
Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer

Abstract
Aberrant activation of the epidermal growth factor receptor (EGFR) is a driving force for cancer growth in a subgroup of non-small cell lung cancer patients. These patients can be identified by the presence of activating EGFR mutations. Currently three generations of EGFR-tyrosine kinase inhibitors (TKIs) have been approved by the Food and Drug Administration and European Medicine Agency. This paper reviews the structure of EGFR and the downstream signaling pathways of EGFR and describes the mechanisms of intrinsic and acquired resistance against EGFR-TKIs. These mechanisms include secondary or tertiary mutations in EGFR, the activation of bypassing signaling pathways or a histological transformation to small cell lung cancer. Moreover, drug efflux transporters will affect the cellular accumulation of EGFR-TKIs and penetration of the first generation of EGFR-TKI into the brain. Lysosomal sequestration of some EGFR-TKIs may also prevent the drugs to reach their target. In conclusion, resistance to EGFR-TKIs is multifactorial, including primary and acquired mutations in the EGFR gene, activation of bypassing pathways and limited uptake of drugs in the cells or target tissues. More pharmacological studies are needed in order to develop new specific compounds targeted to overcome new resistance mechanisms in order to enable a personalized treatment approach.
Keywords
Introduction
Molecular characterization of non-small cell lung cancer (NSCLC) and subsequent identification of targetable activated kinases led to a considerable shift in the treatment of this lethal disease in the past two decades. In patients with oncogene addicted NSCLC, particularly in adenocarcinoma, standard platinum-based chemotherapy has been replaced by molecularly driven strategies significantly improving the outcomes for these patients[1]. Among activated driver kinases suitable of pharmacological inhibition, epidermal growth factor receptor (EGFR) represents the most investigated and targeted one. Under physiological conditions, the EGFR signaling pathway controls cell growth, survival, proliferation, and differentiation by conveying the signal from the cell surface to downstream targets[2]. Oncogenic mutations in EGFR usually increase the kinase activity of EGFR, thus leading to hyperactivation of the pro-survival signaling pathway[3].
Approximately 50% of Asian patients and 11%-16% of patients from Western Countries with NSCLC harbor mutations in EGFR[4]. EGFR mutations are more often found in NSCLC from female never smokers, and tumor histology is mainly adenocarcinoma[5,6]. Three generations of specific EGFR-tyrosine kinase inhibitors (TKIs) have been developed and partially implemented in clinical practice for the treatment of EGFR-driven NSCLC up today: (1) gefitinib and erlotinib (first-generation); (2) afatinib and dacomitinib (second-generation); and (3) osimertinib and rociletinib (third-generation)[1]. Despite initial response and tumor shrinkage, patients experience disease progression due to the onset of a resistance mechanism to targeted treatment. This review will provide a comprehensive overview of current knowledge about EGFR structure and EGFR mutations, focusing on resistance mechanisms to EGFR inhibition and additional strategies to overcome treatment resistance.
Structure and activation of wild-type EGFR
EGFR is a transmembrane receptor protein that belongs to the ErbB family of tyrosine kinases. EGFR is made up of an extracellular domain, also known as ligand binding region, a transmembrane region and an intracellular tyrosine kinase domain[7]. The EGFR kinase domain consists of two lobes: a C-terminal lobe (C-lobe) and an N-terminal lobe (N-lobe). The catalytic site is located in the cleft between these two lobes. As illustrated in Figure 1, the catalytic site contains the activation loop (A-loop) (contributed by the C-lobe), the regulatory C-helix and the nucleotide phosphate binding loop (P-loop) (both contributed by the N-lobe)[8].
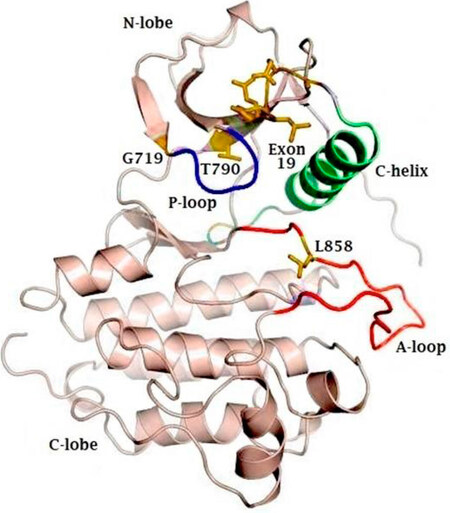
Figure 1. Representative figure of the crystal structure of the EGFR kinase domain, highlighting the N- and C-terminal lobes, as well as the phosphate coordinating P-loop (blue) and activation loop (A-loop). The active site lies in the cleft between the A-loop (red) and the C-lobe. The regulatory C-helix is colored in green. Sites of activating mutations are indicated in yellow. EGFR: epidermal growth factor receptor
For the activation of a tyrosine kinase receptor, two important conditions must be met. Firstly, the critical amino acids need to be positioned correctly to allow transfer of the phosphate group. Secondly, the peptide substrate binding site needs to be accessible[9]. In EGFR, the A-loop occludes the peptide substrate binding site. The C-loop contains a catalytic important glutamate that, together with Lys, is necessary for the coordination of the α- and β-phosphate groups on ATP[10]. The P-loop also contributes to the coordination of ATP through its GXGXXG motif[3].
In wild-type EGFR, the binding of EGF causes dimerization of the EGFR-receptor in a head-to-tail manner[11]. Hereby, the C-lobe of the one receptor pushes the N-lobe of the second receptor away, which leads to a reorientation of the A-loop. This leads to a translocation of the regulatory C-lobe to its active position[11], whereby Glu can interact with Lys. Furthermore, the peptide substrate binding site becomes accessible[10].
EGFR signaling
After binding to its ligand, EGFR frequently heterodimerizes with its family members Her2, Her3 and Her4[12]. Eleven ligands are known within the family, of which EGF binds EGFR with the highest affinity[13]. Signaling through EGFR can be divided in two main categories: kinase-dependent and kinase-independent signaling[14]. Kinase-dependent signaling results in the activation of several downstream pathways. A first activated pathway is Ras-Raf-MEK-ERK and JNK signaling[15,16], that promote cell survival and proliferation. A second signaling is the activation of PI3K/Akt through the association of Her3[17]. Thirdly, activation of EGFR also leads to STAT3 dimerization and translocation to the nucleus[18]. Fourthly, PLCγ signaling is activated through Src[19].
Besides kinase-dependent, EGFR also has kinase-independent functions. These include the stimulation of DNA synthesis[20], activation of mitogen-activated protein kinase (MAPK) signaling through heterodimerization[21], an anti-autophagic effect[22], modulation of protein subcellular trafficking (p53-upregulated modulator of apoptosis[23]) and activation of Akt through clustering in lipid rafts[24,25].
The influence of different mutations on EGFR activation
The vast majority of EGFR activating mutations (> 90%) are L858R point mutations and deletions within exon 19[26].
The residue L858 (exon 21) is part of a hydrophobic cluster on the A-loop. This cluster stabilizes the inactive conformation of EGFR through its hydrophobic interactions. The L858R mutation disrupts this hydrophobic interaction, which leads to destabilization of the inactive form and thus promotes the active conformation of EGFR[3,11]. The L861Q mutation is very close to the L858 residue, and most likely disturbs the inactive conformation in the same way[8].
Exon 19 deletion and exon 20 insertion take place in the C-helix. The exon 19 deletion in the loop of the C-helix “pulls” the regulatory helix in the active conformation. In contrast, the exon 20 insertion on the other end of the helix “pushes” the helix into its active conformation. Detailed crystal structures lack to confirm this hypothesis[3].
The G719 residue (exon 18) is a less frequent point mutation occurring in EGFR. This Glycine is located on the P-loop and is the first Gly in the GXGXXG motif. In the inactive form of EGFR, the P-loop contributes to the hydrophobic interactions that favor the inactive location of the C-helix, together with the cluster of L858. The G719 residue is necessary to cope with the torsion in the P-loop. Mutation to any other residue leads to a displacement of the P-loop, disturbs the hydrophobic interactions and thus destabilizes the inactive conformation of the receptor, resulting in receptor activation[3].
A study by Cho et al.[27] showed that dimerization of EGFR is not always necessary for downstream signaling. The wild-type EGFR receptor and the L858R mutant are dependent on dimerization for their activation, whereas in case of the exon 19 deletion, the exon 20 insertion and the L858R/T790M double mutant, dimerization is not necessary for EGFR-activation. This hypothesis is supported by studies with cetuximab, a monoclonal antibody that prevents EGFR dimerization. Cetuximab has been shown to inhibit EGFR-signaling most efficiently when the L858R mutation is present, whereas for the other mutations it only has a modest effect[27].
First- and second-generation EGFR-TKIs
Several of the above mentioned mutations in EGFR render the tumor sensitive to EGFR-TKIs. Erlotinib and gefitinib are the first generation of EGFR-TKIs that were FDA and EMA-approved. Both erlotinib and gefitinib have anilinoquinazoline-based structures and serve as ATP-competitors[28]. The efficacy of both drugs is dependent on the type of sensitizing mutation[29]. Jiang et al.[29] observed a 100x increased sensitivity of the Ba/F3 variant with the L858R mutation to gefitinib, compared to a 6x increased sensitivity for the G719S mutant. This difference in sensitivity might be related to the increased affinity for ATP of both mutants. In comparison to wild-type EGFR, the L858R mutant has a 50x increased ATP-affinity, in contrast to a 10x increased affinity for the G719S mutant[29]. These differences in affinity and response might be related to the position of both mutants. As discussed above, the L858R mutation is located in the A-loop, whereas the G719S mutation is located in the P-loop of the receptor.
Although there is a difference in affinity of erlotinib and gefitinib for the mutant and wild-type EGFR receptors, the drugs will bind wild-type EGFR as well. This binding results in on-target side effects such as skin rash or diarrhea[30]. Regarding brain activity, the actual concentrations of erlotinib and gefitinib in the central nervous system are too low to be active due to activity of PGP and BCRP drug transporters[31,32].
Afatinib is a second-generation EGFR-TKI. The afatinib structure is also anilinequinazoline-based, and it inhibits both EGFR and ErbB family members Her2, Her3 and Her4. Afatinib is also able to penetrate the blood-brain-barrier (BBB)[33], but it showed moderate activity against brain metastasis. In contrast to the first generation EGFR-TKIs, afatinib binds the receptor irreversibly at the C797 residue[34]. This irreversible binding of the receptor often leads to more severe on-target side effects like skin rash and diarrhea. A meta-analysis on 3000 patients showed that afatinib has a limited effect on the L858R mutation, whereas it has higher efficacy against the exon 19 deletion[35,36]. The exon 20 insertion also leads to innate resistance against afatinib.
Moving to clinical implications of EGFR inhibition, two seminal studies firstly described responsiveness to gefitinib in EGFR-mutated NSCLC[37,38], opening a new landscape for the treatment of oncogene-addicted lung cancer. Phase III trials have further explored the efficacy of first- and second-generation TKIs in this setting: a significant clinical benefit was observed for all the compounds in comparison with chemotherapy, in terms of progression-free survival (PFS), response rates and disease control rates. Indeed, the median PFS of first- and second-generation TKIs ranged from 8.0-13.1 months, which was consistent if compared to 4.6-6.9 months achieved by standard chemotherapy. Moreover, the benefit was independent of the line of treatment for which TKIs were administered, and no difference in efficacy was observed between first- and second-generation inhibitors in available comparative studies, except for the second-generation irreversible TKI dacomitinib[4]. These results allowed first- and second-generation EGFR-TKIs to become the new standard of care for frontline treatment of patients with EGFR-driven NSCLC.
Third-generation EGFR-TKIs
Virtually all patients treated with first- and second-generation TKIs will acquire resistance to this targeted treatment within about one year. The onset of EGFR T790M secondary mutation is responsible for 50% of patients progression[39]. A more detailed description of all resistance mechanism is provided in the paragraphs below. Before the advent of third-generation EGFR-TKIs, patients who harbored T790M mutations had limited treatment options, which usually consisted of standard platinum-based chemotherapy. Even if afatinib demonstrated preclinical activity in T790M positive EGFR-mutated NSCLC[40], it showed no clinical benefit when given to patients who progressed after gefitinib or erlotinib[41]. Development of compounds that can overcome T790M mutation was addressed by the introduction of third-generation EGFR-TKIs.
Osimertinib and rociletinib belong to the category of third-generation EGFR-TKIs. While osimertinib has been approved by the FDA and EMA, further development of rociletinib has been terminated[42]. Osimertinib has a mono-anilino-pyrimidine core[43] and its binding is irreversible to the EGFR receptor via the C797 residue. It is active in the nanomolar range against both the sensitizing EGFR mutations and the T790M mutation. Like all the other inhibitors, it is not active against exon 20 insertions. Since both the insulin growth factor receptor 1 (IGF-1R) and the insulin receptor (IR) contain a gatekeeper, methionine, osimertinib is specifically designed not to inhibit both receptors, to prevent hyperglycemia in treated patients. This is in contrast to rociletinib, where the drug was intended to inhibit the IGF-1R and failed partially because of hyperglycemia[43]. Osimertinib is able to cross the BBB and shows activity against metastases in the central nervous system[44,45].
Since osimertinib was shown to have clinical activity in T790M mutated NSCLC after failure of previous TKIs in multiple studies[46-48], a study of T790M status is mandatory after progression to first- or second-generation inhibitors and it could consist of both blood and tumor tissue sampling analysis[49-51]. Indeed, after progression to gefitinib, erlotinib or afatinib, patients treated with osimertinib obtained a PFS of 10.4 months compared to 4.4 months achieved with the platinum plus pemetrexed doublet (HR 0.30, P < 0.001); the objective response rate (ORR) was 71% vs. 31% in the two arms, respectively[49]. Remarkably, osimertinib was superior to both erlotinib and gefitinib also when administered frontline: PFS was 18.9 months vs. 10.2 months (HR 0.46; P < 0.001) for osimertinib and erlotinib/gefitinib, respectively, and the benefit was independent of T790M mutation[48]. Following these striking results, osimertinib has recently been approved by both the FDA and EMA for the first-line treatment of NSCLC with activating EGFR mutations, irrespective of T790M status[52].
Resistance mechanisms to EGFR-TKIs
Multiple mechanisms can cause primary and secondary resistance to EGFR-TKIs, as depicted in Figure 2. They could be schematically divided into five categories: (1) mutation within EGFR; (2) activation of bypassing pathways; (3) histological transformation; (4) aberrations in drug transporters; and (5) lysosomal sequestration.
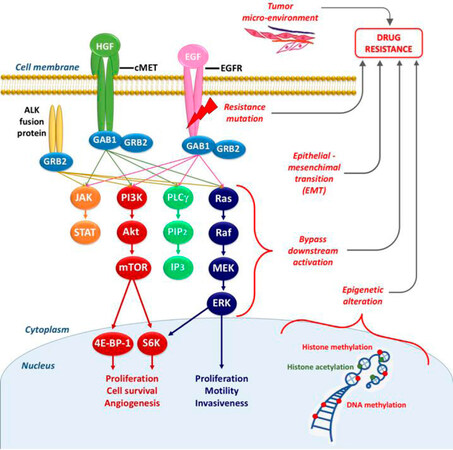
Figure 2. Schematic overview of the signaling through EGFR and other receptor tyrosine kinases (e.g., cMET) underlying the connections within these pathways, and the main mechanisms of resistance (in red). EGFR: epidermal growth factor receptor; HGF: hepatocyte growth factor; EGF: epidermal growth factor; GAB1/GRB2: associated binding protein; GRB2: growth factor receptor bound protein 2; JAK: janus kinase; STAT: signal transducer and activator of transcription; PI3K: phospho-inositide3 kinase; Akt: -protein kinase B; mTOR: mammalian target of rapamycin; 4E-BP-1: eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1; S6K: ribosomal protein S6 kinase; PLCγ: phospholipase gamma; PIP2: phosphatidylinositol-4,5-biphosphate; IP3: inositol trisphosphate; Ras: Ras oncogene; Raf: Raf proto-oncogene; MEK: mitogen activated protein kinase; ERK: extracellular signal-regulated kinase
Resistance mechanisms caused by EGFR mutations
Regarding primary resistance to EGFR-TKIs, the exon 20 insertion confers innate resistance to both reversible and irreversible inhibitors. The exon 20 insertion is a rare mutation in EGFR that occurs in approximately 4% of NSCLC, accounting for 5%-10% of all EGFR mutations in lung adenocarcinoma[53-55]. The patients with exon 20 insertion included in clinical trials testing EGFR-TKI showed a very poor response in comparison with patients carrying an L858R mutation or an exon 19 deletion[54]. However, more recently poziotinib has been identified as an inhibitor of EGFR harboring the ex20 mutation. Due to its relatively small size and flexibility, poziotinib can circumvent steric changes induced by the ex20 insertion and is a potent inhibitor of EGFR ex20[56]. It is currently been tested in a phase II trial and initial data showed an ORR of 64% in 11 patients[55].
Several mutations in EGFR are involved in secondary resistance to EGFR-TKIs, as summarized in Table 1. As stated above, 50%-60% of patients treated upfront with first- and second-generation EGFR-TKIs will experience a disease progression due to acquisition of a T790M mutation. The T790M mutation is located in the ATP binding cleft and is often referred to as “gatekeeper”-mutation[57]. However, crystal structures showed that the mutation does not sterically hinders the binding of the inhibitors. In contrast, it leads to a 4x-decreased affinity for gefitinib and an 18x increased affinity for ATP. This leads to an outcompeting of gefitinib by ATP. Moreover, the T790M mutation by itself stimulates the catalytic activity of the receptor, thus resulting in a survival advantage of these mutant cells[39].
EGFR-TKIs and EGFR mutations that confer drug resistance
| Inhibitor | Target | Acquired mutations conferring resistance | References |
|---|---|---|---|
| Gefitinib | EGFR | T790M, D761Y, T854A, L747S | [57,63-65] |
| Erlotinib | EGFR | T790M, D761Y, T854A, L747S | [57,63-65] |
| Afatinib | EGFR, HER2 | T790M | [34,36] |
| Osimertinib (AZD9291) | EGFR | C797S, G796D | [46,47,59,66] |
| Rociletinib | EGFR | C797S | [42] |
| EAI045 | EGFR | Under investigation | [59,68] |
The C797S mutation is a tertiary mutation that confers sensitivity to irreversible binding compounds like afatinib, osimertinib and rociletinib[58,59]. The C797S mutation targets the gatekeeper methionine residue that is used by the compounds to form a covalent bond with the receptor[60]. We need to discriminate between cells presenting with an L858R or exon 19 deletion in combination with the T790M and C797S mutation. Indeed, triple mutated cells are resistant to all generations of EGFR-TKIs; in this case, C797S mutation is associated to osimertinib resistance and occurs in 40% of patients treated with this third-generation TKI[61]. On the other hand, cells containing an L858R or exon 19 deletion in combination with a C797S mutation alone, are still sensitive to the quinazoline-based inhibitors like erlotinib, gefitinib and afatinib[62]. Among rare EGFR mutations (< 1%), D761Y, T854A and L747S are responsible for acquired resistance to first-generation TKIs; however, the mechanism for how these mutations confer resistance is still unknown[63-66]. Of note, loss of T790M is another mechanism responsible for resistance to third-generation osimertinib[67]. Fourth generation EGFR-TKIs able to overcome T790M and C797S are under development[68].
Resistance mechanisms mediated through bypassing pathways
Another mechanism for lung tumor cells to escape inhibition by EGFR-TKIs is the activation of parallel or downstream pathways. In this section, we will focus on best-known examples of pathways that can confer resistance to EGFR-TKIs. Activation of these pathways is not mutually exclusive with secondary or tertiary resistance mutations in EGFR[69]. Several of these bypassing pathways confer an intrinsic resistance to first-, second- and third-generation EGFR inhibitors, as well as to EGFR directed antibodies such as cetuximab. Some other pathways are activated during the course of treatment and can be considered as acquired resistance.
cMET
An important parallel pathway involved in resistance to EGFR inhibitors is the hepatocyte growth factor (HGF)-cMET pathway. The amplification of cMET plays a role in the activation of cMET-downstream signaling and thus circumvents EGFR-TKI inhibition. Amplification of cMET can be observed in approximately 3% of TKI-naïve NSCLC cases, compared to 10%-20% of EGFR-TKI treated patients[69,70]. The difference in percentage may represent acquired resistance. Secondly, when comparing downstream signaling of EGFR and cMET, these pathways overlap[71]. In vitro studies suggest a role in the reactivation of Her3/ERBB3-PI3K-Akt signaling[17]. This resistance mechanism is relevant for both the first- and third-generation EGFR-TKIs[72-74].
KRAS
KRAS mutation represents another important intrinsic resistance mechanism to EGFR inhibition in NSCLC. KRAS mutations mainly occur in codon 12 (G12C/D/S/V) and codon 13 (G13C)[75]. KRAS is a downstream target of EGFR and normally becomes activated upon stimulation of EGFR. The mutations render KRAS constitutively active, thereby making KRAS independent of EGFR-stimulation. Therefore a patient harboring both sensitizing EGFR mutations concomitantly with KRAS-mutations is unlikely to respond to EGFR-TKIs[76], since EGFR-downstream signaling will continue its signaling due to active KRAS[77]. This has been reported for both the first and third generation of EGFR-TKIs[77,78].
Axl
Upregulation of the receptor tyrosine kinase Axl (anexelekto) is another intrinsic resistance mechanism against erlotinib and gefitinib. For the second- and third- generation EGFR-TKIs no reports on Axl as resistance mechanism are available. The resistance might be caused by either the upregulation of the Axl receptor, or the upregulation of its ligand GAS6[79]. Two effects can be observed that might explain the increased resistance against EGFR-TKIs. Firstly, Axl drives epithelial to mesenchymal transition (EMT). Hence, Axl downregulation leads to a decrease of the EMT markers N-cadherin and vimentin and the upregulation of E-cadherin[80]. Secondly, since PI3K-Akt and MAPK/ERK signaling cascades are downstream from both Axl and EGFR, increased activation of Axl can bypass EGFR-inhibition[81].
Her2
The Her2 receptor contains a kinase domain, but a ligand is still unknown. Her2 frequently dimerizes with EGFR and thus stimulates downstream signaling. Intrinsic aberrations in Her2 include overexpression, an insertion in exon 20 (G776YVMA) and amplification. It has been shown that, in case of Her2 overexpression[82] and Her2 amplification[83,84], EGFR-TKIs are still active in NSCLC patients. Despite of reports in cell line models and mouse models[85], EGFR-TKIs can be active against non-mutated Her2. Mutations in Her2 have been reported both in the kinase domain[86] and in the transmembrane domain[87], however, their concomitance with EGFR mutations is not clear thus far. All generations of EGFR-TKIs can inhibit non-mutant Her2[82,88,89]. Due to the heterodimerization of EGFR with Her2, stimulation of EGFR is often necessary to activate downstream signaling in case of non-mutant Her2. If EGFR is inhibited, downstream signaling is inhibited. In case of mutations in Her2, the receptor often gains in kinase activity and is no longer dependent on EGFR activation to initiate downstream signaling. However, these tumors show responses to Her2-inhibitors like, e.g., trastuzumab or lapatinib[86].
For colorectal cancer (CRC), the situation is different. In case of EGFR mutations in CRC the monoclonal anti-EGFR antibody cetuximab was the most effective treatment for patients. Since this antibody interferes with ligand binding to EGFR, the mechanism of action is different from the EGFR-TKIs administered in NSCLC, that block the kinase activity. Amplification of Her2 is a known resistance mechanism to cetuximab that can be overcome by combining cetuximab with Her2 inhibitors[90].
IGF-1R
The insulin growth factor receptor 1 (IGF-1R) has been shown to induce EMT in NSCLC cells carrying an EGFR exon 19 deletion[91]. In these cells, the activation of IGF-1R or stimulation with TGF-β resulted in a similar phenotype. Moreover, in reaction to EGFR-inhibition, it has been shown that increased EGFR-IGF1R heterodimerization results in activation of mTOR, which in turn leads to de novo synthesis of EGFR and survivin[92,93]. Downstream signaling of IGF-1R results in increased resistance against EGFR-inhibitors. This mechanism of intrinsic resistance is observed with EGFR-TKI of the first[91-93], second[94] and third[95] generation. On the other hand, double inhibition of EGFR and IGF-1R at the same time can overcome this resistance[96,97].
FGFR
The fibroblast growth factor (FGF) family plays a role both in intrinsic and acquired resistance against EGFR-TKIs. In NSCLC, the ligands FGF2, FGF9 and the receptors FGFR1 and FGFR2 have been reported to be overexpressed[98,99]. Expression and activation of FGFR lead to a more mesenchymal phenotype and downstream signaling activates MEK-ERK and PI3K signaling[100]. Upon response to an EGFR inhibitor (erlotinib or gefitinib), FGF ligands and reporters become transcriptionally active[101]. Through the activation of an autocrine loop, this activation leads to EGFR-TKI resistance[102]. This resistance mechanism has also been reported for afatinib[103], but to date not for acquired resistance to osimertinib or rociletinib.
PIK3CA
The PIK3CA gene encodes the 110kDa catalytic subunit of PI3K[104], which is a downstream signaling pathway of EGFR. The PIK3CA pathway generally confers intrinsic resistance to first- and second-line EGFR inhibitors. In case of PIK3CA, both mutations and copy number gains are known aberrations. Mutations that increase the catalytic activity of PIK3CA occur in 1%-10% of EGFR-mutant NSCLC tumors[105,106]. Copy number gain (defined as > 2 copies per cell in over 40% of cells) is reported in approximately 40% of NSCLC patients[107]. Both mutations and copy number gain are correlated with a worse prognosis, overall survival and time to progression[105,107]. In case of EGFR-mutant tumors, 5/6 patients showed progressive disease when treated with EGFR-TKIs, 1 patient had treatment failure after only 4 months of treatment with EGFR-TKIs[105]. A meta-analysis summarized the effects of PIK3CA aberrations on treatment with diverse TKIs, amongst which are erlotinib, gefitinib and afatinib[108]. Reports on the effect of PIK3CA aberrations on osimertinib/rociletinib are not available yet.
BRAF
Overall, BRAF mutations can be considered as an intrinsic resistance mechanism but do not occur frequently in NSCLC. Indeed, BRAF mutations can be found in only 1.5%-3.5% of NSCLC patients and consists of a V600E mutation in about 50% of cases[109]. Concomitant mutations in EGFR and BRAF are even more rare, with reported percentages around or below 1% of EGFR mutant NSCLC patients, dependent on the study[110,111]. Crosstalk between BRAF and EGFR has been reported in a non-EGFR mutant context[112]. Hereby, the activation of BRAF results in the upregulation of EGFR ligands in a c-Jun dependent way. This results in the activation of EGFR-MAPK signaling[112]. It seems a logical step to assume that activation of BRAF can bypass EGFR inhibition. Indeed, case reports described patients harboring a BRAF V600E mutation during treatment with osimertinib[74,113]. In a peculiar case, a combination of osimertinib with the BRAF inhibitor encorafenib resulted in significant inhibition of cell growth. Unfortunately, the patient died before this combination treatment could be administered[113]. Nevertheless, this strategy can be extended to other cases of EGFR-TKI resistance through BRAF mutations.
PTEN
Phosphatase and tensin homologue (PTEN) is a tumor suppressor. Loss of PTEN is reported in about 5% of EGFR mutant NSCLC patients[106] and is linked to intrinsic resistance against EGFR-TKIs[114]. PTEN is a link between EGFR and Akt signaling. When PTEN is lost, EGFR signaling is uncoupled from Akt signaling. Inhibition of EGFR no longer results in inhibition of Akt signaling, resulting in resistance to EGFR-TKIs[115]. This has been demonstrated in cell lines, where resistance to erlotinib or gefitinib was correlated with PTEN levels. Overexpression of PTEN in cell lines with low/no PTEN levels resulted in increased sensitivity to erlotinib or gefitinib[114,115]. Loss of PTEN is both known as a primary mechanism of resistance (present in EGFR-TKI naïve patients)[116] and as acquired mechanism of resistance[114]. For the latter, around 10% of patients without any other known resistance mechanisms are reported to have PTEN loss[114].
TP53
In NSCLC, TP53 mutations occur in approximately 50% of patients. This is also the case in EGFR-mutated NSCLC[117-119]. The influence of TP53 mutations on response and survival of TP53/EGFR double mutated patients is not clear from literature. One report showed a significantly lower progression free survival in case of TP53 exon 8 mutations and exon 19 deletions in EGFR[118]. However, two other studies reported no significant effect in TP53/EGFR concomitant mutated patients[117,119], so the role of TP53 mutations in EGFR-TKI resistance is still unclear.
YAP
Yes-associated protein (YAP) is a part of the HIPPO-pathway, and seems to be associated with both intrinsic and acquired resistance to EGFR-TKI. Several links between EGFR-signaling and YAP have been reported. Firstly, activation of ERK1/2 is linked to activation with YAP. Depletion of ERK1/2 results in degradation of YAP, whereas ERK2 overexpression results in YAP rescue. The MEK1/2 inhibitor trametinib leads to decrease of YAP levels and decreased HIPPO-signaling[120]. Secondly, in cell lines with increased resistance against erlotinib, gefitinib or osimertinib, YAP is found in the nucleus (which is a sign of its activation)[121]. Overexpression of YAP leads to increased resistance to erlotinib[122]. YAP expression is also increased after EGFR-TKI treatment, both in cell line models and human NSCLC samples[123]. Moreover, activation of YAP leads to an increase in Axl expression and activation and a change to EMT-phenotype of the cells[123]. Knock-down of YAP results in resensitization of these cells to EGFR-TKIs. Inhibition of Axl activity also resensitizes to EGFR-TKIs[123].
NF-kB
Recently it was shown that acquired resistance to the novel third-generation EGFR-TKI rociletinib was mediated by activation of the NF-kB pathway[124]. This study focused on H1975 NSCLC cells, which harbor a T790M mutation, and are therefore resistant to gefitinib and erlotinib, but sensitive to rociletinib. Induction of resistance to rociletinib led to NF-kB activation replacing oncogenic EGFR signaling. Conversely, inhibition of this pathway with the proteasome inhibitor bortezomib sensitized the cells again to rociletinib. Since bortezomib showed some activity in drug combinations in NSCLC cells and patients[125-127], this approach seems promising as an alternative to sensitize patients progressive on treatment with third-generation EGFR-TKI.
In conclusion, there is a plethora of bypassing pathways to EGFR [Figure 2]. As our knowledge grows, more bypassing pathways will undoubtedly be discovered.
Resistance mechanisms through histological transformation
A peculiar resistance mechanism to EGFR-TKI is the histological transformation from NSCLC to small cell lung cancer (SCLC), which occurs in approximately 14% of cases[128]. This has been reported for the first time in 2006 by Zakowski et al.[129]. The “transformed” tumor retains the EGFR mutation, but is no longer responsive to EGFR-TKIs[130]. Although several cases of SCLC have been reported, a lot of questions remain on the mechanism and origin of this transformation. However, recent genetic studies[131,132] revealed that all transformed SCLC have an inactivation of RB1 and TP53 in common. Since the treatment regimens for NSCLC and SCLC are very different, this raises some questions about the optimal therapy in the occurrence of a histological transformation. Four patients with a SCLC transformation were switched to a SCLC-oriented platinum-etoposide chemotherapy, which produced marked responses in three patients[128]. Moreover, case reports show good results from “drug-holidays”. For example, a patient originally presenting with an L858R positive adenocarcinoma showed a good initial response to erlotinib. When resistance occurred, a rebiopsy showed a SCLC subtype containing both an L858R and a PIK3CA mutation. This SCLC was resistant to erlotinib. After a couple of months without any EGFR-TKI the tumor had switched back to the adenocarcinoma subtype, retained the L858R mutation but the PIK3CA mutation could not be detected anymore. After a couple of months of response to erlotinib, the patient became resistant again, and rebiopsy showed again the SCLC subtype with both an L858R and PIK3CA mutation[128].
Resistance through aberrations in drug transporters
Several families of drug transporters are currently known which may play a role in resistance to EGFR inhibitors in NSCLC[133,134]. Examples are the organic anion transporters, organic cation transporters (OCT), organic anion transporting polypeptides, equilibrative nucleoside transporters and the ABC-transporter superfamily which contains the Breast Cancer Resistance Protein (BCRP), P-glycoprotein (PGP) and multidrug resistance protein (MRP) family[134]. Gefitinib uptake seems mediated by an OCT and the uptake of erlotinib seems to have a mixed pattern, active and passive. However, literature is not consistent because of the great diversity of drug transporters and their functions on TKIs.
Erlotinib and gefitinib both have been shown to interact with ABCB1/PGP and ABCG2/BCRP; one interesting aspect is their role on trafficking of the transporters, affecting their function[135]. The influence of these efflux pumps has been studied most extensively in relation to gefitinib. Gefitinib acts both as a substrate and as an inhibitor of these pumps. Hereby, the concentration of gefitinib is important. At low concentrations (IC50 values in nmol/L range) gefitinib activates BCRP-ATPase activity in cell lines, whereas at concentrations > 1 µmol/L this stimulatory effect is lower[136,137]. Transduction of BCRP in gefitinib sensitive cell lines in the nanomolar range lead to gefitinib resistance. The BCRP-inhibitor Ko143 could reverse this resistance. However, this effect was much less in cell lines that are relatively insensitive to gefitinib (IC50 values in µmol/L range)[138]. Due to its role as a BCRP-inhibitor, gefitinib is also able to overcome BCRP-mediated resistance in vitro[138,139].
Erlotinib and efflux pumps are studied less well. Erlotinib is a target of PGP and BCRP, but not of MRP2[140]. In the triple knock-out mouse (BCRP1/MDR1a/MDR1b-/-), the oral bioavailability of erlotinib was increased with 66%, which led to increased systemic exposure to erlotinib[140], but in cell lines overexpressing BCRP the sensitivity to erlotinib could not be increased by specific inhibitors of BCRP such as Ko143, questioning the role of BCRP[141].
Afatinib is both a substrate and inhibitor for the efflux pumps PGP and BCRP, however, the therapeutic doses of afatinib are too low to affect the function of these pumps in vivo[142].
Lysosomal sequestration
Lipophilic or amphiphilic compounds can undergo a process called lysosomal sequestration, lysosomotropism or acid trapping[143]. These compounds are characterized by a pKa between 6.5 and 11[144]. Due to this pKa the compounds are neutral in the cytosol, which has a pH of around 7.2-7.4, and can diffuse passively through the membranes. When crossing the membrane of the lysosomes, the pH lowers to around 4.5-5. Hereby the compounds become protonated in the lysosomes and can no longer diffuse through the membranes and become “trapped” in the lysosomes[145].
A study of the physicochemical properties of TKIs[134] showed that several TKIs function as lysosomotropic compounds. This can be experimentally validated by determining the intracellular concentration of these compounds with and without the addition of Bafilomycin A1[146,147]. Bafilomycin A1 is a proton pump inhibitor that causes a disruption in the lysosomal pH, thus eliminating the pH gradient between the cytoplasm and the lysosomes. This prevents the protonation of the compounds and causes a decreased intracellular drug concentration.
Lysosomal trapping has been identified as a resistance mechanism against anticancer drugs by shielding the respective targets from the drugs[148,149]. Furthermore, this drug accumulation causes lysosomal stress, resulting in the activation of the coordinated lysosomal expression and regulation-pathway by transcription factor EB[145] and in lysosomal exocytosis[150].
Focusing on the EGFR-TKIs: erlotinib does not accumulate in the lysosomes by lysosomal sequestration[134], gefitinib on the other hand is a lysosomotropic compound[143]. As mentioned above gefitinib’s uptake is mediated by an active process and subsequently the drug seems to be trapped in the cells[151]. In contrast, erlotinib is likely to be transported into the membrane, binds to the target and is effluxed before entering the cell[151]. For the second- and third-generation EGFR-TKIs, this needs to be assessed.
Future directions
The ultimate goal of every anticancer treatment is to cure cancer, by delaying disease progression and providing a longer overall survival. Unfortunately, in the case of advanced NSCLC, despite the giant steps that have been made in the last decade, a definitive cure is still far from being discovered. The majority of the NSCLC patients lack a targetable driving mutation, and in this case they could be treated with immune checkpoint inhibitors or combinations of cytotoxic drugs, such as gemcitabine-cisplatin (or carboplatin), gemcitabine and a taxane, a taxane and cisplatin (or carboplatin), pemetrexed and cisplatin (for non-squamous NSCLC)[1,152]. Although these combinations have been extended with TKIs, the approach was usually not successful. Therefore, concerning EGFR-mutated NSCLC, a current challenge is to stay ahead of resistance against EGFR-TKIs.
A first strategy is to simultaneously inhibit multiple signaling pathways with combination therapy, such as a dual inhibition of EGFR and cMET signaling. This can prevent the activation of the bypassing cMET pathway upon EGFR inhibition and thus block a possible way-out for the cancer cells. However, given the multiple possible escape strategies, it remains a big challenge to predict the future mechanism of resistance of a specific tumor and target it from the beginning. Moreover, in order to be able to determine the acquired mechanism of resistance, continuous research is needed. Once a novel mechanism of resistance is discovered it is necessary to expand the screening strategies in clinical routine to bring it to the patients. However, sometimes the discovered mechanism of resistance is not fully targetable. A good example is the occurrence of mutations in KRAS: although extensive research has been conducted into Ras-inhibitors, no suitable drug is available to date.
A second strategy is a combination with immunotherapy, although an extensive discussion of these combinations is beyond the scope of this paper. In NSCLC, immunotherapy seems most effective in patients with a high tumor load, but all predictive parameters are still under investigation and need to be validated. A remaining question will be whether immunotherapy should be given before treatment with a TKI or whether the TKI should be given until resistance and then be followed with immunotherapy[153]. Alternatively, TKIs might be combined with immunotherapy, but this approach is in its infancy. However, certain types of therapy, including standard chemotherapy and some TKIs can induce synthesis and excretion of several cytokines and chemokines, which play an important role in the efficacy of immunotherapy.
A third strategy is to reconsider combinations of anti-signaling drugs (including anti-angiogenesis drugs) with standard therapies such as gemcitabine, pemetrexed, cisplatin, taxanes and radiation. These combinations should be designed taking into account the effect of these standard therapies on signaling pathways and the tumor surroundings. These drugs usually activate the AKT pathway as a survival mechanism[154]. Both in experimental systems and in patients it has been demonstrated that inhibition of AKT or EGFR can neutralize the AKT activation and enhance efficacy of standard drugs. For instance in several NSCLC models erlotinib increased the efficacy of pemetrexed[155], while inhibition of AKT with perifosine enhanced the efficacy of the platinum analog satraplatin[156]. MK2206 enhanced the effect of radiation alone[157] or in combination with gemcitabine and radiation (El-Naggar, unpublished results). Unfortunately, the current AKT inhibitors (e.g., MK2206, perifosine) were not sufficiently effective in patients, but several novel inhibitors are under development.
Finally, the implementation of liquid biopsies can be helpful, both for monitoring, e.g., the percentage of mutated cells in a tumor over time and for screening the occurrence of mechanisms of resistance during the treatment (as already approved for T790M detection). This approach can provide the treating physicians with the necessary information to decide to either continue treatment or to switch to another strategy in time (at the earliest sign of resistance).
The influence of drug efflux transporters and lysosomal sequestration on the intracellular concentration and location of small molecule inhibitors should not be overlooked. This is illustrated with erlotinib and gefitinib, which do not reach therapeutic concentrations in the brain, because of rapid efflux. Since osimertinib readily passes the blood-brain barrier and is not effluxed, patients with brain metastases should receive first-line treatment with osimertinib.
Conclusion
Identification of EGFR mutations in NSCLC and development of targeted treatment led to a revolution in the treatment of this peculiar oncogene-driven disease. The TKIs erlotinib, gefitinib, afatinib and osimertinib have been implemented in current clinical practice. However, the successful inhibition of EGFR signaling pathway can be limited by different mechanisms of resistance. A very important role is played by the T790M and C797S mutations that arise in most patients after treatment with EGFR-TKIs. Moreover, the identification of additional resistance mechanisms by activating bypassing pathways has taught us a lot on the intertwining of intracellular signaling pathways and gives us more possibilities to overcome this resistance by inhibiting multiple pathways simultaneously.
The activity of TKIs can also be influenced by other resistance mechanisms such as histological transformation, drug efflux transporters and lysosomal sequestration. The latter mechanisms need to be taken into account for the development and testing of new TKIs, in order to prevent lower than expected activity of these drugs in the clinical practice.
Declarations
Authors’ contributionsOriginal concept and writing: Van Der Steen N
Supervision and revision: Giovannetti E, Peters GJ
Writing and revision: Carbone D, Leonetti A
Supervision: Rolfo CD
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG, Kerr K, Popat S, Reck M, Senan S, Simo GV, Vansteenkiste J, Peters S; ESMO Guidelines Committee. Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27:V1-27.
2. Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 2005;1:2005.0010.
3. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007;11:217-27.
4. Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat Rev Clin Oncol 2018; doi: 10.1038/s41571-018-0081-4.
5. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from " never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306-11.
6. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50.
7. Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002;110:669-72.
8. Eck MJ, Yun C-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta 2010;1804:559-66.
10. Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol 2008;26:1742-51.
11. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006;125:1137-49.
12. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127-37.
13. van Lengerich B, Agnew C, Puchner EM, Huang B, Jura N. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A 2017;114:E2836-45.
14. Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett 2012;318:124-34.
15. Walker F, Kato A, Gonez LJ, Hibbs ML, Pouliot N, Levitzki A, Burgess AW. Activation of the Ras/mitogen-activated protein kinase pathway by kinase-defective epidermal growth factor receptors results in cell survival but not proliferation. Mol Cell Biol 1998;18:7192-204.
16. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal growth factor receptor signaling synergizes with hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res 2009;69:1284-92.
17. Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A 2005;102:3788-93.
18. Park OK, Schaefer TS, Nathans D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci U S A 1996;93:13704-08.
19. Anderson D, Koch C, Grey L, Ellis C, Moran M, Pawson T. Binding of SH2 domains of phospholipase C gamma 1, GAP, and Src to activated growth factor receptors. Science 1990;250:979-82.
20. Coker KJ, Staros J V, Guyer CA. A kinase-negative epidermal growth factor receptor that retains the capacity to stimulate DNA synthesis. Proc Natl Acad Sci U S A 1994;91:6967-71.
21. Deb TB, Su L, Wong L, Bonvini E, Wells a, David M, Johnson GR. Epidermal growth factor (EGF) receptor kinase-independent signaling by EGF. J Biol Chem 2001;276:15554-60.
22. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008;13:385-93.
23. Zhu H, Cao X, Ali-Osman F, Keir S, Lo HW. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Lett 2010;294:101-10.
24. Irwin ME, Bohin N, Boerner JL. Src family kinases mediate epidermal growth factor receptor signaling from lipid rafts in breast cancer cells. Cancer Biol Ther 2011;12:718-26.
25. Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol 2011;226:2316-28.
26. Murray S, Dahabreh IJ, Linardou H, Manoloukos M, Bafaloukos D, Kosmidis P. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol 2008;3:832-9.
27. Cho J, Chen L, Sangji N, Okabe T, Yonesaka K, Francis JM, Flavin RJ, Johnson W, Kwon J, Yu S, Greulich H, Johnson BE, Eck MJ, Jänne PA, Wong KK, Meyerson M. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res 2013;73:6770-9.
28. Rajasekaran R, Sethumadhavan R. In silico identification of significant detrimental missense mutations of EGFR and their effect with 4-anilinoquinazoline-based drugs. Appl Biochem Biotechnol 2010;160:1723-33.
29. Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res 2005;65:8968-74.
30. Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang J-J, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57.
31. Zhao J, Chen M, Zhong W, Zhang L, Li L, Xiao Y, Nie L, Hu P, Wang M. Cerebrospinal fluid concentrations of gefitinib in patients with lung adenocarcinoma. Clin Lung Cancer 2013;14:188-93.
32. de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs 2012;30:443-9.
33. Hoffknecht P1, Tufman A, Wehler T, Pelzer T, Wiewrodt R, Schütz M, Serke M, Stöhlmacher-Williams J, Märten A, Maria Huber R, Dickgreber NJ; Afatinib Compassionate Use Consortium (ACUC). Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated non-small-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol 2015;10:156-63.
34. Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 2012;343:342-50.
35. Jin Y, Shao Y, Shi X, Lou G, Zhang Y, Wu X, Tong X, Yu X. Mutational profiling of non-small-cell lung cancer patients resistant to first-generation EGFR tyrosine kinase inhibitors using next generation sequencing. Oncotarget 2016;7:61755-63.
36. Engle JA, Kolesar JM. Afatinib: a first-line treatment for selected patients with metastatic non-small-cell lung cancer. Am J Health Syst Pharm 2014;71:1933-8.
37. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39.
38. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500.
39. Yun C-H, Mengwasser KE, Toms A V, Woo MS, Greulich H, Wong K-K, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070-5.
40. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma S V, Isselbacher KJ, Settleman J, Haber DA. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci 2005;102:7665-70.
41. Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, Nishio K, Seki Y, Ebisawa R, Shahidi M, Yamamoto N. LUX-lung 4: a phase ii trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol 2013;31:3335-41.
42. Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther 2016;9:6065-74.
43. Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR V, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Brewer MR, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GHP, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046-61.
44. Goss G, Tsai CM, Shepherd FA, Ahn MJ, Bazhenova L, Crinò L, de Marinis F, Felip E, Morabito A, Hodge R, Cantarini M, Johnson M, Mitsudomi T, Jänne PA, Yang JC. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: pooled data from two phase II trials. Ann Oncol 2018;29:687-93.
45. Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, Pickup K, Jordan A, Hickey M, Grist M, Box M, Johnström P, Varnäs K, Malmquist J, Thress KS, Jänne PA, Cross D. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res 2016;22:5130-40.
46. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015;372:1689-99.
47. Floc’h N, Martin MJ, Riess JW, Orme JP, Staniszewska AD, Ménard L, Cuomo ME, O’Neill DJ, Ward RA, Finlay MRV, McKerrecher D, Cheng M, Vang DP, Burich RA, Keck JG, Gandara DR, Mack PC, Cross DAE. Anti-tumor activity of osimertinib, an irreversible mutant-selective EGFR tyrosine kinase inhibitor, in NSCLC harboring EGFR Exon 20 Insertions. Mol Cancer Ther 2018; doi: 10.1158/1535-7163.MCT-17-0758.
48. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, Okamoto I, Zhou C, Cho BC, Cheng Y, Cho EK, Voon PJ, Planchard D, Su WC, Gray JE, Lee SM, Hodge R, Marotti M, Rukazenkov Y, Ramalingam SS; FLAURA Investigators. Osimertinib in untreated EGFR -mutated advanced non–small-cell lung cancer. N Engl J Med 2018;378:113-25.
49. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, Mann H, Marotti M, Ghiorghiu S, Papadimitrakopoulou VA; AURA3 Investigators. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629-40.
50. Mitsudomi T, Ahn M, Bazhenova L, Blackhall F, Hida T, Majem Tarruella M, Vowler SL, Laus G, Jänne PA, Yang JC. Overall survival (OS) in patients (pts) with EGFR T790M-positive advanced non-small cell lung cancer (NSCLC) treated with osimertinib: results from two phase II studies. Ann Oncol 2017;28:v460-96.
51. Goss G, Tsai CM, Shepherd FA, Bazhenova L, Lee JS, Chang GC, Crino L, Satouchi M, Chu Q, Hida T, Han JY, Juan O, Dunphy F, Nishio M, Kang JH, Majem M, Mann H, Cantarini M, Ghiorghiu S, Mitsudomi T. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2016;17:1643-52.
52. Passiglia F, Raez LE, Rolfo C. Moving osimertinib to first-line: the right “strategy” in the chessboard of epidermal growth factor receptor-mutated non-small cell lung cancer? J Thorac Dis 2018;10:S1076-80.
53. Yasuda H, Kobayashi S, Costa DB, Israel B. Review EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13:e23-31.
54. Vanita N, Anuradha C, Vijay M. P, Amit J, Deepa Susan Joy P, Shripad B, Amit D, Kumar P. Epidermal growth factor receptor exon 20 mutation in lung cancer: types, incidence, clinical features and impact on treatment. Oncotargets 2017;10:2903-8.
55. Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, Le AT, Truini A, Nilsson MB, Sun H, Roarty E, Goldberg SB, Brahmer JR, Altan M, Lu C, Papadimitrakopoulou V, Politi K, Doebele RC, Wong KK, Heymach JV. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 2018;24:638-46.
57. Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J, Haber DA. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther 2008;7:874-9.
58. Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, Ladanyi M. Acquired resistance of egfr-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol 2015;1:982-4.
59. Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 2016;9:59.
60. Yu Z, Boggon TJ, Kobayashi S, Jin C, Ma PC, Dowlati A, Kern JA, Tenen DG, Halmos B. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res 2007;67:10417-27.
61. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Jänne PA, Oxnard GR. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560-2.
62. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Jänne PA. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res 2015;21:3913-23.
63. Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA, Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006;12:6494-501.
64. Bean J, Riely GJGJ, Balak M, Marks JLJL, Ladanyi M, Miller VAVA, Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res 2008;14:7519-25.
65. Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol 2008;26:1182-4.
66. Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C, Wang J, Liu L, Wang Z, Ni J, Yang Z, Xu J. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017;8:49671-9.
67. Oxnard G, Hu Y, Mileham K, Tracy P, Feeney N, Sholl L, Paweletz C, Thress K, Jänne P. OA 09.02 osimertinib resistance mediated by loss of EGFR T790M Is associated with early resistance and competing resistance mechanisms. J Thorac Oncol 2017;12:S1767-8.
68. Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett 2017;385:51-4.
69. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104:20932-7.
70. Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, Santoro A, Varella-Garcia M. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 2009;20:298-304.
71. Guo A, Villén J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, Mitchell J, Gygi SP, Rush J, Polakiewicz RD, Comb MJ. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008;105:692-7.
72. Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, Kanteti R, Riehm J, Salgia R, Owonikoko TK, Ramalingam SS, Chen M, Sun SY. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett 2016;380:494-504.
73. Ou SI, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer 2016;98:59-61.
74. Minari R, Bordi P, La Monica S, Squadrilli A, Leonetti A, Bottarelli L, Azzoni C, Lagrasta CAM, Gnetti L, Campanini N, Petronini PG, Alfieri R, Tiseo M. Concurrent acquired BRAF V600E mutation and MET amplification as resistance mechanism of first-line osimertinib treatment in a patient with EGFR-mutated NSCLC. J Thorac Oncol 2018;13:e89-91.
75. Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter. J Clin Oncol 2013;31:1112-21.
76. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73.
77. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic KRAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 2008;9:962-72.
78. Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, Meador CB, Spitzler P, Ichihara E, Ross SJ, Ahdesmaki MJ, Ahmed A, Ratcliffe LE, O’Brien EL, Barnes CH, Brown H, Smith PD, Dry JR, Beran G, Thress KS, Dougherty B, Pao W, Cross DA. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res 2015;75:2489-500.
79. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852-60.
80. Wu F, Li J, Jang C, Wang J, Xiong J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol 2014;7:6653-61.
81. Tian Y, Zhang Z, Miao L, Yang Z, Yang J, Wang Y, Qian D, Cai H, Wang Y. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in non-small cell lung cancer cells. Oncol Res 2016;24:295-303.
82. Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosine kinase inhibitor ZD1839 (“ Iressa ”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res 2001;61:7184-8.
83. Varella-Garcia M, Mitsudomi T, Yatabe Y, Kosaka T, Nakajima E, Xavier AC, Skokan M, Zeng C, Franklin WA, Bunn PA Jr, Hirsch FR. EGFR and HER2 genomic gain in recurrent non-small cell lung cancer after surgery: impact on outcome to treatment with gefitinib and association with EGFR and KRAS mutations in a Japanese cohort. J Thorac Oncol 2009;4:318-25.
84. Cappuzzo F, Varella-Garcia M, Shigematsu H, Domenichini I, Bartolini S, Ceresoli GL, Rossi E, Ludovini V, Gregorc V, Toschi L, Franklin WA, Crino L, Gazdar AF, Bunn PA Jr, Hirsch FR. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol 2005;23:5007-18.
85. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, Ladanyi M, Politi K, Pao W. HER2 amplification: a potential mechanism of acquired resistance to egfr inhibition in EGFR -mutant lung cancers that lack the second-site EGFR T790M mutation. Cancer Discov 2012;2:922-33.
86. Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006;10:25-38.
87. Ou SI, Schrock AB, Bocharov EV, Klempner SJ, Haddad CK, Steinecker G, Johnson M, Gitlitz BJ, Chung J, Campregher PV, Ross JS, Stephens PJ, Miller VA, Suh JH, Ali SM, Velcheti V. HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446-57.
88. Liu S, Li S, Hai J, Wang X, Chen T, Quinn MM, Gao P, Zhang Y, Ji H, Cross DAE, Wong KK. Targeting HER2 aberrations in non-small cell lung cancer with osimertinib. Clin Cancer Res 2018;24:2594-604.
89. Landi L, Tiseo M, Chiari R, Ricciardi S, Rossi E, Galetta D, Novello S, Milella M, D’Incecco A, Minuti G, Tibaldi C, Salvini J, Facchinetti F, Haspinger ER, Cortinovis D, Santo A, Banna G, Catino A, GiajLevra M, Crinò L, de Marinis F, Cappuzzo F. Activity of the EGFR-HER2 dual inhibitor afatinib in EGFR-mutant lung cancer patients with acquired resistance to reversible EGFR tyrosine kinase inhibitors. Clin Lung Cancer 2014;15:411-7.
90. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M, Nakagawa K, Jänne PA. Activation of ERBB2 signaling causes resistance to the EGFR- directed therapeutic antibody cetuximab. Sci Transl Med 2011;3:99ra86.
91. Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyàs E, Bonavia R, Visa J, Martin-Castillo B, Barrajón-Catalán E, Micol V, Bosch-Barrera J, Menendez JA. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci Rep 2013;3:2560.
92. Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006;66:10100-11.
93. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 2007;13:2795-803.
94. Lee Y, Wang Y, James M, Jeong JH, You M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol Carcinog 2016;55:991-1001.
95. Park JH, Choi YJ, Kim SY, Lee JE, Sung KJ, Park S, Kim WS, Song JS, Choi CM, Sung YH, Rho JK, Lee JC. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016;7:22005-15.
96. Choi YJ, Rho JK, Jeon BS, Choi SJ, Park SC, Lee SS, Kim HR, Kim CH, Lee JC. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: a lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother Pharmacol 2010;66:381-8.
97. Hurbin A, Wislez M, Busser B, Antoine M, Tenaud C, Rabbe N, Dufort S, de Fraipont F, Moro-Sibilot D, Cadranel J, Coll JL, Brambilla E. Insulin-like growth factor-1 receptor inhibition overcomes gefitinib resistance in mucinous lung adenocarcinoma. J Pathol 2011;225:8395.
98. Kono SA, Marshall ME, Ware KE, Heasley LE. The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors in non-small cell lung cancer. Drug Resist Updat 2009;12:95-102.
99. Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, McDermott LA, Coldren CD, Nemenoff RA, Merrick DT, Helfrich BA, Bunn PA Jr, Heasley LE. Fibroblast growth factor ( FGF ) and FGF receptor-mediated autocrine signaling in non – small-cell lung cancer cells. Mol Pharmacol 2009;75:196-207.
100. Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis 2008;25:843-54.
101. Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, Helfrich BA, Doebele RC, Heasley LE. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS One 2010;5:e14117.
102. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S, Satomi R, Naoki K, Betsuyaku T. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res 2013;11:759-67.
103. Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, Izumi H, Kage M, Kuwano M, Ono M, Hoshino T. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014;5:5908-19.
104. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Date H, Lam WL, Minna JD, Gazdar AF. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008;68:6913-21.
105. Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, Baldelli E, Iacono D, Mameli MG, Cavaliere A, Crinò L. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol 2011;6:707-15.
106. VanderLaan PA, Rangachari D, Mockus SM, Spotlow V, Reddi HV, Malcolm J, Huberman MS, Joseph LJ, Kobayashi SS, Costa DB. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: correlation with clinical outcomes. Lung Cancer 2017;106:17-21.
107. Fidler MJ, Morrison LE, Basu S, Buckingham L, Walters K, Batus M, Jacobson KK, Jewell SS, Coon J 4th, Bonomi PD. PTEN and PIK3CA gene copy numbers and poor outcomes in non-small cell lung cancer patients with gefitinib therapy. Br J Cancer 2011;105:1920-6.
108. Martín Martorell P, Huerta M, Compañ Quilis A, Abellán R, Seda E, Blesa S, Chaves FJ, Dualde Beltrán D, Roselló Keränen S, Franco J, Insa A. Coexistence of EGFR, KRAS, BRAF, and PIK3CA mutations and ALK rearrangement in a comprehensive cohort of 326 consecutive Spanish nonsquamous NSCLC patients. Clin Lung Cancer 2017;18:e395-402.
109. Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L, Tiseo M, Planchard D. BRAF in non-small cell lung cancer (NSCLC): pickaxing another brick in the wall. Cancer Treat Rev 2018;66:82-94.
110. Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, Viola P, Pullara C, Mucilli F, Buttitta F. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 2011;29:3574-9.
111. Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, Fernandez-Cuesta L, Fidias P, Yang JC, Miller VA, Riely GJ, Kris MG, Engelman JA, Vnencak-Jones CL, Dias-Santagata D, Ladanyi M, Pao W. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci 2012;109:E2127-33.
112. Lin L, Asthana S, Chan E, Bandyopadhyay S, Martins MM, Olivas V, Yan JJ, Pham L, Wang MM, Bollag G, Solit DB, Collisson EA, Rudin CM, Taylor BS, Bivona TG. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci U S A 2014;111:E748-57.
113. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Chih-Hsin Yang J. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol 2017;12:567-72.
114. Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res 2010;70:8715-25.
115. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M, Thomas RK. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009;69:3256-61.
116. Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M, Kudoh S. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br J Cancer 2005;92:1711-9.
117. Shepherd FA, Lacas B, Le Teuff G, Hainaut P, Jänne PA, Pignon JP, Le Chevalier T, Seymour L, Douillard JY, Graziano S, Brambilla E, Pirker R, Filipits M, Kratzke R, Soria JC, Tsao MS; LACE-Bio Collaborative Group. Pooled analysis of the prognostic and predictive effects of TP53 comutation status combined with KRAS or EGFR mutation in early-stage resected non–small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 2017;35:2018-27.
118. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, Capelli L, Casanova C, De Luigi N, Mariotti M, Gamboni A, Chiari R, Bennati C, Calistri D, Ludovini V, Crinò L, Amadori D, Ulivi P. Impact of TP53 mutations on outcome in EGFR -mutated patients treated with first-line tyrosine kinase inhibitors. Clin Cancer Res 2017;23:2195-202.
119. Labbé C, Cabanero M, Korpanty GJ, Tomasini P, Doherty MK, Mascaux C, Jao K, Pitcher B, Wang R, Pintilie M, Leighl NB, Feld R, Liu G, Bradbury PA, Kamel-Reid S, Tsao MS, Shepherd FA. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017;111:23-9.
120. You B, Yang YL, Xu Z, Dai Y, Liu S, Mao JH, Tetsu O, Li H, Jablons DM, You L. Inhibition of ERK1/2 down-regulates the hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015;6:4357-68.
121. McGowan M, Kleinberg L, Halvorsen AR, Helland Å, Brustugun OT. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 2017;8:497-504.
122. Hsu PC, You B, Yang YL, Zhang WQ, Wang YC, Xu Z, Dai Y, Liu S, Yang CT, Li H, Hu B, Jablons DM, You L. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016;7:51922-33.
123. Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S, De Luca E, Gatti G, Volante M, Giordano S. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 2017;19:1012.
124. Galvani E, Sun J, Leon LG, Sciarrillo R, Narayan RS, Sjin RT, Lee K, Ohashi K, Heideman DA, Alfieri RR, Heynen GJ, Bernards R, Smit EF, Pao W, Peters GJ, Giovannetti E. NF-κB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget 2015;6:42717-32.
125. Voortman J, Checińska A, Giaccone G. The proteasomal and apoptotic phenotype determine bortezomib sensitivity of non-small cell lung cancer cells. Mol Cancer 2007;6:73.
126. Voortman J, Pham TV, Knol JC, Giaccone G, Jimenez CR. Prediction of outcome of non-small cell lung cancer patients treated with chemotherapy and bortezomib by time-course MALDI-TOF-MS serum peptide profiling. Proteome Sci 2009;7:34.
127. Ceresa C, Giovannetti E, Voortman J, Laan AC, Honeywell R, Giaccone G, Peters GJ. Bortezomib induces schedule-dependent modulation of gemcitabine pharmacokinetics and pharmacodynamics in non-small cell lung cancer and blood mononuclear cells. Mol Cancer Ther 2009;8:1026-36.
128. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotyping and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
129. Zakowski MF, Ladanyi M, Kris MG; Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group. EGFR mutations in small-cell lung cancers. N Engl J Med 2006;355:213-5.
130. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 2015;16:e165-72.
131. Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, An Y, Keam B, Kim DW, Heo DS, Kim YT, Kim JS, Kim SH, Lee JS, Lee SH, Park K, Ku JL, Jeon YK, Chung DH, Park PJ, Kim J, Kim TM, Ju YS. Clonal history & genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol 2017;35:3065-74.
132. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, Howe E, Fulton LE, Mulvey HE, Bernardo LA, Mohamoud F, Miyoshi N, VanderLaan PA, Costa DB, Jänne PA, Borger DR, Ramaswamy S, Shioda T, Iafrate AJ, Getz G, Rudin CM, Mino-Kenudson M, Engelman JA. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 2015;6:6377.
133. Lemos C, Jansen G, Peters GJ. Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. Br J Cancer 2008;98:857-62.
134. Da Silva CG, Honeywell RJ, Dekker H, Peters GJ. Physicochemical properties of novel protein kinase inhibitors in relation to their substrate specificity for drug transporters. Expert Opin Drug Metab Toxicol 2015;11:703-17.
135. Porcelli L, Lemos C, Peters GJ, Paradiso A, Azzariti A. Intracellular trafficking of MDR transporters and relevance of SNPs. Curr Top Med Chem 2009;9:197-208.
136. Elkind NB, Szentpétery Z, Apáti A, Ozvegy-Laczka C, Várady G, Ujhelly O, Szabó K, Homolya L, Váradi A, Buday L, Kéri G, Német K, Sarkadi B. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor iressa (ZD1839, Gefitinib ) multidrug transporter ABCG2 prevents tumor cell death induced. Cancer Res 2005;65:1770-7.
137. Brózik A, Hegedüs C, Erdei Z, Hegedus T, Özvegy-Laczka C, Szakács G, Sarkadi B. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol 2011;7:623-42.
138. Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein-mediated drug resistance. Mol Cancer Ther 2004;3:1119-25.
139. Nakamura Y, Oka M, Soda H, Shiozawa K, Yoshikawa M, Itoh A, Ikegami Y, Tsurutani J, Nakatomi K, Kitazaki T, Doi S, Yoshida H, Kohno S. Gefitinib (“Iressa”, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2-mediated drug resistance. Cancer Res 2005;65:1541-6.
140. Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JH. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther 2008;7:2280-7.
141. Lemos C, Kathmann I, Giovannetti E, Calhau C, Jansen G, Peters GJ. Impact of cellular folate status and epidermal growth factor receptor expression on BCRP/ABCG2-mediated resistance to gefitinib and erlotinib. Br J Cancer 2009;100:1120-7.
142. Wind S, Schnell D, Ebner T, Freiwald M, Stopfer P. Clinical pharmacokinetics and pharmacodynamics of afatinib. Clin Pharmacokinet 2017;56:235-50.
143. Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos 2013;41:897-905.
144. Nadanaciva S, Lu S, Gebhard DF, Jessen BA, Pennie WD, Will Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol Vitr 2011;25:715-23.
145. Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015;6:1143-56.
146. Honeywell R, Yarzadah K, Giovannetti E, Losekoot N, Smit EF, Walraven M, Lind JS, Tibaldi C, Verheul HM, Peters GJ. Simple and selective method for the determination of various tyrosine kinase inhibitors used in the clinical setting by liquid chromatography tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 2010;878:1059-68.
147. Van Der Steen N, Honeywell RJ, Dekker H, Van Meerloo J, Kole J, Musters R, Ruijtenbeek R, Rolfo C, Pauwels P, Peters GJ, Giovannetti E. Resistance to crizotinib in a cMET gene amplified tumor cell line is associated with impaired sequestration of crizotinib in lysosomes. J Mol Clin Med 2018;1:101-8.
148. Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters RJ, Jansen G, Griffioen AW, Assaraf YG, Pili R, Peters GJ, Verheul HM. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res 2011;17:7337-46.
149. Gotink KJ, Rovithi M, de Haas RR, Honeywell RJ, Dekker H, Poel D, Azijli K, Peters GJ, Broxterman HJ, Verheul HM. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell Oncol (Dordr) 2015;38:119-29.
150. Zhitomirsky B, Assaraf YG. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017;8:45117-32.
151. Honeywell RJ, Hitzerd S, Kathmann I, Peters GJ. Transport of six tyrosine kinase inhibitors: active or passive? ADMET & DMPK 2016;4:23-34.
152. Galvani E, Peters GJ, Giovannetti E. Thymidylate synthase inhibitors for non-small cell lung cancer. Expert Opin Investig Drugs 2011;20:1343-56.
153. Rolfo C, Caglevic C, Santarpia M, Araujo A, Giovannetti E, Gallardo CD, Pauwels P, Mahave M. Immunotherapy in NSCLC: a promising and revolutionary weapon. Adv Exp Med Biol 2017;995:97-125.
154. Avan A, Narayan R, Giovannetti E, Peters GJ. Role of Akt signaling in resistance to DNA-targeted therapy. World J Clin Oncol 2016;7:352-69.
155. Giovannetti E, Lemos C, Tekle C, Smid K, Nannizzi S, Rodriguez JA, Ricciardi S, Danesi R, Giaccone G, Peters GJ. Molecular mechanisms underlying the synergistic interaction of erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor, with the multitargeted antifolate pemetrexed in non-small-cell lung cancer cells. Mol Pharmacol 2008;73:1290-300.
156. Avan A, Adema AD, Hoebe EK, Huijts CM, Avan A, Veal GJ, Ruijtenbeek R, Wosikowski K, Peters GJ. Modulation of signaling enhances the efficacy of the combination of satraplatin and erlotinib. Curr Drug Targets 2014;15:1312-21.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Van Der Steen N, Giovannetti E, Carbone D, Leonetti A, Rolfo CD, Peters GJ. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist 2018;1:230-49. http://dx.doi.org/10.20517/cdr.2018.13
AMA Style
Van Der Steen N, Giovannetti E, Carbone D, Leonetti A, Rolfo CD, Peters GJ. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resistance. 2018; 1(4): 230-49. http://dx.doi.org/10.20517/cdr.2018.13
Chicago/Turabian Style
Van Der Steen, Nele, Elisa Giovannetti, Daniela Carbone, Alessandro Leonetti, Christian D. Rolfo, Godefridus J. Peters. 2018. "Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer" Cancer Drug Resistance. 1, no.4: 230-49. http://dx.doi.org/10.20517/cdr.2018.13
ACS Style
Van Der, Steen N.; Giovannetti E.; Carbone D.; Leonetti A.; Rolfo CD.; Peters GJ. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist. 2018, 1, 230-49. http://dx.doi.org/10.20517/cdr.2018.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 19
likes
Like This Article 19
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.