
Heterotypic signaling of cancer-associated fibroblasts in shaping the cancer cell drug resistance

Abstract
The context-dependent reciprocal interaction between the cancer cells and surrounding fibroblasts is imperative for regulating malignant potential, metabolic reprogramming, immunosuppression, and ECM deposition. However, recent evidence also suggests that cancer-associated fibroblasts induce chemoresistance in cancer cells to various anticancer regimens. Because of the protumorigenic function of cancer-associated fibroblasts, these stromal cell types have emerged as fascinating therapeutic targets for cancer. However, this notion was recently challenged by studies that targeted cancer-associated fibroblasts and highlighted the underlying heterogeneity by identifying a subset of these cells with tumor-restricting functions. Hence, it is imperative to understand the heterogeneity and heterotypic signaling of cancer-associated fibroblasts to target tumor-promoting signaling processes by sparing tumor-restricting ones. In this review, we discuss the heterogeneity and heterotypic signaling of cancer-associated fibroblasts in shaping drug resistance and also list the cancer-associated fibroblast-targeting therapeutics.
Keywords
INTRODUCTION
Accumulation of genetic or epigenetic aberration may be important for the transformation of normal epithelial cells but not sufficient to induce malignant potential. Context-dependent interaction between cancer cells and tumor microenvironment components is imperative for malignant progression[1]. Tumor microenvironments consist of various kinds of non-cancerous cells such as fibroblasts, macrophages, mesenchymal stem cells (MSCs), pericytes, endothelial and immune cells, and extracellular matrix (ECM) known as tumor stroma[2]. Fibroblasts constitute a major component of tumor stroma and exhibit multipronged functions in tumor progression[3,4].
Fibroblasts could be considered cockroaches of the human body as they thrive under severe stress and can even be isolated from decaying/dead tissue. Fibroblasts are quiescent cell types and synthetically and metabolically less active[3]. Upon activation, fibroblasts play a critical role in the wound healing process by remodeling ECM as well as secreting various growth factors and chemoattractant cytokines which ultimately regulate epithelial proliferation and immune cell infiltration[3,5,6]. Dysregulation of their activation leads to the formation of scar and fibrotic diseases. Fibroblasts associated with cancer, termed cancer-associated fibroblasts (CAFs), show functional and molecular differences from normal fibroblasts. Fibroblast activation by the secreted factors, cell-matrix or cell-cell contacts with cancer or other stromal cells leads to the CAF phenotype acquisition[2,7-9]. CAFs have been reported to exhibit higher migratory and contraction potentials along with synthesizing and remodeling ECM, reminiscent of myofibroblasts[8,9]. Several reports show that CAFs secrete a myriad of growth factors and cytokines which are critical for several facets of tumor progression. CAFs were known to regulate several hallmarks of cancer by directly influencing cancer cell proliferation, migration, invasion, and angiogenesis[7-9]. Our earlier study also reported that osteopontin (OPN)-activated CAF-derived CXCL12 promotes epithelial-to-mesenchymal transition (EMT) in breast cancer cells[8]. Moreover, CAFs are known to shape the tumor immune microenvironment through the elevated expression of immunosuppressive cytokines and immune checkpoint proteins that results in immunosuppression and tumor progression[10]. More importantly, CAFs are reported to induce drug resistance and cancer relapse in different cancers by different mechanisms including the induction of EMT, activation of stemness pathways, ECM remodeling, and dysregulated metabolism[11]. Due to their important functions in tumor progression, CAFs have emerged as an intriguing therapeutic target for the clinical control of cancer. However, the studies focused on targeting CAFs for the management of cancer have challenged this dogma. Of note, genetic ablation of the CAF population or fibrosis induces immunosuppressive environment in pancreatic ductal adenocarcinoma (PDAC) which in turn promotes EMT and invasion in cancer cells, leading to tumor progression with poor disease outcomes[12]. In addition, targeting the hedgehog (Hh) pathway in CAFs led to more aggressive and poorly differentiated PDAC with reduced stromal content and survival[13,14]. The above report highlights the presence of a subset of CAFs with tumor-restricting functions. Understanding the heterogeneity of CAFs and their heterotypic signaling might help in tailoring therapeutic intervention that selectively targets tumor-promoting CAF population and spares tumor-restraining ones. This review focuses on CAF heterogeneity and heterotypic signaling in regulating drug resistance to cancer therapies. This review also highlights several current CAF-targeted therapies for the treatment of different cancer types.
NORMAL FIBROBLASTS AND ACTIVATED/CANCER-ASSOCIATED FIBROBLASTS
During the generation of the third germ layer or mesoderm, primitive mesenchymal cells (primary mesenchyme) first appear when the epiblast undergoes EMT[15]. Most of the active mesenchymal cells undergo apoptosis after the completion of tissue development, whereas few cells attain a quiescent phenotype, which was first observed by Virchow[16] and eventually named fibroblasts. Normal fibroblasts are elongated cells with extended cell processes that exhibit a fusiform or spindle-like shape. These are generally present in connected tissues where they are embedded within ECM which consists largely of type I collagen and fibronectin[17]. A specific marker of quiescent fibroblasts is still missing; however, fibroblast-specific protein 1 (FSP1) and vimentin are considered as the closest. Normal fibroblasts also express integrins which are the mediators of the interaction of fibroblasts with their surrounding microenvironment[17]. Additionally, normal fibroblasts are characterized by low metabolic activity and lack of mobility[3].
Fibroblasts can be activated to acquire activated/myofibroblast phenotype, which is associated with enhanced proliferative activity and increased synthesis of ECM proteins such as type I collagen, tenascin C, extra domain A (EDA)-splice variant of fibronectin, and secreted protein acidic and rich in cysteine (SPARC)[17]. Fibroblast activation can be promoted by various stimuli generated from tissue injury or damage, including transforming growth factor beta (TGF-β), epidermal growth factor (EGF), fibroblast growth factor 2 (FGF2), and interferon-γ (IFNγ), interleukin (IL-6), mechano-transductions and enzymes[17-19]. Upon activation, these cells exhibit prolific protein synthesis and higher contraction potential that is crucial for wound healing and the production of connective tissues[3]. In physiological conditions, myofibroblasts play a critical role in wound healing and repairing damaged tissues[19-22]. Upon the completion of their function, these cells are cleared by programmed cell death, apoptosis[23]. However, if the injury is perpetual or dysregulation of the cell death program of these cells, it can lead to hyperproliferation and accumulation of myofibroblasts which culminates in a condition known as fibrosis[24-26].
“Tumors are depicted as wounds that do not heal” as they undergo continuous stromal remodeling and vascular growth, reminiscent of the wound repair program. Similar to wound healing process, activated fibroblasts/myofibroblasts are also present in tumors and are known as CAFs[9]. A diverse set of tumor or stroma-derived factors, including TGF-β1, OPN, and IL-1β, drive the transition of resting fibroblasts to CAFs by regulating Akt, ERK, MAPK, SMAD and NF-κB signaling pathways[8,27-29]. In an activation state, CAFs attain increased contractibility features and migratory potentials, which enables the CAFs to remodel ECM and aid in reciprocal interaction with cancer cells[3,30,31]. Different CAF-specific markers were identified to characterize activated CAFs, such as alpha-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), FSP1 (also known as S100A4), Integrin β1 (CD29), platelet-derived growth factor receptor α or β (PDGFRα/β) or podoplanin (PDPN)[32]. PDGFRs are a class of RTKs, known to be involved in tumor-fibroblast interactions[33]. In contrast to wound healing, but similar to organ fibrosis, the fibroblasts at the tumor site remain perpetually activated and form fibrous growth in the tumor, referred to as desmoplastic reaction/stroma[34]. Moreover, it was observed that senescent fibroblasts, which resemble myofibroblasts, also support preneoplastic tumor growth via secretion of OPN[35,36].
ORIGIN OF CAFS
The expression of different kinds of markers in CAFs indicates the heterogeneous generation and different cellular sources of these cells. CAFs can be originated from epithelial cells through the EMT [Figure 1]. According to a report, epithelial cells undergo specialized EMT by MMP-driven oxidative stress-associated DNA oxidation and mutations that lead to transdifferentiation of these cells into activated myofibroblasts[17,37]. This hypothesis is mainly supported by genetic studies conducted on breast cancers. These studies have reported somatic mutations in TP53 and phosphatase and tensin homolog (PTEN), and gene copy number alteration at other loci in stromal CAFs, similar to mutations in epithelial cells. Moreover, p53 inactivation in stromal fibroblasts and genetic inactivation of PTEN in CAFs promote tumor progression in breast carcinoma models[38-40]. Collectively, these data indicate that the tumor-promoting activity of CAFs may depend on somatic mutations in these tumor suppressor genes. In addition, somatic alterations were frequently detected (> 30%) in tumor cell-surrounding fibroblasts[39,40]. Similarly, CAFs might be generated from cancerous epithelial cells by EMT [Figure 1][41]. The EMT renders cancer cells to acquire mesenchymal phenotype and higher migration and contraction potentials[15]. This EMT program induced by platelet-derived growth factor (PDGF), TGFβ, EGF, etc., and is facilitated by the activation of mesenchymal specific transcription factors like Snail, Slug, Twist and FOXC2[15,42]. Tumor-associated endothelial cells might contribute to the CAF population [Figure 1]. A previous study has shown that endothelial cells are transdifferentiated into CAFs via endothelial to mesenchymal transition (EndMT) by losing expression of CD31 and gaining the expression of FSP-1 and α-SMA under the TGF-β stimulus[43]. In another study, auto/paracrine FGF2 has been shown to regulate the TGF-β-induced EndMT in tumor endothelial cells (TECs) via Elk1[44]. In a similar way, pericytes undergo pericytes to myofibroblast transition (PMT), a mesenchymal-to-mesenchymal transdifferentiation (MMT) process to generate CAFs in a microenvironment [Figure 1]. Hosaka et al. have recently reported that vascular pericytes are converted to CAFs by PDGF-BB to promote tumor growth and metastasis. PDGF-BB binds to PDGFRβ to induce the PMT program in pericytes[45]. CAFs are known to be generated from bone marrow-derived mesenchymal stem cells (MSCs)
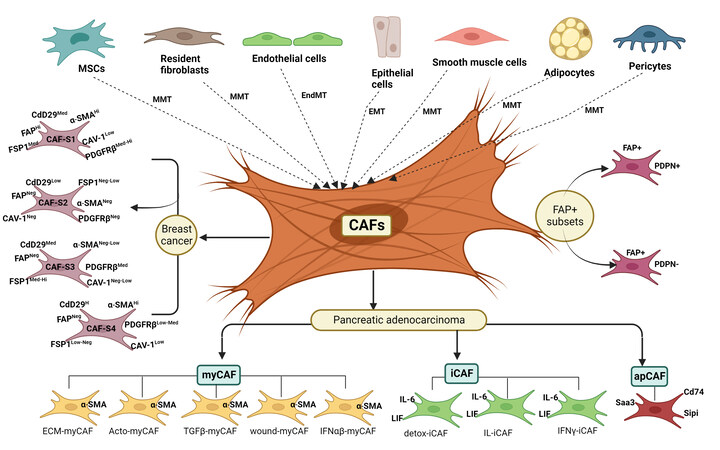
Figure 1. Origin and heterogeneity of cancer-associated fibroblasts. CAFs in the tumor microenvironment can be originated from MSCs, fibroblasts, adipocytes, pericytes, smooth muscle, endothelial and epithelial cells through the different trans-differentiation programs. Varieties of CAF subsets have been identified in cancer types of different tissue origins. The different subsets of CAFs show different functions and molecular features. CAF-S1 to CAF-S4 subsets are present in breast cancer. myCAF, iCAF and apCAF subsets are observed in PDAC. Several cancers exhibit overlapping populations of CAF subsets.
HETEROGENEITY OF CAFS
The multipronged actions of CAFs on tumor cells probably reflect their heterogeneous population with context-dependent functions. Although CAFs are known to originate from resident fibroblasts, MSCs, endothelial cells, pericytes, epithelial cells, and adipocytes through trans-differentiation programs, CAF subsets have been represented as distinct cellular states rather than indicating their different cell origins. Costa et al. have identified four subsets of CAFs (CAF-S1, CAF-S2, CAF-S3 and CAF-S4) in breast cancer by combining the analysis of six CAF markers [Figure 1]. Higher levels of both CAF-S1 (FAPHigh CD29Med SMAMed-High FSP1Med PDGFRβMed-High CAV1Low) and CAF-S4 (FAPNeg-Low CD29High SMAHigh FSP1Low-Med
Givel et al. have demonstrated fibroblast heterogeneity in high-grade serous ovarian cancers (HGSOC) by defining four subsets of CAFs (CAF-S1 to S4) as described in breast cancer[65]. Mesenchymal HGSOC consists of high CAF-S1 fibroblasts, which modulate immunosuppressive functions by increasing infiltration, survival, and differentiation of CD25+FOXP3+ T lymphocytes. SDF-1 β (CXCL12β) is specifically accumulated in the immunosuppressive CAF-S1 subset. Thus, their data highlight a CXCL12β-regulated stromal heterogeneity and immunosuppression in mesenchymal HGSOC[65]. The existence of CAF-S1 and CAF-S4 molecular signatures has been validated in lung cancer[66] and head and neck cancer by leveraging publicly available single-cell data[67]. The presence of these two major CAF-S1/CAF-S4 myofibroblastic subpopulations was validated in different cancer types[68]. These data suggest the existence of both CAF-S1 and CAF-S4 myofibroblastic cells in distinct cancer types and across species.
Two subsets of CAFs were recently reported in pancreatic adenocarcinoma. One subset displays a matrix-synthesizing myofibroblastic phenotype termed myCAF, whereas another exhibits an immunomodulatory phenotype, inflammatory CAFs named iCAF [Figure 1] The CAFs proximal to the cancer cells show a myCAF phenotype with higher expression of α-SMA. Distal CAFs from the cancer cells express high levels of proinflammatory cytokines such as IL-6, G-CSF, CXCL1, and LIF and are defined as iCAFs[69]. IL-1 signaling induces iCAF signature, while TGF-β signaling controls myCAF signature by antagonizing the iCAF phenotype. Another study has demonstrated the two different subpopulations of CAF, named myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs), by employing a 3D co-culture system of PDAC in vitro[70]. Further, Elyada et al. reported the third subtype of CAFs, named antigen-presenting CAFs (apCAFs), using single-cell RNA sequencing (scRNA-seq) in PDAC tissues, and these are characterized by expression of H2-Aa, H2-Ab1 (encoding α, β -chains of MHC II), CD74, secretory leukocyte peptidase inhibitor (SLPI) and serum amyloid A3 (Saa3) genes [Figure 1]. Also, apCAFs possess antioxidant response and are regulated by IFN-γ signaling in vivo[71]. Furthermore, other studies also confirmed the apCAF classification based on the results obtained using scRNA-seq in pancreatic cancer[72,73]. In addition, transcriptomics study in normal pancreatic cells of KPP mice revealed that cells that express mesothelial signature also show the expression of MHC II genes, implicating that apCAF could be of mesothelial origin[74]. Later, apCAFs subtype has also been reported in breast and lung cancer[75-78]. Interestingly, apCAFs activate the CD4 + T lymphocytes, which implies that CAFs have antigen-presenting properties similar to other antigen-presenting cells such as macrophages, dendritic cells and B cells immunomodulatory functions. However,the study on orthotopic murine models of lung cancer showed that lung apCAFs are tumor-suppressive cells[77]. Another report has revealed the presence of two FAP+ subsets on the basis of PDPN expression [Figure 1][79]. The FAP+ PDPN+ fibroblasts show elevated expression of TGF-β signaling proteins and fibrosis-associated genes, whereas FAP+ PDPN- cells displayed less expression of the same genes[68,71,79]. Moreover, a recent study further classified FAPHigh CAFs into eight different clusters. Out of these clusters, five clusters (ECM-myCAF, Acto-myCAF, TGFβ-myCAF, wound-myCAF and IFNαβ-myCAF) belong to the myCAF subgroup and three clusters (detox-iCAF, IL-iCAF, IFNγ-iCAF) fall into the iCAF subgroup[68]. Therefore, CAFs possess multifaceted functions including tumor promotion and prevention based on the gene expression signatures.
CAFS REGULATE DRUG RESISTANCE BY MODULATING CANCER CELL SURVIVAL
Interestingly, cancer cells produce a variety of factors that recruit, activate, and help with the survival of CAFs; nonetheless, CAFs, in return, support cancer cell survival and proliferation by providing appropriate signaling factors which subsequently promote cancer cell resistance. Using mouse models of inflammation-induced gastric cancer, a study reported that at least 20% of CAFs are derived from MSCs of bone marrow, and show the expression of α-SMA, wingless-related integration site 5α (Wnt5α), IL-6, bone morphogenetic protein 4 (BMP4), and DNA hypomethylation. MSC-derived CAFs are recruited to dysplastic stomach in TGF-β and SDF-1α-dependent manner to promote tumor survival[80]. CAFs provide ovarian cancer cells resistance to cisplatin by secreting cisplatin-induced chemokine (C-C motif) ligand 5 (CCL5), which augments the phosphorylation of STAT3 and Akt in cancer cells. Thus, CAFs play a crucial role in promoting ovarian cancer cell growth by regulating STAT3/PI3K/Akt pathway [Figure 2A][81]. Interestingly, a heparin-binding growth factor, midkine (MK), derived from CAFs, provides cisplatin resistance to oral squamous cell carcinoma (OSCC), lung cancer, and ovarian cancer cells by enhancing the expression levels of the lncRNA-ANRIL [Figure 2A]. Therefore, targeting either MK production from CAFs or inhibiting the lncRNA-ANRIL in cancer cells could be the key to cancer treatment[82]. The mir-1-mediated expression of SDF-1 in CAFs induces the proliferation of lung cancer cells and chemoresistance via CXCR4-dependent pathway involving NF-κB and Bcl-xL[83]. CAFs are not only involved in promoting cancer cell viability but also induce EMT in response to drug treatments. An earlier study showed that CAFs promote EMT through the secretion of IGF-1 and HGF. These growth factors enhance the expression and phosphorylation of annexin A2 (ANXA2), which endorse the resistance to the EGFR-TKI (gefitinib) in NSCLC (HCC827 and PC9) cells-harboring EGFR activating mutations [Figure 2A]. Therefore, restricting the CAFs-induced EMT is necessary to subdue TKI-resistance[84]. EMT transcription factors such as Twist1 and Snail regulate the activation of CAFs in cancers[30,85]. Expression of Twist1 and Snail in CAFs also associated with the expression of several cytokines including SDF-1, CXCL1 and CCL2 which can regulate cell proliferation and survival[30,86]. Blanco-Gomez et al. demonstrated that loss of SNAI2 in CAFs limit the production of some cytokines such as SDF-1 and CXCL1, CXCL2, IFN-γ and IL-16, thereby impeding breast cancer cell proliferation and metastasis[86]. Thus, SNAI2 could be considered a therapeutic target to block both proliferation and EMT in tumor cells and cytokine production in CAFs.
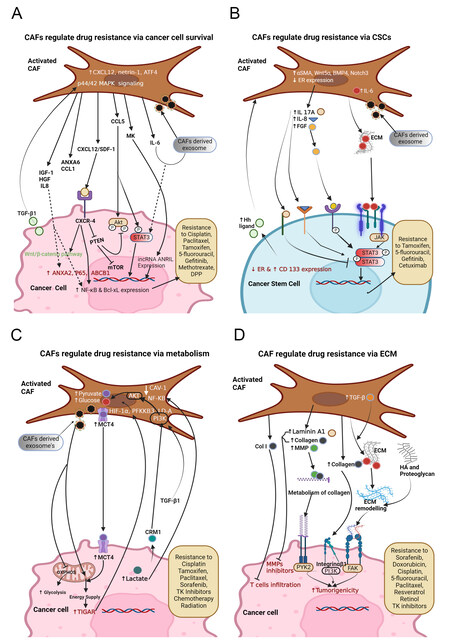
Figure 2. Fibroblast-mediated signaling in shaping the drug resistance in (A) CAFs promote cancer cells survival by secreting ANXA6, CCL1, CXCL 12, CCL5, MK, etc., and the exosomes containing lncRNA ANRIL, miR-196a, miR-103a-3p, miR-24-3p, microRNA-21, microRNA-148b-3p, LPP, CCAL, etc. Also, CAFs show high levels of netrin1, IL-6, and ATF4, which offer cancer cells gemcitabine resistance. TGF-β1 secreted by cancer cells acts on CAFs to attain 5-fluorouracil (5-FU) and tamoxifen (TAM) resistance. Inhibition of PTEN by CXCL12-CXCR4 binding promotes mTOR signaling and cancer proliferation. MK provides cisplatin resistance by ameliorating the expression of lncRNA-ANRIL; (B) CAFs induce stemness in cancer cells through the activation of STAT3 by IL8, IL6, and FGF and secreting exosomes containing miR-221 and H19 that leads to drug resistance, and STAT3 activation can be inhibited by IL-17A. High levels of α-SMA, Wnt5α, BMP4 and Notch3 in CAFs and low expression of ER in both CAFs and cancer cells are associated with enriching CSCs and drug resistance; (C) CAFs provide different nutrients to cancer cells. CAF-secreted factors rewire the cancer cell metabolism by the activation of autophagy, mTOR, and TIGAR and suppression of oxidative phosphorylation that leads to drug resistance; (D) CAFs promote drug resistance by ECM deposition. Activation of CAFs by TGF-β1 or other factors leads to excessive synthesis of ECM proteins such as laminin-A, collagen, and fibronectin. It also induces various MMPs, which leads to ECM modeling that blocks drug effects.
Furthermore, CAFs elicit TGF-β-mediated EMT in ovarian cancer cells via IL-6-regulated JAK2/STAT3 pathway to inhibit cancer cell apoptosis and provide paclitaxel resistance[87]. CAF-secreted SDF-1 stimulates pancreatic cancer progression and aids in gemcitabine resistance by augmenting the expression of
Higher expression of CXCL12 in interstitial CAFs contributes to EMT and cisplatin resistance in epithelial ovarian cancer (EOC) via CXCR4/Wnt/β-catenin pathway[95]. Additionally, this CAF-derived CXCL12 mediates inhibition of PTEN which is crucial for cancer cell proliferation [Figure 2A][96]. Likewise, CAFs are involved in offering cisplatin resistance in HNC cells by exosome-mediated transfer of miR-196a. Upon depletion of CAF-exosomal miR-196a, restoration of cisplatin sensitivity has occurred in HNC cells. Therefore, targeting miR-196a can serve as a better therapeutic approach to overcome cisplatin resistance in HNC cells[97]. Moreover, CAF-derived, highly expressed, exosomal miR-103a-3p accelerates cisplatin resistance and inhibits apoptosis in NSCLC cells by targeting BCL2- antagonist/killer 1 (Bak1)[98]. CAF-derived miR-24-3p containing exosomes promote cancer cell resistance to methotrexate by downregulation of the CDX2/HEPH axis in colon cancer[99]. The CAF-mediated transfer of exosome-containing lncRNA CCAL (colorectal cancer-associated lncRNA) to CRC cells initiates signaling towards gaining resistance to oxaliplatin via the β-catenin pathway. Interaction of CCAL with HuR (human antigen R, an RNA stabilizing protein) leads to an increase in β-catenin, thereby providing oxaliplatin resistance in CRCs[100]. CAFs secreted IL-6/exosomal microRNA-21 (miR-21) induces the activation of STAT3 signaling to generate monocytic myeloid-derived suppressor cells (M-MDSCs) to further accelerate cisplatin (DDP). Therefore, inhibition of STAT3 signaling can restore cancer cells’ drug sensitivity[101]. Transfer of CAF derived exosomes-containing miR-148b-3p to bladder cancer cells enhances tumor proliferation, EMT, metastasis, and drug resistance. Mechanistically, miR-148b-3p induces Wnt/β-catenin pathway by targeting PTEN[102]. Therefore, overexpression of PTEN might lead to suppression of metastasis, EMT, and drug resistance. CAFs respond to tamoxifen treatment by upregulating the expression of high mobility group box 1 (HMGB1) through GPR30/PI3K/AKT signaling. HMGB1 is involved in the induction of autophagy to increase resistance to tamoxifen in MCF-7 cells via an ERK-mediated manner[103]. Overall, the above reports suggest that CAF-secreted growth factors, chemokines and exosomes regulate drug resistance by inducing cell survival in different types of cancer.
CAFS REGULATE DRUG RESISTANCE BY MODULATING CANCER STEM CELLS
Cancer stem cells (CSCs) play a pivotal role in tumorigenesis, progression, and drug resistance. CSCs exhibit self-renewal and tumorigenic properties, which enable them to metastasize to distant sites, offering them a favorable environment. Moreover, the microenvironment around CSCs contributes a lot to fostering tumor growth through the modulation of CSC phenotype. The generation of CSCs through EMT is highly conditional on its surrounding matrix. This underscores the vital role of microenvironmental elements like CAFs and their secreted factors in shaping the renewal and maintenance of CSCs[104].
Stem cell pathways like Wnt signaling are important for maintaining stemness in non-cancerous cells of the colon. An interesting study suggested that cells surrounding the CSCs, especially myofibroblasts, maintain a higher Wnt activity in CSCs and manage to stimulate Wnt signaling in nearby differentiated tumor cells, thereby mending the stemness and tumorigenicity[105]. In response to the chemotherapy, CAFs express
CAFS REGULATE DRUG RESISTANCE BY MODULATING CANCER CELL METABOLISM
The major energy sources for the survival of unconditionally growing tumor cells are glutamine (Gln) and glucose. Rewiring of cancer cell metabolism enables the survival of cancer cells by providing the building blocks/intermediates for the synthesis of nucleic acids, lipids and proteins. Tumor microenvironment (TME) or CAFs-mediated metabolic reprogramming of cancer cells regulates several signaling cascades that also result in drug resistance[119].
Notably, a study has shown that exosomes derived from CAFs can reprogram the metabolic machinery by their uptake into cancer cells[120,121]. These exosomes consist of intact lipids, amino acids, and intermediates of the TCA cycle[121]. Moreover, these exosomes inhibit mitochondrial OXPHOS, leading to increased glycolysis and Gln-dependent reductive carboxylation in cancer cells [Figure 2C]. It has been reported that CAFs predominantly express glucose uptake proteins in non-small cell lung cancer. Among these, glutamine-fructose-6-phosphate transaminase 2 (GFPT2) plays an important role in glycolysis, thus showing its significance in prognosis[122]. A previous study showed that epigenetic changes in CAF instigated a cascade of stromal-epithelial interactions to promote prostate cancer growth and resistance to androgen deprivation therapy (ADT). This study revealed that epigenetic silencing of a Ras inhibitor, RASAL3, in prostatic CAFs leads to oncogenic Ras activity that drives macropinocytosis-mediated glutamine synthesis. Interestingly, ADT further strengthens RASAL3 epigenetic silencing and glutamine secretion by CAFs. Therefore, high levels of glutamine have been found in prostate cancer patients after ADT[123].
Cancer cells, under glucose-deprived states, use aerobic glycolysis as their major energy source, known as the Warburg effect. Pyruvate kinase M2 (PKM2) is overexpressed in NSCLC cell lines and plays a role in mediating the Warburg effect which promotes resistance to cisplatin[124]. In another phenomenon, aerobic glycolysis in the cancer-associated stroma metabolically supports surrounding cancer cells, which is known as the reverse Warburg effect. This stromal-cancer metabolic coupling enables catabolite transfer to cancer cells for the generation of ATP, induction of proliferation, and reduction of cell death[125]. Interestingly, cancer cells educate stromal cells to display aerobic glycolysis that mediates multidrug resistance[126]. Moreover, in the majority of solid tumors, CAFs utilize more glucose and in turn release more lactate in comparison to normal fibroblasts[127]. Notably, cancer cells induce the Warburg effect in CAFs through activation of the PI3K/AKT pathway via translocation of nuclear G-protein-coupled estrogen receptor (GPER) in a chromosomal region maintenance 1 (CRM1)-dependent manner and abnormal activation of the GPER/cAMP/PKA/CREB signaling pathway[126]. Consequently, CAFs delivered lactate transporters to cancer cells, which increases drug resistance [Figure 2C]. In contrast, a study by
CAFS REGULATE DRUG RESISTANCE BY MODULATING ECM
CAFs orchestrate tumor promotion and drug resistance by increasing matrix stiffness via augmenting the expression of ECM components such as hyaluronic acid (HA) and collagens[143]. Both HA and collagen are known to withstand tensile stress and the activity of collagen receptor, integrin α11β1, is associated with matrix stiffness. It has been reported that in NSCLC, CAFs can promote the stiffness of interstitial collagen by enhancing the expression of integrin α11, leading to tumor progression[144]. A recent study demonstrated that collagen secreted by CAFs acts in a paracrine manner to regulate the resistance to microtubule-directed chemotherapeutic drugs through integrinβ1/PI3K/AKT pathway in breast cancer [Figure 2D][145]. The dense number of CAFs was observed to play an important role in desmoplastic reactions in PDAC[146]. In addition, the CAFs-derived intense desmoplastic response in fibrotic tumors builds a dense ECM barrier that reduces the delivery of any drug to the tumor cells. In breast cancer patients, a progressively noncompliant fibrotic stroma limits the chemotherapeutic efficiency of doxorubicin[147]. Also, based on desmoplastic scores, CAFs have been divided into high desmoplastic CAFs (HD-CAFs) and low desmoplastic CAFs (LD-CAFs). The NSCLC patients with HD-CAFs showed a high collagen matrix remodeling rate which played a critical role in tumor progression via regulation of invasion and growth[148]. Likewise, HD-CAFs (alpha-smooth muscle actin positive myofibroblasts) in PDAC are significantly involved in the secretion of type I collagen (Col1) which plays a role in restricting drug delivery and impeding T cell infiltration[149].
Further, CAFs produce metalloproteinases (MMPs) that enhance tumor invasion by matrix remodeling[150]. CAFs employ MMP endopeptidases for the degradation of basement membrane (BM) proteins[151]. A previous study demonstrated the role of CAFs in BM stretching that facilitates the migration of CAFs and tumor cells into the bloodstream and metastasizes to distant organs. Intriguingly, the alternative CAF-dependent mechanism where BM shows a high tendency of stretching due to low expression of type IV collagen and laminin and rendering the head and neck tumor cells resistant to MMP inhibitors[152]. Another study reported that MMPs derived from CAFs are involved in tamoxifen resistance through EGFR and PI3K/AKT pathways in breast cancer [Figure 2D][153].
In addition to this, CAFs also secrete other factors such as caveolin-1 and podoplanin (PDPN), which are associated with wound responses[154]. The expression of a lymphatic vessel marker, PDPN, by stromal CAFs has been reported as a prognostic indicator in different cancer types. For instance, Yoshida et al. have demonstrated that lung adenocarcinoma cells, when co-cultured with the PDPN+ CAFs, show greater drug resistance in comparison to normal cells. Similarly, in postoperative recurrence, PDPN+ patients possess a lower treatment response to EGFR-TKIs compared to PDPN- patients, suggesting the implication of PDPN+ CAFs in regulating drug resistance[155]. The above information indicates that dense ECM produced by CAFs acts as a mechanical barrier for drug delivery and immune cell infiltration, and it also provides the source for matrix remodeling enzymes and signaling molecules that further impede the efficacy of anticancer therapeutics. Therefore, ECM-depletion strategies might pave the way for the development of next-generation anticancer drugs.
TARGETING CAFS WITH NATURAL PRODUCTS AND ANALOGS
The utilization of natural products for the treatment of different diseases is indeed cost-effective and minimally invasive[156-158]. Numerous anticancer products have been isolated and characterized from natural sources. Apart from directly showing anticancer activity, they also provide leads for developing potent therapeutic drugs[159].
CAFs have emerged as an intriguing therapeutic target in cancer due to their indispensable role. CAFs and cancer cells reciprocally crosstalk to regulate several aspects of cancer progression and several growth factors and cytokines act as a messenger in this crosstalk [Table 1]. Hence, targeting CAFs using natural products will be advantageous for reducing the burden of cancer as well as overcoming deleterious effects caused by drug treatments in cancer patients. Polyphenols present in green tea have potential anticancer activities. Treatment with tea polyphenol, epigallocatechin-3-gallate (EGCG), decreases the serum levels of HGF and VEGF in prostate cancer patients. Since HGF and VEGF are mostly secreted by stromal myofibroblasts in the tumor microenvironment, EGCG can prevent myofibroblast differentiation in prostate cancer[160]. Gray et al. have demonstrated that combinational treatment of EGCG and another polyphenol, luteolin, synergistically reduces TGF-β-induced myofibroblast differentiation and fibronectin synthesis by impeding ERK and RhoA signaling in prostate fibroblasts[161]. CAFs are one of the key contributors in introducing drug resistance to various anticancer chemotherapeutic agents. It was shown that CAFs are involved in acquiring the resistance to cisplatin by expressing Wnt16[162]. Quercetin is a member of flavonoids that show antioxidant properties. In this regard, Hu et al. have found that quercetin significantly inhibits Wnt16 expression in activated fibroblasts, thereby improving the anticancer effects of cisplatin[163]. Various reports have shown that curcumin, a phyto-polyphenol pigment found in spice turmeric, exhibits antioxidant, anti-inflammatory, neuroprotective, and anticancer activities against various cancers[164]. In addition, curcumin also regulates the TME of CAFs. An earlier study has shown that curcumin induces DNA damage-independent and safe-senescence in CAFs by upregulating p16. It also decreases the expression of α-SMA and reduces the migration and invasion potentials of CAFs. Furthermore, curcumin abolishes tumorigenic potentials of CAFs by downregulating the expression of IL-6, SDF-1, MMP-2, MMP-9 and TGF-β[165]. In the pancreatic cancer model, curcumin suppresses the expression of α-SMA, vimentin, and secretory factors in CAFs, thereby inhibiting EMT and metastasis of cancer cells[166]. The above reports indicate that curcumin might have therapeutic potential for impeding the crosstalk between cancer cells and CAFs.
The interactions between CAFs and cancer cells
| Source cells | Factors | Recipient cells | Biological effect of released factors | Affected signaling pathways | Reference |
| CAFs | CXCL12 | Breast cancer | OPN-CAF-derived CXCL12 promotes EMT | ERK1/2 and AKT | [8] |
| CAFs | CCL5 | Ovarian cancer | Cisplatin resistance | STAT3/PI3K/Akt pathway | [81] |
| CAFs | SDF-1 | Lung cancer | Chemoresistance | mir-1/SDF-1/CXCR4/NF-κB/Bcl-xL | [83] |
| CAFs | IGF-1, HGF | Lung cancer | EMT in NSCLC | IGF1/HGF/ANXA2 | [84] |
| CAFs | IL-6 | Ovarian cancer | Paclitaxel resistance | TGFβ/JAK2/STAT3/IL6 pathway | [87] |
| CAFs | SDF-1 | Pancreatic cancer | Gemcitabine resistance | SDF-1/CXCR4/SATB-1 pathway | [88] |
| CAFs | CCL1 | Colorectal cancer | Chemoresistance to 5-FU and paclitaxel | TGF-β/NF-κB pathway | [89] |
| CAFs | IL-8 | Gastric cancer | Cisplatin resistance | NF-κB pathway | [91] |
| Breast cancer | TGF-β1 | CAFs | 5-FU and tamoxifen (TAM) resistance | p44/42 MAPK pathway | [94] |
| CAFs | CXCL12 | Ovarian cancer | EMT and cisplatin resistance | CXCR4/Wnt/β-catenin pathway | [95] |
| CAFs | miR-24-3p | Colon cancer | Resistance to methotrexate | CDX2/HEPH axis | [99] |
| CAFs | miR-148b-3p | Bladder cancer | EMT, metastasis, and drug resistance | Wnt/β-catenin pathway | [102] |
| CAFs | IL-17A | Cancer-initiatingcells | Resistance to chemotherapies | IL-17A signaling pathway | [106] |
| CSCs | SHH | CAFs | CSCs expansion | Wnt/β-catenin signaling pathway | [109] |
| CAFs | TGF-β2 | CSCs | Chemoresistance and stemness of CSCs | HIF-1α/TGF-β2-GLI2 pathway | [112] |
| CAFs | H19 | CRC | Elevates stemness in CRCs | miR-675-IGFR signaling circuit & β-catenin pathway | [114] |
Fraxinellone (FRA) is a member of the limonoids family. Several studies have reported the medicinal properties of FRA, including neuroprotective, antifibrotic, anti-inflammatory, and antitumor functions[167]. An earlier study has reported that FRA regulates TGF-β signaling in fibrotic liver disease[168], which hints therapeutic potential of FRA in treating cancer, as both are characterized by the accumulation of myofibroblasts. A recent report demonstrated that FRA-loaded nanoparticle inhibits the CAF phenotype by impeding TGF-β signaling in PDAC[169]. Mangostin (MG) is a xanthone and exhibits several medicinal properties such as antibacterial, antifungal, antioxidant, anti-inflammatory, anticancer, and cardioprotective effects[170]. Studies have demonstrated that MG displays antitumor activities by inducing apoptosis and inhibiting angiogenesis, ECM modification, and EMT[171]. A study has recently demonstrated MG’s effect in regulating the tumor stroma. A nano-formulated MG suppresses TGF-β/Smad signaling leading to CAF inactivation and ECM reduction in pancreatic cancer[172].
Cyclopamine is a steroid alkaloid and the first small-molecule inhibitor of the Hh signaling pathway[173]. Several reports show that Hh signaling displays a critical role in proliferation and tumor-promoting functions indicating the potential of cyclopamine to reprogram CAFs[174,175].
Co-delivery of cyclopamine and paclitaxel nanoparticles in pancreatic cancer modulates tumor stroma by disrupting cancer-stroma crosstalk and reducing ECM stiffness[174]. Chrysin is classified as a member of the flavonoids, which exerts multiple biological effects including antidiabetic, antioxidant, hepatoprotective, anti-inflammatory, and anticancer activities[176]. Chrysin induces apoptosis in colorectal and gastric cancer cells[177,178]. A synthetic analog of chrysin named 8-bromo-7-methoxy chrysin inhibits the activation of hepatic stellate cells to CAFs, thereby reducing the stemness of cancer cells by impeding IL-6 and HGF signaling[179]. We have listed several natural or synthetic drugs for targeting CAFs in Table 2.
Drugs targeting CAFs for management of cancer
| Drug natural | Type of cancer | Target/Interference with | Mechanism | Reference | |
| CAFs | CAFs functions | ||||
| EGCG | Colorectal | ↓Glycolytic activity | ↓PFK | [180] | |
| Conophylline | HCC | ↓α-SMA | ↓IL6, IL8, CCL2, angiogenin, OPN | ↓GPR68 | [181] |
| α-mangostin | Pancreatic | ↓αSMA/FAP/fibronectin | ↓fibronectin/collagen | ↓TGF-β pathway/Smad | [172] |
| Fraxinellone | Pancreatic | ↓αSMA/FAP/fibronectin | - | ↓TGF-β pathway | [169] |
| Triptonide | Gastric | - | ↓IL-6, ↑TIMP2 | ↓MiR-301a ↑MiR-149 | [182] |
| Chrysin | Liver | - | ↓IL-6, HGF | - | [179] |
| Paeoniflorin | Gastric | - | ↓IL-6 | ↑MicroRNA149 | [183] |
| Resveratrol | CCA | - | ↓IL-6 | - | [184] |
| Minnelide | Pancreatic | ↓α-SMA | ↓Collagen/fibronectin/periostin/hyaluronan/ MMP2/MMP9 | ↓TGF-β RAR/RXR pathway | [185] |
| Cyclopamine | Pancreatic | - | ↓LOX/hyaluronan | ↓Hh pathway | [186] |
| Polyphyllin I | Gastric | ↓FAP | ↓HGF | - | [187] |
| Curcumin | Pancreatic | - | ↑E-cadherin, ↓vimentin | - | [166] |
| Astragaloside IV | Gastric | - | ↓M-CSF, ↑TIMP2 | ↑microRNA-214 ↓microRNA-301a | [188] |
| Synthetic drugs | |||||
| Ursolic acid | PTC | - | ↓CXCR4, CXCR7 | - | [189] |
| CFH/OM-L | Hepatic | - | ↑E-cadherin, ↓vimentin, N-cadherin, snail protein | - | [190] |
| Nintedanib | Hepatic | ↓α-SMA | IL-6, IL-8 | - | [191] |
| BTZ and PST | - | - | ↑Caspase-3 mediated apoptosis | [192] | |
| Drugs under clinical trial | |||||
| JNJ-42756493 | NSCLC, Urothelial, Esophageal | ↓FGFR ↓TK | - | - | [193] |
| Plerixafor | Pancreatic, Ovarian and Colorectal | - | ↓CXCR4 | - | [194] |
| PEGPH20 and MK-3475 | PDAC | - | ↓Hyalouronan | - | [195] |
| AT13148 | Advanced solid tumors | - | ↓ROCK | - | [196] |
| IPI-926 and Gemcitabine | Pancreatic | - | - | ↓Hh pathway | [197] |
COMPOUNDS UNDER CLINICAL TRIAL
There are several compounds under clinical trials for targeting the CAFs or CAF-mediated effects for the management of cancer[198]. Notably, losartan, a small molecular inhibitor, sold under the brand name Cozaar, is used for the treatment of diabetic kidney disease, heart failure, and left ventricular enlargement. It inhibits the angiotensin receptor by blocking binding of angiotensin II. It suppresses collagen and hyaluronan levels, which are known to be synthesized by CAFs, and it is currently under clinical trial[199]. Defactinib is a small molecular inhibitor of FAK, available under brand names, VS-6063 and PF-04554878. It is under phase II clinical trial for the treatment of patients with KRAS-mutant NSCLC and is known to target downstream signaling of integrins and interfere with CAF actions[200]. Vitamin D receptor agonist, paricalcitol, is under phase I and II studies to examine the benefit of it in combination with gemcitabine and nab-paclitaxel for the treatment of pancreatic cancer as it is known to normalize pancreatic stellate cells[201]. Galunisertib is a pharmacological small molecule inhibitor of the TGF-β signaling. Treatment with Galunisertib interferes with TGF-β signaling induced activation of CAFs and immunosuppression. Studies of phase I, and phase II trials are underway to compare the overall survival (OS) of patients with pancreatic cancer after the treatment with a combination of Galunisertib and gemcitabine as compared to gemcitabine alone[202,203]. IPI-926 (saridegib) and vismodegib are small molecular inhibitors that target Hh signaling and reduce CAF activation, and are under clinical trials[204,205]. Several compounds for targeting CAFs are under clinical trials [Table 2].
CONCLUSIONS AND FUTURE DIRECTIONS
Stromal fibroblasts constitute a major component of tumor microenvironment. Fibroblasts can thrive in severe adverse conditions because of their intrinsic survival programs and cellular plasticity. Due to this ability, they can withstand insults from anticancer regimens. Simultaneously, these cells activate cell resistance programs in cancer cells in response to inhibitory effects caused by the treatment modalities. Therefore, it is very important to understand the intrinsic survival programs and cellular plasticity that endure these cells from chemotherapy insults. Since the reciprocal interactions between the cancer cells and CAFs through soluble factors play a crucial role in orchestrating drug resistance programs, it is critical to profile CAF-derived secretome in response to chemotherapy to identify druggable targets. The CAF-mediated direct cell-cell and cell-matrix interactions also shape drug resistance in cancer cells. Hence, investigating the ECM-remodeling and changes in cell adhesion molecules triggered during the development of drug resistance might provide insights into target proteins. Moreover, the double-edged sword effect of CAF signaling has been well recognized in tumor progression and drug resistance. Dissecting CAF-signaling and its function enables comprehensive identification of signaling pathways required to instigate drug resistance programs and facilitates targeting drug-resistant inducing cues and sparing the inhibitory cues.
DECLARATIONS
Authors’ contributionsConceptualized, wrote the significant part, and edited manuscript: Butti R
Wrote parts, made figures and edited manuscript: Khaladkar A, Bhardwaj P, Prakasam G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. Bremnes RM, Dønnem T, Al-Saad S, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 2011;6:209-17.
4. Butti R, Kumar TVS, Nimma R, Banerjee P, Kundu IG, Kundu GC. Osteopontin signaling in shaping tumor microenvironment conducive to malignant progression. In: Birbrair A, editor. Tumor Microenvironment. Cham: Springer International Publishing; 2021. pp. 419-41.
5. Strieter R, Wiggins R, Phan S, et al. Monocyte chemotactic protein gene expression by cytokine-treated human fibroblasts and endothelial cells. Biochem Biophys Res Commun 1989;162:694-700.
6. Rollins BJ, Stier P, Ernst T, Wong GG. The human homolog of the JE gene encodes a monocyte secretory protein. Mol Cell Biol 1989;9:4687-95.
7. Kuzet SE, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res 2016;365:607-19.
8. Butti R, Nimma R, Kundu G, et al. Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene 2021;40:2002-17.
9. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res 2012;10:1403-18.
10. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol 2019;10:1835.
11. Wu F, Yang J, Liu J, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther 2021;6:218.
12. Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2015;28:831-3.
13. Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735-47.
14. Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA 2014;111:E3091-100.
15. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420-8.
16. Molenaar JC. [From the library of the Netherlands journal of medicine. Rudolf virchow: die cellularpathologie in ihrer begrundung auf physiologische und pathologische gewebelehre; 1858]. Ned Tijdschr Geneeskd 2003;147:2236-44.
18. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res 2011;1:482-97.
19. Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013;15:637-46.
20. Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmoulière A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 2012;5:S5.
21. Desmoulière A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest 2003;83:1689-707.
22. Darby IA, Laverdet B, Bonté F, Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 2014;7:301-11.
23. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349-63.
24. Hamburg-Shields E, DiNuoscio GJ, Mullin NK, Lafyatis R, Atit RP. Sustained β-catenin activity in dermal fibroblasts promotes fibrosis by up-regulating expression of extracellular matrix protein-coding genes. J Pathol 2015;235:686-97.
25. Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 2011;108:E1475-83.
26. LeBleu VS, Taduri G, O'Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 2013;19:1047-53.
27. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an nf-kappab-dependent manner. Cancer Cell 2010;17:135-47.
28. Li Q, Zhang D, Wang Y, et al. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci Rep 2013;3:2038.
29. Goulet C, Bernard G, Tremblay S, Chabaud S, Bolduc S, Pouliot F. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Mol Cancer Res 2018;16:1196-204.
30. Lee KW, Yeo SY, Sung CO, Kim SH. Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Res 2015;75:73-85.
31. Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 2007;9:1392-400.
32. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: cancer-associated fibroblasts and their markers. Int J Cancer 2020;146:895-905.
33. Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer 2018;17:34.
34. Nissen NI, Karsdal M, Willumsen N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res 2019;38:115.
35. Pazolli E, Luo X, Brehm S, et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res 2009;69:1230-9.
36. Butti R GP, Kumar Totakura KVS, Venkata RNN, Nimma R, Kundu GC. Role of osteopontin in tumor microenvironment: a new paradigm in cancer therapy. in: multi-targeted approach to treatment of cancer. Adis, Cham; 2015. pp. 113–25.
37. Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005;436:123-7.
38. Tuhkanen H, Anttila M, Kosma VM, et al. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int J Cancer 2004;109:247-52.
39. Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet 2002;32:355-7.
40. Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res 2000;60:2562-6.
41. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 2007;101:830-9.
42. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 2008;19:4875-87.
43. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007;67:10123-8.
44. Akatsu Y, Takahashi N, Yoshimatsu Y, et al. Fibroblast growth factor signals regulate transforming growth factor-β-induced endothelial-to-myofibroblast transition of tumor endothelial cells via Elk1. Mol Oncol 2019;13:1706-24.
45. Hosaka K, Yang Y, Seki T, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci USA 2016;113:E5618-27.
46. Hung SC, Deng WP, Yang WK, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res 2005;11:7749-56.
47. Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009;27:2614-23.
48. Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. In: Kauser K, Zeiher A, editors. Bone Marrow-Derived Progenitors. Berlin: Springer Berlin Heidelberg; 2007. pp. 263-83.
49. Dwyer RM, Potter-Beirne SM, Harrington KA, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res 2007;13:5020-7.
50. Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther 2008;15:730-8.
51. Feng B, Chen L. Review of mesenchymal stem cells and tumors: executioner or coconspirator? Cancer Biother Radiopharm 2009;24:717-21.
52. Spaeth EL, Dembinski JL, Sasser AK, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 2009;4:e4992.
53. Weber CE, Kothari AN, Wai PY, et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015;34:4821-33.
54. Kojima Y, Acar A, Eaton EN, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA 2010;107:20009-14.
55. Procopio MG, Laszlo C, Al Labban D, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 2015;17:1193-204.
56. Shimoda M, Principe S, Jackson HW, et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol 2014;16:889-901.
57. Li Z, Zhang J, Zhou J, et al. Nodal facilitates differentiation of fibroblasts to cancer-associated fibroblasts that support tumor growth in melanoma and colorectal cancer. Cells 2019;8:538.
58. Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 2014;25:15-22.
59. Avgustinova A, Iravani M, Robertson D, et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat Commun 2016;7:10305.
60. Albrengues J, Bertero T, Grasset E, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 2015;6:10204.
61. Costa A, Kieffer Y, Scholer-Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018;33:463-479.e10.
62. Bonneau C, Eliès A, Kieffer Y, et al. A subset of activated fibroblasts is associated with distant relapse in early luminal breast cancer. Breast Cancer Res 2020;22:76.
63. Li H, Courtois ET, Sengupta D, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 2017;49:708-18.
64. Pelon F, Bourachot B, Kieffer Y, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun 2020;11:404.
65. Givel AM, Kieffer Y, Scholer-Dahirel A, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun 2018;9:1056.
66. Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277-89.
67. Puram SV, Tirosh I, Parikh AS, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017;171:1611-1624.e24.
68. Kieffer Y, Hocine HR, Gentric G, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 2020;10:1330-51.
69. Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019;9:282-301.
70. Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579-96.
71. Elyada E, Bolisetty M, Laise P, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 2019;9:1102-23.
72. Hosein AN, Huang H, Wang Z, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019;5:129212.
73. Bernard V, Semaan A, Huang J, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res 2019;25:2194-205.
74. Dominguez CX, Müller S, Keerthivasan S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov 2020;10:232-53.
75. Friedman G, Levi-Galibov O, David E, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer 2020;1:692-708.
76. Sebastian A, Hum NR, Martin KA, et al. Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers 2020;12:1307.
77. Kerdidani D, Aerakis E, Verrou K, et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J Exp Med 2022;219:e20210815.
78. Bartoschek M, Oskolkov N, Bocci M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 2018;9:5150.
79. Cremasco V, Astarita JL, Grauel AL, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res 2018;6:1472-85.
80. Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011;19:257-72.
81. Zhou B, Sun C, Li N, et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int J Oncol 2016;48:2087-97.
82. Zhang D, Ding L, Li Y, et al. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci Rep 2017;7:16231.
83. Li J, Guan J, Long X, Wang Y, Xiang X. Mir-1-mediated paracrine effect of cancer-associated fibroblasts on lung cancer cell proliferation and chemoresistance. Oncol Rep 2016;35:3523-31.
84. Yi Y, Zeng S, Wang Z, et al. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim Biophys Acta Mol Basis Dis 2018;1864:793-803.
85. Stanisavljevic J, Loubat-Casanovas J, Herrera M, et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res 2015;75:284-95.
86. Blanco-Gómez A, Hontecillas-Prieto L, Corchado-Cobos R, et al. Stromal SNAI2 is required for ERBB2 breast cancer progression. Cancer Res 2020;80:5216-30.
87. Wang L, Zhang F, Cui JY, Chen L, Chen YT, Liu BW. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol Rep 2018;39:2081-90.
88. Wei L, Ye H, Li G, et al. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis 2018;9:1065.
89. Li Z, Chan K, Qi Y, et al. Participation of CCL1 in snail-positive fibroblasts in colorectal cancer contribute to 5-fluorouracil/paclitaxel chemoresistance. Cancer Res Treat 2018;50:894-907.
90. Leung CS, Yeung TL, Yip KP, et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J Clin Invest 2018;128:589-606.
91. Zhai J, Shen J, Xie G, et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett 2019;454:37-43.
92. Uchihara T, Miyake K, Yonemura A, et al. Extracellular vesicles from cancer-associated fibroblasts containing annexin A6 induces FAK-YAP activation by stabilizing β1 integrin, enhancing drug resistance. Cancer Res 2020;80:3222-35.
93. Wei L, Lin Q, Lu Y, et al. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-β1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis 2021;12:334.
94. Chandra Jena B, Kanta Das C, Banerjee I, et al. Paracrine TGF-β1 from breast cancer contributes to chemoresistance in cancer associated fibroblasts via upregulation of the p44/42 MAPK signaling pathway. Biochem Pharmacol 2021;186:114474.
95. Zhang F, Cui JY, Gao HF, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol 2020;16:2619-33.
96. Martelli AM, Evangelisti C, Chappell W, et al. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 2011;25:1064-79.
97. Qin X, Guo H, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol 2019;20:12.
98. Wang H, Huang H, Wang L, et al. Cancer-associated fibroblasts secreted miR-103a-3p suppresses apoptosis and promotes cisplatin resistance in non-small cell lung cancer. Aging (Albany NY) 2021;13:14456-68.
99. Zhang HW, Shi Y, Liu JB, et al. Cancer-associated fibroblast-derived exosomal microRNA-24-3p enhances colon cancer cell resistance to MTX by down-regulating CDX2/HEPH axis. J Cell Mol Med 2021;25:3699-713.
100. Deng X, Ruan H, Zhang X, et al. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int J Cancer 2020;146:1700-16.
101. Zhao Q, Huang L, Qin G, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett 2021;518:35-48.
102. Shan G, Zhou X, Gu J, et al. Downregulated exosomal microRNA-148b-3p in cancer associated fibroblasts enhance chemosensitivity of bladder cancer cells by downregulating the Wnt/β-catenin pathway and upregulating PTEN. Cell Oncol 2021;44:45-59.
103. Liu L, Liu S, Luo H, et al. GPR30-mediated HMGB1 upregulation in CAFs induces autophagy and tamoxifen resistance in ERα-positive breast cancer cells. Aging (Albany NY) 2021;13:16178-97.
104. Butti R, Gunasekaran VP, Kumar TVS, Banerjee P, Kundu GC. Breast cancer stem cells: biology and therapeutic implications. Int J Biochem Cell Biol 2019;107:38-52.
105. Vermeulen L, De Sousa E Melo F, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010;12:468-76.
106. Lotti F, Jarrar AM, Pai RK, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med 2013;210:2851-72.
107. Hu Y, Yan C, Mu L, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One 2015;10:e0125625.
108. Hu YB, Yan C, Mu L, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019;38:1951-65.
109. Sansone P, Berishaj M, Rajasekhar VK, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res 2017;77:1927-41.
110. Valenti G, Quinn HM, Heynen GJJE, et al. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res 2017;77:2134-47.
111. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun 2018;9:2897.
112. Tang YA, Chen YF, Bao Y, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci USA 2018;115:E5990-9.
113. Su S, Chen J, Yao H, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018;172:841-856.e16.
114. Ren J, Ding L, Zhang D, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018;8:3932-48.
115. Du Y, Shao H, Moller M, Prokupets R, Tse YT, Liu ZJ. Intracellular notch1 signaling in cancer-associated fibroblasts dictates the plasticity and stemness of melanoma stem/initiating cells. Stem Cells 2019;37:865-75.
116. Phi LTH, Sari IN, Yang YG, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int 2018;2018:5416923.
117. Maeda M, Takeshima H, Iida N, et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut 2020;69:243-51.
118. Sung PJ, Rama N, Imbach J, et al. Cancer-associated fibroblasts produce netrin-1 to control cancer cell plasticity. Cancer Res 2019;79:3651-61.
119. Bu L, Baba H, Yasuda T, Uchihara T, Ishimoto T. Functional diversity of cancer-associated fibroblasts in modulating drug resistance. Cancer Sci 2020;111:3468-77.
120. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci 2010;15:166-79.
121. Zhao H, Yang L, Baddour J, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016;5:e10250.
122. Zhang W, Bouchard G, Yu A, et al. GFPT2-Expressing cancer-associated fibroblasts mediate metabolic reprogramming in human lung adenocarcinoma. Cancer Res 2018;78:3445-57.
123. Mishra R, Haldar S, Placencio V, et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J Clin Invest 2018;128:4472-84.
124. Suzuki A, Puri S, Leland P, et al. Subcellular compartmentalization of PKM2 identifies anti-PKM2 therapy response in vitro and in vivo mouse model of human non-small-cell lung cancer. PLoS One 2019;14:e0217131.
125. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the Reverse Warburg Effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol 2017;44:198-203.
126. Yu T, Yang G, Hou Y, et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017;36:2131-45.
127. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res 2015;34:111.
128. Apicella M, Giannoni E, Fiore S, et al. Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab 2018;28:848-865.e6.
129. Vukovic V, Tannock IF. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. Br J Cancer 1997;75:1167-72.
130. Kugeratski FG, Atkinson SJ, Neilson LJ, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal 2019:12.
131. Ziani L, Buart S, Chouaib S, Thiery J. Hypoxia increases melanoma-associated fibroblasts immunosuppressive potential and inhibitory effect on T cell-mediated cytotoxicity. Oncoimmunology 2021;10:1950953.
132. Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol 2008;295:G709-17.
133. Guido C, Whitaker-Menezes D, Capparelli C, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012;11:3019-35.
134. Bartrons R, Simon-Molas H, Rodríguez-García A, et al. Fructose 2, 6-bisphosphate in cancer cell metabolism. Front Oncol 2018;8:331.
135. Martinez-Outschoorn UE, Trimmer C, Lin Z, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 2010;9:3515-33.
136. Zhu Y, Lu L, Qiao C, et al. Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor. Oncogene 2018;37:2837-49.
137. Semenza GL. Signal transduction to hypoxia-inducible factor 1. Biochemical Pharmacology 2002;64:993-8.
138. Hu Y, Liu J, Huang H. Recent agents targeting HIF-1α for cancer therapy. J Cell Biochem 2013;114:498-509.
139. Pranzini E, Pardella E, Paoli P, Fendt SM, Taddei ML. Metabolic reprogramming in anticancer drug resistance: a focus on amino acids. Trends Cancer 2021;7:682-99.
140. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and furious: lipid metabolism in antitumoral therapy response and resistance. Trends Cancer 2021;7:198-213.
141. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021;11:8322-36.
142. Lee U, Cho EY, Jho EH. Regulation of Hippo signaling by metabolic pathways in cancer. Biochim Biophys Acta Mol Cell Res 2022;1869:119201.
143. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev 2016;99:186-96.
144. Navab R, Strumpf D, To C, et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016;35:1899-908.
145. Li X, Li Q, Yu X, Li H, Huang G. Reverse of microtubule-directed chemotherapeutic drugs resistance induced by cancer-associated fibroblasts in breast cancer. Onco Targets Ther 2019;12:7963-73.
146. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423-37.
147. Joyce MH, Lu C, James ER, et al. Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front Oncol 2018;8:337.
148. Hao J, Zeltz C, Pintilie M, et al. Characterization of distinct populations of carcinoma-associated fibroblasts from non-small cell lung carcinoma reveals a role for ST8SIA2 in cancer cell invasion. Neoplasia 2019;21:482-93.
149. Chen Y, Kim J, Yang S, et al. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021;39:548-565.e6.
150. Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiology 2015;82:142-52.
151. Gonzalez-Avila G, Sommer B, Mendoza-Posada DA, Ramos C, Garcia-Hernandez AA, Falfan-Valencia R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit Rev Oncol Hematol 2019;137:57-83.
152. Glentis A, Oertle P, Mariani P, et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun 2017;8:924.
153. Pontiggia O, Sampayo R, Raffo D, et al. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res Treat 2012;133:459-71.
154. Folgueira MA, Maistro S, Katayama ML, et al. Markers of breast cancer stromal fibroblasts in the primary tumour site associated with lymph node metastasis: a systematic review including our case series. Biosci Rep 2013:33.
155. Yoshida T, Ishii G, Goto K, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin Cancer Res 2015;21:642-51.
156. Ibrahim N', Wong SK, Mohamed IN, et al. Wound healing properties of selected natural products. Int J Environ Res Public Health 2018;15:2360.
157. Porwal A, Kundu GC, Bhagwat G, Butti R. Polyherbal formulation Anoac-H suppresses the expression of RANTES and VEGF for the management of bleeding hemorrhoids and fistula. Mol Med Rep 2021;24:736.
158. Santana FP, Pinheiro NM, Mernak MI, et al. Evidences of herbal medicine-derived natural products effects in inflammatory lung diseases. Mediators Inflamm 2016;2016:2348968.
159. Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 2013;1830:3670-95.
160. McLarty J, Bigelow RL, Smith M, Elmajian D, Ankem M, Cardelli JA. Tea polyphenols decrease serum levels of prostate-specific antigen, hepatocyte growth factor, and vascular endothelial growth factor in prostate cancer patients and inhibit production of hepatocyte growth factor and vascular endothelial growth factor in vitro. Cancer Prev Res 2009;2:673-82.
161. Gray AL, Stephens CA, Bigelow RL, Coleman DT, Cardelli JA. The polyphenols (-)-epigallocatechin-3-gallate and luteolin synergistically inhibit TGF-β-induced myofibroblast phenotypes through RhoA and ERK inhibition. PLoS One 2014;9:e109208.
162. Sun Y, Campisi J, Higano C, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 2012;18:1359-68.
163. Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin remodels the tumor microenvironment to improve the permeation, retention, and antitumor effects of nanoparticles. ACS Nano 2017;11:4916-25.
164. Wilken R, Veena MS, Wang MB, Srivatsan ES. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol Cancer 2011;10:12.
165. Hendrayani SF, Al-Khalaf HH, Aboussekhra A. Curcumin triggers p16-dependent senescence in active breast cancer-associated fibroblasts and suppresses their paracrine procarcinogenic effects. Neoplasia 2013;15:631-40.
166. Wang Q, Qu C, Xie F et al. Curcumin suppresses epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells by inhibiting cancer-associated fibroblasts. Am J Cancer Res 2017;7:125-33.
167. Xing Y, Mi C, Wang Z, et al. Fraxinellone has anticancer activity in vivo by inhibiting programmed cell death-ligand 1 expression by reducing hypoxia-inducible factor-1α and STAT3. Pharmacol Res 2018;135:166-80.
168. Wu X, Wu X, Ma Y, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun 2016;7:13498.
169. Pei Y, Chen L, Huang Y, et al. Sequential targeting TGF-β signaling and KRAS mutation increases therapeutic efficacy in pancreatic cancer. Small 2019;15:e1900631.
170. Abate M, Pagano C, Masullo M, et al. Mangostanin, a xanthone derived from garcinia mangostana fruit, exerts protective and reparative effects on oxidative damage in human keratinocytes. Pharmaceuticals 2022;15:84.
171. Zhang KJ, Gu QL, Yang K, Ming XJ, Wang JX. Anticarcinogenic effects of α-mangostin: a review. Planta Med 2017;83:188-202.
172. Feng J, Xu M, Wang J, et al. Sequential delivery of nanoformulated α-mangostin and triptolide overcomes permeation obstacles and improves therapeutic effects in pancreatic cancer. Biomaterials 2020;241:119907.
173. Chen JK. I only have eye for ewe: the discovery of cyclopamine and development of Hedgehog pathway-targeting drugs. Nat Prod Rep 2016;33:595-601.
174. Zhang B, Jiang T, Shen S, et al. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016;103:12-21.
175. Zhao J, Wu C, Abbruzzese J, Hwang RF, Li C. Cyclopamine-loaded core-cross-linked polymeric micelles enhance radiation response in pancreatic cancer and pancreatic stellate cells. Mol Pharm 2015;12:2093-100.
176. Kasala ER, Bodduluru LN, Madana RM, V AK, Gogoi R, Barua CC. Chemopreventive and therapeutic potential of chrysin in cancer: mechanistic perspectives. Toxicol Lett 2015;233:214-25.
177. Lin YM, Chen CI, Hsiang YP, et al. Chrysin Attenuates cell viability of human colorectal cancer cells through autophagy induction unlike 5-fluorouracil/oxaliplatin. Int J Mol Sci 2018;19:1763.
178. Chen L, Li Q, Jiang Z, et al. Chrysin induced cell apoptosis through H19/let-7a/COPB2 axis in gastric cancer cells and inhibited tumor growth. Front Oncol 2021;11:651644.
179. Wen Q, Xu C, Zhou J, et al. 8-bromo-7-methoxychrysin suppress stemness of SMMC-7721 cells induced by co-culture of liver cancer stem-like cells with hepatic stellate cells. BMC Cancer 2019;19:224.
180. Chen S, Nishi M, Morine Y, et al. Epigallocatechin-3-gallate hinders metabolic coupling to suppress colorectal cancer malignancy through targeting aerobic glycolysis in cancer associated fibroblasts. Int J Oncol 2022;60:19.
181. Yamanaka T, Harimoto N, Yokobori T, et al. Conophylline inhibits hepatocellular carcinoma by inhibiting activated cancer-associated fibroblasts through suppression of G protein-coupled receptor 68. Mol Cancer Ther 2021;20:1019-28.
182. Wang Z, Ma D, Wang C, et al. Triptonide inhibits the pathological functions of gastric cancer-associated fibroblasts. Biomed Pharmacother 2017;96:757-67.
183. Wang ZF, Ma DG, Wang L, et al. Paeoniflorin inhibits migration- and invasion-promoting capacities of gastric cancer associated fibroblasts. Chin J Integr Med 2019;25:837-44.
184. Thongchot S, Ferraresi A, Vidoni C, et al. Erratum to “resveratrol interrupts the pro-invasive communication between cancer associated fibroblasts and cholangiocarcinoma cells” [Cancer Letters 430C (2018) 160-171]. Cancer Lett 2018;434:206-7.
185. Dauer P, Zhao X, Gupta VK, et al. Inactivation of cancer-associated-fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res 2018;78:1321-33.
186. Zhao J, Wang H, Hsiao CH, et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018;159:215-28.
187. Dong R, Guo J, Zhang Z, Zhou Y, Hua Y. Polyphyllin I inhibits gastric cancer cell proliferation by downregulating the expression of fibroblast activation protein alpha (FAP) and hepatocyte growth factor (HGF) in cancer-associated fibroblasts. Biochem Biophys Res Commun 2018;497:1129-34.
188. Wang ZF, Ma DG, Zhu Z, et al. Astragaloside IV inhibits pathological functions of gastric cancer-associated fibroblasts. World J Gastroenterol 2017;23:8512-25.
189. Cao X, He Q. Ursolic acid inhibits proliferation, migration and invasion of human papillary thyroid carcinoma cells via CXCL12/CXCR4/CXCR7 axis through cancer-associated fibroblasts. Hum Exp Toxicol 2022;41:9603271221111333.
190. Guo J, Zeng H, Shi X, et al. A CFH peptide-decorated liposomal oxymatrine inactivates cancer-associated fibroblasts of hepatocellular carcinoma through epithelial-mesenchymal transition reversion. J Nanobiotechnology 2022;20:114.
191. Yamanaka T, Harimoto N, Yokobori T, et al. Nintedanib inhibits intrahepatic cholangiocarcinoma aggressiveness via suppression of cytokines extracted from activated cancer-associated fibroblasts. Br J Cancer 2020;122:986-94.
192. Lee HM, Lee E, Yeo SY, et al. Drug repurposing screening identifies bortezomib and panobinostat as drugs targeting cancer associated fibroblasts (CAFs) by synergistic induction of apoptosis. Invest New Drugs 2018;36:545-60.
193. ClinicalTrials.gov. A study to evaluate the clinical efficacy of JNJ-42756493 (Erdafitinib), a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in asian participants with advanced non-small-cell lung cancer, urothelial cancer, esophageal cancer or cholangiocarcinoma. Available from: https://ClinicalTrials.gov/show/NCT02699606 [Last accessed on 2 Feb 2023].
194. ClinicalTrials.gov. Plerixafor and cemiplimab in metastatic pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT04177810 [Last accessed on 2 Feb 2023].
195. ClinicalTrials.gov. Second-line study of PEGPH20 and pembro for HA high metastatic PDAC. Available from: https://ClinicalTrials.gov/show/NCT03634332 [Last accessed on 2 Feb 2023].
196. ClinicalTrials.gov. Phase I study of AT13148, a novel AGC kinase inhibitor. Available from: https://ClinicalTrials.gov/show/NCT01585701 [Last accessed on 2 Feb 2023].
197. ClinicalTrials.gov. FOLFIRINOX plus IPI-926 for advanced pancreatic adenocarcinoma. Available from: https://ClinicalTrials.gov/show/NCT01383538 [Last accessed on 2 Feb 2023].
198. Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174-86.
199. Murphy JE, Wo JY, Ryan DP, et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol 2019;5:1020-7.
200. Shimizu T, Fukuoka K, Takeda M, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 2016;77:997-1003.
201. ClinicalTrials.gov. Paricalcitol plus gemcitabine and nab-paclitaxel in metastatic pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT03520790 [Last accessed on 2 Feb 2023].
202. ClinicalTrials.gov. A study in metastatic cancer and advanced or metastatic unresectable pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT01373164 [Last accessed on 2 Feb 2023].
203. ClinicalTrials.gov. A study of galunisertib (LY2157299) and durvalumab (MEDI4736) in participants with metastatic pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT02734160 [Last accessed on 2 Feb 2023].
204. ClinicalTrials.gov. A study evaluating IPI-926 in combination with gemcitabine in patients with metastatic pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT01130142 [Last accessed on 2 Feb 2023].
205. ClinicalTrials.gov. Vismodegib and gemcitabine hydrochloride in treating patients with advanced pancreatic cancer. Available from: https://ClinicalTrials.gov/show/NCT01195415 [Last accessed on 2 Feb 2023].
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Butti R, Khaladkar A, Bhardwaj P, Prakasam G. Heterotypic signaling of cancer-associated fibroblasts in shaping the cancer cell drug resistance. Cancer Drug Resist 2023;6:182-204. http://dx.doi.org/10.20517/cdr.2022.72
AMA Style
Butti R, Khaladkar A, Bhardwaj P, Prakasam G. Heterotypic signaling of cancer-associated fibroblasts in shaping the cancer cell drug resistance. Cancer Drug Resistance. 2023; 6(1): 182-204. http://dx.doi.org/10.20517/cdr.2022.72
Chicago/Turabian Style
Butti, Ramesh, Ashwini Khaladkar, Priya Bhardwaj, Gopinath Prakasam. 2023. "Heterotypic signaling of cancer-associated fibroblasts in shaping the cancer cell drug resistance" Cancer Drug Resistance. 6, no.1: 182-204. http://dx.doi.org/10.20517/cdr.2022.72
ACS Style
Butti, R.; Khaladkar A.; Bhardwaj P.; Prakasam G. Heterotypic signaling of cancer-associated fibroblasts in shaping the cancer cell drug resistance. Cancer Drug Resist. 2023, 6, 182-204. http://dx.doi.org/10.20517/cdr.2022.72
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 31
likes
Like This Article 31
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.