
Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer

Abstract
Cellular homeostasis crucially relies on the correct nucleocytoplasmic distribution of a vast number of proteins and RNA molecules, which are shuttled in and out of the nucleus by specialized transport receptors. The nuclear export receptor XPO1, also called CRM1, mediates the translocation of hundreds of proteins and several classes of RNA to the cytoplasm, and thus regulates critical signaling pathways and cellular functions. The normal function of XPO1 appears to be often disrupted in malignant cells due to gene mutations or, most commonly, aberrant overexpression. Due to its important physiological roles and its frequent alteration in human tumors, XPO1 is a promising target for cancer therapy. XPO1 inhibitors have undergone extensive testing as therapeutic agents in preclinical models of cancer, with promising results. One of these inhibitors, Selinexor, is currently being evaluated in multiple clinical trials of different types of solid tumors and hematological malignancies. Here, we review several key aspects of XPO1 function, as well as the mechanisms that may lead to its alteration in cancer, and provide an update on the status of XPO1 inhibitors being developed as drugs for cancer therapy, including the definitive results of the first clinical trials with Selinexor that have been recently published.
Keywords
Introduction
In 1997, a 120 kDa protein called CRM1, known to function as a chromosome region maintenance factor in yeast, was identified as the first receptor for the nuclear export of proteins, and it was consequently renamed exportin 1 (XPO1)[1-4]. In these initials reports, XPO1/CRM1 (hereafter referred to as XPO1) was found to be the cellular target for a potent inhibitor of nuclear export termed leptomycin B (LMB), and to bind short amino acid sequences (so-called nuclear export signals or NESs) in proteins that were actively exported from the nucleus. Over the last two decades, many aspects of XPO1 physiopathology have been elucidated. Thus, XPO1 has been shown to mediate the nuclear export of not only hundreds of cellular and viral proteins, but also of different types of RNA molecules[5,6]. In fact, crucial signaling pathways, such as the NF-κB pathway, and essential cellular processes, such as cell cycle progression, have been shown to involve XPO1-dependent nuclear export steps[7]. In addition, export-independent functions of XPO1 in mitosis have also been identified[8]. The normal function of XPO1 appears to be often disrupted in malignant cells. Thus, overexpression of the XPO1 mRNA or protein has been frequently reported in a variety of tumor types and recurrent XPO1 gene mutations have been detected in certain hematological malignancies, suggesting that XPO1 may represent a therapeutic target in cancer[9,10]. Importantly, the results of multiple cellular, biochemical and structural analyses have led to a detailed mechanistic understanding of XPO1 function[11-13], paving the way for the development of clinically useful inhibitors of XPO1. Several compounds targeting XPO1 have been extensively tested in preclinical studies, and one of them, Selinexor, is now undergoing clinical trials, with promising results in patients with different types of cancer.
The physiological roles of XPO1
An overview of nucleocytoplasmic transport of proteins
In eukaryotic cells, the nuclear envelope establishes a physical separation between the two major cellular compartments: the nucleus and the cytoplasm. Cellular homeostasis requires continuous communication between these compartments through the bidirectional trafficking of molecules. This trafficking may occur by diffusion in the case of small molecules, or by budding of nuclear envelope-derived vesicles for a minority of specific proteins[14]. However, the vast majority of proteins can only enter and exit the nucleus through proteinaceous channels embedded in the nuclear envelope termed nuclear pore complexes (NPCs)[15,16]. For most proteins, nucleocytoplasmic transport is an active, energy-dependent process that requires a specialized transport machinery with three crucial components: (1) the NPCs; (2) a family of soluble transport receptors (karyopherins) that recognize and bind specific transport signals in the cargo proteins; (3) a gradient of the small GTPase Ran (bound to either GTP or GDP) across the nuclear envelope, which confers directionality to the transport [Figure 1A][17-19].
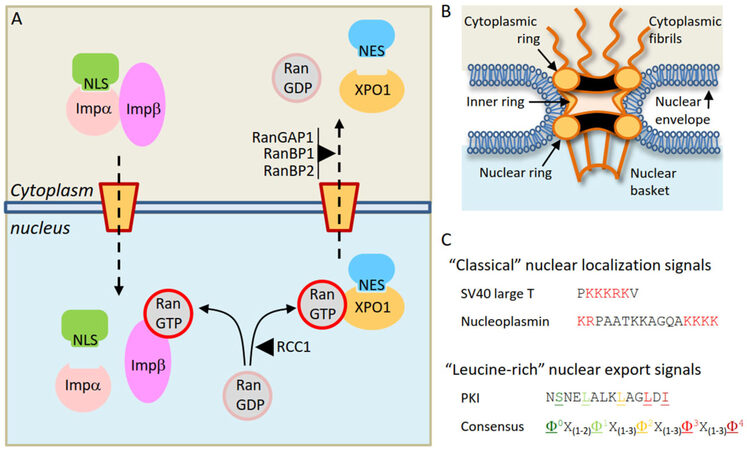
Figure 1. Receptor-mediated nucleocytoplasmic transport of proteins. A: Illustrative overview of the nuclear import of a cargo protein bearing a "classical" NLS mediated by the Importina/Importinb heterodimer (left) and the nuclear export of a cargo protein bearing a "leucine-rich" NES mediated by XPO1 (right); B: schematic depiction of the nuclear pore complex, illustrating its main structural features; C: examples of nucleocytoplasmic transport signals. The NLSs of SV40 large T antigen (monopartite) and nucleoplasmin (bipartite) are shown, with the basic residues that characterize "classical" NLSs highlighted in red. Below, the NES of PKI and a general consensus sequence of "leucine-rich" NESs are shown. The characteristic hydrophobic residues (represented by Φ in the consensus) are highlighted in colors and underlined. NLS: nuclear localization signal; NES: nuclear export signal
NPCs, recently reviewed by Knockenhauer and Schwartz[15] and Pemberton and Paschal[17], are very large complexes (over 120 MDa in size) formed by the assembly of several copies of each of approximately 30 different proteins called nucleoporins (NUPs). NPCs present a characteristic eight-fold rotational symmetry and are composed by three stacked rings inserted into the nuclear envelope, with a series of filaments emanating to the cytoplasmic side of the NPC and a basket-like structure protruding to the nucleoplasmic side [Figure 1B]. NUPs in the inner channel of the pore contain intrinsically disordered domains rich in phenyalanine-glycine (FG) repeats. These so-called FG-nucleoporins constitute a barrier that efficiently prevents proteins above a certain size from freely diffusing across the NPC. This threshold size for exclusion has long been believed to be relatively sharp (30-60 kDa), but a recent study suggests that the NPC lacks such a firm size threshold[20]. The selective barrier of the NPC can be overcome by large proteins (and even by very large nucleoprotein complexes, such as ribosomal subunits) through binding to karyopherins[21].
The human genome codes for approximately 20 different karyopherins[22]. While some of these receptors can mediate bidirectional transport of cargos in and out of the nucleus, most of them function exclusively as either import receptors (importins) or export receptors (exportins), such as XPO1. Karyopherins can recognize and bind specific peptide sequences in the cargo protein, which function as transport signals, and can be broadly classified as nuclear localization signals (NLSs, recognized by importins) or nuclear export signals (NESs, recognized by exportins) [Figure 1C]. The best-characterized import receptor is the Importinα/Importinβ heterodimer, which mediates nuclear import of cargos bearing a "classical" NLS (a peptide sequence characterized by the presence of one or two clusters of basic residues)[23]. XPO1, the first nuclear export receptor to be identified, is also the best-characterized exportin. XPO1 mediates export of proteins bearing “leucine-rich” NESs, short peptides with a characteristic spacing of hydrophobic residues [Figure 1C]. Of note, some proteins possess both an NLS and an NES and can undergo cyclic shuttling between the nucleus and the cytoplasm[24]. Importinα/β and XPO1 mediate the nucleocytoplasmic transport of hundreds of different proteins. Other karyopherins, which are less well characterized, seem to have a more limited repertoire of cargos, and the transport signals that may mediate their binding remain, in most cases, yet to be identified.
Binding and release of a protein in the nucleus or the cytoplasm establishes the direction of its transport, and the key factor that regulates cargo binding and release by karyopherins is the small GTPase Ran, which can be bound to either GDP (RanGDP) or GTP (RanGTP)[17-19]. There is a RanGDP/RanGTP gradient across the nuclear envelope. This gradient (a high concentration of RanGDP in the cytoplasm and a high concentration of RanGTP in the nucleus) is maintained by the Ran cofactors RanGAP1 (a cytoplasmic GTPase activating protein) and RCC1 (a chromatin-bound nucleotide exchange factor). RanGTP promotes disassembly of the Importin/cargo complexes, leading to release of import cargos in the nucleus. Conversely, RanGTP stabilizes the interaction between XPO1 and export cargos in the nucleus by forming a trimeric XPO1/RanGTP/cargo complex. This complex is disassembled upon GTP hydrolysis in the cytoplasmic side of the NPC, leading to release of the export cargo in the cytoplasm. Thus, by regulating receptor/cargo interactions, the RanGTP/RanGDP gradient determines the directionality of nucleocytoplasmic transport. In fact, it has been shown that, by artificially raising the concentration of RanGTP in the cytoplasm, the direction of the transport can be inverted[25].
Beyond the basic transport machinery described above, multiple additional mechanisms may contribute to regulate the nucleocytoplasmic distribution of a given protein in a dynamic and finely-tuned manner. These mechanisms include post-translational modifications, such as phosphorylation[26,27] or ubiquitination (reviewed by Rodríguez[28]), as well as masking/unmasking of the transport signals by homo/heterodimerization[29,30].
XPO1-mediated protein nuclear export: cargos, mechanisms and signals
XPO1 has a wide repertoire of cargos, including not only cellular proteins, but also viral proteins expressed in infected cells (reviewed by Ding et al.[31]).
Identification of XPO1 cargos has been greatly facilitated by the use of LMB as an inhibitor. In cellular experiments, cytoplasmic XPO1 cargos often relocate to the nucleus in the presence of LMB. This experimental approach cannot be used to demonstrate XPO1-mediated export of proteins that are constitutively located to the nucleus. An alternative approach in this case could be ectopic overexpression of the receptor, which promotes export of NES-containing nuclear cargos to the cytoplasm[32]. Besides LMB-based experiments, the identification, validation and characterization of XPO1 cargos often involve biochemical analyses to demonstrate RanGTP-dependent binding, as well as mutagenesis to map the NES. In over 15 years of research, hundreds of individual proteins were studied using these approaches and around 200 bona-fide XPO1-exported cargos were identified[33]. More recently, the introduction of tandem mass spectrometry (MS/MS)-based high throughput analyses has expanded the repertoire of potential XPO1 cargos (the so-called “XPO1 exportome”) to above 1000 cellular proteins[34], although many of them still need to be further validated and their NESs identified.
The search for novel cargos is still on-going, and continues to provide further insight into the physiological relevance of XPO1. For example, it has been recently found that the NES-containing protein POST and the ubiquitin-binding protein UBIN form a complex that mediates XPO1-dependent nuclear export of polyubiquitinated proteins[35], a process that seems to be exacerbated in cancer cells treated with the proteasome inhibitor bortezomib[36]. These findings reveal a novel role for XPO1 in nuclear protein homeostasis that might also have important implications for cancer therapy.
From a mechanistic point of view, XPO1-mediated nuclear export consists essentially in the binding of an NES-containing protein in the nucleus and its release in the cytoplasm. The XPO1/NES interaction has low affinity, and needs to be stabilized by the cooperative binding of nuclear RanGTP, facilitated by the cofactor RanBP3[37-39]. Structural and biochemical studies carried out over the last decade have contributed to dissecting the series of molecular events that underlie the cycle of assembly and disassembly of the XPO1/RanGTP/NES complex (reviewed by Koyama and Matsuura[11], Fung and Chook[12] and Monecke et al.[13]). As schematically illustrated in Figure 2A, XPO1 is a ring-shaped protein with a concave inner surface and a convex outer surface. RanGTP binds to the inner surface, and NESs dock into a hydrophobic groove in the outer surface of the receptor. The open/close state of the NES binding groove is allosterically regulated by conformational rearrangements of two additional XPO1 structural elements, termed the H9 loop and the C-terminal extension, which play a crucial role in the cycle of NES binding and release.
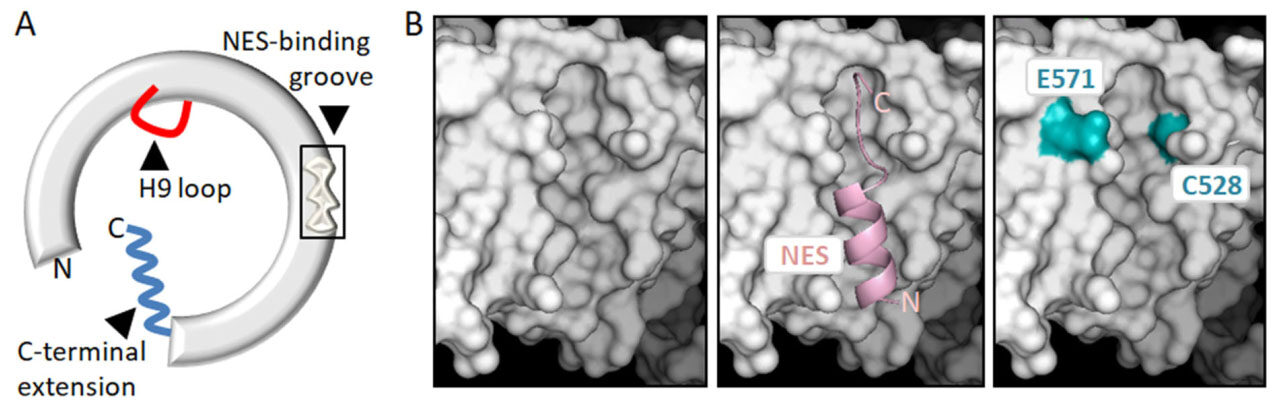
Figure 2. Structural features of XPO1 related to its nuclear export function, its role in cancer and its potential as a therapeutic target. A: Schematic representation of XPO1 protein illustrating its general ring-shaped conformation, and showing the three structural motifs that are crucial for its function as a nuclear export receptor: the NES-binding groove, the H9 loop and the C-terminal extension; B: detailed views of the NES-binding groove on the molecular surface of XPO1. The UCSF Chimera package[206] and XPO1 structure 3GJX[207] were used to generate the images. The left panel shows the empty groove, the middle panel shows a “leucine-rich” NES peptide (pink) bound to the groove. The right panel shows residues E571 and C528 highlighted in blue. E571 is a mutational hotspot in several hematological malignancies. C528 is the residue targeted by XPO1-inhibiting drugs, such as LMB or Selinexor. These compounds attach covalently to C528 and physically occupy the groove, blocking NES binding. NES: nuclear export signal; LMB: leptomycin B
As illustrated in Figure 1C, "leucine-rich" NESs conform to a loose consensus sequence with a characteristic spacing of hydrophobic residues[40,41]. Hundreds of different amino acid sequences have been experimentally validated as bona-fide NESs that bind the receptor with different affinity, and may be exported with different efficiency[33,42,43]. This high variability can be explained by the recent finding that NESs with different backbone conformations can bind the receptor, and that not all export signals occupy the XPO1 NES-binding groove to the same extent[44].
“Leucine-rich” NESs dock into the groove and engage in predominantly hydrophobic interactions with several XPO1 residues. Two non-hydrophobic amino acids (C528 and E571) located in or near the NES-binding groove [Figure 2B] are of particular interest regarding the targeting of XPO1 and its potential role in cancer. On one hand, the amino acid E571 is recurrently mutated in certain hematological malignancies (see below), suggesting that mutation of this particular residue can be a driver event in some types of cancer. On the other hand, C528 is the crucial target for the effect of LMB and more clinically relevant XPO1 inhibitors, which covalently bind to this residue and block NES binding by physically occupying the groove. In fact, experimental mutation of C528 renders cells resistant to these inhibitors[45].
Role of XPO1 in RNA nuclear export
Following transcription in the nucleus, active export to the cytoplasm is an essential step during the biogenesis of many classes of RNA, and/or a critical requirement for their function (recently reviewed by Williams et al.[46]). Thus, messenger RNAs (mRNAs) need to be exported to undergo translation into proteins, while ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) need to be transported to the cytoplasm in order to be processed or to carry out their cellular activities. Nuclear export of RNA is a tightly regulated process that involves the coordinated function of many different factors, including a large array of RNA-binding adaptor proteins as well as dedicated export receptors[6]. XPO1 plays a pervasive role in this process, mediating the nuclear export of several different classes of RNA.
XPO1 plays a prominent role in the export of 40S and 60S ribosomal subunits, containing rRNA, to the cytoplasm, which is a necessary step for their final maturation. The NES-containing protein Nmd3 functions as the adaptor for 60S subunit export, while the adaptor involved in the export of the 40S subunit remains to be identified[47].
In contrast to rRNAs, the vast majority of cellular mRNAs are exported to the cytoplasm by a receptor unrelated to karyopherins, called NXF1, but XPO1 mediates nuclear export of a subset of mRNAs[48]. Since XPO1 does not bind mRNA directly, NES-containing RNA-binding proteins that act as adaptors to bridge the interaction between XPO1 and mRNA are essential in this process. These adaptors include NXF3[49], and the HuR/APRIL/pp32[50] or the eIF4E/LRPPRC[51] complexes. Interestingly, several mRNAs exported by XPO1 code for proteins that are involved in tumorigenesis-related processes, such as invasion and metastasis[46].
Finally, some RNA species with an important role in the regulation of gene expression (snRNA, lncRNA and miRNA) can also be exported by XPO1[52-54]. For example, although the major exporter of miRNAs is not XPO1 but another exportin called XPO5, XPO1 mediated-export plays a role in the export and biogenesis of specific subsets of miRNAs[55,56]. Intriguingly, XPO1 has also been reported to mediate the nuclear import of mature miRNAs[57].
Nuclear export-independent role of XPO1 as a key regulator of mitosis
Besides mediating the export of proteins and RNA to the cytoplasm, XPO1 also plays a role in processes that do not directly involve nuclear export, such as intranuclear trafficking of small nucleolar RNAs (snoRNAs) from Cajal bodies to the nucleolus[58]. A particularly relevant aspect of cell physiology where XPO1 carries out export-independent functions is mitosis[8]. This role of XPO1 has been reviewed by Forbes et al.[59].
In eukaryotic cells undergoing open mitosis, the breakdown of the nuclear envelope at the onset of prometaphase dramatically disrupts the nucleocytoplasmic compartmentalization. With no physical separation between nucleus and cytoplasm, the nuclear transport machinery, including certain transport receptors, NUPs and the Ran GTPase, is "repurposed" to carry out transport-independent mitotic functions, such as regulating the assembly of the mitotic spindle[59]. In this context, XPO1 has been shown to function as a "mitotic effector" of Ran, mediating RanGTP-dependent targeting of key mitotic proteins to specific spindle structures, such as the centrosomes or the kinetochore. Thus, the NES-containing protein pericentrin, a crucial scaffold for microtubule nucleation at the spindle poles, is recruited to the centrosomes by XPO1 in a RanGTP-containing trimeric complex that resembles the nuclear export complexes described above[60]. On the other hand, the stable microtubule-kinetochore interactions necessary for proper chromosome segregation appear to require XPO1-mediated recruitment of a protein complex containing RanGTP, RanGAP1 and the nucleoporin RanBP2 to the kinetochores[8].
The mitotic functions of XPO1, like its nuclear export activity, seem to be the subject of careful regulation through mechanisms that include phosphorylation[61] and competition with importins[62].
In summary, although its primary role may be in protein nuclear export, XPO1 is a multifaceted protein with roles in other processes. This functional complexity should be taken into account when interpreting the results of XPO1 inhibition studies.
Pathological alteration of XPO1 in cancer
Altered nucleocytoplasmic localization of proteins in cancer
Normal cell function relies on the correct subcellular distribution of thousands of proteins. The presence of a critical protein in the wrong cellular compartment may have severe pathological consequences. For example, aberrant cytoplasmic localization of a physiologically nuclear tumor suppressor protein may render this protein inactive, and thus contribute to tumorigenesis. In fact, mislocalization of cancer-related proteins, including the products of prominent oncogenes and tumor suppressor genes, has been often demonstrated in human tumors[63,64-66].
Nucleocytoplasmic localization of proteins can be disrupted by different mechanisms in cancer cells. On one hand, the trafficking of a specific protein can be altered by mutations that either interfere with the activity of its transport signals (NLSs or NESs) or that create a novel signal in the mutant protein. For example, aberrant localization of tumor suppressors BRCA2[67] and PALB2[68] to the cytoplasm can result from mutations that unmask normally hidden NESs, whereas cytoplasmic mislocalization of certain NPM1 mutants is the result of a frameshift mutation that creates a novel strong NES, not present in the wild-type protein[69].
On the other hand, a general defect in the nucleocytoplasmic localization of proteins (and RNA) may arise in tumor cells, if elements of the transport machinery themselves are genetically altered or aberrantly expressed[70]. Examples of genetic alterations targeting the nuclear transport machinery include chromosome rearrangements involving nucleoporin genes (e.g., NUP98 and NUP214) in hematologic malignancies[71]. The abnormal fusion proteins resulting from these translocations have been reported to disrupt XPO1-mediated export[72,73]. Examples of nuclear transport factors abnormally expressed in tumors include the nuclear import receptors Importin β (see Dickmanns et al.[64] and references therein) and Importina1 (see Christiansen and Dyrskjøt[74], and references therein).
In the case of XPO1, both aberrant expression and genetic alterations have been detected in different types of cancer, as detailed below. The abnormal XPO1 function that may result from these alterations would, in turn, hinder the normal nucleocytoplasmic localization of hundreds of XPO1 cargo proteins. In the context of the present review, those XPO1 cargos with a known role in the development of human tumors are of particular interest. In this regard, we note that the extended list of potential XPO1 cargos identified in HeLa cells by a recent high throughput analysis[34] includes 136 members of the protein class "cancer-related genes" registered in the Human Protein Atlas initiative (https://www.proteinatlas.org/) [Figure 3]. The set of cancer-related proteins exported by XPO1 (which could be referred to as the "XPO1 cancer exportome") includes prominent tumor suppressors, such as p53[75] and BRCA1[76], as well as protooncogenes, such as c-abl[77]. A more extensive account of cancer-related proteins that undergo XPO1-mediated nuclear export can be found in previous reviews[10,65,66,78,79].
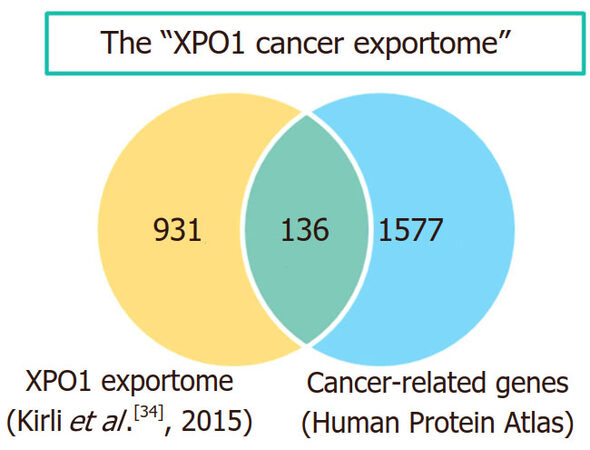
Figure 3. The "XPO1 cancer exportome". The Venn diagram shows the overlap between the list of potential XPO1 cargos identified in HeLa cells[34] and the group of "cancer related genes" defined in the Human Protein Atlas (v.18). The 136 overlapping proteins represent what could be referred to as the “XPO1 cancer exportome”. The diagram was created using the jvenn web tool[208]
Altered XPO1 expression in human tumors
The expression level of XPO1 at either the mRNA or protein level has been analyzed in many different cancer types. As summarized in Table 1, XPO1 is frequently overexpressed in tumor samples with respect to the corresponding normal tissue samples[80-99]. In fact, XPO1 overexpression was observed in all solid tumor types and hematologic malignances examined, with the exception of liver cancer[91].
Expression of XPO1 in human cancer
| Type of cancer | Number of samples (technique) | Relationship to prognosis | Remarks | Ref. |
|---|---|---|---|---|
| AML | 511 patients (Reverse-phase protein array) | High XPO1 expression associated with poor prognosis | [80] | |
| MCL | Data from public expression array data sets plus 3 patients and 8 cell lines (qRT-PCR) | NA | Higher expression in mantle cell lymphoma cells | [81] |
| MM | Data from public expression array data sets | NA | XPO1 expression increases with disease progression | [82] |
| 351 patients (data from public expression array data sets) plus 8 patients (IB) | High XPO1 expression associated with poor prognosis | [83] | ||
| Glioma | 273 patients (data collected from public expression array data sets) plus 12 patients (IB) | High XPO1 expression associated with poor prognosis | [84] | |
| 70 patients (IHC + IB) | High XPO1 expression associated with poor prognosis | XPO1 expression level inversely correlated with p27 level | [85] | |
| Non-small cell lung cancer | 148 patients (microarray) plus 291 patients (qRT-PCR) | High XPO1 expression associated with poor prognosis | XPO1, BRCA1, HIF1A and DLC1 constitute a robust prognostic classifier in stage I lung adenocarcinoma patients | [86] |
| Gastric cancer | 240 patients (IHC) | Low XPO1 expression associated with poor prognosis | [87] | |
| 120 patients (IHC) | High XPO1 expression associated with poor prognosis | [88] | ||
| Thymic epithelial tumors | 118 patients (IHC) | High XPO1 expression associated with poor prognosis | [89] | |
| Ovarian cancer | 88 patients (IHC) | XPO1 is expressed in ovarian carcinomas with aggressive behavior and is related to poor patient survival | XPO1 expression correlated with cyclooxygenase-2 expression | [90] |
| Liver cancer | 154 cases (IHC) | NA | XPO1 expression assessed in tumor and adjacent normal tissues | [91] |
| Esophageal squamous cell carcinoma | 220 patients (IHC+IB+IF) | High XPO1 expression associated with shorter survival | XPO1 siRNA causes apoptosis in esophageal cancer cell lines | [92] |
| 56 patients (IHC+qRT-PCR) | Trend for shorter survival of patients with higher XPO1 expression | XPO1 localization also altered in cancer cells | [93] | |
| Pancreas cancer | 69 patients (IB) | High XPO1 expression associated with poor prognosis | [94] | |
| Renal cancer | (IHC) | NA | Higher XPO1 expression in higher grade tumors | [95] |
| Breast carcinoma | 280 patients (IHC + IB) | High XPO1 expression associated with poor prognosis | [96] | |
| Ewing sarcoma | 37 patients (IHC) plus data from public expression array data sets | NA | XPO1 is highly expressed in Ewing sarcoma | [97] |
| Osteosarcoma | 57 patients (IHC) | High XPO1 expression associated with shorter survival | [98] | |
| Melanoma | 83 patients (31 primary, 52 metastatic) (MA) | NA | XPO1 overexpressed in metastatic melanoma | [99] |
In several of these studies, the potential prognostic significance of XPO1 expression has been evaluated. Higher XPO1 expression was associated with poorer patient prognosis in patients with ovarian tumors[90], pancreatic tumors[94], esophageal tumors[92], gliomas[84,85], thymic epithelial tumors[89], and breast tumors[96]. In contrast, high XPO1 expression was related to better prognosis in osteosarcoma patients[98]. Finally, contradictory findings on the prognostic value of XPO1 expression in gastric cancer have been reported[87,88].
The molecular mechanisms responsible for XPO1 overexpression in cancer cells are still poorly characterized. Copy number gains at chromosomal region 2p, affecting the XPO1 locus, have been found to correlate with high XPO1 mRNA expression in lymphomas[100] and chronic lymphocytic leukemia (CLL)[101]. In addition, XPO1 transcription has been reported to be regulated by cMyc and p53[102,103], two proteins that are frequently altered in cancer. Conceivably, disruption of this regulation may contribute to aberrant XPO1 expression in some tumors, although further studies are required to test this possibility.
A recurrent XPO1 gene mutation in hematological malignancies
Missense mutations of the glutamic residue E571 (mostly E571K) have been detected in around 25% of patients with two specific types of hematological malignancies: primary mediastinal B-cell lymphoma (PMBL)[100], and classical Hodgkin’s lymphoma (HL)[104]. The E571 "hotspot" mutations in XPO1 were first detected by whole genome sequencing analysis of CLL samples[105]. A large number of targeted studies[100,101,104-124][Table 2] have subsequently confirmed the presence of E571 missense mutations in around 5% of CLL patients.
Recurrent XPO1 mutations in hematological malignancies. More than 90% of the reported mutations are missense changes affecting XPO1 “hotspot” residue E571
| Type of malignancy | Samples with XPO1 mutations | Ref. |
|---|---|---|
| Chronic lymphocytic leukemia | 4/165 (2.4%) | [105] |
| 2/105 (1.9%) | [106] | |
| 6/192 (3.1%) | [107] | |
| 7/160 (4.4%) | [108] | |
| 33/969 (3.4%) | [109] | |
| 1/10 (10%) | [110] | |
| 6/24 (25%) | [111] | |
| 4/159 (2.5%) | [112] | |
| 2/12 (16.7%) | [113] | |
| 13/136 (9.5%) | [114] | |
| 25/538 (4.6%) | [115] | |
| 17/114 (14.9%) | [116] | |
| 2/25 (8%) | [117] | |
| 14/180 (7.8%) | [118] | |
| 7/61 (11.5%) | [119] | |
| 38/486 (7,8%) | [120] | |
| 25/436 (5.7%) | [101] | |
| 4/56 (7.1%) | [121] | |
| 28/288 (9.7%) | [122] | |
| Aggregate chronic lymphocytic leukemia | 238/4116 (5.8%) | |
| Primary mediastinal B-cell lymphoma | 28/117 (24%) | [100] |
| 7/18 (38.9%) | [123] | |
| Aggregate primary mediastinal B-cell lymphoma | 35/135 (25.9%) | |
| Hodgkin’s lymphoma | 22/91 (24.2%) | [104] |
| 5/19 (26%) | [100] | |
| 6/34 (18%) | [124] | |
| Aggregate Hodgkin’s lymphoma | 33/144 (22.9%) | |
| Other diffuse large B-cell lymphoma | 1%-3% | [123] |
| 0%-1.5% | [100] | |
In CLL, the presence of XPO1 mutations is often associated with unmutated IGHV status[105,109,120], but does not seem to be a marker of poor patient prognosis[120]. Similarly, HL patients with XPO1 mutations do not appear to have a worse prognosis than patients with wild type XPO1[104]. In contrast, a shorter progression-free survival was reported for PMBL patients bearing XPO1 mutations[100]. Interestingly, it has been suggested that XPO1 mutations could represent useful biomarkers to evaluate minimal residual disease in HL and PMBL[125].
Isolated instances of mutant XPO1 have been reported in esophageal[126] and thyroid cancer[127], but XPO1 genetic alterations seem to be a very rare event in solid tumors.
It is still unclear why XPO1 E571 mutations are particularly common in certain types of cancer, and why they may have different prognostic significance in different types of hematological malignancies. In fact, the molecular mechanisms that may be responsible for the pathogenic effect of XPO1 mutations remain to be established. Consistent with the location of the mutational "hotspot" proximal to the NES-binding site [Figure 2B], it has been reported that the E571K mutation subtly increases the affinity of the receptor for some NESs with a negatively charged carboxy-terminal end[32]. Conceivably, this could lead to altered export of one or more cargos, whose mislocalization might in turn contribute to tumorigenesis.
Given its frequent alteration in human tumors, and its crucial cellular roles described above, XPO1 has long been regarded as a potentially relevant target in cancer therapy.
XPO1 inhibition in cancer therapy
Development and preclinical evaluation of selective inhibitors of nuclear export
Even before XPO1 was identified as its cellular target, LMB (also called elactocin) had been found to possess antitumor activity, and it had been tested in a clinical trial[128].
LMB was found to have severe toxicities when administered to patients, precluding its development as a clinically useful drug[128]. Nevertheless, the availability of this potent and specific inhibitor made it possible to carry out proof-of-concept experiments testing the effect of XPO1 inhibition in different tumor settings. As an illustrative example, we will briefly describe some early data regarding the effect of LMB treatment in chronic myelogenous leukemia (CML) cells expressing the BCR-ABL oncoprotein. Shortly after the identification of XPO1 as a nuclear export receptor, the c-ABL kinase was identified as one of its cargos, bearing a C-terminal NES that is also present in the BCR-ABL fusion protein[77]. At that time, treatment of BCR-ABL-positive CML patients was undergoing a dramatic improvement with the introduction of the kinase inhibitor imatinib[129,130]. In this context, it was reported that the sequential combination of imatinib plus LMB led to the nuclear entrapment of BCR-ABL, which selectively induced apoptosis of CML cells[131]. Furthermore, subsequent experiments showed that the combination with LMB could overcome imatinib resistance due to BCR-ABL amplification[132].
These and other encouraging findings in different tumor types (reviewed by Turner and Sullivan[7]) suggested that XPO1 inhibition might represent a valid strategy for cancer treatment, fostering the search for other XPO1 inhibitors. Over the next years, several natural and synthetic inhibitors of XPO1 were reported (reviewed by Tan et al.[10] and Senapedis et al.[133]). Similar to LMB, these compounds bind covalently to XPO1 residue C528, and occupy the NES-binding groove, blocking access to NESs. However, unlike LMB, some of these novel inhibitors, such as CBS9106 or S109, bind to XPO1 in a reversible manner, which was associated to less severe toxicity in preclinical in vivo models[84,134]. Studies with these compounds further validated XPO1 inhibition as a relevant strategy for cancer treatment. For example, blocking nuclear export of topoisomerase II with the XPO1 inhibitor Ratjadone C was found to sensitize multiple myeloma (MM) cells to doxorubicin and etoposide[135].
While most XPO1 inhibitors have only been tested in vitro or in mouse xenograft, there is a series of compounds, termed selective inhibitors of nuclear export (SINEs) that are undergoing development as potential anticancer drugs, and some of these compounds are already being evaluated in clinical trials[133].
SINEs were developed in 2012 using structure-assisted relationship methodology combined with a novel computational approach termed consensus-induced fit docking[136,137], a strategy that relied crucially on the recently solved structures of NES-bound XPO1. The “first-generation” series of SINEs included a relative large number of slowly reversible XPO1 inhibitors, such as KPT-127, KPT-185, KPT-205, KPT-227, KPT-249, KPT-251, KPT-276 and KPT-330 (Selinexor). As summarized in Tables 3 and 4, SINE compounds have been extensively tested in preclinical models of many different hematological malignancies[80,82,83,100,101,137-157] and solid tumors[89,95,97,158-188]. In these models, SINEs have demonstrated potent in vitro and in vivo activity against cancer cells (including growth inhibition, induction of apoptosis, and cell cycle arrest), with only minor toxic effects on normal cells. Importantly, several SINEs (most prominently Selinexor) have been shown to increase the sensitivity of cancer cells to currently used drugs, such as doxorubicin or the proteasome inhibitors bortezomib and carfilzomib, and also to synergize with other targeted therapeutic agents, such as ibrutinib (an inhibitor of Bruton tyrosine kinase) or linsitinib (an inhibitor of insulin-like growth factor receptor-1). A more extensive and detailed discussion of the preclinical results obtained with SINEs in specific tumor settings can be found in recent reviews[9,10,79,133,189].
Summary of preclinical studies with "first-generation" SINEs in hematological malignancies
| Malignancy type | Preclinical model(s) | SINE(s) | Cellular effects | Remarks | Ref. |
|---|---|---|---|---|---|
| MM | In vitro/patient-derived cells/xenograft/mouse model | KPT-276 | Growth inhibition, apoptosis | [82] | |
| In vitro/patient-derived cells | Selinexor KPT-127 KPT-185 KPT-249 KPT-276 | Reduced viability, apoptosis | SINEs sensitize MM cells to doxorubicin, bortezomib, and carfilzomib. Overcome stroma cell-promoted drug resistance | [138] | |
| In vitro/xenograft | Selinexor KPT-185 | Growth inhibition, apoptosis | SINEs inhibit MM-induced bone lysis. Mechanism related to NF-κB pathway and NFATc1 | [83] | |
| In vitro/patient-derived cells/xenograft | Selinexor | Apoptosis | Synergizes with carfilzomib. Apoptosis mediated by caspase 10 | [139] | |
| In vitro/xenograft/ patient biopsies | Selinexor | Apoptosis, DNA damage | Restores sensitivity to doxorubicin. Synergizes with doxorubicin | [140] | |
| In vitro/xenograft/ patient biopsies | Selinexor | Growth inhibition | Restores sensitivity to bortezomib and carfilzomib. Mechanism related to NF-κB pathway | [141] | |
| In vitro/xenograft | Selinexor | Delay in tumor initiation and progression, apoptosis | Overcomes hypoxia-induced bortezomib resistance | [142] | |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis | Synergizes with dexamethasone. Mechanism related to glucocorticoid receptor and mTOR pathway | [143] | |
| AML | In vitro/xenograft | KPT-185 KPT-276 | Reduced proliferation, apoptosis, cell cycle arrest, myeloid differentiation | [144] | |
| In vitro/xenograft | KPT-251 | Apoptosis | [145] | ||
| In vitro/patient-derived cells | KPT-185 | Reduced proliferation, apoptosis | Synergizes with MDM2 inhibitor. p53-dependent apoptosis | [80] | |
| In vitro/xenograft | Selinexor | Priming with decitabine enhances Selinexor activity | [146] | ||
| In vitro/xenograft | Apoptosis, myeloid differentiation | Synergizes with sorafenib in xenografts of FLT3-mutated AML | [147] | ||
| AML and ALL | In vitro/xenograft | Selinexor | Apoptosis | Little toxicity to normal haematopoietic cells | [148] |
| CLL | In vitro/xenograft | KPT-185 KPT-251 | Apoptosis | SINEs counteract protective effects of the microenvironment | [137] |
| In vitro/mouse model | Selinexor | Reduced proliferation | [149] | ||
| Patient-derived cells/mouse models | Selinexor | Synergizes with ibrutinib | [150] | ||
| In vitro/patient-derived cells | Selinexor | Apoptosis | XPO1 mutation or chromosome 2p gains decrease Selinexor sensitivity | [101] | |
| CML and ALL | In vitro/mouse model | Selinexor | Apoptosis | Compassionate use in a patient with TKI-resistant CML reduced disease signs. Mechanism related to reactivation of PP2A | [151] |
| CML | In vitro/xenograft | Selinexor | Sensitizes CML cells to imatinib | [152] | |
| MCL | In vitro/xenograft | KPT-185 KPT-276 | Growth inhibition, apoptosis | [153] | |
| In vitro | KPT-185 | Growth inhibition, repression of ribosomal biogenesis, alterations in cell metabolism | [154] | ||
| NHL | In vitro/xenograft | KPT-185 KPT-251 KPT-276 | Growth inhibition, apoptosis, cell cycle arrest | Cellular effects dependent on p53 and p73 | [155] |
| In vitro/xenograft | KPT-185 KPT-276 | Growth inhibition, apoptosis, cell cycle arrest | [156] | ||
| In vitro/mouse model | Selinexor KPT-251 KPT-276 | Apoptosis | Dexamethasone or everolimus enhance Selinexor activity | [157] | |
| PMBL | In vitro | Selinexor KPT-185 | Reduced proliferation, apoptosis | E571K mutation does not affect SINE activity | [100] |
Summary of preclinical studies with “first-generation” SINEs in human solid tumors
| Tumor type | Preclinical model(s) | SINE(s) | Cellular effects | Remarks | Ref. |
|---|---|---|---|---|---|
| Renal cancer | In vitro/xenograft | KPT-185, KPT-251 | Growth inhibition, apoptosis, cell cycle arrest | SINEs compare favorably to sorafenib | [95] |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis | [158] | ||
| Prostate cancer | In vitro/xenograft | Selinexor KPT-185, KPT-251 | Reduced proliferation, apoptosis (no cell cycle arrest) | SINEs synergize with doxorubicin | [159] |
| In vitro/xenograft | Selinexor, KPT-251 | SINEs reduce bone metastasis | [160] | ||
| In vitro/xenograft | Selinexor, KPT-251 | Growth inhibition, apoptosis, increased DNA damage | SINEs sensitize cells to docetaxel | [161] | |
| Breast cancer | In vitro/xenograft | Selinexor, KPT-185, KPT-251, KPT-276 | Growth inhibition, apoptosis | Mechanism related to STAT3 and survivin | [162] |
| In vitro/xenograft | Selinexor | Restores sensitivity to tamoxifen | [163] | ||
| In vitro/xenograft | Selinexor | Growth inhibition | Effective as single agent and synergizes with chemotherapy in triple-negative BC | [164] | |
| Ovarian cancer | In vitro | Selinexor, KPT-185 | Apoptosis | SINEs synergize with chemotherapy. Mechanism related to IGF2BP1 | [165] |
| In vitro/xenograft | Selinexor, KPT-185 | Apoptosis (p53-dependent and p53-independent) | Overcomes resistance to platinum compounds | [166] | |
| In vitro | Selinexor | Reduced proliferation, apoptosis | Synergizes with cisplatin. Mechanism related to FoxO1 | [167] | |
| Colorectal cancer | In vitro/xenograft | Selinexor | Reduced proliferation, apoptosis | Synergizes with radiotherapy | [168] |
| Liver cancer | In vitro/xenograft | Selinexor | Growth inhibition, apoptosis cell cycle arrest | [169] | |
| Pancreatic cancer | In vitro/xenograft | KPT-185, KPT-127, KPT-205, KPT-227 | Reduced proliferation, apoptosis | Mechanism related to PAR-4 | [170] |
| In vitro/xenograft | KPT-185 | Reduced proliferation and migration, apoptosis | Mechanism related to Fbw7 and Notch-1 | [171] | |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis | Synergizes with gemcitabine | [172] | |
| In vitro | Selinexor | Reduced proliferation and migration | Mechanism related to miR-145 micro RNA | [173] | |
| Non-small cell lung cancer | In vitro/xenograft | Selinexor | Reduced proliferation, apoptosis, cell cycle arrest | Synergizes with cisplatin. Effective against NSCLC cells with different molecular alterations | [174] |
| In vitro/xenograft | KPT-185, KPT-276 | Reduced viability, apoptosis, cell cycle arrest | KPT-185 active against NSCLC cell lines resistant to EGFR-TKIs | [175] | |
| In vitro/xenograft | Selinexor, KPT-185 | Synthetic-lethal interaction with K-Ras. Mechanism related to NF-κB pathway | [176] | ||
| Thyroid cancer | In vitro/xenograft | Selinexor | Growth inhibition, apoptosis, cell cycle arrest | Synergizes with doxorubicin | [177] |
| Thymic epitelial tumors | In vitro/xenograft | Selinexor | Reduced proliferation, apoptosis | p53-dependent and independent effect | [89] |
| Sarcoma | In vitro/xenograft | Selinexor | Growth inhibition, apoptosis, cell cycle arrest | Study on liposarcoma | [178] |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis | Synergizes with proteasome inhibitors. Mechanism related to NF-κB pathway | [179] | |
| In vitro/xenograft | Selinexor | Cell cycle arrest | Activity against a wide variety of sarcoma models including liposarcoma and gastrointestinal stromal tumor | [180] | |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis | Marfilzomib increases sensitivity to Selinexor. Mechanism related to NF-κB pathway and survivin | [181] | |
| In vitro/xenograft | Selinexor | Growth inhibition, apoptosis, cell-cycle arrest | Study on Ewing sarcoma. Synergizes with the IGF-1R inhibitor linsitinib | [97] | |
| Mesothelioma | In vitro/xenograft | Selinexor, KPT-251, KPT-276 | Growth inhibition, apoptosis, cell-cycle arrest | SINEs synergize with survivin inhibitor YM155 | [182] |
| Glioma | Patient-derived cells/xenograft | Selinexor, KPT-251, KPT-276 | Growth inhibition, apoptosis | [183] | |
| In vitro/xenograft | Selinexor | Enhances radiosensitivity of glioblastoma cells | [184] | ||
| Melanoma | In vitro/xenograft | Selinexor, KPT-185, KPT-251, KPT-276 | Growth inhibition, apoptosis, cell cycle arrest | SINEs synergize with BRAF inhibitors. Independent of BRAF mutational status | [185] |
| In vitro/xenograft | Selinexor, KPT-276 | Growth inhibition, apoptosis | Independent of BRAF mutational status | [186] | |
| Multiple types (solid) | In vitro | Selinexor | DNA damage (double-strand breaks) | Synergizes with DNA-damaging agents | [187] |
| Pediatric cancer (solid and hematological) | In vitro/xenograft | Selinexor | Activity against a broad range of pediatric cancer types. Independent of TP53 mutation status | [188] |
In general terms, the anticancer effect of XPO1 inhibition is thought to rely on the relocation of mislocalized XPO1 cargos with tumor-suppressive and growth-regulatory functions (e.g., p53) to the nucleus, where they carry out their normal activity. In our opinion, this may be an overly simplistic view. Given the large number of potential XPO1 cargos with a role in cancer, the export-independent roles of XPO1, and the complex nature of the tumorigenesis process, the specific molecular and cellular mechanisms underlying the anticancer effect of SINEs may differ in different tumor settings. In this regard, as indicated in Tables 3 and 4, preclinical studies are providing important information on tumor context-specific proteins and signaling pathways that may mediate SINE activity, such as the BCR-ABL oncoprotein in CML mentioned above, or the NF-κB pathway in lung cancer[176].
Intriguingly, there is emerging evidence that, in addition to cancer, other conditions, such as demyelinating diseases[190] or viral infections[191] might be amenable to treatment with SINEs.
Evaluation of Selinexor in clinical trials
In preclinical studies, Selinexor compared favorably to other “first-generation” SINEs in terms of the balance between potency and tolerability. Selinexor, an orally available drug, is the only compound of the series that has advanced into clinical development for human cancer.
The ClinicalTrials.gov site (https://clinicaltrials.gov/, accessed on 11 Jun 2018) registers 60 clinical studies on different tumor types with Selinexor as single agent or in combination with other drugs. In addition, there are isolated clinical trials registered for other XPO1 inhibitors, such as the “second-generation” SINE compound KPT-8602 (Eltanexor) or the non-SINE compound SL-801.
Interim data from some clinical studies with Selinexor have been reported as meeting proceedings (some of these data are reviewed by Mahipal and Malafa[192]). Here, we will limit our discussion to the results of phase I and II trials that have undergone full peer-reviewed publication as PubMed-indexed articles (summarized in Table 5).
Summary of the results of published clinical studies of Selinexor in human malignancies
| Tumor type | Phase | Remarks | Reference/ID |
|---|---|---|---|
| Advanced solid tumors | Phase I | Selinexor single agent. 189 patients enrolled. Most common grade ≥ 3 adverse events: thrombocytopenia, fatigue and hyponatremia. RP2D 35 mg/m2 given twice weekly. 157 evaluable patients. 1 CR and 5 PR | [193] ID: NCT01607905 |
| Sarcoma | Phase I | Selinexor single agent. 54 patients enrolled. Most common grade ≥ 3 adverse events: fatigue, thrombocytopenia, anemia, lymphopenia, and leucopenia. 52 evaluable patients. SD: 33% | [194] ID: NCT01896505 |
| Pediatric refractory acute leukemia | Phase I | Selinexor combined with Fludarabine and Cytarabine. 18 patients enrolled. Selinexor tolerable at doses up to 55 mg/m2 in pediatric patients. 15 evaluable patients. CR: 47% | [195] ID: NCT02212561 |
| Non-Hodgkin lymphoma | Phase I | Selinexor single agent. 79 patients enrolled. Most common grade ≥ 3 adverse events: thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, and hyponatremia. RP2D 60 mg. 70 evaluable patients. OR: 31% (including 4 CR and 18 PR) | [196] ID: NCT01607892 |
| Acute myeloid leukemia | Phase I | Selinexor single agent. 95 patients enrolled. Most common grade ≥ 3 nonhematological adverse event: fatigue. No reported dose-limiting toxicities. RP2D 60 mg. 81 evaluable patients. OR: 14% | [197] ID: NCT01607892 |
| Phase I | Selinexor combined with cytarabine and mitoxantrone. 20 patients enrolled. Serious adverse events 30%, including one fatal adverse event. RP2D: 80 mg. 20 evaluable patients. Overall response rate 70% (including 10 CR) | [198] ID: NCT02573363 | |
| Multiple myeloma | Phase I | Dose-escalation phase: Selinexor as single agent in 25 patients enrolled. Dose-expansion phase: Selinexor as single agent or combined with dexamethasone. 59 patients enrolled. Most common grade ≥ 3 adverse event: thrombocytopenia. RP2D: 80 mg plus 20 mg dexamethasone given twice weekly. Objective response rate: 4% Selinexor as single agent, 50% when combined with dexamethasone | [199] ID: NCT01607892 |
| Phase II | Selinexor combined with dexamethasone. 79 patients (multi-refractory disease) enrolled. Most common grade ≥ 3 adverse events: thrombocytopenia, anemia, neutropenia, hyponatremia, leukopenia, and fatigue. Overall response rate 21% | [200] | |
| Castration-resistant prostate cancer | Phase II | Selinexor as single agent. 14 patients (refractory to anti-androgenic therapy) enrolled. Some activity (PR 25%), but poor tolerability. | [201] ID: NCT02215161 |
Abdul Razak et al.[193] evaluated the safety, pharmacokinetics, pharmacodynamics, and efficacy of Selinexor in 189 patients with advanced solid tumors, testing several doses and administration schedules. The most common grade 3 or 4 toxicities in this series were thrombocytopenia, fatigue, and hyponatremia. One hundred and fifty seven patients were evaluable for response. Seven patients achieved partial or complete response, and 27 patients achieved stable disease for ≥ 4 months. The authors concluded that Selinexor is a safe therapeutic with broad antitumor activity, and proposed a recommended phase II dose (RP2D) of 35 mg/m2 with a twice-a-week dosing schedule.
Gounder et al.[194] carried out a phase I study on 54 patients with advanced soft tissue or bone sarcoma. Selinexor was administered twice per week at doses of 30 mg/m2, 50 mg/m2, or 60 mg flat dose. The most common grade 3 or 4 toxicities in this series were fatigue, thrombocytopenia, anemia, lymphopenia, and leucopenia. In 52 evaluable patients, no objective responses were seen, but 17 patients achieved stable disease for ≥ 4 months. The authors concluded that Selinexor shows preliminary evidence of anticancer activity in sarcoma, and is well tolerated at a 60 mg flat dose.
Alexander et al.[195] evaluated the combination of Selinexor with fludarabine and cytarabine in 18 pediatric patients with relapsed or refractory leukemia. Selinexor was administered twice per week at several doses between 30 mg/m2 and 70 mg/m2. Dose-limiting reversible cerebellar toxicity was experienced by some patients treated with the 70 mg/m2 dose. Seven of the 15 patients that were evaluable achieved complete response. The authors concluded that Selinexor combined with fludarabine and cytarabine shows promising response rates in pediatric patients with relapsed or refractory leukemia. A 55 mg/m2 dose Selinexor is tolerable in this combination.
Kuruvilla et al.[196] evaluated Selinexor in 79 patients with different subtypes of non-Hodgkin lymphoma (NHL). In the dose-expansion phase of the study, Selinexor was administered at doses of 35 mg/m2 or 60 mg/m2. The most common grade 3 or 4 toxicities in this series were thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, and hyponatremia. Twenty-two objective responses (4 of them complete responses) were observed in 70 evaluable patients. The authors concluded that Selinexor shows encouraging activity in NHL patients, and proposed 35 mg/m2 Selinexor (60 mg flat dose) as the RP2D.
Garzon et al.[197] carried out a phase I dose-escalation study in 95 patients with relapsed or refractory acute myeloid leukemia (AML). Several doses and administration schedules were tested. The most common grade 3 or 4 toxicities were thrombocytopenia, anemia, fatigue, and neutropenia. Objective responses were observed in 11 of the 81 evaluable patients. The authors concluded that Selinexor is a safe therapy in AML patients, and established the RP2D at 35 mg/m2 (60 mg flat dose) given twice weekly. Another phase I dose-escalation trial in AML evaluated the combination of Selinexor with high-dose cytarabine and mitoxantrone in 20 patients[198]. Selinexor doses of 60 mg or 80 mg were administered. Serious toxicities, including one fatal adverse event, occurred in 30% of the patients. The overall response rate was 70%, including 10 patients achieving complete remission. The authors concluded that Selinexor combined with high-dose cytarabine and mitoxantrone is a feasible and tolerable treatment in AML patients, and proposed 80 mg Selinexor twice weekly as the RP2D in this combination.
Chen et al.[199] evaluated Selinexor in 84 patients with heavily pretreated MM (81 patients) or Waldenstrom macroglobulinemia (3 patients). Single agent Selinexor was given to 25 patients in the dose-escalation phase. In the dose-expansion phase, Selinexor was administered in combination with dexamethasone to 59 patients. The most commonly reported grade 3 or 4 toxicity in this series was thrombocytopenia. Although the efficacy of Selinexor as single agent was modest, its combination with dexamethasone resulted in a significantly increased activity, with an objective response rate of 50%. The authors proposed a RP2D of 80 mg Selinexor plus 20 mg dexamethasone given twice weekly. This treatment regimen was administered to 79 patients with multi-refractory MM in a recently reported phase II study[200]. The overall response rate (the primary endpoint of the study) was 21% and the most frequent grade 3 or 4 toxicity was thrombocytopenia. In relation to these studies, a more extensive and detailed discussion on the clinical implementation of Selinexor in MM has been recently published[189].
The last clinical study on Selinexor published to date (May 2018) is a phase II trial that evaluated its efficacy and tolerability in 14 patients with metastatic, castration-resistant, prostate cancer[201]. Selinexor was administered twice weekly at a dose of 65 mg/m2 that had to be subsequently reduced to a flat dose of 60 mg to improve tolerability. In fact, although Selinexor showed some evidence of clinical activity (reduction in prostate-specific antigen levels, and radiographic response), it was poorly tolerated in this patient population.
In summary, the results of these studies show that Selinexor, as single agent or in combination with other drugs, has broad clinical activity in multiple types of solid tumors and hematological malignancies and is generally well tolerated by patients. One of the most common high-grade toxicities experienced by patients treated with Selinexor is thrombocytopenia. The mechanism underlying this adverse event has been recently elucidated. Machlus et al.[202] have shown that thrombocytopenia results from reduced maturation of megakaryocyte progenitors due to Selinexor-mediated inhibition of thrombopoietin signaling. Importantly, the severity of thrombocytopenia could be reduced by temporary interruption of Selinexor treatment and administration of thrombopoietin mimetics to patients[202].
Altogether, these clinical data support the view that inhibition of XPO1 represents a valid therapeutic strategy in cancer.
Conclusion and future directions
Twenty years after its identification as a receptor that mediates the nuclear export of proteins, there is compelling evidence that XPO1 represents a relevant target in cancer. Further basic, preclinical and clinical investigations are required to address several salient questions on the use of XPO1 inhibition as a therapeutic approach.
On one hand, novel XPO1 inhibitors with more favorable clinical properties are being developed. In this regard, a "second-generation" SINE (KPT-8602 or Eltanexor) has demonstrated improved tolerability in preclinical models[203-205] and is currently undergoing clinical evaluation.
On the other hand, it is necessary to further elucidate the mechanisms that mediate the oncogenic role of XPO1 alterations (overexpression or mutation) in different types of cancer and to better characterize the molecular and cellular mechanisms underlying the effect of XPO1 inhibitors. This basic mechanistic information, which is still rather limited, would be crucial to successfully implement XPO1-targeting drugs in the clinic, as it could help to design rational combinations with other agents, to identify subsets of patients that may benefit more from the treatment and to improve the clinical management of adverse effects.
Declarations
AcknowledgmentsWe thank our colleagues at the UPV/EHU Dr. Sonia Bañuelos (Dept. of Biochemistry and Molecular Biology), Dr. Gorka Prieto (Dept. of Communication Engineering) and Dr. Asier Fullaondo (Dept. of Genetics, Physical Anthropology and Animal Physiology) for their help in preparing Figures 2 and 3. Supported by grants from the Spanish Government MINECO-FEDER (SAF2014-57743-R), the Basque Country Government (IT634-13) and the University of the Basque Country (UFI11/20), as well as a fellowship from the Basque Country Government (to MS).
Authors’ contributionsConception of the work, draft and revision of the work: Sendino M, Omaetxebarria MJ, Rodríguez JA
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997;390:308-11.
2. Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 1997;90:1051-60.
3. Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997;90:1041-50.
4. Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 1997;278:141-4.
5. Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 2007;17:193-201.
6. Okamura M, Inose H, Masuda S. RNA export through the NPC in Eukaryotes. Genes (Basel) 2015;6:124-49.
7. Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem 2008;15:2648-55.
8. Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T, McNally J, Dasso M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol 2005;7:626-32.
9. Gravina GL, Senapedis W, McCauley D, Baloglu E, Shacham S, Festuccia C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol 2014;7:85.
10. Tan DS, Bedard PL, Kuruvilla J, Siu LL, Razak AR. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov 2014;4:527-37.
11. Koyama M, Matsuura Y. Mechanistic insights from the recent structures of the CRM1 nuclear export complex and its disassembly intermediate. Biophysics (Nagoya-shi) 2012;8:145-50.
12. Fung HY, Chook YM. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol 2014;27:52-61.
13. Monecke T, Dickmanns A, Ficner R. Allosteric control of the exportin CRM1 unraveled by crystal structure analysis. FEBS J 2014;281:4179-94.
14. Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 2012;149:832-46.
15. Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell 2016;164:1162-71.
16. Schmidt HB, Görlich D. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem Sci 2016;41:46-61.
17. Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005;6:187-98.
18. Tran EJ, King MC, Corbett AH. Macromolecular transport between the nucleus and the cytoplasm: advances in mechanism and emerging links to disease. Biochim Biophys Acta 2014;1843:2784-95.
19. Cautain B, Hill R, de Pedro N, Link W. Components and regulation of nuclear transport processes. FEBS J 2015;282:445-62.
20. Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP. Simple rules for passive diffusion through the nuclear pore complex. J Cell Biol 2016;215:57-76.
21. Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 2017;18:73-89.
23. Soniat M, Chook YM. Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem J 2015;468:353-62.
24. Gama-Carvalho M, Carmo-Fonseca M. The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett 2001;498:157-63.
25. Nachury MV, Weis K. The direction of transport through the nuclear pore can be inverted. Proc Natl Acad Sci U S A 1999;96:9622-7.
26. Nardozzi JD, Lott K, Cingolani G. Phosphorylation meets nuclear import: a review. Cell Commun Signal 2010;8:32.
27. Panayiotou R, Miralles F, Pawlowski R, Diring J, Flynn HR, Skehel M, Treisman R. Phosphorylation acts positively and negatively to regulate MRTF-A subcellular localisation and activity. Elife 2016;5:e15460.
28. Rodríguez JA. Interplay between nuclear transport and ubiquitin/SUMO modifications in the regulation of cancer-related proteins. Semin Cancer Biol 2014;27:11-9.
29. Rodriguez JA, Schüchner S, Au WW, Fabbro M, Henderson BR. Nuclear-cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene 2004;23:1809-20.
30. Engelsma D, Rodriguez JA, Fish A, Giaccone G, Fornerod M. Homodimerization antagonizes nuclear export of survivin. Traffic 2007;8:1495-502.
31. Ding Q, Zhao L, Guo H, Zheng AC. The nucleocytoplasmic transport of viral proteins. Virol Sin 2010;25:79-85.
32. García-Santisteban I, Arregi I, Alonso-Mariño M, Urbaneja MA, Garcia-Vallejo JJ, Bañuelos S, Rodríguez JA. A cellular reporter to evaluate CRM1 nuclear export activity: functional analysis of the cancer-related mutant E571K. Cell Mol Life Sci 2016;73:4685-99.
33. Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell 2012;23:3677-93.
34. Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Görlich D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife 2015;4:e11466.
35. Hirayama S, Sugihara M, Morito D, Iemura SI, Natsume T, Murata S, Nagata K. Nuclear export of ubiquitinated proteins via the UBIN-POST system. Proc Natl Acad Sci U S A 2018;115:E4199-208.
36. Wu T, Chen W, Zhong Y, Hou X, Fang S, Liu CY, Wang G, Yu T, Huang YY, Ouyang X, Li HQ, Cui L, Yang Y. Nuclear export of ubiquitinated proteins determines the sensitivity of colorectal cancer to proteasome inhibitor. Mol Cancer Ther 2017;16:717-28.
37. Englmeier L, Fornerod M, Bischoff FR, Petosa C, Mattaj IW, Kutay U. RanBP3 influences interactions between CRM1 and its nuclear protein export substrates. EMBO Rep 2001;2:926-32.
38. Lindsay ME, Holaska JM, Welch K, Paschal BM, Macara IG. Ran-binding protein 3 is a cofactor for Crm1-mediated nuclear protein export. J Cell Biol 2001;153:1391-402.
39. Koyama M, Shirai N, Matsuura Y. Structural insights into how Yrb2p accelerates the assembly of the Xpo1p nuclear export complex. Cell Rep 2014;9:983-95.
40. Kosugi S, Hasebe M, Tomita M, Yanagawa H. Nuclear export signal consensus sequences defined using a localization-based yeast selection system. Traffic 2008;9:2053-62.
41. Güttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Görlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol 2010;17:1367-76.
42. Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res 2000;256:213-24.
43. Fu SC, Huang HC, Horton P, Juan HF. ValidNESs: a database of validated leucine-rich nuclear export signals. Nucleic Acids Res 2013;41:D338-43.
44. Fung HY, Fu SC, Chook YM. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. Elife 2017;6:e23961.
45. Neggers JE, Vercruysse T, Jacquemyn M, Vanstreels E, Baloglu E, Shacham S, Crochiere M, Landesman Y, Daelemans D. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol 2015;22:107-16.
46. Williams T, Ngo LH, Wickramasinghe VO. Nuclear export of RNA: different sizes, shapes and functions. Semin Cell Dev Biol 2018;75:70-7.
47. Thomson E, Ferreira-Cerca S, Hurt E. Eukaryotic ribosome biogenesis at a glance. J Cell Sci 2013;126:4815-21.
49. Yang J, Bogerd HP, Wang PJ, Page DC, Cullen BR. Two closely related human nuclear export factors utilize entirely distinct export pathways. Mol Cell 2001;8:397-406.
50. Brennan CM, Gallouzi IE, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol 2000;151:1-14.
51. Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, Piñol-Roma S, Borden KL. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J 2009;28:1087-98.
52. Ohno M, Segref A, Bachi A, Wilm M, Mattaj IW. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell 2000;101:187-98.
53. Xie M, Li M, Vilborg A, Lee N, Shu MD, Yartseva V, Šestan N, Steitz JA. Mammalian 5'-capped microRNA precursors that generate a single microRNA. Cell 2013;155:1568-80.
54. Noh JH, Kim KM, Abdelmohsen K, Yoon JH, Panda AC, Munk R, Kim J, Curtis J, Moad CA, Wohler CM, Indig FE, de Paula W, Dudekula DB, De S, Piao Y, Yang X, Martindale JL, de Cabo R, Gorospe M. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev 2016;30:1224-39.
55. Martinez I, Hayes KE, Barr JA, Harold AD, Xie M, Bukhari SIA, Vasudevan S, Steitz JA, DiMaio D. An Exportin-1-dependent microRNA biogenesis pathway during human cell quiescence. Proc Natl Acad Sci U S A 2017;114:E4961-70.
56. Sheng P, Fields C, Aadland K, Wei T, Kolaczkowski O, Gu T, Kolaczkowski B, Xie M. Dicer cleaves 5'-extended microRNA precursors originating from RNA polymerase II transcription start sites. Nucleic Acids Res 2018;46:5737-52.
57. Castanotto D, Lingeman R, Riggs AD, Rossi JJ. CRM1 mediates nuclear-cytoplasmic shuttling of mature microRNAs. Proc Natl Acad Sci U S A 2009;106:21655-9.
58. Pradet-Balade B, Girard C, Boulon S, Paul C, Azzag K, Bordonné R, Bertrand E, Verheggen C. CRM1 controls the composition of nucleoplasmic pre-snoRNA complexes to licence them for nucleolar transport. EMBO J 2011;30:2205-18.
59. Forbes DJ, Travesa A, Nord MS, Bernis C. Reprint of "Nuclear transport factors: global regulation of mitosis". Curr Opin Cell Biol 2015;34:122-34.
60. Liu Q, Jiang Q, Zhang C. A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochem Biophys Res Commun 2009;384:383-8.
61. Wu Z, Jiang Q, Clarke PR, Zhang C. Phosphorylation of Crm1 by CDK1-cyclin-B promotes Ran-dependent mitotic spindle assembly. J Cell Sci 2013;126:3417-28.
62. Gilistro E, de Turris V, Damizia M, Verrico A, Moroni S, De Santis R, Rosa A, Lavia P. Importin-β and CRM1 control a RANBP2 spatiotemporal switch essential for mitotic kinetochore function. J Cell Sci 2017;130:2564-78.
63. Wang X, Li S. Protein mislocalization: mechanisms, functions and clinical applications in cancer. Biochim Biophys Acta 2014;1846:13-25.
64. Dickmanns A, Monecke T, Ficner R. Structural basis of targeting the exportin CRM1 in cancer. Cells 2015;4:538-68.
66. Hill R, Cautain B, de Pedro N, Link W. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget 2014;5:11-28.
67. Jeyasekharan AD, Liu Y, Hattori H, Pisupati V, Jonsdottir AB, Rajendra E, Lee M, Sundaramoorthy E, Schlachter S, Kaminski CF, Ofir-Rosenfeld Y, Sato K, Savill J, Ayoub N, Venkitaraman AR. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat Struct Mol Biol 2013;20:1191-8.
68. Pauty J, Couturier AM, Rodrigue A, Caron MC, Coulombe Y, Dellaire G, Masson JY. Cancer-causing mutations in the tumor suppressor PALB2 reveal a novel cancer mechanism using a hidden nuclear export signal in the WD40 repeat motif. Nucleic Acids Res 2017;45:2644-57.
69. Mariano AR, Colombo E, Luzi L, Martinelli P, Volorio S, Bernard L, Meani N, Bergomas R, Alcalay M, Pelicci PG. Cytoplasmic localization of NPM in myeloid leukemias is dictated by gain-of-function mutations that create a functional nuclear export signal. Oncogene 2006;25:4376-80.
70. Mor A, White MA, Fontoura BM. Nuclear trafficking in health and disease. Curr Opin Cell Biol 2014;28:28-35.
71. Takeda A, Yaseen NR. Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 2014;27:3-10.
72. Takeda A, Sarma NJ, Abdul-Nabi AM, Yaseen NR. Inhibition of CRM1-mediated nuclear export of transcription factors by leukemogenic NUP98 fusion proteins. J Biol Chem 2010;285:16248-57.
73. Saito S, Cigdem S, Okuwaki M, Nagata K. Leukemia-associated Nup214 fusion proteins disturb the XPO1-mediated nuclear-cytoplasmic transport pathway and thereby the NF-κB signaling pathway. Mol Cell Biol 2016;36:1820-35.
74. Christiansen A, Dyrskjøt L. The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer. Cancer Lett 2013;331:18-23.
75. Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 1999;18:1660-72.
76. Rodríguez JA, Henderson BR. Identification of a functional nuclear export sequence in BRCA1. J Biol Chem 2000;275:38589-96.
77. Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, Hope TJ. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci U S A 1998;95:7457-62.
78. Fabbro M, Henderson BR. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp Cell Res 2003;282:59-69.
79. Conforti F, Wang Y, Rodriguez JA, Alberobello AT, Zhang YW, Giaccone G. Molecular pathways: anticancer activity by inhibition of nucleocytoplasmic shuttling. Clin Cancer Res 2015;21:4508-13.
80. Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, Qiu YH, Burks JK, Kantarjian H, Shacham S, Kauffman M, Andreeff M. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013;121:4166-74.
81. Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, Ford RJ, Pham LV. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol 2013;41:67-78.
82. Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, Tiedemann RE, Palmer SE, Garbitt VM, McCauley D, Kauffman M, Shacham S, Chesi M, Bergsagel PL, Stewart AK. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013;27:2357-65.
83. Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA, Senapedis W, Saint-Martin JR, Kashyap T, Shacham S, Kauffman M, Gu Y, Wu L, Ghobrial I, Zhan F, Kung AL, Schey SA, Richardson P, Munshi NC, Anderson KC. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia 2014;28:155-65.
84. Liu X, Chong Y, Tu Y, Liu N, Yue C, Qi Z, Liu H, Yao Y, Liu H, Gao S, Niu M, Yu R. CRM1/XPO1 is associated with clinical outcome in glioma and represents a therapeutic target by perturbing multiple core pathways. J Hematol Oncol 2016;9:108.
85. Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009;65:153-9.
86. Akagi I, Okayama H, Schetter AJ, Robles AI, Kohno T, Bowman ED, Kazandjian D, Welsh JA, Oue N, Saito M, Miyashita M, Uchida E, Takizawa T, Takenoshita S, Skaug V, Mollerup S, Haugen A, Yokota J, Harris CC. Combination of protein coding and noncoding gene expression as a robust prognostic classifier in stage I lung adenocarcinoma. Cancer Res 2013;73:3821-32.
87. Sun YQ, Xie JW, Xie HT, Chen PC, Zhang XL, Zheng CH, Li P, Wang JB, Lin JX, Cao LL, Huang CM, Lin Y. Expression of CRM1 and CDK5 shows high prognostic accuracy for gastric cancer. World J Gastroenterol 2017;23:2012-22.
88. Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu S, Lv J, Yue L. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med Oncol 2013;30:726.
89. Conforti F, Zhang X, Rao G, De Pas T, Yonemori Y, Rodriguez JA, McCutcheon JN, Rahhal R, Alberobello AT, Wang Y, Zhang YW, Guha U, Giaccone G. Therapeutic effects of XPO1 inhibition in thymic epithelial tumors. Cancer Res 2017;77:5614-27.
90. Noske A, Weichert W, Niesporek S, Röske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008;112:1733-43.
91. Xie QL, Liu Y, Zhu Y. Chromosome region maintenance 1 expression and its association with clinical pathological features in primary carcinoma of the liver. Exp Ther Med 2016;12:59-68.
92. Yang X, Cheng L, Yao L, Ren H, Zhang S, Min X, Chen X, Zhang J, Li M. Involvement of chromosome region maintenance 1 (CRM1) in the formation and progression of esophageal squamous cell carcinoma. Med Oncol 2014;31:155.
93. van der Watt PJ, Zemanay W, Govender D, Hendricks DT, Parker MI, Leaner VD. Elevated expression of the nuclear export protein, Crm1 (exportin 1), associates with human oesophageal squamous cell carcinoma. Oncol Rep 2014;32:730-8.
94. Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH, Sun YJ. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med 2009;32:E315.
95. Inoue H, Kauffman M, Shacham S, Landesman Y, Yang J, Evans CP, Weiss RH. CRM1 blockade by selective inhibitors of nuclear export attenuates kidney cancer growth. J Urol 2013;189:2317-26.
96. Yue L, Sun ZN, Yao YS, Shen Z, Wang HB, Liu XP, Zhou F, Xiang JY, Yao RY, Niu HT. CRM1, a novel independent prognostic factor overexpressed in invasive breast carcinoma of poor prognosis. Oncol Lett 2018;15:7515-22.
97. Sun H, Lin DC, Cao Q, Guo X, Marijon H, Zhao Z, Gery S, Xu L, Yang H, Pang B, Lee VK, Lim HJ, Doan N, Said JW, Chu P, Mayakonda A, Thomas T, Forscher C, Baloglu E, Shacham S, Rajalingam R, Koeffler HP. CRM1 Inhibition promotes cytotoxicity in ewing sarcoma cells by repressing EWS-FLI1-dependent IGF-1 signaling. Cancer Res 2016;76:2687-97.
98. Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen P, Sun YJ, Tang LN, Zheng SE. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep 2009;21:229-35.
99. Pathria G, Wagner C, Wagner SN. Inhibition of CRM1-mediated nucleocytoplasmic transport: triggering human melanoma cell apoptosis by perturbing multiple cellular pathways. J Invest Dermatol 2012;132:2780-90.
100. Jardin F, Pujals A, Pelletier L, Bohers E, Camus V, Mareschal S, Dubois S, Sola B, Ochmann M, Lemonnier F, Viailly PJ, Bertrand P, Maingonnat C, Traverse-Glehen A, Gaulard P, Damotte D, Delarue R, Haioun C, Argueta C, Landesman Y, Salles G, Jais JP, Figeac M, Copie-Bergman C, Molina TJ, Picquenot JM, Cornic M, Fest T, Milpied N, Lemasle E, Stamatoullas A, Moeller P, Dyer MJ, Sundstrom C, Bastard C, Tilly H, Leroy K. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am J Hematol 2016;91:923-30.
101. Cosson A, Chapiro E, Bougacha N, Lambert J, Herbi L, Cung HA, Algrin C, Keren B, Damm F, Gabillaud C, Brunelle-Navas MN, Davi F Merle-Béral H, Le Garff-Tavernier M, Roos-Weil D, Choquet S, Uzunov M, Morel V, Leblond V, Maloum K, Lepretre S, Feugier P, Lesty C, Lejeune J, Sutton L, Landesman Y, Susin SA, Nguyen-Khac F. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: analysis of the central role of XPO1. Leukemia 2017;31:1625-29.
102. Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, Volarevic S, Oren M. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell 2012;45:222-32.
103. van der Watt PJ, Leaner VD. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim Biophys Acta 2011;1809:316-26.
104. Camus V, Stamatoullas A, Mareschal S, Viailly PJ, Sarafan-Vasseur N, Bohers E, Dubois S, Picquenot JM, Ruminy P, Maingonnat C, Bertrand P, Cornic M, Tallon-Simon V, Becker S, Veresezan L, Frebourg T, Vera P, Bastard C, Tilly H, Jardin F. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016;101:1094-101.
105. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M, Bassaganyas L, Baumann T, Juan M, López-Guerra M, Colomer D, Tubío JM, López C, Navarro A, Tornador C, Aymerich M, Rozman M, Hernández JM, Puente DA, Freije JM, Velasco G, Gutiérrez-Fernández A, Costa D, Carrió A, Guijarro S, Enjuanes A, Hernández L, Yagüe J, Nicolás P, Romeo-Casabona CM, Himmelbauer H, Castillo E, Dohm JC, de Sanjosé S, Piris MA, de Alava E, San Miguel J, Royo R, Gelpí JL, Torrents D, Orozco M, Pisano DG, Valencia A, Guigó R, Bayés M, Heath S, Gut M, Klatt P, Marshall J, Raine K, Stebbings LA, Futreal PA, Stratton MR, Campbell PJ, Gut I, López-Guillermo A, Estivill X, Montserrat E, López-Otín C, Campo E. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101-5.
106. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Giné E, Hernández JM, González-Díaz M, Puente DA, Velasco G, Freije JM, Tubío JM, Royo R, Gelpí JL, Orozco M, Pisano DG, Zamora J, Vázquez M, Valencia A, Himmelbauer H, Bayés M, Heath S, Gut M, Gut I, Estivill X, López-Guillermo A, Puente XS, Campo E, López-Otín C. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2011;44:47-52.
107. Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, Pekarsky Y, Croce CM. NOTCH1 mutations in CLL associated with trisomy 12. Blood 2012;119:329-31.
108. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, Vartanov A, Fernandes SM, Saksena G, Cibulskis K, Tesar B, Gabriel S, Hacohen N, Meyerson M, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013;152:714-26.
109. Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V, Alpermann T, Roller A, Kohlmann A, Haferlach T, Kern W, Schnittger S. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014;28:108-17.
110. Messina M, Del Giudice I, Khiabanian H, Rossi D, Chiaretti S, Rasi S, Spina V, Holmes AB, Marinelli M, Fabbri G, Piciocchi A, Mauro FR, Guarini A, Gaidano G, Dalla-Favera R, Pasqualucci L, Rabadan R, Foà R. Genetic lesions associated with chronic lymphocytic leukemia chemo-refractoriness. Blood 2014;123:2378-88.
111. Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, Diop M, Scourzic L, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Kikushige Y, Davi F, Lambert J, Gautheret D, Merle-Béral H, Sutton L, Dessen P, Solary E, Akashi K, Vainchenker W, Mercher T, Droin N, Ogawa S, Nguyen-Khac F, Bernard OA. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov 2014;4:1088-101.
112. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495-501.
113. Ojha J, Ayres J, Secreto C, Tschumper R, Rabe K, Van Dyke D, Slager S, Shanafelt T, Fonseca R, Kay NE, Braggio E. Deep sequencing identifies genetic heterogeneity and recurrent convergent evolution in chronic lymphocytic leukemia. Blood 2015;125:492-8.
114. Vollbrecht C, Mairinger FD, Koitzsch U, Peifer M, Koenig K, Heukamp LC, Crispatzu G, Wilden L, Kreuzer KA, Hallek M, Odenthal M, Herling CD, Buettner R. Comprehensive analysis of disease-related genes in chronic lymphocytic leukemia by multiplex PCR-based next generation sequencing. PLoS One 2015;10:e0129544.
115. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter SL, Cibulskis K, Mertens D, Sougnez CL, Rosenberg M, Hess JM, Edelmann J, Kless S, Kneba M, Ritgen M, Fink A, Fischer K, Gabriel S, Lander ES, Nowak MA, Döhner H, Hallek M, Neuberg D, Getz G, Stilgenbauer S, Wu CJ. Mutations driving CLL and their evolution in progression and relapse. Nature 2015;526:525-30.
116. Guièze R, Robbe P, Clifford R, de Guibert S, Pereira B, Timbs A, Dilhuydy MS, Cabes M, Ysebaert L, Burns A, Nguyen-Khac F, Davi F, Véronèse L, Combes P, Le Garff-Tavernier M, Leblond V, Merle-Béral H, Alsolami R, Hamblin A, Mason J, Pettitt A, Hillmen P, Taylor J, Knight SJ, Tournilhac O, Schuh A. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood 2015;126:2110-7.
117. Hernández JA, Hernández-Sánchez M, Rodríguez-Vicente AE, Grossmann V, Collado R, Heras C, Puiggros A, Martín AÁ, Puig N, Benito R, Robledo C, Delgado J, González T, Queizán JA, Galende J, de la Fuente I, Martín-Núñez G, Alonso JM, Abrisqueta P, Luño E, Marugán I, González-Gascón I, Bosch F, Kohlmann A, González M, Espinet B, Hernández-Rivas JM; Grupo Cooperativo Español de Citogenética Hematológica (GCECGH) and Grupo Español de Leucemia Linfática Crónica (GELLC). A low frequency of losses in 11q chromosome is associated with better outcome and lower rate of genomic mutations in patients with chronic lymphocytic leukemia. PLoS One 2015;10:e0143073.
118. Sutton LA, Ljungström V, Mansouri L, Young E, Cortese D, Navrkalova V, Malcikova J, Muggen AF, Trbusek M, Panagiotidis P, Davi F, Belessi C, Langerak AW, Ghia P, Pospisilova S, Stamatopoulos K, Rosenquist R. Targeted next-generation sequencing in chronic lymphocytic leukemia: a high-throughput yet tailored approach will facilitate implementation in a clinical setting. Haematologica 2015;100:370-6.
119. Amin NA, Seymour E, Saiya-Cork K, Parkin B, Shedden K, Malek SN. A quantitative analysis of subclonal and clonal gene mutations before and after therapy in chronic lymphocytic leukemia. Clin Cancer Res 2016;22:4525-35.
120. Jain P, Kanagal-Shamanna R, Wierda W, Keating M, Sarwari N, Rozovski U, Thompson P, Burger J, Kantarjian H, Patel KP, Medeiros LJ, Luthra R, Estrov Z. Clinical and molecular characteristics of XPO1 mutations in patients with chronic lymphocytic leukemia. Am J Hematol 2016;91:E478-9.
121. Quijada-álamo M, Hernández-Sánchez M, Robledo C, Hernández-Sánchez JM, Benito R, Montaño A, Rodríguez-Vicente AE, Quwaider D, Martín AÁ, García-álvarez M, Vidal-Manceñido MJ, Ferrer-Garrido G, Delgado-Beltrán MP, Galende J, Rodríguez JN, Martín-Núñez G, Alonso JM, García de Coca A, Queizán JA, Sierra M, Aguilar C, Kohlmann A, Hernández JÁ, González M, Hernández-Rivas JM. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J Hematol Oncol 2017;10:83.
122. Takahashi K, Hu B, Wang F, Yan Y, Kim E, Vitale C, Patel KP, Strati P, Gumbs C, Little L, Tippen S, Song X, Zhang J, Jain N, Thompson P, Garcia-Manero G, Kantarjian H, Estrov Z, Do KA, Keating M, Burger JA, Wierda WG, Futreal PA, Ferrajoli A. Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 2018;131:1820-32.
123. Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, Molina TJ, Desmots F, Fest T, Haioun C, Lamy T, Copie-Bergman C, Brière J, Petrella T, Canioni D, Fabiani B, Coiffier B, Delarue R, Peyrade F, Bosly A, André M, Ketterer N, Salles G, Tilly H, Leroy K, Jardin F. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res 2016;22:2919-28.
124. Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, Wang Y, Rosseto A, Venanzi A, Vlasevska S, Pacini R, Piattoni S, Tabarrini A, Pucciarini A, Bigerna B, Santi A, Gianni AM, Viviani S, Cabras A, Ascani S, Crescenzi B, Mecucci C, Pasqualucci L, Rabadan R, Falini B. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018;131:2454-65.
125. Camus V, Miloudi H, Taly A, Sola B, Jardin F. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol 2017;10:47.
126. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, Yin D, Shi ZZ, Jiang YY, Gu WY, Gong T, Zhang Y, Xu X, Kalid O, Shacham S, Ogawa S, Wang MR, Koeffler HP. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet 2014;46:467-73.
127. Pitt SC, Hernandez RA, Nehs MA, Gawande AA, Moore FD Jr, Ruan DT, Cho NL. Identification of novel oncogenic mutations in thyroid cancer. J Am Coll Surg 2016;222:1036-43.
129. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561-6.
130. le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E, Giardini R, Formelli F, Gambacorti-Passerini C. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst 1999;91:163-8.
131. Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med 2001;7:228-34.
132. Kancha RK, von Bubnoff N, Miething C, Peschel C, Götze KS, Duyster J. Imatinib and leptomycin B are effective in overcoming imatinib-resistance due to Bcr-Abl amplification and clonal evolution but not due to Bcr-Abl kinase domain mutation. Haematologica 2008;93:1718-22.
133. Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol 2014;27:74-86.
134. Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, Kufe DW, Vonhoff DD, Iwami T, Kawabe T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood 2011;118:3922-31.
135. Turner JG, Marchion DC, Dawson JL, Emmons MF, Hazlehurst LA, Washausen P, Sullivan DM. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res 2009;69:6899-905.
136. Kalid O, Toledo Warshaviak D, Shechter S, Sherman W, Shacham S. Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J Comput Aided Mol Des 2012;26:1217-28.
137. Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S, Farmer A, Mani R, Johnson AJ, Lucas D, Mo X, Daelemans D, Sandanayaka V, Shechter S, McCauley D, Shacham S, Kauffman M, Chook YM, Byrd JC. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012;120:4621-34.
138. Turner JG, Dawson J, Emmons MF, Cubitt CL, Kauffman M, Shacham S, Hazlehurst LA, Sullivan DM. CRM1 inhibition sensitizes drug resistant human myeloma cells to topoisomerase II and proteasome inhibitors both in vitro and ex vivo. J Cancer 2013;4:614-25.
139. Rosebeck S, Alonge MM, Kandarpa M, Mayampurath A, Volchenboum SL, Jasielec J, Dytfeld D, Maxwell SP, Kraftson SJ, McCauley D, Shacham S, Kauffman M, Jakubowiak AJ. Synergistic myeloma cell death via novel intracellular activation of caspase-10-dependent apoptosis by carfilzomib and selinexor. Mol Cancer Ther 2016;15:60-71.
140. Turner JG, Dawson JL, Grant S, Shain KH, Dalton WS, Dai Y, Meads M, Baz R, Kauffman M, Shacham S, Sullivan DM. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J Hematol Oncol 2016;9:73.
141. Turner JG, Kashyap T, Dawson JL, Gomez J, Bauer AA, Grant S, Dai Y, Shain KH, Meads M, Landesman Y, Sullivan DM. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget 2016;7:78896-909.
142. Muz B, Azab F, de la Puente P, Landesman Y, Azab AK. Selinexor overcomes hypoxia-induced drug resistance in multiple myeloma. Transl Oncol 2017;10:632-40.
143. Argueta C, Kashyap T, Klebanov B, Unger TJ, Guo C, Harrington S, Baloglu E, Lee M, Senapedis W, Shacham S, Landesman Y. Selinexor synergizes with dexamethasone to repress mTORC1 signaling and induce multiple myeloma cell death. Oncotarget 2018;9:25529-44.
144. Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012;120:1765-73.
145. Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2013;27:66-74.
146. Ranganathan P, Yu X, Santhanam R, Hofstetter J, Walker A, Walsh K, Bhatnagar B, Klisovic R, Vasu S, Phelps MA, Devine S, Shacham S, Kauffman M, Marcucci G, Blum W, Garzon R. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood 2015;125:2689-92.
147. Zhang W, Ly C, Ishizawa J, Mu H, Ruvolo V, Shacham S, Daver N, Andreeff M. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: from concept to clinical trial. Haematologica 2018; doi: 10.3324/haematol.2017.185082.
148. Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT, Christie AL, McCauley D, Rodig SJ, Kauffman M, Shacham S, Stone R, Letai A, Kung AL, Thomas Look A. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol 2013;161:117-27.
149. Zhong Y, El-Gamal D, Dubovsky JA, Beckwith KA, Harrington BK, Williams KE, Goettl VM, Jha S, Mo X, Jones JA, Flynn JM, Maddocks KJ, Andritsos LA, McCauley D, Shacham S, Kauffman M, Byrd JC, Lapalombella R. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells. Leukemia 2014;28:1158-63.
150. Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, Williams K, Johnson AJ, Lehman AM, Byrd JC, Woyach JA, Lapalombella R. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood 2015;125:3128-32.
151. Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld AK, Gabrail NY, Smith CL, Caligiuri MA, Hokland P, Roy DC, Reid A, Milojkovic D, Goldman JM, Apperley J, Garzon R, Marcucci G, Shacham S, Kauffman MG, Perrotti D. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood 2013;122:3034-44.
152. Nie D, Huang K, Yin S, Li Y, Xie S, Ma L, Wang X, Wu Y, Xiao J, Wang J, Yang W, Liu H. KPT-330 inhibition of chromosome region maintenance 1 is cytotoxic and sensitizes chronic myeloid leukemia to Imatinib. Cell Death Discov 2018;4:48.
153. Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, Ford RJ, Pham LV. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol 2013;41:67-78.e4.
154. Sekihara K, Saitoh K, Han L, Ciurea S, Yamamoto S, Kikkawa M, Kazuno S, Taka H, Kaga N, Arai H, Miida T, Andreeff M, Konopleva M, Tabe Y. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017;8:34552-64.
155. Azmi AS, Al-Katib A, Aboukameel A, McCauley D, Kauffman M, Shacham S, Mohammad RM. Selective inhibitors of nuclear export for the treatment of non-Hodgkin's lymphomas. Haematologica 2013;98:1098-106.
156. Han X, Wang J, Shen Y, Zhang N, Wang S, Yao J, Shi Y. CRM1 as a new therapeutic target for non-Hodgkin lymphoma. Leuk Res 2015;39:38-46.
157. Muqbil I, Aboukameel A, Elloul S, Carlson R, Senapedis W, Baloglu E, Kauffman M, Shacham S, Bhutani D, Zonder J, Azmi AS, Mohammad RM. Anti-tumor activity of selective inhibitor of nuclear export (SINE) compounds, is enhanced in non-Hodgkin lymphoma through combination with mTOR inhibitor and dexamethasone. Cancer Lett 2016;383:309-17.
158. Wettersten HI, Landesman Y, Friedlander S, Shacham S, Kauffman M, Weiss RH. Specific inhibition of the nuclear exporter exportin-1 attenuates kidney cancer growth. PLoS One 2014;9:e113867.
159. Mendonca J, Sharma A, Kim HS, Hammers H, Meeker A, De Marzo A, Carducci M, Kauffman M, Shacham S, Kachhap S. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014;5:6102-12.
160. Gravina GL, Tortoreto M, Mancini A, Addis A, Di Cesare E, Lenzi A, Landesman Y, McCauley D, Kauffman M, Shacham S, Zaffaroni N, Festuccia C. XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J Hematol Oncol 2014;7:46.
161. Gravina GL, Mancini A, Colapietro A, Marampon F, Sferra R, Pompili S, Biordi LA, Iorio R, Flati V, Argueta C, Landesman Y, Kauffman M, Shacham S, Festuccia C. Pharmacological treatment with inhibitors of nuclear export enhances the antitumor activity of docetaxel in human prostate cancer. Oncotarget 2017;8:111225-45.
162. Cheng Y, Holloway MP, Nguyen K, McCauley D, Landesman Y, Kauffman MG, Shacham S, Altura RA. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol Cancer Ther 2014;13:675-86.
163. Wrobel K, Zhao YC, Kulkoyluoglu E, Chen KL, Hieronymi K, Holloway J, Li S, Ray T, Ray PS, Landesman Y, Lipka AE, Smith RL, Madak-Erdogan Z. ERα-XPO1 cross talk controls tamoxifen sensitivity in tumors by altering ERK5 cellular localization. Mol Endocrinol 2016;30:1029-45.
164. Arango NP, Yuca E, Zhao M, Evans KW, Scott S, Kim C, Gonzalez-Angulo AM, Janku F, Ueno NT, Tripathy D, Akcakanat A, Naing A, Meric-Bernstam F. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res 2017;19:93.
165. Miyake T, Pradeep S, Wu SY, Rupaimoole R, Zand B, Wen Y, Gharpure KM, Nagaraja AS, Hu W, Cho MS, Dalton HJ, Previs RA, Taylor ML, Hisamatsu T, Kang Y, Liu T, Shacham S, McCauley D, Hawke DH, Wiktorowicz JE, Coleman RL, Sood AK. XPO1/CRM1 inhibition causes antitumor effects by mitochondrial accumulation of eIF5A. Clin Cancer Res 2015;21:3286-97.
166. Chen Y, Camacho SC, Silvers TR, Razak AR, Gabrail NY, Gerecitano JF, Kalir E, Pereira E, Evans BR, Ramus SJ, Huang F, Priedigkeit N, Rodriguez E, Donovan M, Khan F, Kalir T, Sebra R, Uzilov A, Chen R, Sinha R, Halpert R, Billaud JN, Shacham S, McCauley D, Landesman Y, Rashal T, Kauffman M, Mirza MR, Mau-Sørensen M, Dottino P, Martignetti JA. Inhibition of the nuclear export receptor XPO1 as a therapeutic target for platinum-resistant ovarian cancer. Clin Cancer Res 2017;23:1552-63.
167. Corno C, Stucchi S, De Cesare M, Carenini N, Stamatakos S, Ciusani E, Minoli L, Scanziani E, Argueta C, Landesman Y, Zaffaroni N, Gatti L, Perego P. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem Pharmacol 2018;147:93-103.
168. Ferreiro-Neira I, Torres NE, Liesenfeld LF, Chan CH, Penson T, Landesman Y, Senapedis W, Shacham S, Hong TS, Cusack JC. XPO1 inhibition enhances radiation response in preclinical models of rectal cancer. Clin Cancer Res 2016;22:1663-73.
169. Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 2014;74:487-95.
170. Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M, Shacham S, Mohammad RM. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013;144:447-56.
171. Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB, Mohammad RM. Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget 2014;5:3444-54.
172. Kazim S, Malafa MP, Coppola D, Husain K, Zibadi S, Kashyap T, Crochiere M, Landesman Y, Rashal T, Sullivan DM, Mahipal A. Selective nuclear export inhibitor KPT-330 enhances the antitumor activity of gemcitabine in human pancreatic cancer. Mol Cancer Ther 2015;14:1570-81.
173. Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman MG, Philip PA, Mohammad RM. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget 2017;8:82144-55.
174. Sun H, Hattori N, Chien W, Sun Q, Sudo M, E-Ling GL, Ding L, Lim SL, Shacham S, Kauffman M, Nakamaki T, Koeffler HP. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer 2014;111:281-91.
175. Wang S, Han X, Wang J, Yao J, Shi Y. Antitumor effects of a novel chromosome region maintenance 1 (CRM1) inhibitor on non-small cell lung cancer cells in vitro and in mouse tumor xenografts. PLoS One 2014;9:e89848.
176. Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, Villalobos P, Neggers JE, Mendiratta S, Wei S, Landesman Y, Senapedis W, Baloglu E, Chow CB, Frink RE, Gao B, Roth M, Minna JD, Daelemans D, Wistuba II, Posner BA, Scaglioni PP, White MA. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016;538:114-7.
177. Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, Sneha S, Nagare RP, Said JW, Doan NB, Ding LW, Baloglu E, Shacham S, Kauffman M, Koeffler HP. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep 2017;7:9749.
178. Garg M, Kanojia D, Mayakonda A, Said JW, Doan NB, Chien W, Ganesan TS, Chuang LS, Venkatachalam N, Baloglu E, Shacham S, Kauffman M, Koeffler HP. Molecular mechanism and therapeutic implications of selinexor (KPT-330) in liposarcoma. Oncotarget 2017;8:7521-32.
179. Kashyap T, Argueta C, Aboukameel A, Unger TJ, Klebanov B, Mohammad RM, Muqbil I, Azmi AS, Drolen C, Senapedis W, Lee M, Kauffman M, Shacham S, Landesman Y. Selinexor, a selective inhibitor of nuclear export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016;7:78883-95.
180. Nakayama R, Zhang YX, Czaplinski JT, Anatone AJ, Sicinska ET, Fletcher JA, Demetri GD, Wagner AJ. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget 2016;7:16581-92.
181. Nair JS, Musi E, Schwartz GK. Selinexor (KPT-330) induces tumor suppression through nuclear sequestration of IκB and downregulation of Survivin. Clin Cancer Res 2017;23:4301-11.
182. De Cesare M, Cominetti D, Doldi V, Lopergolo A, Deraco M, Gandellini P, Friedlander S, Landesman Y, Kauffman MG, Shacham S, Pennati M, Zaffaroni N. Anti-tumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: the role of survivin. Oncotarget 2015;6:13119-32.
183. Green AL, Ramkissoon SH, McCauley D, Jones K, Perry JA, Hsu JH, Ramkissoon LA, Maire CL, Hubbell-Engler B, Knoff DS, Shacham S, Ligon KL, Kung AL. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol 2015;17:697-707.
184. Wahba A, Rath BH, O'Neill JW, Camphausen K, Tofilon PJ. The XPO1 inhibitor selinexor inhibits translation and enhances the radiosensitivity of glioblastoma cells grown in vitro and in vivo. Mol Cancer Ther 2018;17:1717-26.
185. Salas-Fragomeni RA, Chung HW, Landesman Y, Senapedis W, Saint-Martin JR, Tsao H, Flaherty KT, Shacham S, Kauffman M, Cusack JC. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther 2013;12:1171-9.
186. Yang J, Bill MA, Young GS, La Perle K, Landesman Y, Shacham S, Kauffman M, Senapedis W, Kashyap T, Saint-Martin JR, Kendra K, Lesinski GB. Novel small molecule XPO1/CRM1 inhibitors induce nuclear accumulation of TP53, phosphorylated MAPK and apoptosis in human melanoma cells. PLoS One 2014;9:e102983.
187. Burke RT, Marcus JM, Orth JD. Inhibition of exportin-1 function results in rapid cell cycle-associated DNA damage in cancer cells. Oncotarget 2017;8:39460-75.
188. Attiyeh EF, Maris JM, Lock R, Reynolds CP, Kang MH, Carol H, Gorlick R, Kolb EA, Keir ST, Wu J, Landesman Y, Shacham S, Lyalin D, Kurmasheva RT, Houghton PJ, Smith MA. Pharmacodynamic and genomic markers associated with response to the XPO1/CRM1 inhibitor selinexor (KPT-330): a report from the pediatric preclinical testing program. Pediatr Blood Cancer 2016;63:276-86.
189. Gandhi UH, Senapedis W, Baloglu E, Unger TJ, Chari A, Vogl D, Cornell RF. Clinical implications of targeting XPO1-mediated nuclear export in multiple myeloma. Clin Lymphoma Myeloma Leuk 2018;18:335-45.
190. Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, Chook YM, Kuhlmann T, Kidd GJ, Shacham S, Casaccia P. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci 2015;18:511-20.
191. Pickens JA, Tripp RA. Verdinexor targeting of CRM1 is a promising therapeutic approach against RSV and influenza viruses. Viruses 2018;10: pii: E48.
192. Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther 2016;164:135-43.
193. Abdul Razak AR, Mau-Soerensen M, Gabrail NY, Gerecitano JF, Shields AF, Unger TJ, Saint-Martin JR, Carlson R, Landesman Y, McCauley D, Rashal T, Lassen U, Kim R, Stayner LA, Mirza MR, Kauffman M, Shacham S, Mahipal A. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol 2016;34:4142-50.
194. Gounder MM, Zer A, Tap WD, Salah S, Dickson MA, Gupta AA, Keohan ML, Loong HH, D'Angelo SP, Baker S, Condy M, Nyquist-Schultz K, Tanner L, Erinjeri JP, Jasmine FH, Friedlander S, Carlson R, Unger TJ, Saint-Martin JR, Rashal T, Ellis J, Kauffman M, Shacham S, Schwartz GK, Abdul Razak AR. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol 2016;34:3166-74.
195. Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol 2016;34:4094-101.
196. Kuruvilla J, Savona M, Baz R, Mau-Sorensen PM, Gabrail N, Garzon R, Stone R, Wang M, Savoie L, Martin P, Flinn I, Jacoby M, Unger TJ, Saint-Martin JR, Rashal T, Friedlander S, Carlson R, Kauffman M, Shacham S, Gutierrez M. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017;129:3175-83.
197. Garzon R, Savona M, Baz R, Andreeff M, Gabrail N, Gutierrez M, Savoie L, Mau-Sorensen PM, Wagner-Johnston N, Yee K, Unger TJ, Saint-Martin JR, Carlson R, Rashal T, Kashyap T, Klebanov B, Shacham S, Kauffman M, Stone R. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 2017;129:3165-74.
198. Wang AY, Weiner H, Green M, Chang H, Fulton N, Larson RA, Odenike O, Artz AS, Bishop MR, Godley LA, Thirman MJ, Kosuri S, Churpek JE, Curran E, Pettit K, Stock W, Liu H. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol 2018;11:4.
199. Chen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N, Baz R, Mau-Sorensen M, Berdeja JG, Savona M, Savoie L, Trudel S, Areethamsirikul N, Unger TJ, Rashal T, Hanke T, Kauffman M, Shacham S, Reece D. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood 2018;131:855-63.
200. Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D, Zonder J, Baz R, Nooka A, Richter J, Cole C, Vij R, Jakubowiak A, Abonour R, Schiller G, Parker TL, Costa LJ, Kaminetzky D, Hoffman JE, Yee AJ, Chari A, Siegel D, Fonseca R, Van Wier S, Ahmann G, Lopez I, Kauffman M, Shacham S, Saint-Martin JR, Picklesimer CD, Choe-Juliak C, Stewart AK. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol 2018;36:859-66.
201. Wei XX, Siegel AP, Aggarwal R, Lin AM, Friedlander TW, Fong L, Kim W, Louttit M, Chang E, Zhang L, Ryan CJ. A phase II trial of selinexor, an oral selective inhibitor of nuclear export compound, in abiraterone- and/or enzalutamide-refractory metastatic castration-resistant prostate cancer. Oncologist 2018;23:656.
202. Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E, Unger TJ, Kashyap T, Klebanov B, Sola-Visner M, Crochiere M, Italiano JE Jr, Landesman Y. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood 2017;130:1132-43.
203. Hing ZA, Fung HY, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA, Williams K, Goettl VM, Smith J, Yu X, Meng X, Sun Q, Cagatay T, Lehman AM, Lucas DM, Baloglu E, Shacham S, Kauffman MG, Byrd JC, Chook YM, Garzon R, Lapalombella R. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016;30:2364-72.
204. Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, Senapedis W, Landesman Y, Kauffman M, Shacham S, Wang JC, Look AT. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017;31:143-50.
205. Vercruysse T, De Bie J, Neggers JE, Jacquemyn M, Vanstreels E, Schmid-Burgk JL, Hornung V, Baloglu E, Landesman Y, Senapedis W, Shacham S, Dagklis A, Cools J, Daelemans D. The second-generation exportin-1 inhibitor KPT-8602 demonstrates potent activity against acute lymphoblastic leukemia. Clin Cancer Res 2017;23:2528-41.
206. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605-12.
207. Dong X, Biswas A, Chook YM. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol 2009;16:558-560.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sendino M, Omaetxebarria MJ, Rodríguez JA. Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resist 2018;1:139-63. http://dx.doi.org/10.20517/cdr.2018.09
AMA Style
Sendino M, Omaetxebarria MJ, Rodríguez JA. Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resistance. 2018; 1(3): 139-63. http://dx.doi.org/10.20517/cdr.2018.09
Chicago/Turabian Style
Sendino, Maria, Miren Josu Omaetxebarria, Jose Antonio Rodríguez. 2018. "Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer" Cancer Drug Resistance. 1, no.3: 139-63. http://dx.doi.org/10.20517/cdr.2018.09
ACS Style
Sendino, M.; Omaetxebarria MJ.; Rodríguez JA. Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resist. 2018, 1, 139-63. http://dx.doi.org/10.20517/cdr.2018.09
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 16
likes
Like This Article 16
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.