in drug resistance represent different manifestations of the same phenotype. One possible mechanism is that EMT cells share many similarities in signaling pathways with cancer stem cells (CSCs), such as Wnt, Notch and Hedgehog pathways[123]. Therefore, EMT enables tumor cells to gain resistance to anticancer drugs and evade drug induced cell death. For example, TGF-β is a well-studied key cytokine in EMT, the signaling pathways of which are correlated with gain of drug resistance[124,125]. Inhibition of TGF-β can reverse the process of EMT and remarkably increased the sensitivity of cancer cells to chemotherapies[126,127]. Wnt and Hedgehog pathways are also reported to be related with drug resistance[128,129].
Furthermore, accumulating evidence shows that the CSCs rely on the EMT program as a critical regulator when mediating drug resistance. The CSC state of carcinoma cells requires epigenetic changes resulted from activation of EMT. Understanding the mechanistic linkage between EMT, CSC and drug resistance would significantly contribute to anticancer therapeutics[130].
EMT inducing transcriptional factors (EMT-TFs) also play roles in promoting drug resistance. Overexpression of EMT-TFs like Twist, Snail, Slug, ZEB and FOXC2 are known to induce drug resistance[131-135]. One recent study reported that suppressing EMT by knocking out EMT transcription factors Twist1 or Snail1 enhanced sensitivity to gemcitabine and increased survival rate in pancreatic ductal adenocarcinoma-bearing mice treated with the drug[136]. Some of these EMT-TFs promote resistance by enhancing drug efflux by ABC transporters. Promoters of genes coding for ABC transporters were found to have EMT-TF binding sites[137]. Overexpression of Twist, ZEB1/2, Slug, and Snail enhances expression and activity of ABCB1, thus inducing drug resistance[138-140]. ABCG2, another ABC transporter closely linked to MDR, is known to be regulated by Snail, MSX2, SOX2 and ZEB1[141-144]. Other ABC transporters involved in MDR, such as ABCC1, ABCC2, ABCC4, and ABCC5, also have been shown under the regulation of EMT-TFs[145-147]. For example, overexpression of ABCC5 is correlated with FOXM1 in paclitaxel-resistant nasopharyngeal carcinoma cells. Depletion of either FOXM1 or ABCC5 decreases drug efflux and increases cell death induced by paclitaxel[148]. Knockdown of these EMT-TFs sensitize cancer cells to chemotherapeutic drugs by suppressing ABC transporters[142,143,148]. Targeting these EMT-TFs may inhibit metastasis and drug resistance at the same time.
Besides EMT-TFs, miRNAs are believed to be essential molecules that link EMT and ABC transporters[149]. MiRNAs are small endogenous RNA of 20-24 nucleotides, which have a complex network and can regulate expression of genes that are associated with EMT and are of ABC family[149]. Haenisch et al.[150] have summarized miRNA-mediated regulations of ABC transporters. MiRNA network regulates ABC transporters at different levels, with the majority of miRNAs acting at post-transcriptional levels via binding sites at the three prime untranslated region (3’-UTR) and some miRNAs acting at transcriptional levels by binding to the gene promoter region[151]. miRNAs can not only regulate expression of ABC transporters but also regulate EMT markers. For example, miR-200c is reported to both down-regulate multiple ABC transporters including ABCB1 and ABCG2[152,153], and negatively regulate EMT by directly targeting 3’-UTR regions of ZEB1 and ZEB2, and retain the epithelial phenotype[154]. Some miRNAs, such as miR-200c and miR-145, inhibit ABC transporters and suppress EMT[155]; other miRNAs like miR-27a positively regulate ABC transporters and promote EMT[156,157].
MiRNAs also regulate drug resistance by other mechanisms including regulating apoptosis and autophagy, controlling anti-cancer drug metabolism, modulating drug targets and DNA repair, and regulating GSH biosynthesis, which has been summarized by An et al.[158]. One recent study revealed that over expression of miR-134, miR-487b, and miR-655 promotes TGF-β induced EMT and drug resistance to gefitinib in NSCLC[159]. This miRNA cluster induces resistance to EGFR-TKI by directly inhibiting MAGI2, leading to reduced PTEN activity. Reduction of PTEN and upregulation of PI3K-Akt-pathway is associated with acquired EGFR-TKI resistance[160].
All these discussed drug resistance mechanisms are schematically shown in Figure 1.
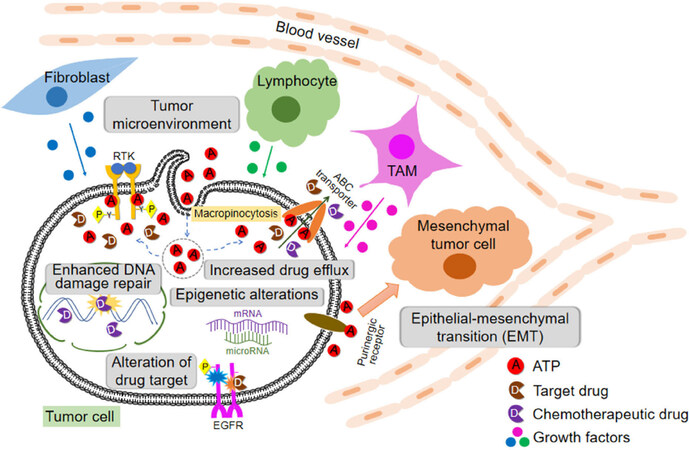
Figure 1. Cells, protein factors, and mechanisms involved in drug resistance in cancer described in this review including extracellular ATP-induced resistance. ABC: ATP binding cassette; RTK: receptor tyrosine kinases; EGFR: epidermal growth factor receptor; TAM: tumor-associated macrophage
Drug resistance can be intrinsic or extrinsic in nature. Drug resistance is induced at levels of DNA (enhanced DNA damage repair and epigenetics alternations), RNA (microRNA), and proteins [concentration and activity changes of receptor tyrosine kinases (RTK in general and EGFR in specific, and also ABC transporters)]. In addition, different stromal cells and stressed/lysed cancer cells in a tumor release ATP into intratumoral space, creating a high ATP concentration TME. Intratumoral extracellular ATP (eATP) works as a messenger outside of cancer cells through purinergic signaling cascade to induce EMT, which contributes to drug resistance. eATP is also internalized by cancer cells via macropinocytosis, leading to greatly elevated intracellular ATP (iATP) levels. The higher iATP levels in turn enhance the efflux activity of ABC transporters for pumping anticancer drugs out of cancer cells, increase competition between iATP with ATP analog anticancer drugs at the intracellular ATP binding domain of RTKs located on cancer cell plasma membrane and increase RTK phosphorylation. All these mechanisms work together dependent or independent of ATP to augment drug resistance by reducing intracellular drug concentration, increasing cell survival signaling and inducing EMT (cancer stem cell-like features). More studies are needed for the final validation of ATP-mediated mechanisms of drug resistance.
ATP and ATP-mediated drug resistance
Intracellular ATP promotes drug resistance
ATP, either intracellular or extracellular, plays significant roles in cancer cell growth, survival and resistance. It is known that the intracellular ATP level in cancer cells is higher than that in normal tissues of the same origin, likely due to the upregulated glucose transport and aerobic glycolysis in cancer cells, a process also called the Warburg effect[161-167]. Furthermore, even higher intracellular ATP levels were found in acquired resistant cancer cells lines compared to their parental cell lines[168,169]. In one study using colon cancer cells lines, intracellular ATP levels were found to have a two-fold increase in the chemo-resistant cell lines compared to those in their drug-sensitive parental cell lines[168]. The authors demonstrated that ATP levels played a pivotal role in multiple drug resistance by showing that artificial delivery of ATP into the drug-sensitive cells resulted in drug resistance while depleting intracellular ATP with a glycolysis inhibitor sensitized resistant cancer cells. A study from another group also showed that intracellular ATP contributed to drug resistance to cisplatin in ovarian adenocarcinoma cells[169]. In contrast to the more glycolysis-related ATP elevation in the previous study, the authors showed that the resistant cell line had an increased intracellular ATP level as a consequence of enhanced mitochondrial ATP synthesis that was induced during the development of resistance. They suggested that the resistant cell line may have increased their energy storage for the purpose of protecting the cells from xenobiotics and environmental stress. These studies showed the metabolic- (energetic-) role of intracellular ATP in acquired resistance to chemotherapeutic drugs. Elevated intracellular ATP levels is likely to be a necessary condition for cancer cells particularly resistant cancer cells.
For all the reasons mentioned above, inhibitors targeting glycolysis related proteins (GAPDH and LDH) like 3-bromopyruvate, oxamate and FX11 that significantly deplete intracellular ATP levels might sensitize cancer cells to anticancer drugs[170-174].
Extracellular ATP induces drug resistance
eATP levels of various cancer types have been reported to be 103 to 104 times higher than those in their corresponding normal tissues[175-178]. The functions of this high eATP have been under investigation. In our recent study, eight anticancer drugs, including both targeted and chemotherapeutic drugs, were tested in five cancer cell lines of five different organ origins, and extracellular ATP was found to promote intracellular ATP increase and cancer cell survival in most cases[179]. When studying ATP promoted drug resistance to sunitinib in NSCLC A549 cells, we found that extracellular ATP can be internalized by cancer cells through macropinocytosis and other endocytic mechanisms, resulting in substantially elevated intracellular ATP levels from 150 to 200% of the original intracellular ATP concentrations[161,179,180]. Thus, macropinocytosis, and other endocytosis-mediated extracellular ATP internalization, and the resulting intracellular ATP level elevation is responsible, at least in part, for the observed drug resistance.
One of the possible drug resistance mechanisms induced by the internalization of extracellular ATP is that more abundant intracellular ATP molecules compete with tyrosine kinase inhibitors which are ATP competitors, for the ATP binding site located on RTKs, leading to increased phosphorylation and activation of downstream signaling pathways. Meanwhile, the internalized ATP molecules also enhance efflux of TKIs and chemo drugs by ABC transporters, resulting in decreased drug accumulation and increased cell survival. The reduced intracellular drug concentrations and elevated intracellular ATP levels further enhance the ATP binding and reduce TKI binding on RTKs, leading to even more RTK-mediated signaling and drug resistance. These were all observed in the study[179], which suggested the dual roles of ATP as both an energy molecule facilitating drug efflux and signal-transduction (phosphorylation) molecule activating cell survival signaling pathways.
In the five cell lines studied, increased intracellular ATP was found to correlate with drug resistance status when the ABC transporters expressed by the cell line matched those required for the efflux of a given drug[179]. Extracellular ATP was also shown to alter expression levels of ABC transporters, indicating the profound effects of ATP on modulating ABC transporter activities at both transporting activity rate and transporter expression levels to potentially efflux anticancer drugs and enhance drug resistance[179]. Prescreening of tumors based on their ability of ATP internalization (macropinocytosis) and expression of specific set of ABC transporters will provide valuable information for the selection of proper anticancer drugs and prediction of patient’s response to therapeutics, therefore reducing drug resistance and enhancing the efficacy of drug treatment. Our lab’s findings in extracellular ATP’s roles in cancer cell growth, survival and drug resistance were reviewed in a 2018 Nature Reviews Cancer paper[181].
The drug resistance observed here is intrinsic in nature and could last during later treatments. Reducing intratumoral extracellular ATP concentration and/or blocking ATP internalization may increase drug efficacy at both initial and later stages of cancer treatment.
ATP might also contribute to cancer drug resistance from outside of the cell through purinergic receptor signaling. Purinergic signaling has been known to promote cell growth and proliferation[182,183]. Purinergic receptors such as members of the P2X and P2Y families were reported to be involved in cancer drug resistance[184-186]. In one recent study, researchers demonstrated that ATP promoted resistance to chemotherapeutic drugs in colorectal cancer cells through P2Y-mediated upregulation of MRP2 and concurrent drug pumping[187]. Another study showed that P2X7 receptor, when activated by ATP, exhibited anti-apoptotic activity in methoxyestradiol-treated melanoma cells[185]. Extracellular ATP has also been found to upregulate expression of glucose transporter 1, possibly via the P2X7 induced PI3K-AKT pathway and hypoxia-inducible factor 1α-dependent signaling[181]. These changes are also likely to increase cancer cell survival and drug resistance. Therefore, reducing the extracellular ATP concentration could increase drug efficacy by reducing specific purinergic receptor signaling in addition to reducing ATP internalization. An ATPase, apyrase, was shown to reduce the growth of glioblastoma when injected in a rat glioma model[187], suggesting the feasibility of this strategy.
Extracellular ATP also plays significant role in immunoregulation in TME and potentially affects therapeutic results. ATP can be secreted into extracellular environment by cancer cells undergoing autophagy or apoptosis induced by chemotherapy or radiotherapy, generating a chemotactic gradient around dying cells and recruiting myeloid cells through purinergic signaling[177,188-191]. Defects in the molecular machinery for autophagy in cancer cells or purinergic receptors in immune cells lead to poor response to stimuli that would cause immunogenically induced cell death[189,192]. Furthermore, immune cells in TME can release and use extracellular ATP, potentially creating an ATP-rich and tumor-friendly environment. For example, lymphocytes are known to release large amounts of ATP as a signaling molecule into the extracellular space when stimulated, serving as a messenger in cellular interactions of T lymphocytes[193]. Stimulated monocytes can release ATP as an autocrine signal molecule[194]. ATP released from these immune cells as well as dying or stressed cells can be sequentially converted to AMP and adenosine by cell surface enzymes CD39 (ecto-nucleoside triphosphate diphosphohydrolase 1, E-NTPDase1) and CD73 (ecto-5’-nucleotidase, Ecto5’NTase)[195,196]. Adenosine mediated signaling would lead to the establishment of an immunosuppressive environment[197,198]. Combined administration of an inhibitor of extracellular ATPase and a synthetic TLR4 ligand was reported to restore the infiltration of necroptosis-deficient tumors by APCs and CD8+ T cells, and re-establish normal sensitivity to mitoxantrone-based chemotherapy[199]. These findings suggest that extracellular ATP is essential to anti-tumor immune response while its hydrolysis products contribute to immunosuppression. Therefore, the anti-resistance strategy through ATP degradation by ATPase is perhaps not appropriate to use together with therapies designed to boost immune system. However, this may be an extracellular ATP concentration-dependent phenomenon. There will be a concentration window for extracellular ATP where extracellular ATP concentrations are sufficiently low not to generate significant drug resistance but sufficiently high to maintain anti-tumor immune responses. Significantly more studies need to be done before we fully understand the different functions and different concentrations of extracellular ATP on cancer and immune cells in tumors.
Conclusion: strategies for fighting against drug resistance
Due to the high heterogeneity among tumors growing in patients and high complexity of evolution of tumor progression, identifying the best strategy to overcome drug resistance will be very challenging. On the other hand, with the development of high throughput cancer genomics, cancer proteomics, and cancer metabolomics analyses, it is now possible to identify driver genes and major components that contribute the drug resistance the most at any specific stage of tumorigenesis in a patient. Due to the nature of individual differences in mutations in multiple and different cancer-causing genes, combinational and personalized therapies are required. Combinational therapies are strongly preferred since tumors are almost always multi-clonal and genetically heterogeneous. Therapeutic strategies using single drugs are most likely to lead to eventual treatment failure due to drug resistance as the treatment kills sensitive cancer cells but leaves resistant cancer cells to survive and proliferate. In comparison, combinational therapy using two or more drugs is likely to target multiple driver genes simultaneously, not only inhibiting more clones in a tumor but also making new cancer mutations resistant to multi-drug treatment much more difficult to be selected and grow up.
Current strategies to deal with drug resistance depend on continuous monitoring of patients and treatment with a cocktail of chemotherapeutic/target drugs, each targeting one or more proteins encoded by driver genes responsible for drug resistance pathways operating in cancer patients. Like recent trends of successful target drug therapy, simultaneous multi-targeting will be more effective in combating drug resistance, thereby enhancing anticancer efficacy of therapies and prolong patients’ lives. However, the outcomes of therapies inevitably depend on the composition/unique resistance profile of tumors and the toxicity tolerance of patients, so that the therapeutic results are hard to predict. Since tumor cells could always develop alternative mechanisms to circumvent current therapy, fighting against drug resistance seems to be an endless game.
Conventionally, cancers were treated with chemo or targeted drugs at the highest dosage that the patients can tolerate. In recent years, it was realized that such a treatment strategy may lead to drug resistance more rapidly, since such treatment puts constant pressure on tumors to select those cancer cells that are strongly resistant to the drugs. New treatment strategies of “on and off” or “high dose followed by low dose” resulted in longer survival and delayed drug resistance, because this intermittent or adaptive dosing may interrupt the growth of drug-dependent resistant cells and allow the competition of sensitive and resistant cells[200]. In one study, researchers found that melanoma cells that acquired resistance to combined BRAF- and MEK- targeted therapy displayed robust drug addiction and were exquisitely sensitive to acute drug withdrawal[201]. This drug sensitivity is also detected in other cancer types such as lymphoma cells treated with an ALK kinase inhibitor that became dependent on it, suggesting that intermittent dosing may prolong control of ALK+ tumors[202]. These findings encouraged testing of this pulsatile dosing regimen in clinical trials (ClinicalTrials.gov Identifiers: NCT02196181).
However, these new treatment strategies produced inconsistent results in that improved survival was achieved in some cases but no change or even worse outcomes occurred in others. These indicate that the new strategies only work in some specific cases but cannot be used as a general strategy. We still need to know more about drug-cancer interactions at a personalized level before we can develop treatment plans that fit individual needs.
One possible strategy of circumventing resistance is to block energy supply of tumor cells. Tumor might bypass any mechanism, but could not evade the need for energy to support their growth, proliferation and other activities such as drug resistance and cell migration. Combinational therapy with an addition of blockage of energy may increase efficacy of therapeutic reagents. Although normal cells are more versatile and flexible in using various energy supply molecules, cancer cells appear to be more rigid in using energy sources. For example, it is well known from PET-scans that aggressive tumors with poor prognosis are almost always glucose/energy metabolism active tumors[203,204]. Some cancer cells strongly prefer to use glucose as the energy and carbon sources. These cancer cells are “addicted” to glucose and are more sensitive to glucose concentration changes than normal cells, dying significantly faster than normal cells under glucose deprivation[99,166,167,205]. In those cancer cases, using a glucose transport inhibitor or a glycolysis inhibitor in combination with another target drug may be particularly effective in inducing cancer cell death.
New cancer research indicates that TME plays very important roles in tumorigenesis and drug resistance. Any new therapies that expect to significantly improve the therapeutic outcomes must consider TME - tumor interactions. Intratumoral extracellular ATP has emerged as one of TME molecules that exert profound impacts on tumor cells regarding cell growth, survival, drug resistance and even metastasis[161,179,181]. Although different molecules can be used as the energy source for synthesizing ATP, the ultimate energy molecule used in all cells is ATP. Cancer cells appear to have to have higher ATP levels for survival and drug resistance. This difference between cancer and normal cells can be explored to combat cancer growth. If a method, such as tumor-selective targeting, can be developed and used to deliver ATP synthesis-inhibitor or extracellular ATP degrader resulting in reduction of ATP internalization inside tumors, tumors will be deficient in ATP, forcing them to stop growing or even undergo cell death. Extracellular ATP degradation or the inhibition of extracellular ATP internalization can be considered for combinational therapy with chemotherapeutics to enhance the anticancer efficacy of TKIs and chemo drugs. It is also conceivable that alteration of extracellular ATP levels may further enhance cancer immunotherapy, as immune cells in tumors are sensitive to ATP levels[188-193].
Finally, the earlier the tumor is detected, the lower the heterogeneity of tumor cells would be, and the less drug resistance and the more successful the therapy should be. Early prevention and early detection should be considered with at least equal importance as cancer treatment at advanced stages.
Declarations
AcknowledgmentsWe thank David Cottrill for proofreading the manuscript.
Authors’ contributionsWrote the manuscript and drew the figure: Wang X
Wrote the manuscript and provided technical support: Zhang H
Finalized manuscript and figure: Chen X
Data source and availabilityCorresponding author may be contacted for any data inquiries.
Financial support and sponsorshipThis work was partially supported by John Kopchick MCB awards to Wang X and Chen X.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. National Center for Health Statistics. Health, United States, 2015: with special feature on racial and ethnic health disparities. Hyattsville, MD: 2016.
2. American Cancer Society. Cancer Facts & Figures 2017. Atlanta: American Cancer Society; 2017.
3. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6:1769-92.
4. Rueff J, Rodrigues AS. Cancer drug resistance: a brief overview from a genetic viewpoint. Methods Mol Biol 2016:1395:1-18.
5. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26.
6. Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol 2012;2:120066.
7. Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int 2015;15:71.
8. Urruticoechea A, Alemany R, Balart J, Villanueva A, Viñals F, et al. Recent advances in cancer therapy: an overview. Curr Pharm Des 2010;16:3-10.
9. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci 2012;9:193-9.
10. Damin DC, Lazzaron AR. Evolving treatment strategies for colorectal cancer: a critical review of current therapeutic options. World J Gastroenterol 2014;20:877.
11. Lumachi F, Luisetto G, Basso SMM, Basso U, Brunello A, et al. Endocrine therapy of breast cancer. Curr Med Chem 2011;18:513-22.
12. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016;13:273-90.
13. Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl lung cancer Res 2014;3:242-9.
14. Castells M, Thibault B, Delord JP, Couderc B. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci 2012;13:9545-71.
15. O’Connor D, Sibson K, Caswell M, Connor P, Cummins M, et al. Early UK experience in the use of clofarabine in the treatment of relapsed and refractory paediatric acute lymphoblastic leukaemia. Br J Haematol 2011;154:482-5.
16. Lippert T, Ruoff HJ, Volm M. Intrinsic and acquired drug resistance in malignant tumors. Arzneimittelforschung 2008;58:261-4.
17. Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol 2014;8:1132-9.
18. Huang D, Duan H, Huang H, Tong X, Han Y, et al. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep 2016;6:20502.
19. Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, et al. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009;27:2059-68.
20. Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006;66:944-50.
21. Sayan AE, Griffiths TR, Pal R, Browne GJ, Ruddick A, et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci U S A 2009;106:14884-9.
22. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013;501:338-45.
24. Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol 2012;13:e178-85.
27. Kuczynski EA, Sargent DJ, Grothey A, Kerbel RS. Drug rechallenge and treatment beyond progression--implications for drug resistance. Nat Rev Clin Oncol 2013;10:571-87.
28. Frank NY, Schatton T, Frank MH, Geles K, Grindley J, et al. The therapeutic promise of the cancer stem cell concept. J Clin Invest 2010;120:41-50.
29. Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009;457:51-6.
30. Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 2006;13:1238-41.
31. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313-23.
32. Gillet JP, Calcagno AM, Varma S, Davidson B, Bunkholt Elstrand M, et al. Multidrug resistance-linked gene signature predicts overall survival of patients with primary ovarian serous carcinoma. Clin Cancer Res 2012;18:3197-206.
33. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013;2013:972913.
34. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012;481:506-10.
35. Quintás-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control 2009;16:122-31.
36. Jabbour EJ, Cortes JE, Kantarjian HM. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: a clinical perspective and emerging treatment options. Clin Lymphoma Myeloma Leuk 2013;13:515-29.
37. Kimura S, Ando T, Kojima K. BCR-ABL point mutations and TKI treatment in CML patients. J Hematol Transfus 2014;2.
38. Challagundla KB, Wise PM, Neviani P, Chava H, Murtadha M, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst 2015;107:djv135.
39. Vadlapatla R, Vadlapudi A, Pal D, Mitra A. Mechanisms of drug resistance in cancer chemotherapy: coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr Pharm Des 2013;19:7126-40.
40. Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett 2014;347:159-66.
41. Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics 2009;3:281-90.
43. Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1). J Bioenerg Biomembr 2007;39:481-7.
44. Lagas JS, Fan L, Wagenaar E, Vlaming ML, van Tellingen O, et al. P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 determine the pharmacokinetics of etoposide. Clin Cancer Res 2010;16:130-40.
45. Lal S, Wong ZW, Sandanaraj E, Xiang X, Ang PC, et al. Influence of ABCB1 and ABCG2 polymorphisms on doxorubicin disposition in Asian breast cancer patients. Cancer Sci 2008;99:816-23.
46. Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 2016;115:431-41.
47. Satake K, Tsukamoto M, Mitani Y, Regasini LO, da Silva Bolzani V, et al. Human ABCB1 confers cells resistance to cytotoxic guanidine alkaloids from Pterogyne nitens. Biomed Mater Eng 2015;25:249-56.
48. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105-27.
49. Fojo AT, Ueda K, Slamon DJ, Poplack DG, Gottesman MM, et al. Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A 1987;84:265-9.
50. Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, et al. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat 2012;15:62-9.
51. Baudis M, Prima V, Tung YH, Hunger SP. ABCB1 over-expression and drug-efflux in acute lymphoblastic leukemia cell lines with t(17;19) and E2A-HLF expression. Pediatr Blood Cancer 2006;47:757-64.
52. Peng XX, Tiwari AK, Wu HC, Chen ZS. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin J Cancer 2012;31:110-8.
53. Yin J, Zhang J. Multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphism: from discovery to clinical application. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011;36:927-38.
54. Müller M, Meijer C, Zaman GJ, Borst P, Scheper RJ, Mulder NH, et al. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc Natl Acad Sci U S A 1994;91:13033-7.
55. Cole SPC, Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci 2006;27:438-46.
56. Cole SP. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem 2014;289:30880-8.
57. Munoz M, Henderson M, Haber M, Norris M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life 2007;59:752-7.
58. Cho S, Lu M, He X, Ee PL, Bhat U, et al. Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proc Natl Acad Sci U S A 2011;108:20778-83.
59. Stacy AE, Jansson PJ, Richardson DR. Molecular pharmacology of ABCG2 and Its role in chemoresistance. Mol Pharmacol 2013;84:655-69.
60. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport--an update. AAPS J 2015;17:65-82.
61. Horsey AJ, Cox MH, Sarwat S, Kerr ID. The multidrug transporter ABCG2: still more questions than answers Multidrug transporters in biology. Biochem Soc Trans 2016;44:824-30.
62. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol 2016;43:723-37.
63. Folmer Y, Schneider M, Blum HE, Hafkemeyer P. Reversal of drug resistance of hepatocellular carcinoma cells by adenoviral delivery of anti-ABCC2 antisense constructs. Cancer Gene Ther 2007;14:875-84.
64. Balaji SA, Udupa N, Chamallamudi MR, Gupta V, Rangarajan A. Role of the drug transporter ABCC3 in breast cancer chemoresistance. PLoS One 2016;11:e0155013.
65. Zhao Y, Lu H, Yan A, Yang Y, Meng Q, et al. ABCC3 as a marker for multidrug resistance in non-small cell lung cancer. Sci Rep 2013;3:3120.
66. Wang Y, Schmid-Bindert G, Zhou C. Erlotinib in the treatment of advanced non-small cell lung cancer: an update for clinicians. Ther Adv Med Oncol 2012;4:19-29.
67. Gridelli C, De Marinis F, Di Maio M, Cortinovis D, Cappuzzo F, et al. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: review of the evidence. Lung Cancer 2011;71:249-57.
68. Tang J, Salama R, Gadgeel SM, Sarkar FH, Ahmad A. Erlotinib resistance in lung cancer: current progress and future perspectives. Front Pharmacol 2013;4:15.
69. Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 2005;37:1315-6.
70. Ma C, Wei S, Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis 2011;3:10-8.
71. Chan BA, Hughes BGM. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl lung cancer Res 2015;4:36-54.
72. Liao BC, Lin CC, Yang JC. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol 2015;27:94-101.
73. Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol 2016;9:34.
74. Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 2016;9:59.
75. Jia Y, Yun CH, Park E, Ercan D, Manuia M, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016;534:129-32.
76. Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett 2017;385:51-4.
77. MacGregor Schafer J, Liu H, Bentrem DJ, Zapf JW, Jordan VC. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Res 2000;60:5097-105.
78. Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol 2006;20:3120-32.
79. Avvaru SP, Noolvi MN, Aminbhavi TM, Chkraborty S, Dash A, et al. Aromatase inhibitors evolution as potential class of drugs in the treatment of postmenopausal breast cancer women. Mini Rev Med Chem 2018;18:609-21.
80. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008;8:193-204.
81. de Angelis PM, Fjell B, Kravik KL, Haug T, Tunheim SH, et al. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol 2004;24:1279-88.
82. De Angelis PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer 2006;5:20.
84. Gordon RR, Nelson PS. Cellular senescence and cancer chemotherapy resistance. Drug Resist Updat 2012;15:123-31.
85. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002;109:335-46.
86. Demaria M, O’Leary MN, Chang J, Shao L, Liu S, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 2017;7:165-76.
87. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005;65:2795-803.
88. Sabisz M, Skladanowski A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: implications for drug resistance and in vitro drug screening models. Cell Cycle Georget Tex 2009;8:3208-17.
89. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553:96-100.
90. Lantermann AB, McCutcheon KJ, Sharma SV. The role of epigenetics in drug resistance in cancer. Epigenetic Diagnosis Ther 2016;1:106-19.
91. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014;54:716-27.
92. Wilting RH, Dannenberg JH. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat 2012;15:21-38.
93. Hoey T. Drug resistance, epigenetics, and tumor cell heterogeneity. Sci Transl Med 2010;2:28ps19.
94. Zeller C, Brown R. Therapeutic modulation of epigenetic drivers of drug resistance in ovarian cancer. Ther Adv Med Oncol 2010;2:319-29.
95. Ohata Y, Shimada S, Akiyama Y, Mogushi K, Nakao K, et al. Acquired resistance with epigenetic alterations under long-term anti-angiogenic therapy for hepatocellular carcinoma. Mol Cancer Ther 2017;16:1155-65.
96. Chen QN, Wei CC, Wang ZX, Sun M. Long non-coding RNAs in anti-cancer drug resistance. Oncotarget 2017;8:1925-36.
97. Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget 2014;5:8027-38.
98. Fan Y, Shen B, Tan M, Mu X, Qin Y, et al. Long non-coding RNA UCA1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J 2014;281:1750-8.
99. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med 2015;79:253-63.
100. Bashashati A, Ha G, Tone A, Ding J, Prentice LM, et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol 2013;231:21-34.
101. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883-92.
102. Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, et al. Inferring tumor progression from genomic heterogeneity. Genome Res 2010;20:68-80.
103. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013;152:714-26.
104. Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol 2014;8:1095-111.
105. Chen W, Liu X, Lv M, Chen L, Zhao JH, et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One 2014;9:e95240.
107. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309-22.
108. Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 2010;11:50-61.
109. Swietach P, Vaughan-Jones RD, Harris AL, Hulikova A. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc Lond B Biol Sci 2014;369:20130099.
110. Sharma M, Astekar M, Soi S, Manjunatha B, Shetty D, et al. pH gradient reversal: an emerging hallmark of cancers. Recent Pat Anticancer Drug Discov 2015;10:244-58.
111. Taylor S, Spugnini EP, Assaraf YG, Azzarito T, Rauch C, et al. Microenvironment acidity as a major determinant of tumor chemoresistance: proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat 2015;23:69-78.
112. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011;11:671-7.
113. Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm 2011;8:2032-8.
114. Azzarito T, Venturi G, Cesolini A, Fais S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett 2015;356:697-703.
115. Quail DF, Bowman RL, Akkari L, Akkari L, Quick ML, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016;352:aad3018.
116. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013;23:277-86.
117. Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med 2015;373:428-37.
118. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264-72.
119. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014;25:846-59.
120. Reynolds TY, Rockwell S, Glazer PM. Genetic instability induced by the tumor microenvironment. Cancer Res 1996;56:5754-7.
121. Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis. Mutat Res Mol Mech Mutagen 2005;569:75-85.
122. Fischer KR, Durrans A, Lee S, Sheng J, Li F, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527:472-6.
123. Du B, Shim J. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 2016;21:965.
124. Brunen D, Willems SM, Kellner U, Midgley R, Simon I, et al. TGF-β: an emerging player in drug resistance. Cell Cycle 2013;12:2960-8.
125. Oshimori N, Oristian D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015;160:963-76.
126. Li J, Liu H, Yu J, Yu H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor β signaling in HCT116 colon cancer cells. Mol Med Rep 2015;12:192-8.
127. Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 2013;123:1348-58.
128. Wu Y, Ginther C, Kim J, Mosher N, Chung S, et al. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol cancer Res MCR 2012;10:1597-606.
129. Della Corte CM, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin Cancer Res 2015;21:4686-97.
130. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611-29.
131. Deng JJ, Zhang W, Xu XM, Zhang F, Tao WP, et al. Twist mediates an aggressive phenotype in human colorectal cancer cells. Int J Oncol 2016;48:1117-24.
132. Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012;12:91.
133. Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 2013;5:1196-212.
134. Lazarova D, Bordonaro M. ZEB1 mediates drug resistance and EMT in p300-deficient CRC. J Cancer 2017;8:1453-9.
135. Zhou Z, Zhang L, Xie B, Wang X, Yang X, et al. FOXC2 promotes chemoresistance in nasopharyngeal carcinomas via induction of epithelial mesenchymal transition. Cancer Lett 2015;363:137-45.
136. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525-30.
137. Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2011;2:e179.
138. Zhu K, Chen L, Han X, Wang J, Wang J. Short hairpin RNA targeting Twist1 suppresses cell proliferation and improves chemosensitivity to cisplatin in HeLa human cervical cancer cells. Oncol Rep 2012;27:1027-34.
139. Tsou SH, Chen TM, Hsiao HT, Chen YH. A critical dose of doxorubicin is required to alter the gene expression profiles in MCF-7 cells acquiring multidrug resistance. PLoS One 2015;10:e0116747.
140. Li W, Liu C, Tang Y, Li H, Zhou F, et al. Overexpression of Snail accelerates adriamycin induction of multidrug resistance in breast cancer cells. Asian Pac J Cancer Prev 2011;12:2575-80.
141. Chen WJ, Wang H, Tang Y, Liu CL, Li HL, et al. Multidrug resistance in breast cancer cells during epithelial-mesenchymal transition is modulated by breast cancer resistant protein. Chin J Cancer 2010;29:151-7.
142. Hamada S, Satoh K, Hirota M, Kanno A, Umino J, et al. The homeobox gene MSX2 determines chemosensitivity of pancreatic cancer cells via the regulation of transporter gene ABCG2. J Cell Physiol 2012;227:729-38.
143. Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, et al. SOX2 regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. Br J Cancer 2014;111:2122-30.
144. Mato E, González C, Moral A, Pérez JI, Bell O, et al. ABCG2/BCRP gene expression is related to epithelial-mesenchymal transition inducer genes in a papillary thyroid carcinoma cell line (TPC-1). J Mol Endocrinol 2014;52:289-300.
145. Sun L, Ke J, He Z, Chen Z, Huang Q, et al. HES1 promotes colorectal cancer cell resistance to 5-Fu by inducing of EMT and ABC transporter proteins. J Cancer 2017;8:2802-8.
146. Uchibori K, Kasamatsu A, Sunaga M, Yokota S, Sakurada T, et al. Establishment and characterization of two 5-fluorouracil-resistant hepatocellular carcinoma cell lines. Int J Oncol 2012;40:1005-10.
147. Bhuvanalakshmi G, Arfuso F, Millward M, Dharmarajan A, Warrier S. Secreted frizzled-related protein 4 inhibits glioma stem-like cells by reversing epithelial to mesenchymal transition, inducing apoptosis and decreasing cancer stem cell properties. PLoS One 2015;10:e0127517.
148. Hou Y, Zhu Q, Li Z, Peng Y, Yu X, et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis 2017;8:e2659.
149. Jiang ZS, Sun YZ, Wang SM, Ruan JS. Epithelial-mesenchymal transition: potential regulator of ABC transporters in tumor progression. J Cancer 2017;8:2319-27.
150. Haenisch S, Werk AN, Cascorbi I. MicroRNAs and their relevance to ABC transporters. Br J Clin Pharmacol 2014;77:587-96.
151. Lopes-Rodrigues V, Seca H, Sousa D, Sousa E, Lima RT, et al. The network of P-glycoprotein and microRNAs interactions. Int J Cancer 2014;135:253-63.
152. Liu S, Tetzlaff MT, Cui R, Xu X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of Bmi-1. Am J Pathol 2012;181:1823-35.
153. Sui H, Cai GX, Pan SF, Deng WL, Wang YW, et al. miR200c attenuates P-gp-mediated MDR and metastasis by targeting JNK2/c-Jun signaling pathway in colorectal cancer. Mol Cancer Ther 2014;13:3137-51.
154. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008;22:894-907.
155. Hu J, Qiu M, Jiang F, Zhang S, Yang X, et al. MiR-145 regulates cancer stem-like properties and epithelial-to-mesenchymal transition in lung adenocarcinoma-initiating cells. Tumour Biol 2014;35:8953-61.
156. Zhang Z, Liu S, Shi R, Zhao G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet 2011;204:486-91.
157. Zhang H, Li M, Han Y, Hong L, Gong T, et al. Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Dig Dis Sci 2010;55:2545-51.
158. An X, Sarmiento C, Tan T, Zhu H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm Sin B 2017;7:38-51.
159. Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, et al. MiR-134/487b/655 cluster regulates TGF-β-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther 2014;13:444-53.
160. Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, et al. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br J Cancer 2005;92:1711-9.
161. Qian Y, Wang X, Liu Y, Li Y, Colvin RA, et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett 2014;351:242-51.
162. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001;292:504-7.
163. Pecqueur C, Oliver L, Oizel K, Lalier L, Vallette FM. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol 2013;2013:805975.
164. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 2012;11:1672-82.
166. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011;11:325-37.
167. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029-33.
168. Zhou Y, Tozzi F, Chen J, Fan F, Xia L, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res 2012;72:304-14.
169. Schneider V, Krieger ML, Bendas G, Jaehde U, Kalayda GV. Contribution of intracellular ATP to cisplatin resistance of tumor cells. J Biol Inorg Chem 2013;18:165-74.
170. Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, et al. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun 2004;324:269-75.
171. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes Taxol-resistant cancer cells to Taxol. Mol Cancer 2010;9:33.
172. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci 2010;107:2037-42.
173. Qian Y, Wang X, Chen X. Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics. World J Transl Med 2014;3:37.
174. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 2013;12:152.
175. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, et al. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 2008;3:e2599.
176. Falzoni S, Donvito G, Di Virgilio F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013;3:20120101.
177. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011;334:1573-7.
178. Wilhelm K, Ganesan J, Müller T, Dürr C, Grimm M, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 2010;16:1434-8.
179. Wang X, Li Y, Qian Y, Cao Y, Shriwas P, et al. Extracellular ATP, as an energy and phosphorylating molecule, induces different types of drug resistances in cancer cells through ATP internalization and intracellular ATP level increase. Oncotarget 2017;8:87860-77.
180. Qian Y, Wang X, Li Y, Cao Y, Chen X. Extracellular ATP a new player in cancer metabolism: NSCLC cells internalize ATP in vitro and in vivo using multiple endocytic mechanisms. Mol Cancer Res 2016;14:1087-96.
181. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer 2018;18:601-18.
182. Di Virgilio F, Ferrari D, Adinolfi E. P2X(7): a growth-promoting receptor-implications for cancer. Purinergic Signal 2009;5:251-6.
183. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal 2013;9:491-540.
184. Vinette V, Placet M, Arguin G, Gendron FP. Multidrug resistance-associated protein 2 expression is upregulated by adenosine 5’-triphosphate in colorectal cancer cells and enhances their survival to chemotherapeutic drugs. PLoS One 2015;10:e0136080.
185. Deli T, Varga N, Ádám A, Kenessey I, Rásó E, et al. Functional genomics of calcium channels in human melanoma cells. Int J Cancer 2007;121:55-65.
186. Limami Y, Pinon A, Leger DY, Pinault E, Delage C, et al. The P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie 2012;94:1754-63.
187. Morrone FB, Oliveira DL, Gamermann P, Stella J, Wofchuk S, et al. In vivo glioblastoma growth is reduced by apyrase activity in a rat glioma model. BMC Cancer 2006;6:226.
188. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009;461:282-6.
189. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97-111.
190. Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ 2014;21:92-9.
191. Galluzzi L, Kepp O, Kroemer G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J 2012;31:1055-7.
192. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med 2009;15:1170-8.
193. Filippini A, Taffs RE, Sitkovsky MV. Extracellular ATP in T-lymphocyte activation: possible role in effector functions. Proc Natl Acad Sci U S A 1990;87:8267-71.
194. Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, et al. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1 and IL-18 secretion in an autocrine way. Proc Natl Acad Sci 2008;105:8067-72.
195. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med 2013;19:355-67.
196. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol 2013;171:1-7.
197. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010;29:5346-58.
198. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 2016;7:109.
199. Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016;5:e1149673.
201. Moriceau G, Hugo W, Hong A, Shi H, Kong X, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015;27:240-56.
202. Amin AD, Rajan SS, Liang WS, Pongtornpipat P, Groysman MJ, et al. Evidence suggesting that discontinuous dosing of ALK kinase inhibitors may prolong control of ALK+ tumors. Cancer Res 2015;75:2916.
203. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Publ Gr 2016;16:635-49.
204. Zhu A, Lee D, Shim H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol 2011;38:55-69.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019;2:141-60. http://dx.doi.org/10.20517/cdr.2019.10
AMA Style
Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance. 2019; 2(2): 141-60. http://dx.doi.org/10.20517/cdr.2019.10
Chicago/Turabian Style
Wang, Xuan, Haiyun Zhang, Xiaozhuo Chen. 2019. "Drug resistance and combating drug resistance in cancer" Cancer Drug Resistance. 2, no.2: 141-60. http://dx.doi.org/10.20517/cdr.2019.10
ACS Style
Wang, X.; Zhang H.; Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141-60. http://dx.doi.org/10.20517/cdr.2019.10
About This Article
Special Issue
Copyright

Data & Comments
Data



 Cite This Article 545 clicks
Cite This Article 545 clicks

 Like This Article 37
likes
Like This Article 37
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.