
Dysregulation of calcium homeostasis in cancer and its role in chemoresistance
 0
0 , ...
, ... Abstract
Globally, cancer, as a major public health concern, poses a severe threat to people’s well-being. Advanced and specialized therapies can now cure the majority of people with early-stage cancer. However, emerging resistance to traditional and novel chemotherapeutic drugs remains a serious issue in clinical medicine. Chemoresistance often leads to cancer recurrence, metastasis, and increased mortality, accounting for 90% of chemotherapy failures. Thus, it is important to understand the molecular mechanisms of chemoresistance and find novel therapeutic approaches for cancer treatment. Among the several factors responsible for chemoresistance, calcium (Ca2+) dysregulation plays a significant role in cancer progression and chemoresistance. Therefore, targeting this derailed Ca2+ signalling for cancer therapy has become an emerging research area. Of note, the Ca2+ signal and its proteins are a multifaceted and potent tool by which cells achieve specific outcomes. Depending on cell survival needs, Ca2+ is either upregulated or downregulated in both chemosensitive and chemoresistant cancer cells. Consequently, the appropriate treatment should be selected based on Ca2+ signalling dysregulation. This review discusses the role of Ca2+ in cancer cells and the targeting of Ca2+ channels, pumps, and exchangers. Furthermore, we have emphasised the role of Ca2+ in chemoresistance and therapeutic strategies. In conclusion, targeting Ca2+ signalling is a multifaceted process. Methods such as site-specific drug delivery, target-based drug-designing, and targeting two or more Ca2+ proteins simultaneously may be explored; however, further clinical studies are essential to validate Ca2+ blockers’ anti-cancer efficacy.
Keywords
INTRODUCTION
Despite advances in technology and available treatments, cancer remains the second leading global cause of death[1]. With improved therapeutic approaches, most patients with early-stage cancer are now curable. There are several treatment options, including conventional and advanced treatments, based on cancer stage, histology, and biomarkers. Conventional treatments are chemotherapy, radiation, and surgery, while advanced treatment options include hyperthermia, ferroptosis-based therapy, immunotherapy, stem cell transplant, targeted therapy, hormone therapy, gene therapy, phototherapy, and others[2,3]. Of these, chemotherapy is the most preferred mode of treatment by oncologists as a primary treatment to reduce the cancer burden. Abraxane, bortezomib, cisplatin, docetaxel, doxorubicin, paclitaxel, and temozolomide are some examples of anti-cancer drugs used in chemotherapy[4]. However, chemotherapy has many challenges, such as non-specific drugs, side effects, rapid metabolism of drugs, and ineffective drugs. Furthermore, 90% of the major factor contributing to chemotherapy failure is chemoresistance[5]. Intrinsic or acquired chemoresistance is a major hurdle in successful cancer treatment as it often leads to cancer relapse and metastasis, which, in turn, reduces the efficacy of cancer treatment and causes patient death. This resistance can arise either to a single anti-cancer drug or to multiple anti-cancer drugs that differ structurally and/or functionally[6,7]. This is due to underlying genetic mutations, tumour heterogeneity, activated intrinsic pathways, pharmacological factors, and the tumour microenvironment (TME)[3,7,8]. Moreover, it is vital to find novel therapeutic strategies to overcome chemoresistance. For this, it is necessary to understand the molecular mechanisms of chemoresistance, which include tumour suppressor genes, transporters, pumps, oncogenes, mitochondrial alteration, autophagy, epithelial-mesenchymal transition (EMT), DNA repair, cancer stemness, and exosome[9-11].
Several studies have emphasised the role of calcium (Ca2+) in cancer progression and chemoresistance[12]. Being a secondary messenger, Ca2+ plays a crucial role in maintaining cellular functions, apoptosis, and regulating the network of Ca2+ signalling in cells. Disrupted Ca2+ signalling networks in cancer cells implicate a critical role of Ca2+ in assisting cancer cells in growing and overcoming the anti-cancer effects of drugs. In the past decade, there have been significant breakthroughs in our understanding of how the Ca2+ signal may influence or even drive pathways that are crucial for therapeutic resistance in cancer. Since it is not possible to review the large number of studies done in this area, cancer studies targeting the Ca2+ signalling proteins are briefly described here. In addition, studies targeting Ca2+ signalling and its proteins to overcome chemoresistance are also summarised. Thus, in this review, we aim to understand the role and potential therapeutic strategies of targeting Ca2+ proteins in chemoresistant cancer cells.
Ca2+ SIGNALLING VIA Ca2+ CHANNELS, PUMPS, AND EXCHANGERS
Ca2+ is a multifaceted tool that plays a significant role as a secondary messenger in several cellular functions, such as cell growth and division, migration, absorption, secretion, transcellular transport, signalling, and apoptosis[13]. In cells in the resting phase, the cytoplasmic Ca2+ concentration ([Ca2+]cyt) is 100 nM and the extracellular Ca2+ concentration ([Ca2+]ext) is 1-2 mM. In intracellular Ca2+ stores, such as endoplasmic reticulum (ER) ([Ca2+]ER), the Ca2+ concentration is > 100 µM. In mitochondria ([Ca2+]mit) and the nucleus, the Ca2+ concentration remains the same as in the cytoplasm. Upon activation of the Ca2+ signalling proteins, the [Ca2+]cyt can exceed 1 µM[14-18]. This changed Ca2+ level initiates several signalling cascades. Sustained elevation of Ca2+ levels can lead to cell death; hence, a controlled Ca2+ level is maintained by Ca2+ channels, pumps, and exchangers. As illustrated in Figure 1, these Ca2+-related proteins are located on both the plasma membrane and cellular organelles, including the ER, mitochondria, Golgi apparatus (GA), and lysosomes[18-20]. Ca2+ binding proteins present in cells are calmodulin (CaM), parvalbumin, calcineurin, S100 protein, and several other proteins that are present in both extracellular and intracellular regions[21].
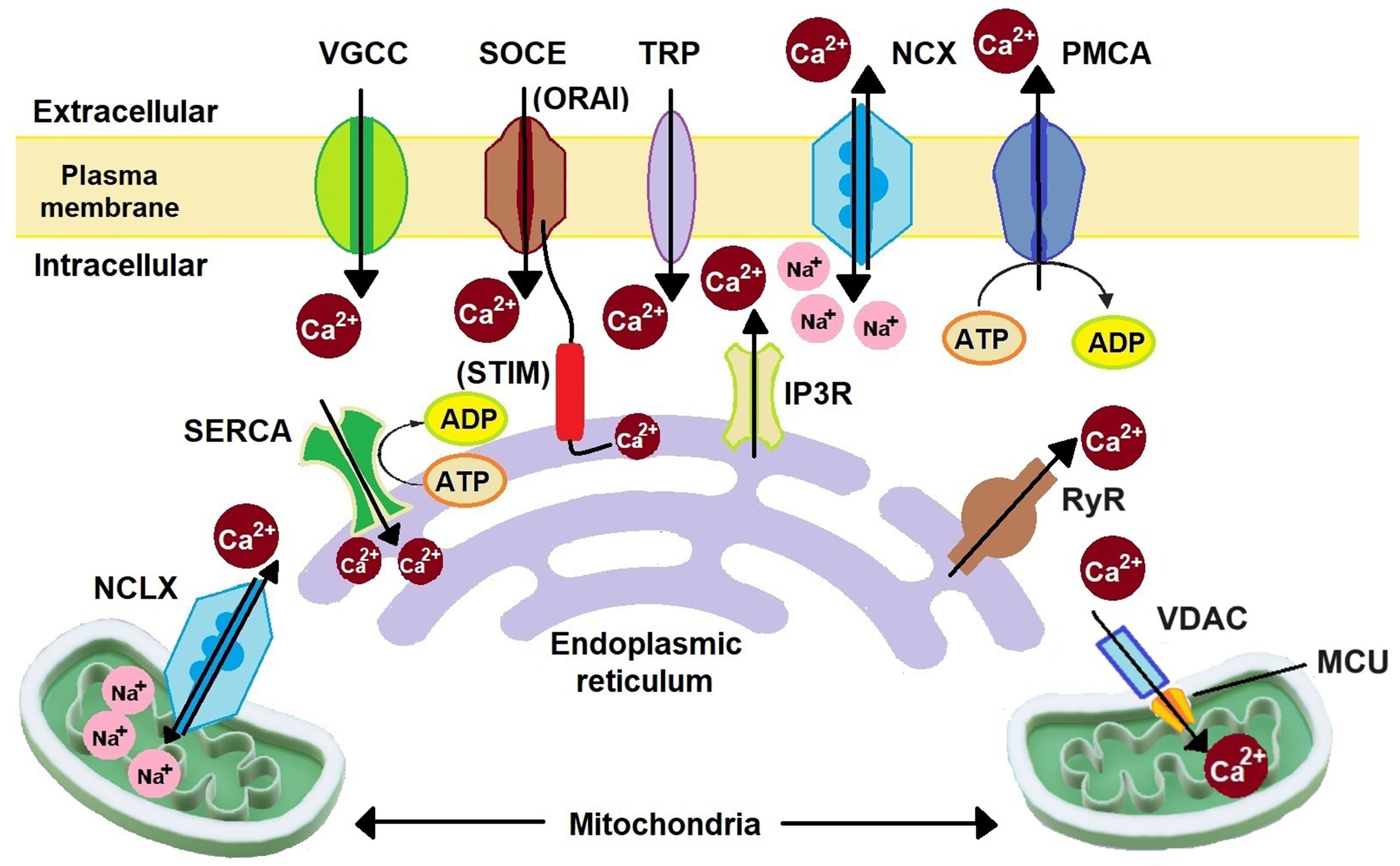
Figure 1. Schematic representation of major Ca2+ signalling proteins. Extracellular Ca2+ is transported to the cytoplasm through VGCCs, SOCC (ORAI), and TRP channels. Organelle Ca2+ is transported to the cytoplasm through mitochondrial NCLX, IP3R, and RyR. Cytoplasmic Ca2+ is removed by NCX, PMCA, SOCC (STIM), SERCA, and MCU. NCLX: Na+/Ca2+/Li+ exchanger; SERCA: sarco/endoplasmic reticulum Ca2+ ATPase; VGCCs: voltage-gated Ca2+ channels; SOCE: store-operated Ca2+ entry; STIM: stromal interacting molecule; TRP: transient receptor potential; IP3R: inositol-1,4,5-trisphosphate receptor; NCX: Na+/Ca2+ exchanger; PMCA: plasma membrane Ca2+ ATPase; RyR: ryanodine receptor; VDAC: voltage-dependent anion channel; MCU: mitochondrial Ca2+ uniporter; SOCC: store-operated Ca2+ channels.
On the plasma membrane, voltage-gated Ca2+ channels (VGCC), store-operated Ca2+ channels (SOCC), transient receptor potential (TRP) channels, Na+/Ca2+ exchanger (NCX), and plasma membrane Ca2+ ATPase (PMCA) are present. Depending on the electrophysiological properties and the genes encoding them, there are five types of VGCC: L-type (LTCC), P/Q type (P/QTCC), N-type (NTCC), R-type (RTCC), and T-type (TTCC). LTCC has four isoforms: Cav1.1-1.4. P/QTCC, NTCC, and RTCC each have one isoform: Cav2.1, Cav2.2, and Cav2.3, respectively. TTCC has three isoforms: Cav3.1-3.2[22]. These channels are activated upon reaching their respective voltage threshold during the voltage change in the membrane. LTCC, P/QTCC, NTCC, and RTCC are high-voltage-activated (HVA) channels, while TTCC is a low-voltage-activated (LVA) channel. Therefore, HVA channels are mostly found in excitable cells, and TTCC is found mostly in non-excitable cells. However, HVA channels are also found to be expressed in some non-excitable cells[18]. SOCC has two subunits: ORAI and stromal interacting molecule (STIM). ORAI is present on the plasma membrane, while STIM is located on the ER. ORAI has three isoforms: ORAI1-3. STIM has two isoforms: STIM1 and STIM2[23]. Upon Ca2+ depletion in the ER, ORAI and STIM proteins are relocalized to plasma membrane-ER junctions and interact afterwards with each other to activate Ca2+ influx[18,24-26]. This process is termed store-operated Ca2+ entry (SOCE). ORAI channels can also lead to Ca2+ influx driven by arachidonic acid without interacting with STIM proteins[18]. TRP has 28 different families divided into six subfamilies encoded by TRP genes such as TRPC (canonical), TRPV (vanilloids), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystins), and TRPML (mucolipins)[27,28]. TRPC7 enhances cell growth and migration with the activation of Ca2+/CaM-dependent protein kinase II (CAMKII), phosphatidylinositol 3-kinase (PI3K)/Akt, and extracellular signal-regulated kinase (ERK)[29]. Three isoforms of NCX are found in cells: NCX1-3[30]. NCX functions in both forward and reverse modes. In the forward mode, it transfers three Na+ ions inside and two Ca2+ ions outside the cells and thus acts as an anti-apoptotic channel. In the reverse mode, it transfers three Na+ ions outside and two Ca2+ ions inside and thus acts as a pro-apoptotic channel by increasing the Ca2+ accumulation in the cells[31]. PMCA is an ATP-based Ca2+ pump, helping to maintain low [Ca2+]cyt. PMCA has four isoforms: PMCA1-4[32,33]. This pump removes one Ca2+ ion per ATP molecule utilized[33].
On the ER membrane, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), STIM, inositol-1,4,5-trisphosphate receptor (IP3R), and ryanodine receptor (RyR) are present. Unlike PMCA, SERCA removes two Ca2+ ions from the cytosol to the ER[33]. Ca2+ release from the ER takes place with the help of IP3R and RyR. IP3R has three isoforms: IP3R1-3[26]. IP3R is involved in SOCE by reducing [Ca2+]ER and thereby stimulating the activation of STIM1 and ORAI1 to replenish the ER Ca2+ level[26]. RyR has three isoforms: RyR1-3[34]. Upon VGCC activation, the influx of Ca2+ from the extracellular region increases [Ca2+]cyt, which results in the inactivation of VGCC in a negative feedback mechanism. The resulting increased Ca2+ ions in the cytosol then induce activation of RyR, which in turn releases Ca2+ from the ER, and thus the process is called “Ca2+-induced-Ca2+-release” (CICR)[34-36]. On mitochondria, the mitochondrial Ca2+ uniporter (MCU) complex and Na+/Ca2+/Li+ exchanger (NCLX) are present. Cytosolic Ca2+ enters mitochondria through the MCU complex, controlled by a voltage-dependent anion channel (VDAC)[37]. Like NCX, NCLX also functions in forward and reverse modes. In the forward mode, NCLX transfers Ca2+ from mitochondria to the cytosol, and in the reverse mode, it transfers Ca2+ from the cytosol to mitochondria[38]. In the GA, along with IP3R, RyR, and SERCA, the secretory-pathway Ca2+ ATPase (SPCA) is also expressed. SPCA has two isoforms, SPCA1 and SPCA2. It transfers one Ca2+ ion at a time inside GA[39]. Lysosomes are also known to function as Ca2+ stores, though less extensively than the ER. Endocytosis, Ca2+/H+ exchanger, TRP channels, and multiple Ca2+ sensors including CaM help in Ca2+ homeostasis in lysosomes[18,20].
During the cell cycle, Ca2+ is required in the early G1 phase and in the transition from G1 to S, G2 to M, and metaphase to anaphase. CaM and its downstream targets, CaMKII, calcineurin, and protein kinase C (PKC), are direct effectors of Ca2+ signalling. In turn, they regulate transcription factors such as the nuclear factor of activated T cells (NFATs), cyclic adenosine monophosphate (cAMP)-responsive element-binding (CREB) protein, and nuclear factor-κB (NFκB) for cell-cycle progression[12,13,40]. CaM assists in the progression through the G1 and M phases. Calcineurin is required for the transition from the G1 to S phases and cyclin D1 expression during the G1 phase[12]. Ca2+ and CaMKII also regulate centrosome duplication and the separation of chromosomes into daughter cells[12].
Ca2+ signalling in cancer cells
In pathological conditions like cancer, Ca2+ signalling is frequently disrupted due to the dysregulation of various Ca2+ channels, pumps, exchangers, and binding/storage proteins[13,41]. This dysregulation is caused by mutations, changes in expression, regulation, and/or subcellular targeting of the Ca2+ signalling proteins[13]. Excessive Ca2+ levels are toxic to cells, thus requiring tight regulation of Ca2+ signalling. Despite this, cancer cells can thrive in high-Ca2+ environments due to their additional need for Ca2+ to support increased cell division, cell signalling, and tissue invasion[18]. In contrast to this, some studies have reported that cancer cells have a reduced dependence on Ca2+ for proliferation[12] and evading apoptosis[13].
Henceforth, dysregulated Ca2+ signalling proteins either increase or decrease Ca2+ levels in cancer cells depending on their perceived benefit to survival. Reduced expression of some VGCCs in cancer tissue compared to normal tissue is also seen, indicating the possible role of those Ca2+ channels as tumour suppressor gene markers[42]. Studies have also shown that monomeric and heteromeric channels exhibit different advantages or disadvantages to cancer cells. For example, ORAI1-ORAI3 heteromers induce apoptosis resistance in prostate cancer cells compared to ORAI1 monomers through reduced SOCE because the number of ORAI1 monomers is reduced. Similarly, because of the increase in the number of this heteromer, there is an increased proliferation signalling via arachidonic acid[40,43].
In cancer cells, increased production of reactive oxygen species (ROS) leads to the accumulation of ROS at the plasma membrane. This leads to post-translational modification of the plasma membrane channels, thereby promoting Ca2+ entry into the cells to counteract the ROS activities[18]. Furthermore, disrupted Ca2+ signalling promotes the activation of Ca2+-dependent transcription factors such as NFAT and G1 cyclins such as c-Myc, c-Jun, and c-Fos. These factors promote cancer growth by inducing the expression of G1 and G1/S phase transition cyclins and associated cyclin-dependent kinases (CDK4 and CDK2)[12,13]. Ca2+ is also involved in pathways such as mitogen-activated protein kinases (MAPK) and PI3K/Akt that promote malignancy in cancer cells[29].
Excessive Ca2+ stimulates Ca2+-sensitive catabolic enzymes such as proteases and endonucleases[44]. As shown in Figure 2, elevated [Ca2+]cyt, facilitated by channels such as TRP and ORAI1-ORA3 heteromers, activates CaMKII, ultimately inhibiting caspases 8 and 9 and promoting cell survival[43]. However, cancer cells show reduced Ca2+ signalling from the ER required for apoptosis. Therefore, blockage of SERCA by Cartilage Oligomeric Matrix Protein (COMP1) reduces Ca2+ uptake in the ER. Furthermore, ORAI1 monomers are responsible for ER-induced apoptosis. Thus, downregulation of ORAI1 further inhibits apoptosis[43]. B-cell lymphoma-2 (Bcl-2), an anti-apoptotic protein, modulates ER Ca2+ signalling to regulate the apoptosis pathway. Several studies have shown that Bcl-2 is overexpressed in cancer cells[45]. Oncogenes such as rat sarcoma (RAS), Akt, and miR-25 also regulate Ca2+ signalling or proteins responsible for Ca2+-dependent apoptosis to prevent apoptosis[46]. Increased [Ca2+]mit is also associated with apoptosis. Increased Ca2+ levels in mitochondria reduce its membrane integrity by opening the mitochondrial permeability transition pore. Subsequently, pro-apoptotic factors are released from mitochondria[40]. However, regulation of oncogenes such as Kristen rat sarcoma (KRAS) 2 viral oncogene homolog and tumour suppressor proteins such as p53 by Ca2+ signalling subsequently reduces [Ca2+]mit and thereby promotes cancer cell survival[25,40]. Mitochondrial-induced apoptosis is also inhibited by downregulating IP3R. IP3R is downregulated by COMP1 and F-box protein (FBXL2). Phosphatase and tensin homolog (PTEN) (deleted on chromosome 10) inhibits the activity of FBXL2. However, its mutation or inactivation leads to the upregulation of FBXL2, which targets IP3R3 for proteasome degradation[17,43]. IP3R downregulation reduces [Ca2+]mit and thereby prevents mitochondrial-induced apoptosis. Apart from apoptosis, Ca2+ also regulates other cell death modes such as autophagy, necrosis, anoikis, entosis, and ferroptosis, reviewed in other papers[40,44,47,48]. Although there is still a lack of clear understanding regarding how Ca2+ regulates different cell death modes, increased and sustained [Ca2+]cyt is often associated with necrosis and other death modes such as necroptosis[40].

Figure 2. Apoptosis inhibition in cancer cells. Increased Ca2+ influx through upregulated VGCC, TRP, and ORAI1-ORAI3 heterodimer leads to activation of CAMKII, which inhibits pro-apoptotic caspases 8 and 9. Downregulation of ORAI1 monomer and SERCA reduces
Ca2+ signalling is known to regulate many immune-associated pathways in cancer cells[27]. Examples include the activation of STAT3, a transcription factor in cancer, as well as MAPK pathway, Akt pathway, and Wnt pathway[27]. TME is also remodelled due to Ca2+ signalling[27]. Ca2+ also induces chemoresistance by facilitating cancer cell invasion and metastasis[25]. Metastasis accounts for more than 90% of cancer-related deaths. Overexpression of several Ca2+ proteins is associated with metastasis[25]. Prerequisite steps in metastasis such as pro-migratory signals, movement rate, directional control, and extracellular matrix (ECM) degradation are dependent on Ca2+ signalling[40]. Channels like ORAI1 and TRPV2 play roles in cancer cell invasion through the ECM and formation of invadopodia. Other pathways such as ROS production and cAMP pathways are also modulated by Ca2+, contributing to cancer cell migration and invasion. However, Ca2+ proteins involved in cancer cell migration and invasion vary among cancer types and stimuli[40].
Furthermore, Ca2+ signalling is also known to regulate epigenetic mechanisms[49]. Changes in the DNA methylation and histone post-translational modifications are in good relation with the altered gene expression of Ca2+ signalling proteins such as the cadherin and VGCCs in all breast cancer subtypes[50]. Targeting Ca2+ signalling can reactivate tumour suppressor genes silenced by cancer cell epigenetics. In colon cancer, altered Ca2+ signalling results in activated Ca2+ CaM kinase which plays a central role in tumour-suppressor genes reactivation and suppressing cancer cell growth[51]. Program death ligand 1
TARGETING Ca2+ SIGNALLING PROTEINS INVOLVED IN CANCER
Ca2+ proteins present in the plasma membrane
The plasma membrane serves as a crucial interface for Ca2+ signalling. Targeting the plasma membrane Ca2+ signalling proteins can significantly impact [Ca2+]cyt by modulating the entry and exit of Ca2+ ions through VGCCs, ORAI, TRPs, NCX, and PMCA. This modulation can either decrease or increase [Ca2+]cyt, thereby placing a burden on intracellular Ca2+ signalling proteins to maintain [Ca2+]cyt. However, sustained modulation is impractical due to the interconnected activities of plasma membrane and intracellular proteins.
Dysregulation of various channels, pumps, transporters, and proteins occurs in different cancer types, leading to either upregulation or downregulation depending on their survival benefits[42,55]. For instance, in a clinical study on lung adenocarcinoma, TRPC7 expression varied among patients[29]. Consequently, studies have explored the modulation of Ca2+ levels as a therapeutic target. In hepatocarcinoma cells, fluorouracil (5FU) induced cell death by inhibiting Ca2+ influx through ORAI1 channels[56], while in colon carcinoma cells, it exerted cytotoxic effects by elevating [Ca2+]cyt level to activate CaM, triggering apoptosis via p53 phosphorylation[57].
Overexpression of Ca2+ channels, pumps, or exchangers is associated with cancer prognosis. For example, NTCC overexpression correlates with poor survival in patients with non-small cell lung cancer (NSCLC)[58] and adult adrenocortical carcinoma (ACC)[59], while it predicts good prognosis in gliomas[60]. Similarly, TRPM2 expression negatively correlates with prognosis in hepatocellular carcinoma patients[61]. In lung adenocarcinoma patients, overexpression of TRPC7 is associated with poor prognosis and lower survival rates[29]. The study by Wang et al. has shown that overexpression of PMCA4 is associated with good prognosis in gastric cancer patients[62].
Targeting these proteins induces cancer cell death through various modes, including apoptosis, autophagy, necrosis, entosis, and ferroptosis. Different channels are associated with different modes of cell death. For instance, ORAI1 plays a role in entosis in cancer development via the septin-ORAI1-Ca2+/CaM-myosin light chain kinase (MLCK)-actomyosin axis[63]. The study by Lee and Park showed that when blocking or knocking out the ORAI1 channel, there was reduced entosis in the MCF7 breast cancer cells[63]. Notably, the role of entosis in cancer remains unclear[64], but both invading and engulfing cells require intracellular Ca2+ signalling for entosis.
Ca2+ proteins present in intracellular organelles
Similar to plasma membrane Ca2+ signalling proteins, Ca2+ signalling proteins within intracellular organelles are associated with different modes of cell death and cancer prognosis. Reduced [Ca2+]ER protects cancer cells from apoptosis and resistance to cisplatin and taxol[58,65]; increased [Ca2+]ER, facilitated by upregulated IP3R and RyR, promotes proliferation, migration, invasion, malignancy, and apoptosis in various cancer cells[66].
Increased [Ca2+]mit is associated with apoptosis and necrosis[58,65]. MCU promotes ferroptosis under cystine deprivation, while inhibiting ferroptosis induced by glutathione peroxidase 4 (GPX4) inhibition[67,68]. Accumulation of lipid ROS, either due to cysteine deprivation or inactivated GPX4 (the enzyme required to remove lipid ROS), leads to ferroptosis[68]. Cells overexpressing MCU upregulate cysteine consumption to remove lipid ROS. This leads to cysteine deprivation, which in turn induces the transcription of “Ferroptosis Signature” genes[67]. This ferroptosis due to cysteine deprivation is inhibited on MCU knockout[67]. In contrast, in a study by Marmolejo-Garza et al., it is shown that MICU1 from the MCU complex has a protective role against ferroptosis. On knocking out MICU1 in HT22 (mouse hippocampal neuronal cell line) and mouse embryonic fibroblasts (MEF) cells, they showed increased sensitivity of these cells toward ferroptosis[69]. Moreover, altered expression of the MCU complex is associated with poor prognosis, increased metastasis, migration, invasion, and metabolic stress resistance in cancers[67,70].
Furthermore, SPCA1 and SPCA2 overexpression in the GA is associated with a pro-survival role in breast cancer[71-73]. Knocking down SPCA1[73] and SPCA2[72] has shown to reduce MDA-MB-231[73] and MCF-7[72] breast cancer cells proliferation, respectively. Knockdown of SPCA2 has also shown anchorage-independent growth of MCF-7 cells and tumour formation in mice models[72,73]. However, the role of SPCA2 in tumour progression is indirect by increasing Ca2+ influx across the plasma membrane by activating ORAI1 channels after SPCA2 is localized at the plasma membrane on its overexpression[73].
Further, it is reported that lysosomal Ca2+ signalling through TRPML1 and TPC2 regulates autophagy[20,74]. Targeting these channels has been shown to impair autophagy in non-cancer cells such as ARPE-19 (retinal pigment epithelium cell line)[74], thereby indicating the possibility of deregulating autophagy in cancer cells by targeting TRPM channels. In addition to autophagy, lysosomal Ca2+ signalling is also involved in proliferation, metastasis, angiogenesis, and multidrug resistance[74].
Table 1 summarizes the channels, pumps, and exchangers targeted in various studies and their effects on the subjects. Data reveal that each protein exhibits cell-specific, isoform-specific, and treatment-specific effects.
List of channels, pumps, and exchangers targeted in various studies and the effects on the targeted subjects
| Ca2+proteins | Isoforms | ||
| LTCC | Not defined[75], Cav1.2[76] | Subjects | Colon cancer cell lines (AZ-97 and HT-29)[75], ameloblastoma primary cells and cell line (AM-1)[76], mice[76] |
| Treatment | BayK 8644[75,76], Verapamil[75,76], siRNA[76] | ||
| Effects | ApoptosisBayK8644[75], apoptosis inhibitionVerapamil[75], suppressed cell aggregation and collective migration[76], reduced invasion[76] | ||
| P/QTCC | Cav2.1 | Subjects | Neuroblastoma cells (SH-SY5Y)[77] |
| Treatment | Cav2.1 mutants[77] | ||
| Effects | Reduced proliferation[77], apoptosis[77], decreased Bcl-2/Bax ratio[77] | ||
| NTCC | Cav2.2 | Subjects | Neuroblastoma glioma cells (NG108-15)[78] |
| Treatment | Cannabinoid[78] | ||
| Effects | Inhibition of Ca2+ current[78] | ||
| RTCC | Cav2.3 | Subjects | Neuroendocrine tumour BON cells[79] |
| Treatment | SNX-482[79] | ||
| Effects | Decreased chromogranin A secretion[79], reduced resting [Ca2+]cyt[79] | ||
| TTCC | Cav3.1[80], Cav3.2[80,81], Cav3.3[80], not defined[82-85] | Subjects | Breast cancer cell line (T-47D)[80], glioblastoma cell lines[81], mice[82], lung cancer cell line (A549)[83,84], leukaemia cell lines[86] |
| Treatment | NNC-55-0396[80,82,86], mibefradil[81,86], RNA interference[81,82], KYS05090[83], TTA-A2[84,85] | ||
| Effects | Reduced proliferation[80-82,84,86], apoptosis[81,83,85,86], autophagy[83], increased sensitivity to anti-cancer drug[81,82], no effect on tumour size[82], inhibition of survivin[81], activation of BAX, Caspase 9, PARP, p27, and Rb[81], cell cycle arrest[81] at G1 phase[82,86], inhibition of stemness marker CD133[81], increased expression of astrocytic marker GFAP[81], increased ROS level[83], reduced viability[84], reduced wound healing[84], altered cell and spheroid morphology[84,85], reduced colony formation efficiency[85] | ||
| SOCC | Subjects | Breast cancer cell line (MCF-7)[63,89], prostate cancer cell line[90], NSCLC cells[88], thyroid cancer cell line[87] | |
| Treatment | RNA interference[89,90], AnCoA4[63], knockdown[63,87], Afatinib[88] | ||
| Effects | Apoptosis[89,90], autophagy[88], reduced proliferation[87,89,90], cell cycle arrest at G1 phase[87,89]/G2-M phase[90], altered cell cycle proteins[89], reduced entosis[63], reduced cell viability[88], reduced invasion[87] | ||
| STIM1[87,88] | Subjects | Non-small cell lung cancer cells[88], thyroid cancer cells[87], zebrafish[87], patient samples[87] | |
| Treatment | Afatinib[88], knockdown[87] | ||
| Effects | Autophagy[88], reduced cell viability[88], reduced invasion[87], reduced proliferation[87], increased sensitivity to anti-cancer drug[87], reduced tumour growth[87], apoptosis[87], cell cycle arrest at G1 phase[87] | ||
| TRP | TRPC7[29], TRPM2[61,91] | Subjects | Patient-derived lung adenocarcinoma tissue[29], lung adenocarcinoma cells[29], lung squamous cell carcinoma cells[29], HCC cell lines[61], patient-derived HCC tissue[61], human adrenal gland and neuroblastoma tissues[91], neuroblastoma cell line (SH-SY5Y)[91], athymic female mice[91] |
| Treatment | siRNA[29], A10[61], N-(p-amylcinnamoyl) ACA[61], lentivirus-coated shRNA[61], clotrimazole[91], TRPM2-S (short length of TRPM2)[91] | ||
| Effects | Reduced proliferation[29,61], reduced cell migration[29], reduced invasion[29], altered cell cycle proteins[61], slowed growth of patient-derived xenografts and Huh-7 xenografts in mice[61], cell cycle arrest at G0/G1[29] or G1/S phase[61], increased sensitivity to anti-cancer drug[91], reduced tumour volume[91], reduced autophagy/mitophagy[91] | ||
| NCX | Not defined[30] | Subjects | Glioblastoma cell line (U251)[30] |
| Treatment | Nickel[30], bepridil[30] | ||
| Effects | Reduced wound healing[30], reduced proliferation[30] | ||
| PMCA | Not defined[92], PMCA2[93], PMCA1[32,62], PMCA4[32,62] | Subjects | Prostate cancer cell line (PC-3)[92], breast cancer cell line (MDA-MB 231)[32,93], gastric cancer cell lines[62], mice[62] |
| Treatment | trans-resveratrol[92], siRNA[32,62,93] | ||
| Effects | Apoptosis[92], increased [Ca2+]cyt[92], decreased cell viability[92], cell death in the presence of a stimulus[93], increased sensitivity to anti-cancer drug[93] PMCA1: no effect on cell viability[32], high ionomycin (3 µM) induced necrosis[32], no effect on ABT-263-induced apoptosis[32], no effect on E-cadherin and vimentin expressions[62], no effect on wound healing rate[62] PMCA4: no effect on cell viability[32], no effect on ionomycin (3 µM) induced necrosis[32], augmented ABT-263-induced apoptosis[32], inhibition of NFκB translocation[32], increased metastasis[62], reduced expression of E-cadherin, GRHL2, and OVOL1[62], increased expression of vimentin[62], increased wound healing[62], increased [Ca2+]cyt[62], increased nuclear NFATc1 accumulation[62] | ||
| IP3R | Not defined[94] | Subjects | T-ALL[94,95], breast adenocarcinoma cell lines[95] |
| Treatment | Xestospongin B[94], 2APB[95] | ||
| Effects | Reduced mitochondrial respiration[94], cell death[94], increased NAD+/NADH ratio[94], inhibition of | ||
| SERCA | SERCA2[88] | Subjects | NSCLC cells[88] |
| Treatment | Afatinib[88] | ||
| Effects | Autophagy[88], reduced cell viability[88] | ||
| RyR | Not defined[95], RyR2[96] | Subjects | Breast adenocarcinoma cell lines[95], T-ALL cells[95], insulinoma cell line (INS-1)[96] |
| Treatment | Dantrolene[95], knockout[96] | ||
| Effects | Inhibition of ER-Ca2+ release[95], inhibition of ROS production[95], inhibition of immunepotent | ||
| MCU | Not defined[37,67,70] | Subjects | MCU-knockout transformed fibroblast cells[37], mice[37], Pancreatic ductal adenocarcinoma tissue[67], mice[67], CRC[70] |
| Treatment | Knockout[37,67], knockdown[70] | ||
| Effects | Small tumour size[37], reduced proliferation[37,70], cell cycle arrest at S phase[37], increased glycolysis[37], inhibited cell migration[67], inhibited invasion[37,67], inhibited soft agar colony formation[67], reduced tumour weight[67], reduced ROS level[67], ferroptosis inhibition[67], Nrf2 upregulation[67], decreased mitochondrial content[70], decreased mtDNA copy number[70], decreased ATP production[70], decreased expression levels of oxidative phosphorylation-related proteins[70] | ||
| NCLX | Not defined[67,97] | Subjects | Pancreatic ductal adenocarcinoma tissue[67], colorectal cancer cell lines[97], knockout mice[97] |
| Treatment | CGP37157[67], si/shRNA[97] | ||
| Effects | Increased ferroptosis and lipid peroxidation in the absence of cysteine[67], no effect on cell viability in presence of cystine[67], 50% less tumours[97], smaller tumour[97], reduced dysplasia[97], increased metastasis[97], reduced proliferation[97], apoptosis[97], increased invasion[97], mitochondrial perturbations[97], increased mitochondrial ROS[97], stem cell-like phenotype[97], chemoresistance of CRC cells[97] | ||
| SPCA | SPCA2[71,72] | Subjects | Breast cancer cell lines[71,72], mice[72] |
| Treatment | Knockdown[71,72] | ||
| Effects | Reduced cell growth[71,72], cell cycle arrest at G0/G1 phase[71], diminished SICE[71], DNA damage[71], increased sensitivity to anti-cancer therapy[71], increased ROS level[71], delayed tumour formation[72] |
INTERSECTION BETWEEN Ca2+ SIGNALLING AND CHEMORESISTANCE: FROM PATHWAYS TO THERAPEUTIC INTERVENTIONS
Chemotherapy often produces encouraging responses in cancer patients. However, patient life expectancy is ultimately constrained by chemotherapy resistance throughout treatment. Ca2+ signalling also significantly contributes to chemoresistance in cancer cells. Of note, [Ca2+]cyt may play a role in the development of intrinsic and acquired chemoresistance[98,99].
The Ca2+ channel TRPC5 has been shown to induce resistance in non-resistant breast cancer cells by increasing Ca2+ influx. This typically occurs after the transfer of TRPC5, present in extracellular vesicles of resistant breast cancer cells, to non-resistant breast cancer cells. Subsequently, the Ca2+ influx induces the expression of multidrug resistant ATPase 1 (MDR ATPase) via the Ca2+-dependent transcription factor, NFATc3. Increased MDR ATPase then removes several types of anti-cancer agents from the cells, leading to chemoresistance[17,100,101].
The complexity of Ca2+ signalling and the multitude of Ca2+ signalling proteins make overcoming chemoresistance a multifaceted process. Therefore, various approaches can be employed to address intrinsic or prevent acquired chemoresistance. Apart from the possible role of Ca2+ pharmacological modulators as standalone anti-cancer drugs, several studies have demonstrated their role as adjuvants in enhancing the anticarcinogenic effects of anti-cancer drugs. However, considering the vast number of research in this field, encompassing all studies is beyond the scope of this review. Consequently, this section briefly discusses the role of Ca2+ in the TME, cellular pathways, cancer stem cells (CSCs), utilization of combination and sequential treatments, and relevant clinical studies.
Ca2+ signalling in the TME
The TME is one of the crucial elements in cancer progression, where various cell types, including endothelial cells, mesenchymal cells, immune cells, adipocytes, and pericytes, coexist alongside cancer cells[102]. These diverse cell populations help cancer cells by providing mechanical support and releasing cytokines. Tumour development is mediated by anti- or proinflammatory cytokines that shape the TME[28]. These changes in the TME pose challenges to successful treatment and help cancer cells evade immune responses[102]. Ca2+ plays a critical role in supporting cancer cell migration and invasion by phosphorylating contractile proteins, inducing matrix metalloproteinases (MMPs), and remodelling peripheral and focal adhesions[17].
Disrupted Ca2+ signalling regulates immune responses, contributing to chemoresistance in cancer cells[103,104]. Increased ROS in mitochondria, triggered by Ca2+ signalling, phosphorylates and translocates STAT3, a transcription factor, into the nucleus. There, STAT3 activates genes related to immunosuppression[27]. Inflammation induced by these mechanisms reduces the efficacy of anti-cancer drugs such as cisplatin[41]. Therefore, targeting Ca2+ signalling to reduce inflammatory mediators represents a therapeutic approach in cancer treatment. Alhamed et al. demonstrated that although BTP2 (a SOCE inhibitor) treatment alone showed no effect on inflammatory mediators when administered in combination with cisplatin, it reversed cisplatin’s effect on increasing the gene expression of inflammatory mediators such as COX2, IL-8, and Tumour Necrosis Factor-α (TNF-α)[41].
Hypoxic conditions within tumours induce EMT, allowing cancer cells to transition from an epithelial to a mesenchymal phenotype, facilitating migration to other locations within the body. Additionally, angiogenesis, another hallmark of cancer, promotes tumour growth and invasiveness[105]. Both EMT and angiogenesis are regulated by Ca2+ channels such as VGCC and TRP channels. Consequently, chelating cytosolic Ca2+ results in the blockage of several EMT markers such as vimentin, Twist, and N-cadherin, potentially inhibiting angiogenesis[17,27,106].
Ca2+ signalling pathways in chemoresistance
Chemoresistance also arises from the activation of various cell signalling pathways including MAPK, CaMKKβ/AMPKα/mTOR, PI3K/Akt, and Wnt/β-catenin pathways[17,27]. These pathways are important in diverse cancer cell processes such as tumourigenesis, proliferation, protein synthesis, metastasis, and invasion[27]. G-protein coupled receptors (GPCRs) play a central role in translating extracellular signals and regulating Ca2+ signalling by coupling to
Various G-proteins modulate Ca2+ signalling[108], initiating a cascade of intracellular events. While the complexity of cellular pathways involves numerous molecules and signalling cues, providing a detailed description of each pathway is beyond the scope of this review. In brief, GPCRs have three types of
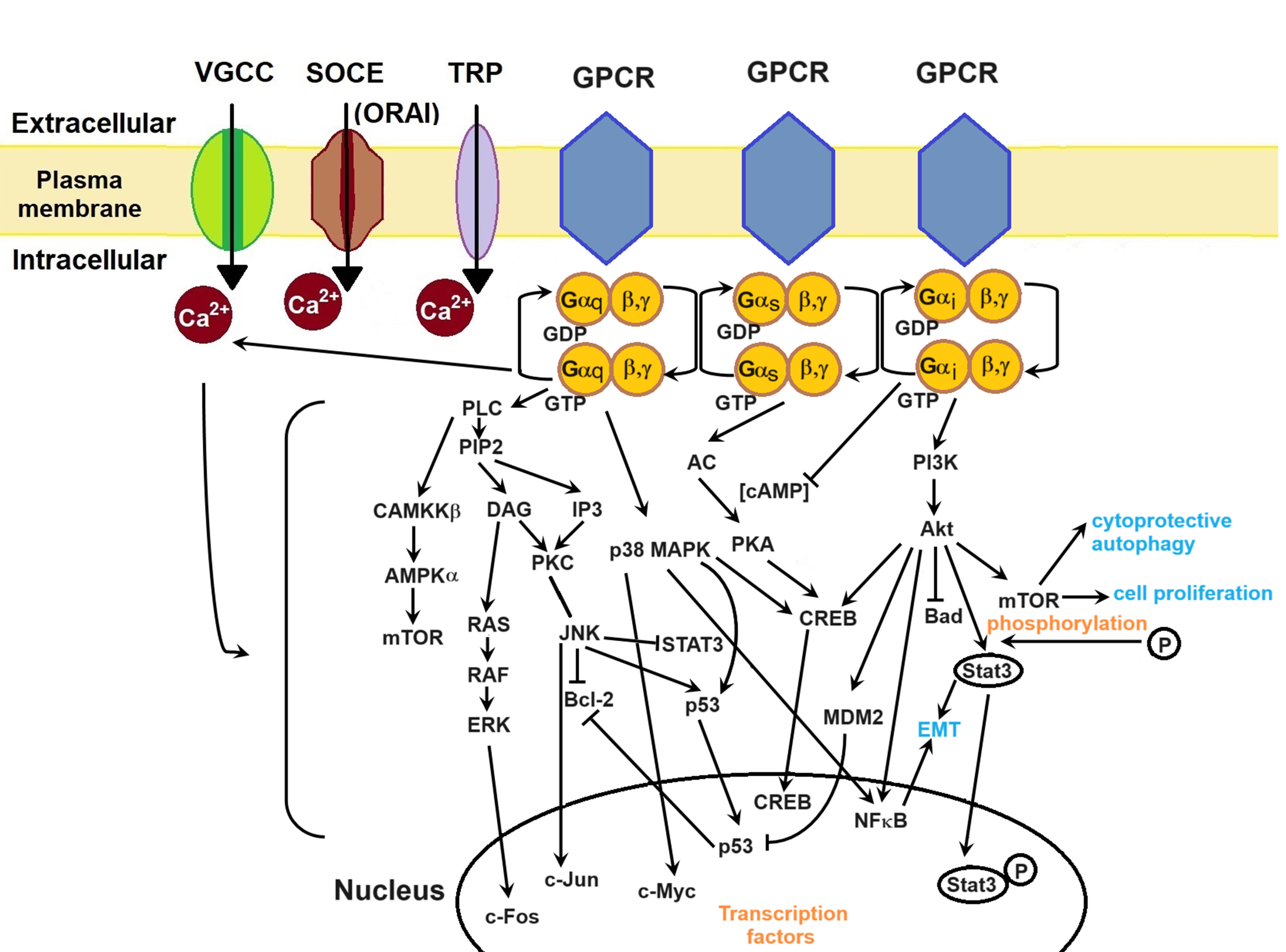
Figure 3. Ca2+ signalling pathways in cancer cells. GPCRs mediate cellular pathways such as MAPK, CaMKKβ/AMPKα/mTOR, and PI3K/Akt, thereby exerting regulatory control over Ca2+ channels. Consequently, Ca2+ ions modulate these cell pathways. The generation and regulation of transcription factors lead to cancer cell proliferation, EMT, metastasis, invasion, and survival. Akt and Murine Double Minute 2 (MDM2) inhibit Bad and p53, respectively, thereby supporting evasion from apoptosis. Inhibition of p53 releases Bcl-2, which in turn inhibits apoptosis. STAT3 and NFκB promote EMT, while mTOR promotes cell proliferation and cytoprotective autophagy. Transcription factors such as c-Fos, c-Jun, c-Myc, and CREB play pivotal roles in gene transcription crucial for cellular growth. VGCC: voltage-gated Ca2+ channel; SOCE: store-operated Ca2+ entry; TRP: transient receptor potential; GPCR: G-protein coupled receptor; PLC: phospholipase C; PIP2: phosphatidylinositol 4,5-bisphosphate; DAG: diacylglycerol; IP3: inositol-1,4,5-trisphosphate; PKC: protein kinase C; RAS: rat sarcoma; RAF: rapidly accelerated fibrosarcoma; ERK: extracellular signal-regulated kinase; JNK: c-Jun N-terminal kinase; STAT3: signal transducer and activator of transcription 3; MAPK: mitogen-activated protein kinases; NFκB: nuclear factor-κB; CREB: cyclic adenosine monophosphate-responsive element-binding; AC: adenylyl cyclase; PKA: protein kinase A; cAMP: cyclic adenosine monophosphate; PI3K: phosphatidylinositol 3-kinase; EMT: epithelial-mesenchymal transition.
Activation of Gαs leads to AC activation, initiating cAMP production. cAMP activates protein kinase A (PKA), which phosphorylates proteins regulating excitation-contraction coupling through VGCCs, RyRs, and myosin-binding protein C[108]. Upon Gαi activation, the Gαi subunit inhibits cAMP production and VGCCs[108] while activating the PI3K/Akt pathway. PI3K/Akt promotes cytoprotective autophagy, cell proliferation, EMT, and apoptosis inhibition through the degradation of several molecules such as mTOR, STAT3, NFκB, and p53. For instance, in prostate cancer cells (PCa cells), Ca2+ influx through TRPM8 leads to Ca2+/CaM activation of Akt kinase, thereby increasing cell proliferation[43].
In a study by Wang et al., it was shown that in the human ovarian cancer cell line A2780 (resistant to paclitaxel), human lung adenocarcinoma cell line A549 (resistant to paclitaxel), human breast cancer cell line MCF7 (resistant to paclitaxel and cisplatin), and human immortalized myelogenous leukaemia cell line K562 (resistant to paclitaxel, cisplatin, and Adriamycin), [Ca2+]cyt was reduced. Successful results were obtained in reversing the chemoresistance by chelating Ca2+ ions using Ca2+-chelated self-assembled nanoparticles (CSNPs). This intervention altered the expression of a chemoresistant gene and induced apoptosis by inhibiting the NFκB signalling pathway[6]. TRPC7 has been implicated in mediating CaMKII, Akt, and MAPK signalling, which are crucial in cancer progression. Knockdown or inhibition of TRPC7 has been shown to suppress the activation of CaMKII, Akt, and ERK[29].
Targeting Ca2+ machinery in CSCs
CSCs are robust chemoresistant cells present in a dormant state in various cancer types[109-111]. Upon activation and subsequent differentiation into cancer cells, CSCs contribute significantly to cancer relapse. Similar to cancer cells, CSCs also use abnormally activated cell pathways, including Notch, Hedgehog, Wnt/β-catenin, NFκB, PI3K/Akt, and PTEN pathways[112]. Therefore, targeting both CSCs and cancer cells would ensure effective cancer treatment and reduce cancer relapse. Studies suggest that Ca2+ channels play crucial roles in CSC functioning, with dysregulated Ca2+ signalling proteins promoting their differentiation into cancer cells and enhancing chemoresistance[113,114]. Dysregulation of various Ca2+ channels, including VGCCs[81,115], RyRs[116,117], IP3R[118], SOCE[119], or TRPs[120,121], has been observed in CSCs across different cancer types. Targeting these dysregulated Ca2+ channels induces terminal differentiation of CSCs and sensitizes them to chemotherapy.
LTCCs are associated with stem cell differentiation, while TTCCs play a role in cell cycle progression and the self-renewal capacity of undifferentiated stem cells. Additionally, IP3Rs are implicated in maintaining CSC properties such as self-renewal, proliferation, and differentiation[113]. Some studies have shown that targeting Ca2+ channels inhibits proliferation and induces apoptosis in CSCs. For example, Zhang et al. demonstrated that blocking TTCC with mibefradil inhibited the proliferation, survival, and stemness of glioblastoma stem-like cells (GSCs) and sensitized them to temozolomide chemotherapy. This effect was attributed to the inhibition of the pro-survival Akt/mTOR pathway and stimulation of the pro-apoptotic Survivin and Bax pathways. Moreover, TTCC inhibition reduced the expression of oncogenes and increased the expression of tumour suppressor genes[81]. In another study, CCBs (manidipine, lacidipine, benidipine, and lomerizine) inhibited stemness and PI3K/Akt and ERK pathways in ovarian CSCs[112].
Therapeutic interventions targeting Ca2+ signalling
Combination therapy utilizing both anti-cancer drugs and Ca2+ protein blockers is often studied. Anti-cancer drugs change Ca2+ signalling within cancer cells, by targeting pathways related to cell growth or inducing cell death, compromising cellular integrity, reducing ATP production, or blocking specific growth factors[17]. The effect of anti-cancer agents on Ca2+ signalling depends on the exposure time. During shorter exposure, for up to 8 h, there is an increase in Ca2+ influx, ER Ca2+ release, and ER-mitochondria Ca2+ transfer; however, longer exposure, spanning from hours to weeks, leads to heightened Ca2+ efflux, altered ER Ca2+ regulation, and elevated activity of the ER Ca2+ release channel[17]. For example, short-term exposure to 5FU increases Ca2+ influx via plasma membrane channels, while long-term exposure increases Ca2+ efflux via PMCA[17]. Conversely, inhibiting Ca2+ channels results in sensitized or increased anti-cancer effects of some drugs by lowering [Ca2+]cyt. For example, Andrographis sensitizes cancer cells synergistically to gemcitabine[122], cisplatin[123], 5-FU[124], and doxorubicin[125]in vitro. Co-treatment of Andrographis and gemcitabine decreases [Ca2+]cyt in gemcitabine-resistant cells[122]. Co-treatment of cisplatin and SOCE inhibitors increases cisplatin cytotoxicity in cisplatin-resistant breast cancer cells[41] and NSCLC[126]. Furthermore, BTP2 treatment reverses cisplatin-induced upregulation of SOCE components (ORAI1 and STIM1) genes[41]. Knockdown of ORAI1 and STIM1 has also been shown to sensitize cancer cells to anti-cancer drugs[87,126,127]. However, in one study, it was shown that cisplatin cytotoxicity was reduced upon knockdown of ORAI1 and STIM1 in NSCLC and prostate cancer cells[128,129].
Silencing PMCA2 in MDA-MB-231 cells promoted the anti-cancer effects of doxorubicin[130]. Blocking TTCC with mibefradil sensitized platinum-resistant ovarian tumours to carboplatin in a mouse model[131] and increased the sensitivity of melanoma cells to MAPK inhibitors[132]. In diffuse large B-cell lymphoma (DLBCL) patients resistant to rituximab, co-treatment of rituximab with Bay K8644 (an LTCC agonist) significantly induced apoptosis in DLBCL cell lines and markedly reduced tumour volume and weight in DLBCL patient-derived xenografted mice[133].
This increased sensitization to anti-cancer drugs through co-treatment with CCBs is further enhanced by sequential treatment. Blocking or knocking down Ca2+ channels arrest cells at specific phases of the cell cycle. For example, blocking or knockdown of TTCCs can arrest cancer cells at the G0/G1 phase[134]. Similarly, in one study, TRPC7 knockdown arrested lung adenocarcinoma cells, H1299, at the G0/G1 phase[29], synchronizing cells to a single phase. Consequently, when an anti-cancer drug such as Taxol, which inhibits microtubule depolymerization and stops cell division[135], is administered, a large number of cells are killed. Another advantage of sequential treatment is the requirement for smaller drug doses compared to single or combination treatments. This was evidenced in one of our studies with A549 3D spheroids[136]. Sequential treatment of A549 3D spheroids with a TTCC blocker (TTA-A2) and paclitaxel led to a significant reduction in spheroid viability compared to combination treatment. Sequential treatment not only synchronizes cells to a single phase but also prevents or reduces nonspecific interactions of the second drug. For example, paclitaxel, in addition to microtubules, can also interact with IP3R[137] and other Ca2+ channels such as TTCC[136]. Consequently, in combination treatment, paclitaxel may compete for binding sites with the TTCC blocker on TTCC. Since the paclitaxel interaction is not as strong as that of specific TTCC blockers, this results in weaker inhibition of these channels by paclitaxel and reduced anti-cancer effects[136].
Clinical trials with Ca2+ protein blockers
The effects of targeting Ca2+ proteins in humans are relatively unexplored, as indicated by the limited number of clinical studies[138-142]. In a clinical trial by Fu et al., TRPV6 was targeted in individuals with advanced epithelial-origin tumours, revealing that SOR-C13, an inhibitor of TRPV6, exhibit anti-cancer effects without any typical chemotherapy-related complications[138]. This study highlights the potential of targeted therapy in managing cancer, and mitigating adverse effects associated with traditional chemotherapy.
Carboxyamido-triazole (CAI), a synthetic inhibitor of non-VGCCs, has been studied in clinical phase IB for recurrent and newly diagnosed glioblastoma and anaplastic gliomas[141], as well as phase II trials for glioblastoma[139] and ovarian cancer[142]. In phase IB trials, CAI combined with temozolomide or chemoradiation, showed promising results in terms of safety and efficacy in challenging cases[141]. However, in a phase II trial by Mikkelsen et al., CAI was tested along with radiation on patients with newly diagnosed glioblastoma multiforme. Results showed that even though CAI was safe to administer, it was ineffective in improving the survival rate[139]. In patients with relapsed ovarian cancer, CAI exhibited safety with limited toxicity and stabilized the cancer for over six months[142].
Additionally, a phase I study by Holdhoff et al. demonstrated the safety of sequential treatment involving mibefradil followed by temozolomide in glioblastoma patients, with some patients showing partial to complete responses to the regimen[140].
These varying results emphasize the complexity of targeting Ca2+ proteins for effective cancer treatments. While inhibitors such as SOR-C13 and CAI exhibit promising results in certain cancer types, their efficacy and impact on survival rates vary across cancer stages and types. Positive results from in vitro and in vivo studies, coupled with the lack of adequate clinical data, indicate the need for more extensive clinical studies to elucidate the role of targeting Ca2+ proteins as an effective cancer treatment option.
CONCLUSION
Although many novel drugs exhibit promising efficacy against chemoresistant cancers, Ca2+ signalling-driven chemoresistance remains a major obstacle. The ubiquitous and varying role of Ca2+ in normal and cancer cells complicates targeting approaches and also raises safety concerns. A deeper understanding of Ca2+ disruptions across various cancers will facilitate the development of effective therapeutic approaches. Researchers worldwide are exploring innovative strategies, including site-specific drug delivery that can target cancer cells without harming normal cells by taking advantage of cancer biomarkers and designing drugs for cancer-specific Ca2+ protein splice variants or their proteins expressed in cancer cells. Simultaneously targeting of multiple dysregulated proteins and exploring intracellular organelles interactions, such as lysosome-mitochondria, using advanced models like 3D cell cultures or patient-derived organoids, are promising approaches. Furthermore, an extensive increase in clinical studies is required to establish the anti-cancer efficacy of Ca2+ protein blockers, which have demonstrated positive in vitro and in vivo results.
DECLARATIONS
Authors’ contributions
Conceptualization, literature search, study design, writing, editing, and review: Kumari N
Literature search, writing, editing, and review: Pullaguri N
Manuscript review and editing: Rath SN, Bajaj A
Conceptualization and manuscript review: Sahu V, Ealla KKR
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Cancer. World Health Organization. Available from: https://www.who.int/health-topics/cancer. [Last accessed on 13 March 2024].
2. Types of cancer treatment. National Cancer Institute. Available from: https://www.cancer.gov/about-cancer/treatment/types. [Last accessed on 13 March 2024].
3. Ramos A, Sadeghi S, Tabatabaeian H. Battling chemoresistance in cancer: root causes and strategies to uproot them. Int J Mol Sci 2021;22:9451.
4. Cancer drugs A to Z list. Cancer Research UK. Available from: https://www.cancerresearchuk.org/about-cancer/treatment/drugs. [Last accessed on 13 March 2024].
5. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull 2017;7:339-48.
6. Wang C, Xu X, Xiong S, et al. Calcium-chelated nanosystem reversing cancer chemoresistance via replenishing intracellular calcium ions. Chem Eng J 2022;448:137500.
7. Büsselberg D, Florea AM. Targeting intracellular calcium signaling ([Ca2+]i) to overcome acquired multidrug resistance of cancer cells: a mini-overview. Cancers 2017;9:48.
8. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol 2013;4:28.
10. Brasseur K, Gévry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017;8:4008-42.
12. Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 2008;8:361-75.
13. Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y. Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 2014;369:20130097.
14. Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 2007;7:519-30.
15. Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta 2015;1848:2502-11.
17. Bong AHL, Monteith GR. Calcium signaling and the therapeutic targeting of cancer cells. Biochim Biophys Acta Mol Cell Res 2018;1865:1786-94.
19. Tajada S, Villalobos C. Calcium permeable channels in cancer hallmarks. Front Pharmacol 2020;11:968.
20. Wu Y, Huang P, Dong XP. Lysosomal calcium channels in autophagy and cancer. Cancers 2021;13:1299.
21. Yáñez M, Gil-Longo J, Campos-Toimil M. Calcium binding proteins. Adv Exp Med Biol 2012;740:461-82.
23. Čendula R, Chomaničová N, Adamičková A, Gažová A, Kyselovič J, Máťuš M. Altered expression of ORAI and STIM isoforms in activated human cardiac fibroblasts. Physiol Res 2021;70:S21-30.
24. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 2007;7:690-702.
25. Zheng S, Wang X, Zhao D, Liu H, Hu Y. Calcium homeostasis and cancer: insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol 2023;33:312-23.
26. Prole DL, Taylor CW. Structure and function of IP3 receptors. Cold Spring Harb Perspect Biol 2019;11:a035063.
27. Wu L, Lian W, Zhao L. Calcium signaling in cancer progression and therapy. FEBS J 2021;288:6187-205.
28. Bujak JK, Kosmala D, Szopa IM, Majchrzak K, Bednarczyk P. Inflammation, cancer and immunity-implication of TRPV1 channel. Front Oncol 2019;9:1087.
29. Liang JL, Tsai MH, Hsieh YC, et al. TRPC7 facilitates cell growth and migration by regulating intracellular Ca2+ mobilization in lung adenocarcinoma cells. Oncol Lett 2023;25:92.
30. Brandalise F, Ramieri M, Pastorelli E, et al. Role of Na+/Ca2+ exchanger (NCX) in glioblastoma cell migration (in vitro). Int J Mol Sci 2023;24:12673.
31. Loeck T, Schwab A. The role of the Na+/Ca2+-exchanger (NCX) in cancer-associated fibroblasts. Biol Chem 2023;404:325-37.
32. Curry MC, Luk NA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J Biol Chem 2012;287:28598-608.
33. Stafford N, Wilson C, Oceandy D, Neyses L, Cartwright EJ. The plasma membrane calcium ATPases and their role as major new players in human disease. Physiol Rev 2017;97:1089-125.
35. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000;1:11-21.
36. Kumari N, Gaur H, Bhargava A. Cardiac voltage gated calcium channels and their regulation by β-adrenergic signaling. Life Sci 2018;194:139-49.
37. Fernandez Garcia E, Paudel U, Noji MC, et al. The mitochondrial Ca2+ channel MCU is critical for tumor growth by supporting cell cycle progression and proliferation. Front Cell Dev Biol 2023;11:1082213.
38. Samanta K, Mirams GR, Parekh AB. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat Commun 2018;9:156.
39. Pizzo P, Lissandron V, Capitanio P, Pozzan T. Ca2+ signalling in the Golgi apparatus. Cell Calcium 2011;50:184-92.
40. Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer 2017;17:367-80.
41. Alhamed AS, Alqinyah M, Alsufayan MA, et al. Blockade of store-operated calcium entry sensitizes breast cancer cells to cisplatin therapy via modulating inflammatory response. Saudi Pharm J 2023;31:245-54.
42. Phan NN, Wang CY, Chen CF, Sun Z, Lai MD, Lin YC. Voltage-gated calcium channels: novel targets for cancer therapy. Oncol Lett 2017;14:2059-74.
43. Ardura JA, Álvarez-Carrión L, Gutiérrez-Rojas I, Alonso V. Role of calcium signaling in prostate cancer progression: effects on cancer hallmarks and bone metastatic mechanisms. Cancers 2020;12:1071.
44. Danese A, Leo S, Rimessi A, et al. Cell death as a result of calcium signaling modulation: a cancer-centric prospective. Biochim Biophys Acta Mol Cell Res 2021;1868:119061.
45. Um HD. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016;7:5193-203.
46. Patergnani S, Danese A, Bouhamida E, et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int J Mol Sci 2020;21:8323.
47. Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 2011;50:211-21.
48. Pedrera L, Ros U, García-Sáez AJ. Calcium as a master regulator of ferroptosis and other types of regulated necrosis. Cell Calcium 2023;114:102778.
49. Hernández-Oliveras A, Zarain-Herzberg A. The role of Ca2+-signaling in the regulation of epigenetic mechanisms. Cell Calcium 2024;117:102836.
50. Hernández-Oliveras A, Zarain-Herzberg Á. Expression and associated epigenetic mechanisms of the Ca2+-signaling genes in breast cancer subtypes and epithelial-to-mesenchymal transition. J Cell Commun Signal 2022;16:461-74.
51. Raynal NJ, Lee JT, Wang Y, et al. Targeting calcium signaling induces epigenetic reactivation of tumor suppressor genes in cancer. Cancer Res 2016;76:1494-505.
52. Alam MR, Rahman MM, Li Z. The link between intracellular calcium signaling and exosomal PD-L1 in cancer progression and immunotherapy. Genes Dis 2024;11:321-34.
53. Savina A, Vidal M, Colombo MI. The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci 2002;115:2505-15.
54. Chalmin F, Ladoire S, Mignot G, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 2010;120:457-71.
55. Wang CY, Lai MD, Phan NN, Sun Z, Lin YC. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS One 2015;10:e0125766.
56. Tang BD, Xia X, Lv XF, et al. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J Cell Mol Med 2017;21:904-15.
57. Can G, Akpinar B, Baran Y, Zhivotovsky B, Olsson M. 5-Fluorouracil signaling through a calcium-calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2013;32:4529-38.
58. Zhou X, Wang W, Zhang S, et al. CACNA1B (Cav2.2) overexpression and its association with clinicopathologic characteristics and unfavorable prognosis in non-small cell lung cancer. Dis Markers 2017;2017:6136401.
59. Passaia BDS, Kremer JL, Fragoso MCV, Lotfi CFP. N-type calcium channel v2.2 is a target of TCF21 in adrenocortical carcinomas. Neoplasma 2022;69:899-908.
60. Li G, Roy B, Huang X, et al. High expression of N-type calcium channel indicates a favorable prognosis in gliomas. Medicine 2022;101:e29782.
61. Cai X, Yu X, Yang J, et al. TRPM2 regulates cell cycle through the Ca2+-CaM-CaMKII signaling pathway to promote HCC. Hepatol Commun 2023;7:e0101.
62. Wang T, Li N, Jin L, Qi X, Zhang C, Hua D. The calcium pump PMCA4 prevents epithelial-mesenchymal transition by inhibiting NFATc1-ZEB1 pathway in gastric cancer. Biochim Biophys Acta Mol Cell Res 2020;1867:118833.
63. Lee AR, Park CY. Orai1 is an entotic Ca2+ channel for non-apoptotic cell death, entosis in cancer development. Adv Sci 2023;10:e2205913.
64. Kianfar M, Balcerak A, Chmielarczyk M, Tarnowski L, Grzybowska EA. Cell death by entosis: triggers, molecular mechanisms and clinical significance. Int J Mol Sci 2022;23:4985.
65. Rupert C, Dell’ Aversana C, Mosca L, et al. Therapeutic targeting of P2X4 receptor and mitochondrial metabolism in clear cell renal carcinoma models. J Exp Clin Cancer Res 2023;42:134.
66. Zhai X, Sterea AM, Hiani YE. Lessons from the endoplasmic reticulum Ca2+ transporters-a cancer connection. Cells 2020;9:1536.
67. Wang X, Li Y, Li Z, et al. Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer Res 2022;82:2254-68.
69. Marmolejo-Garza A, Krabbendam IE, Luu MDA, et al. Negative modulation of mitochondrial calcium uniporter complex protects neurons against ferroptosis. Cell Death Dis 2023;14:772.
70. Liu Y, Jin M, Wang Y, et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct Target Ther 2020;5:59.
71. Makena MR, Ko M, Mekile AX, et al. Secretory pathway Ca2+-ATPase SPCA2 regulates mitochondrial respiration and DNA damage response through store-independent calcium entry. Redox Biol 2022;50:102240.
72. Feng M, Grice DM, Faddy HM, et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010;143:84-98.
73. Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem 2012;287:31666-73.
74. Scotto Rosato A, Montefusco S, Soldati C, et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat Commun 2019;10:5630.
75. Zawadzki A, Liu Q, Wang Y, Melander A, Jeppsson B, Thorlacius H. Verapamil inhibits L-type calcium channel mediated apoptosis in human colon cancer cells. Dis Colon Rectum 2008;51:1696-702.
76. Li S, Kim HY, Lee DJ, et al. Inhibition of L-type voltage-gated calcium channel-mediated Ca2+ influx suppresses the collective migration and invasion of ameloblastoma. Cell Prolif 2022;55:e13305.
77. Sun J, Sun X, Li Z, Ma D, Lv Y. An elongated tract of polyQ in the carboxyl-terminus of human α1A calcium channel induces cell apoptosis by nuclear translocation. Oncol Rep 2020;44:156-64.
78. Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A 1992;89:3825-9.
79. Mergler S, Wiedenmann B, Prada J. R-type Ca2+-channel activity is associated with chromogranin A secretion in human neuroendocrine tumor BON cells. J Membr Biol 2003;194:177-86.
80. Sekar S, Subbamanda Y, Pullaguri N, et al. Isoform-specific expression of T-type voltage-gated calcium channels and estrogen receptors in breast cancer reveals specific isoforms that may be potential targets. Curr Res Biotechnol 2022;4:459-67.
81. Zhang Y, Cruickshanks N, Yuan F, et al. Targetable T-type calcium channels drive glioblastoma. Cancer Res 2017;77:3479-90.
82. Pottle J, Sun C, Gray L, Li M. Exploiting MCF-7 cells’ calcium dependence with interlaced therapy. J Cancer Ther 2013;4:32-40.
83. Rim HK, Cho S, Shin DH, et al. T-type Ca2+ channel blocker, KYS05090 induces autophagy and apoptosis in A549 cells through inhibiting glucose uptake. Molecules 2014;19:9864-75.
84. Kumari N, Bhargava A, Rath SN. T-type calcium channel antagonist, TTA-A2 exhibits anti-cancer properties in 3D spheroids of A549, a lung adenocarcinoma cell line. Life Sci 2020;260:118291.
85. Kumari N, Giri PS, Rath SN. Adjuvant role of a T-type calcium channel blocker, TTA-A2, in lung cancer treatment with paclitaxel. Cancer Drug Resist 2021;4:996-1007.
86. Huang W, Lu C, Wu Y, Ouyang S, Chen Y. T-type calcium channel antagonists, mibefradil and NNC-55-0396 inhibit cell proliferation and induce cell apoptosis in leukemia cell lines. J Exp Clin Cancer Res 2015;34:54.
87. Asghar MY, Lassila T, Paatero I, et al. Stromal interaction molecule 1 (STIM1) knock down attenuates invasion and proliferation and enhances the expression of thyroid-specific proteins in human follicular thyroid cancer cells. Cell Mol Life Sci 2021;78:5827-46.
88. Kim MS, Kim SH, Yang SH, Kim MS. Afatinib mediates autophagic degradation of ORAI1, STIM1, and SERCA2, which inhibits proliferation of non-small cell lung cancer cells. Tuberc Respir Dis 2022;85:147-54.
89. Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol 2011;226:542-51.
90. Kouba S, Buscaglia P, Guéguinou M, et al. Pivotal role of the ORAI3-STIM2 complex in the control of mitotic death and prostate cancer cell cycle progression. Cell Calcium 2023;115:102794.
91. Chen SJ, Hoffman NE, Shanmughapriya S, et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2α. J Biol Chem 2014;289:36284-302.
92. Peterson JA, Crowther CM, Andrus MB, Kenealey JD. Resveratrol derivatives increase cytosolic calcium by inhibiting plasma membrane ATPase and inducing calcium release from the endoplasmic reticulum in prostate cancer cells. Biochem Biophys Rep 2019;19:100667.
93. Curry M, Roberts-Thomson SJ, Monteith GR. PMCA2 silencing potentiates MDA-MB-231 breast cancer cell death initiated with the Bcl-2 inhibitor ABT-263. Biochem Biophys Res Commun 2016;478:1792-7.
94. Cruz P, Ahumada-Castro U, Bustos G, et al. Inhibition of insP3R with xestospongin B reduces mitochondrial respiration and induces selective cell death in T cell acute lymphoblastic leukemia cells. Int J Mol Sci 2021;22:651.
95. Lorenzo-Anota HY, Reyes-Ruiz A, Calvillo-Rodríguez KM, et al. IMMUNEPOTENT CRP increases intracellular calcium through ER-calcium channels, leading to ROS production and cell death in breast cancer and leukemic cell lines. EXCLI J 2023;22:352-66.
96. Harvey KE, Tang S, LaVigne EK, Pratt EPS, Hockerman GH. RyR2 regulates store-operated Ca2+ entry, phospholipase C activity, and electrical excitability in the insulinoma cell line INS-1. PLoS One 2023;18:e0285316.
97. Pathak T, Gueguinou M, Walter V, et al. Dichotomous role of the human mitochondrial Na+/Ca2+/Li+ exchanger NCLX in colorectal cancer growth and metastasis. Elife 2020;9:e59686.
98. Pera E, Kaemmerer E, Milevskiy MJG, et al. The voltage gated Ca2+-channel Cav3.2 and therapeutic responses in breast cancer. Cancer Cell Int 2016;16:24.
99. VanHouten J, Sullivan C, Bazinet C, et al. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc Natl Acad Sci U S A 2010;107:11405-10.
100. Ma X, Chen Z, Hua D, et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc Natl Acad Sci U S A 2014;111:6389-94.
101. Ma X, Cai Y, He D, et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc Natl Acad Sci U S A 2012;109:16282-7.
102. Ni Y, Zhou X, Yang J, et al. The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front Cell Dev Biol 2021;9:637675.
103. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 2019;51:27-41.
104. Bergmeier W, Weidinger C, Zee I, Feske S. Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels 2013;7:379-91.
105. Ribatti D, Tamma R, Annese T. Epithelial-mesenchymal transition in cancer: a historical overview. Transl Oncol 2020;13:100773.
106. Davis FM, Azimi I, Faville RA, et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014;33:2307-16.
107. Klabunde T, Hessler G. Drug design strategies for targeting g-protein-coupled receptors. ChemBioChem 2002;3:928-44.
108. Dhyani V, Gare S, Gupta RK, Swain S, Venkatesh KV, Giri L. GPCR mediated control of calcium dynamics: a systems perspective. Cell Signal 2020;74:109717.
109. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983-8.
110. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106-10.
111. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396-401.
112. Lee H, Kim JW, Kim DK, et al. Calcium channels as novel therapeutic targets for ovarian cancer stem cells. Int J Mol Sci 2020;21:2327.
114. Cheng Q, Chen A, Du Q, et al. Novel insights into ion channels in cancer stem cells (review). Int J Oncol 2018;53:1435-41.
115. Sui X, Geng JH, Li YH, Zhu GY, Wang WH. Calcium channel α2δ1 subunit (CACNA2D1) enhances radioresistance in cancer stem-like cells in non-small cell lung cancer cell lines. Cancer Manag Res 2018;10:5009-18.
116. Forostyak O, Forostyak S, Kortus S, Sykova E, Verkhratsky A, Dayanithi G. Physiology of Ca2+ signalling in stem cells of different origins and differentiation stages. Cell Calcium 2016;59:57-66.
117. Lu H, Chen I, Shimoda LA, et al. Chemotherapy-induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep 2017;18:1946-57.
118. Yanagida E, Shoji S, Hirayama Y, et al. Functional expression of Ca2+ signaling pathways in mouse embryonic stem cells. Cell Calcium 2004;36:135-46.
119. Lee SH, Rigas NK, Lee CR, et al. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016;7:43239-55.
120. Shiozaki A, Kudou M, Ichikawa D, et al. Esophageal cancer stem cells are suppressed by tranilast, a TRPV2 channel inhibitor. J Gastroenterol 2018;53:197-207.
121. Morelli MB, Nabissi M, Amantini C, et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int J Cancer 2012;131:E1067-77.
122. Okuno K, Xu C, Pascual-Sabater S, et al. Andrographis reverses gemcitabine resistance through regulation of ERBB3 and calcium signaling pathway in pancreatic ductal adenocarcinoma. Biomedicines 2023;11:119.
123. Yunos NM, Mutalip SSM, Jauri MH, Yu JQ, Huq F. Anti-proliferative and pro-apoptotic effects from sequenced combinations of andrographolide and cisplatin on ovarian cancer cell lines. Anticancer Res 2013;33:4365-71.
124. Sharma P, Shimura T, Banwait JK, Goel A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020;41:1385-94.
125. Zhou J, Ong CN, Hur GM, Shen HM. Inhibition of the JAK-STAT3 pathway by andrographolide enhances chemosensitivity of cancer cells to doxorubicin. Biochem Pharmacol 2010;79:1242-50.
126. Li W, Zhang M, Xu L, Lin D, Cai S, Zou F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp Toxicol Pathol 2013;65:1073-81.
127. Kondratska K, Kondratskyi A, Yassine M, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta 2014;1843:2263-9.
128. Gualdani R, de Clippele M, Ratbi I, Gailly P, Tajeddine N. Store-operated calcium entry contributes to cisplatin-induced cell death in non-small cell lung carcinoma. Cancers 2019;11:430.
129. Flourakis M, Lehen’kyi V, Beck B, et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis 2010;1:e75.
130. Peters AA, Milevskiy MJ, Lee WC, et al. The calcium pump plasma membrane Ca2+-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci Rep 2016;6:25505.
131. Dziegielewska B, Casarez EV, Yang WZ, Gray LS, Dziegielewski J, Slack-Davis JK. T-type Ca2+ channel inhibition sensitizes ovarian cancer to carboplatin. Mol Cancer Ther 2016;15:460-70.
132. Granados K, Hüser L, Federico A, et al. T-type calcium channel inhibition restores sensitivity to MAPK inhibitors in de-differentiated and adaptive melanoma cells. Br J Cancer 2020;122:1023-36.
133. Zhang JY, Zhang PP, Zhou WP, et al. L-type cav 1.2 calcium channel-α-1C regulates response to rituximab in diffuse large B-cell lymphoma. Clin Cancer Res 2019;25:4168-78.
134. Das A, Pushparaj C, Bahí N, et al. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res 2012;25:200-12.
135. Gornstein E, Schwarz TL. The paradox of paclitaxel neurotoxicity: mechanisms and unanswered questions. Neuropharmacology 2014;76:175-83.
136. Kumari N, Dalal V, Kumar P, Rath SN. Antagonistic interaction between TTA-A2 and paclitaxel for anti-cancer effects by complex formation with T-type calcium channel. J Biomol Struct Dyn 2022;40:2395-406.
137. Boehmerle W, Zhang K, Sivula M, et al. Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc Natl Acad Sci U S A 2007;104:11103-8.
138. Fu S, Hirte H, Welch S, et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Invest New Drugs 2017;35:324-33.
139. Mikkelsen T, Lush R, Grossman SA, et al. Phase II clinical and pharmacologic study of radiation therapy and carboxyamido-triazole (CAI) in adults with newly diagnosed glioblastoma multiforme. Invest New Drugs 2007;25:259-63.
140. Holdhoff M, Ye X, Supko JG, et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro Oncol 2017;19:845-52.
141. Omuro A, Beal K, McNeill K, et al. Multicenter phase IB trial of carboxyamidotriazole orotate and temozolomide for recurrent and newly diagnosed glioblastoma and other anaplastic gliomas. J Clin Oncol 2018;36:1702-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kumari N, Pullaguri N, Rath SN, Bajaj A, Sahu V, Ealla KKR. Dysregulation of calcium homeostasis in cancer and its role in chemoresistance. Cancer Drug Resist 2024;7:11. http://dx.doi.org/10.20517/cdr.2023.145
AMA Style
Kumari N, Pullaguri N, Rath SN, Bajaj A, Sahu V, Ealla KKR. Dysregulation of calcium homeostasis in cancer and its role in chemoresistance. Cancer Drug Resistance. 2024; 7: 11. http://dx.doi.org/10.20517/cdr.2023.145
Chicago/Turabian Style
Kumari, Neema, Narasimha Pullaguri, Subha Narayan Rath, Ashish Bajaj, Vikas Sahu, Kranti Kiran Reddy Ealla. 2024. "Dysregulation of calcium homeostasis in cancer and its role in chemoresistance" Cancer Drug Resistance. 7: 11. http://dx.doi.org/10.20517/cdr.2023.145
ACS Style
Kumari, N.; Pullaguri N.; Rath SN.; Bajaj A.; Sahu V.; Ealla KKR. Dysregulation of calcium homeostasis in cancer and its role in chemoresistance. Cancer Drug Resist. 2024, 7, 11. http://dx.doi.org/10.20517/cdr.2023.145
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 17
likes
Like This Article 17
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.