
Cancer drug resistance: a new perspective


Introduction
Cancer drug resistance has been and unfortunately still is a major problem in cancer therapy. Almost any therapy (except surgery) that is being used in the treatment of cancer can result in resistance. Unfortunately there is a large group of patients that will either not respond to the applied therapy (intrinsic resistance) or will become resistant during therapy (acquired resistance). Sometimes patients can become resistant to one specific drug and remain sensitive to other drugs (one-drug resistance); another group of patients may become resistant to one drug and will be resistant to other unrelated drugs as well (multiple drug resistance, MDR).
Types of resistance
There is a difference in the response between various tumor types. Some tumors, such as pancreatic cancer have a limited survival[1], most likely due to a combination of failures, such as to surgery and the subsequent adjuvant chemotherapy, consisting of either a gemcitabine based therapy or a 5-fluorouracil (5FU) based combination such as FOLFIRINOX[2]. Although the latter therapy is more effective, this is at the cost of serious toxicity. Hence, pancreatic cancer is a disease for which resistance is intrinsic. In contrast, the majority of breast cancer patients will be cured, due to a combination of effective screening, improved surgery and radiation, and effective adjuvant therapy[3]. Even triple negative patients have a > 70% 5-year survival. In this disease and in stage III and IV patients acquired resistance is a major problem. For a subpopulation of breast cancer patients, specific causes for resistance such as BRCA have been identified, and fortunately, for a subgroup of patients effective new therapies are available.
Cancer Drug Resistance: why a new journal?
Although all major journals publish papers on resistance, no specific journal on resistance in cancer exists and we felt that this gap should be closed. With Cancer Drug Resistance, we aim to establish a forum for papers dealing with all aspects of drug resistance. In Figure 1, a schematic overview of potential resistance mechanisms is given. Unfortunately, novel drug development by cell and molecular biologists, often neglects the pharmacology of a drug. However, poor pharmacology is a major type of resistance, although poor pharmacology can sometimes be turned into an advantage[4]. A drug that simply does not reach a tumor will not be effective. This holds for both oral and intravenous administration. At intravenous administration, the drug may be catabolized by either phase I or phase II enzymes, before it reaches the tumor. An extensive clearance, e.g., renal clearance, may also prevent a drug reaching a tumor. At oral administration an additional hurdle can be expected, since the gut epithelial cells are designed to prevent uptake of xenobiotics, which include anticancer drugs. This can lead to a poor gut uptake and low plasma and tumor drug concentrations. Next the first pass effect of the liver can prevent further distribution through the body leading to a low tumor exposure.
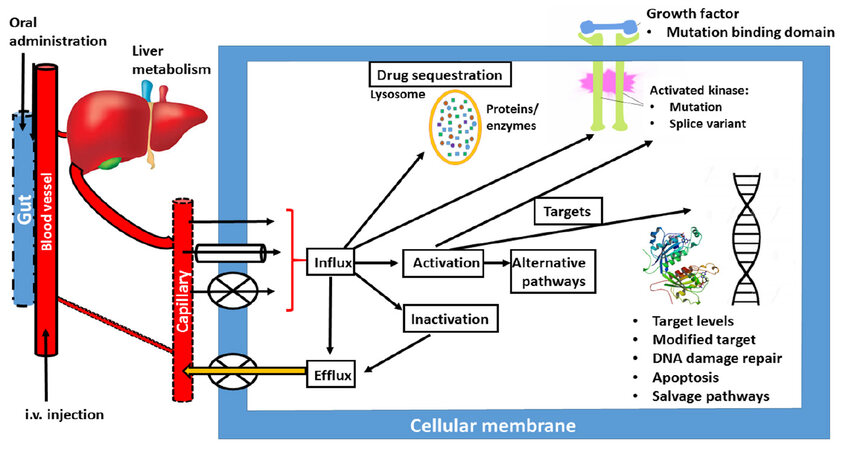
Figure 1. Simplified scheme, summarizing various possibilities for drug resistance: (1) at oral administration gut uptake can be poor; (2) i.v. administration can be associated with extensive renal clearance or liver metabolic clearance by phase I and II enzymes; (3) from the tumor blood capillaries the drug can be taken up by either diffusion (single arrow), facilitated transport (tube) or active transport (cross); (4) in the cell the drug can be activated or inactivated; (5) these metabolites can be effluxed; (6) the drug can be sequestered (e.g., in the lysosome, where it can be protonated); (7) the drug can hit a protein kinase; (8) or DNA; (9) or other targets. The cell can respond to changes in any of these processes (either increase or decrease), activate alternative signaling pathways, modify the target, or repair the damage
What is special in the first issue?
When a drug reaches a tumor, the cancer cell has an extensive arsenal of defense mechanism. A drug has to be taken up by the cancer cells and defective transport is a major mechanism of resistance. Uptake can be mediated by diffusion, a facilitated transporter or a concentrative energy dependent transporter. When a drug manages to be taken up, it can almost immediately being kicked out by one of the ATP-binding cassette transporters, as described by Jaramillo et al.[5] in this issue. In this paper, approaches to bypass increased efflux are summarized, with emphasis on novel developments since the landmark paper by Szakács et al.[6]. When a drug manages to be taken up into a cancer cell, the drug will encounter more hurdles leading to resistance, such as defective activation, or extensive degradation. The paper by Dominijanni and Gmeiner[7] describes a dual approach to bypass resistance. By using a prodrug complex, the drug can bypass the usual transporters, does not need activation and can immediately hit its target, thymidylate synthase (TS). This novel application of an old drug is ready to be tested in patients. When a drug, in this case FdUMP, the active metabolite of 5FU, hits its target TS, the cell can respond by modifying the target, e.g., by increasing the gene expression or the activity of the target, modify the drug binding or enabling the cell to increase the concentration of its target[8], making it less sensitive. In the case of TS, this means an increased expression of TS, which has been associated by resistance to 5FU. 5FU is often given in combination with leucovorin which prevents this increase in TS[9]. Another approach to bypass resistance to this drug (and basically any drug) is to develop clever combinations in which one drug sometimes uses the resistance mechanism in order to be effective[10]. Therefore 5FU is often combined with cisplatin in the treatment of head & neck cancer, and with oxaliplatin in colon cancer (FOLFOX regimens)[11]. Novel combinations include bevacizumab which neutralizes the vascular endothelial growth factor[12], and inhibits angiogenesis. On the other hand cetuximab inhibits epidermal growth factor (EGFR) mediated signaling. Small molecule inhibitors of EGFR such as erlotinib and gefitinib, are only active in adenocarcinoma of non-small cell lung cancer when the tumor has activating mutations, making the tumor cell dependent on (or addicted to) EGFR signaling. Additional mutations (e.g., T790M) lead to resistance, but these cells are sensitive to the 3rd generation of EGFR inhibitors[13]. From these data it should be noted, that dependent on the target a high expression (or mutated enzyme) can lead to either resistance or increased sensitivity. Additional mutations can lead to resistance, but sensitivity to other inhibitors designed to hit the mutated site[14].
Drugs, such as cisplatin can hit their target DNA by forming either intrastrand or inter strand adducts. The cell subsequently dies or finds ways to bypass its death, for cisplatin this is usually mediated by apoptosis, as described in the paper by Ballestreri et al.[15]. Aberrations in the apoptotic machinery may prevent a cancer cell from dying, leading to resistance. Another protection mechanism of the cell is autophagy, which is either another form of cell kill or a survival pathway[16]. It can also provide the cell with additional nutrients, such as amino acids, so that the cells survive.
Enzymes that repair the DNA damage form an important resistance mechanism and are a major hurdle in treatment. Treatment often induces repair mechanism in the cell. The paper by Sharaf Eldin et al.[17] describes the role of apoptosis in resistance to another alkylating drug, but surprisingly RAS expression is also involved in resistance. They describe a novel approach to reverse resistance by the use of siRNA, which suppresses the survival pathway in which RAS plays a role. Uckun and Qazi[18] also used siRNA (encapsulated in nanoparticles) to downregulate the CD22∆ E12 in drug-resistant B-precursor ALL. Unfortunately siRNA application is still limited to model systems, although carriers such as nanoparticles are being investigated to solve this problem.
A not very well known resistance mechanism involves drug sequestration, which means that drugs accumulate in cellular organelles or vesicles such as lysosomes, preventing that the drugs do not reach their targets. Gotink et al.[19] characterized the mechanism of lysosomal uptake of sunitinib, which involves protonation of the drug, so that it cannot efflux. This effect seems to be widely distributed for several tyrosine kinase inhibitors[20]. A sudden release of the lysosomal content will lead to immediate cell death as was demonstrated elegantly by Adar et al.[21] for certain acridones that can also accumulate in lysosomes.
New challenges
A major challenge in studying drug resistance is how to identify this in patients and how to bypass resistance. Fortunately novel technology, such as imaging and positron emission tomography help to identify in vivo mechanisms of resistance in animal models and patients[22]. Novel medicinal chemistry technology enables to synthesize new drugs using scaffolds such as molecular modeling[23]. Another approach is to use pharmacogenetics to identify patients that express a resistance marker before treatment[24].
Another new challenge is how to prevent resistance to immunotherapy. Currently immunotherapy is restricted to antibodies, but small molecules that affect the defective immune system are being developed[25]. Since these drugs do not directly target a cancer cell, resistance mechanisms in immune cells need to be taken care of, e.g., some types of differentiated macrophages express a high level of BCRP[26].
Treatment modalities such as radiation are often combined with standard chemotherapy or with one of the novel tyrosine kinase inhibitors. Radiation often induces one of the survival pathways such as the AKT pathway[27], which can be inhibited by specific drugs such as MK2206[28] or perifosine[29]. Hyperthermia is based on an altered expression of activating or repair enzymes at a higher temperature, which may bypass resistance as well[30].
Cancer Drug Resistance aims to provide a forum for all types of drug resistance, in order to identify or elucidate the mechanism of resistance and importantly, how to bypass the resistance. Novel targeted drugs, usually consisting of a multitarget tyrosine kinase inhibitor (TKI) or a serine-threonine protein kinase inhibitor, were originally developed with the assumption, that being targeted would not lead to resistance, since the cell would die rapidly or cell proliferation would be controlled. Unfortunately, this wishful thinking was completely incorrect, and TKIs are subject to similar resistance mechanisms as the standard cytotoxic drugs, as indicated in Figure 1 as well. Additionally, these drugs often have a poor bioavailability, because of an effective transport from the gut epithelium back to the gut lumen[31,32].
This first issue provides a high standard, with a diversity of targets and drugs. We are looking forward to receive papers covering one or more of these aspects.
Declarations
Authors’ contributionsPeters GJ contributed solely to the editorial.
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Coppola S, Carnevale I, Danen EHJ, Peters GJ, Schmidt T, Assaraf YG, Giovannetti E. A mechanopharmacology approach to overcome chemoresistance in pancreatic cancer. Drug Resist Updat 2017;31:43-51.
2. Caparello C, Meijer LL, Garajova I, Falcone A, Le Large TY, Funel N, Kazemier G, Peters GJ, Vasile E, Giovannetti E. FOLFIRINOX and translational studies: towards personalized therapy in pancreatic cancer. World J Gastroenterology 2016;22:6987-7005.
3. Shachar SS, Jolly TA, Jones E, Muss HB. Management of triple-negative breast cancer in older patients: how is it different? Oncology (Williston Park) 2018;32:58-63.
4. Phillips RM, Hendriks HR, Peters GJ; EORTC-Pharmacology and Molecular Mechanism Group. EO9 (Apaziquone): from the clinic to the laboratory and back again. Brit J Pharmacol 2013;168:11-8.
5. Jaramillo AC, Al Saig F, Cloos J, Jansen G, Peters GJ. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist 2018;1:6-29.
6. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219-34.
7. Dominijanni A, Gmeiner WH. Improved potency of F10 relative to 5-fluorouracil in colorectal cancer cells with p53 mutations. Cancer Drug Resist 2018;1:48-58.
8. Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL, Smid K, Lunec J, Calvert AH, Marsh S, McLeod HL, Bloemena E, Meijer S, Jansen G, van Groeningen CJ, Pinedo HM. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta 2002;1587:194-205.
9. Danenberg PV, Gustavsson B, Johnston PJ, Lindgren P, Moser R, Odin E, Peters GJ, Petrelli N. Folates as adjuvants to anticancer agents: chemical rationale and mechanism of action. Crit Rev Oncol Hematol 2016;106:118-31.
10. Peters GJ, van der Wilt CL, van Moorsel CJ, Kroep JR, Bergman AM, Ackland SP. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther 2000;87:227-53.
11. Grivicich I, Mans DR, Peters GJ, Schwartsmann G. Irinotecan and oxaliplatin: an overview of the novel chemotherapeutic options for the treatment of advanced colorectal cancer. Braz J Med Biol Res 2001;34:1087-103.
12. Strickler JH, Hurwitz HI. Bevacizumab-based therapies in the first-line treatment of metastatic colorectal cancer. Oncologist 2012;17:513-24.
13. Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E. New developments in the management of non-small cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther 2016;9:6065-74.
14. Muller IB, De Langen AJ, Honeywell RJ, Giovannetti E, Peters GJ. Overcoming crizotinib resistance in ALK-rearranged NSCLC with the second-generation ALK-inhibitor ceritinib. Expert Rev Anticancer Ther 2016;16:147-57.
15. Ballestreri É, Simon D, de Souza AP, Grott CS, Nabinger DD, Dihl RR, Grivicich I. Resistance mechanism to cisplatin in NCI-H460 non-small cell lung cancer cell line: investigating apoptosis, autophagy, and cytogenetic damage. Cancer Drug Resist 2018;1:72-81.
16. Gómez VE, Giovannetti E, Peters GJ. Unraveling the complexity of autophagy: potential therapeutic applications in pancreatic ductal adenocarcinoma. Semin Cancer Biol 2015;35:11-9.
17. Sharaf Eldin O, Fouda AM, Youssef AR, Hamilton P, Maxwell P, Williamson KE. Reduction of mitomycin C resistance in human bladder cancer T24 cells by knocking-down ras oncogene. Cancer Drug Resist 2018;1:59-71.
18. Uckun FM, Qazi S. Identification and targeting of CD22ΔE12 as a molecular RNAi target to overcome drug resistance in high-risk B-lineage leukemias and lymphomas. Cancer Drug Resist 2018;1:30-47.
19. Gotink KG, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters RJ, Jansen G, Griffioen AW, Assaraf YG, Pili R, Peters GJ, Verheul HM. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res 2011;17:7337-46.
20. Da Silva CG, Honeywell RJ, Dekker H, Peters GJ. Physicochemical properties of novel protein kinase inhibitors in relation to their substrate specificity for drug transporters. Expert Opin Drug Metab Toxicol 2015;11:703-17.
21. Adar Y, Stark M, Bram EE, Nowak-Sliwinska P, van den Bergh H, Szewczyk G, Sarna T, Skladanowski A, Griffioen AW, Assaraf YG. Imidazoacridinone-dependent lysosomal photodestruction: a pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis 2012;3:e293.
22. Direcks WGE, Berndsen SC, Proost N, Peters GJ, Balzarini J, Spreeuwenberg MD, Lammertsma AA, Molthoff CFM. [18F]FDG and [18F]FLT uptake in human breast cancer cells in relation to the effects of chemotherapy: an in vitro study. Brit J Cancer 2008;99:481-7.
23. Gade DR, Makkapati A, Yarlagadda RB, Peters GJ, Sastry BS, Rajendra Prasad VVS. Elucidation of chemosensitization effect of acridones in cancer cell lines: combined pharmacophore modelling, 3D QSAR and molecular dynamics studies. Comp Biol Chem 2018;74:63-75.
24. Robert J, Le Morvan V, Giovannetti E, Peters GJ; PAMM Group of EORTC. On the use of pharmacogenetics in cancer treatment and clinical trials. Eur J Cancer 2014;50:2532-43.
25. Azijli K, Stelloo E, Peters GJ, van den Eertwegh AJ. New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res 2014;34:1493-505.
26. van de Ven R, Oerlemans R, van der Heijden JW, Scheffer GL, de Gruijl TD, Jansen G, Scheper RJ. ABC drug transporters and immunity: novel therapeutic targets in autoimmunity and cancer. J Leukoc Biol 2009;86:1075-87.
27. Avan A, Narayan R, Giovannetti E, Peters GJ. Role of Akt signaling in resistance to DNA-targeted therapy. World J Clin Oncol 2016;7:352-69.
28. Narayanan RS, Fedrigo CA, Brands E, Dik R, Stalpers LJ, Baumert BG, Slotman BJ, Westerman BA, Peters GJ, Sminia P. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017;17:204.
29. Massihnia D, Avan A, Funel N, Maftouh M, Van Krieken A, Granchi C, Raktoe R, Boggi U, Aicher B, Minutolo F, Russo A, Leon LG, Peters GJ, Giovannetti E. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of AKT inhibitors with gemcitabine in pancreatic cancer. J Hematol Oncol 2017;10:9.
30. Kwakman R, De Cuba E, De Winter J, De Hingh I, Delis-Van Diemen P, Tijssen M, Rooimans MA, Krijgsman O, Carvalho B, Peters GJ, Bonjer HJ, Meijer GA, Te Velde EA. Tailoring heated intraperitoneal Mitomycin C for peritoneal metastases originating from colorectal carcinoma: a translational approach to improve survival. Br J Cancer 2015;112:851-6.
31. Honeywell RJ, Hitzerd S, Kathmann I, Peters GJ. Transport of six tyrosine kinase inhibitors: active or passive? ADMET & DMPK 2016;4:23-34.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Peters GJ. Cancer drug resistance: a new perspective. Cancer Drug Resist 2018;1:1-5. http://dx.doi.org/10.20517/cdr.2018.03
AMA Style
Peters GJ. Cancer drug resistance: a new perspective. Cancer Drug Resistance. 2018; 1(1): 1-5. http://dx.doi.org/10.20517/cdr.2018.03
Chicago/Turabian Style
Peters, Godefridus J.. 2018. "Cancer drug resistance: a new perspective" Cancer Drug Resistance. 1, no.1: 1-5. http://dx.doi.org/10.20517/cdr.2018.03
ACS Style
Peters, GJ. Cancer drug resistance: a new perspective. Cancer Drug Resist. 2018, 1, 1-5. http://dx.doi.org/10.20517/cdr.2018.03
About This Article
Copyright

Data & Comments
Data


 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 23
likes
Like This Article 23
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.