
Targeting regulated cell death pathways in acute myeloid leukemia

 , ...
, ... Abstract
The use of the BCL2 inhibitor venetoclax has transformed the management of patients with acute myeloid leukemia (AML) who are ineligible for intensive chemotherapy. By triggering intrinsic apoptosis, the drug is an excellent illustration of how our greater understanding of molecular cell death pathways can be translated into the clinic. Nevertheless, most venetoclax-treated patients will relapse, suggesting the need to target additional regulated cell death pathways. To highlight advances in this strategy, we review the recognized regulated cell death pathways, including apoptosis, necroptosis, ferroptosis and autophagy. Next, we detail the therapeutic opportunities to trigger regulated cell death in AML. Finally, we describe the main drug discovery challenges for regulated cell death inducers and their translation into clinical trials. A better knowledge of the molecular pathways regulating cell death represents a promising strategy to develop new drugs to cure resistant or refractory AML patients, particularly those resistant to intrinsic apoptosis.
Keywords
INTRODUCTION
Regulated cell death (RCD) is a biologically controlled process that differs from accidental cell death (ACD) by its reliance on defined molecular signaling pathways and tight regulation. Its well-defined nature implies that it can be modulated by pharmacological or genetic interventions contrary to ACD[1-3]. Schweichel and Merker were the first to report the presence of three distinct cell death morphologies: type I (apoptosis), type II cell (cell death associated with autophagy) and type III (necrosis)[4]. While apoptosis is the most well-known RCD, many other pathways and molecular characteristics have subsequently been described[2]. A better understanding of the mechanisms driving RCD may lead to the discovery of new anticancer drugs or the repositioning of older drugs to treat aggressive cancers.
Acute myeloid leukemia (AML) is a severe hematological malignancy driven by various molecular alterations and mainly occurring in adults > 60 years old. Treatment modalities of newly diagnosed AML depend on age, general conditions, comorbidities, and molecular risk factors based on cytogenetics and the presence of molecular alterations. These mutations are integrated together with cytogenetic abnormalities in the widely used and recently updated ELN 2022 classifications[5,6]. More than 60% of younger patients will be cured by an intensive cytotoxic therapy (ICT) induction based on the association of anthracyclin and cytarabine (7 + 3), followed by consolidations with high doses of cytarabine and/or allogeneic stem cell transplantation[7]. Half of the patients > 65 years cannot receive ICT because of age, poor general status, or comorbidities. In this context, the hypomethylating agents (HMA) decitabine and azacytidine (AZA) have been associated with complete response rates of 20-30% and 10-month survival[8,9], highlighting the need for more effective treatments for older and not adequately fit patients.
BCL-2 inhibition is an excellent illustration of how the molecular understanding of RCD has led to the rapid development of a drug that has transformed the therapeutic treatment landscape for older AML patients. Venetoclax (VEN) in combination with the hypomethylating agent AZA has become part of standard frontline therapy for patients not eligible for ICT by improving the rates of response and overall survival compared with AZA monotherapy[10,11]. However, 10% to 50% of newly diagnosed patients with AML will not respond to VEN-AZA. In addition, half of the patients have relapsed by 18 months and no plateau is seen on overall survival curves. In the population of VEN-AZA refractory or relapsed patients, response rate and overall survival are poor (20% and 2.4 months, respectively)[12,13]. The combination of VEN with ICT is also associated with high response, but 3% to 15% of patients do not respond to the treatment[14]. Altogether, these data show that targeting RCD is a valuable strategy and has already improved the efficacy of the current AML therapeutic strategies. However, there is a real need to develop new drugs to go beyond BCL2 inhibition in AML[13]. In this review, we will discuss the main RCD pathways, describe their therapeutic targeting and discuss the main challenges for translating preclinical results into the clinic.
REGULATED CELL DEATH PATHWAYS
Intrinsic apoptotic pathway
Proapoptotic and antiapoptotic members of the BCL2 protein family share one to four BCL2 homology (BH) domains and control intrinsic apoptosis[15,16]. Under physiological conditions, BCL2-associated X (BAX) resides in the cytosol in an inactive conformation while BCL2 antagonist/killer 1 (BAK) is inserted at the outer mitochondrial membrane (OMM) via an α9 helix that connects with voltage-dependent anion channel 2 (VDAC2)[17]. In response to apoptotic stimuli, BAX and BAK associate to form pores in the OMM, inducing mitochondrial outer membrane permeabilization (MOMP, Figure 1).
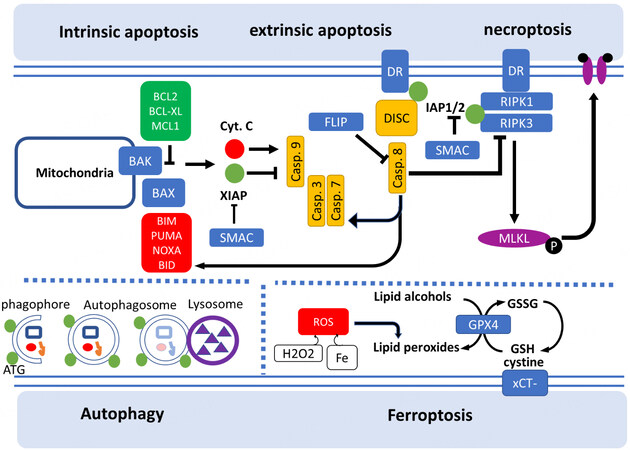
Figure 1. Mechanisms of regulated cell death pathways.
MOMP is regulated by the members of the BCL2 protein family containing a single BH3 domain named BH3-only proteins. The main representatives of this class are p53-upregulated modulator of apoptosis (PUMA), BCL2-like 11 (BCL2L11), also called BCL2-interacting mediator of cell death (BIM), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also called NOXA) and BH3 interacting domain death agonist (BID)[18]. All these proteins have a proapoptotic role by directly interacting with BAX and/or BAK to induce MOMP. Conversely, MOMP is blocked by a series of proteins that have an antiapoptotic role. This includes BCL2, BCL2-like 1 (BCL2L1, also known as BCL-XL), MCL1, BCL2 like 2 (BCL2L2, also known as BCL-W), and BCL2 related protein A1 (BCL2A1, also known as BFL-1)[19]. Other BH3-only proteins, including BCL2-associated agonist of cell death (BAD), BCL2 modifying factor (BMF), or harakiri, BCL2 interacting protein (HRK), have the ability to induce MOMP in the absence of direct interaction with BAX or BAK by limiting the ability of the antiapoptotic BCL2 family members to sequester BAX BAK, or BH3-only activators[20].
MOMP promotes the cytosolic release of cytochrome c (Cyt c) and diablo IAP-binding mitochondrial protein (DIABLO), also known as second mitochondrial activator of caspases, SMAC[16]. The cytosolic pool of Cyt c binds to apoptotic peptidase activating factor 1 (APAF1) and pro-caspase 9 to form the multiprotein complex called the “apoptosome” responsible for caspase 9 activation. Activated caspase 9 catalyzes the proteolytic activation of the executioner caspase 3 and caspase 7, inducing apoptotic cell death. Blocking post-mitochondrial caspase activation with Z-VAD-fmk and Q-VD-OPh caspase inhibitors delays but does not completely rescue apoptosis in vitro and in vivo as it induces a switch to other types of RCD[2].
Extrinsic apoptosis
The extrinsic apoptosis pathway is initiated by cell membrane proteins known as death receptors (DR). Proapoptotic death receptors include FAS, also known as APO1 and CD95, the tumor necrosis factor (TNF) receptors TNFR1 and TNFR2 and the TNF-related apoptosis-inducing ligand (TRAIL) receptors DR4 and DR5. DR ligation allows for the assembly of the “death-inducing signaling complex” (DISC), a multiprotein complex that activates caspase 8. At this point, the execution of extrinsic apoptosis driven by DR follows two distinct pathways. In most cancer cells, caspase 8-mediated cleavage and activation of BID converges with intrinsic apoptosis by triggering a BAX/BAK-dependent MOMP[21]. However, in some cells, (for instance, mature lymphocytes), the caspase 8-dependent activation of caspase 3 and caspase 7 is sufficient to promote cell death independently of mitochondria and BCL2-family of proteins[22]. In DR-induced apoptotic intrinsic pathway, caspases are the main executioner of cell death, causing rapid proteolysis, DNA fragmentation and chromatin condensation, which are the hallmarks of apoptosis.
Intrinsic and extrinsic apoptosis are both regulated by the class of inhibitors of apoptosis proteins (IAPs). Among the 8 IAP family members described in humans, the best known is XIAP which inhibits caspases 3, 7, and 9[23]. IAP proteins are antagonized by SMAC/DIABLO, which is released by mitochondria during MOMP[24].
Necroptosis
Tumor necrosis factor alpha (TNF-α) is a potent trigger for apoptosis, but it was observed that cells treated with TNF-α and the caspase inhibitor zVAD-fmk were still dying without caspase activation, suggesting the presence of an alternative RCD pathway[25]. In 2005, Degterev et al. coined the term necroptosis by demonstrating that this unique RCD distinct from necrosis and apoptosis[26] is initiation by the receptor-interacting protein RIPK1 and could be inhibited by the pharmacological agent necrostatin-1. Indeed, necroptosis is initiated by the activation of the cell death receptors such as TNFR1 and RIPK1 (if caspase 8 is inactive) and depends on the subsequent activation of RIPK3 and the protein complex termed mixed lineage kinase domain-like pseudokinase (MLKL), resulting in the formation of pore on the cell membrane followed by cell death. Nevertheless, several of the upstream signaling elements of extrinsic apoptosis and necroptosis are shared, and sensitivity to each death pathway is regulated by the same set of regulatory molecules, including FLIP, the cellular inhibitors of apoptosis proteins cIAP1 and SMAC/DIABLO[27].
Ferroptosis
The term ferroptosis was coined in 2012 to describe a cell death that can be triggered by inactivation of the cystine/glutamate antiporter (also known as SLC7A11), leading to depletion of intracellular glutathione (GSH) or direct inhibition of glutathione peroxidase 4 (GPX4). GPX4 inhibition induces an accumulation of reactive oxygen species (ROS), ultimately leading to lipid peroxidation via the iron-dependent Fenton reaction, where H2O2 and iron react to generate hydroxyl radicals[28,29]. Inactivation of GPX4 through depletion of GSH with Erastin or with the direct GPX4 inhibitor RSL3 ultimately results in overwhelming lipid peroxidation that can be rescued by the use of antioxidant ferrostatin-1 or liprostatin-1 that block lipid peroxidation[30,31]. Ferroptosis is regulated at several levels, including amino acids, lipids (particularly polyunsaturated fatty acids (PUFA)) and iron metabolism[32-36]. In addition, ferroptosis is regulated by the suppressor protein 1 (FSP1) and dihydroorotate dehydrogenase (DHODH) that reduce ubiquinone (CoQ) to ubiquinol (CoQH2) on the plasma membrane and inner mitochondrial membrane, respectively. CoQH2 acts as a hyperoxide radical-trapping antioxidant and decreases lipid peroxidation, resulting in ferroptosis suppression[37,38]. Finally, GTP cyclohydrolase-1 (GCH1) and its metabolic derivatives tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) are also potent antioxidants protecting against ferroptosis[39,40]. Ferroptosis is also regulated by the non-canonical activities of TP53 on cellular metabolism in a context-specific manner[41]. TP53 enhances ferroptosis by inhibiting SLC7A11 expression or increasing expression of spermidine/spermine N1-acetyltransferase 1 (SAT1) and glutaminase 2 (GLS2). Likewise, TP53 can inhibit ferroptosis by reducing dipeptidyl peptidase 4 (DPP4) activity or inducing cyclin-dependent kinase inhibitor 1 A (CDKN1A/p21) expression. Unlike the other RCDs, it remains to be determined whether ferroptosis is a consequence of physiological processes or necessary for development as opposed to only pathophysiological situations[42].
Autophagy
Macroautophagy (hereafter autophagy) is a ubiquitous catabolic process that involves the degradation of cytoplasmic components, including intracellular organelles, via the lysosomal pathway. The first step of autophagy is the formation of phagophores, followed by the generation of double-membrane autophagosomes regulated by a series of well-conserved autophagy-regulated genes (ATG). Late steps involved the fusion of autophagosomes to lysosomes, leading to cargo degradation by lysosomal enzymes and the recycling of intracellular contents providing fuels for cell growth[43]. The autophagic response provides cytoprotective effects, as indicated by the fact that blocking autophagy with pharmacological or genetic interventions generally induces cell death through the accumulation of toxic proteins, damaged organelles or undigested autophagosomes toxic for tumor cells[43]. Autophagy is often activated alongside other RCD, such as ferroptosis, which can be promoted by the autophagic degradation of ferritin, extrinsic apoptosis or necroptosis[2]. The main features of RCD are summarized in Table 1.
Summary of the main regulated cell death mechanisms
| Type of RCD | Trigger | Facilitator | Inhibitor | Executioner |
| Intrinsic apoptosis | Mitochondrial outer membrane permeabilization by BAX/BAK macropores and cytochrome c release | Proapoptotic factors from the BCL2 family | Antiapoptotic factors from the BCL2 family, Inhibitors of apoptosis (IAPs) | Proteolysis by executioner caspases |
| Extrinsic apoptosis | Activation of membrane cell death receptors | Death-inducing signaling complex (DISC). SMAC/DIABLO | Inhibitor of apoptosis (IAPs), FLIP | Proteolysis by executioner caspases |
| Necroptosis | Activation of membrane cell death receptors | RIPK1, RIPK3, SMAC/DIABLO | Caspase 8 | Plasma membrane disruption by MLKL |
| Ferroptosis | Inhibition of GPX4, decrease of cystine uptake | hydroxyl radicals generation through Fenton reaction | Glutathione (GSH), antioxidant defense | Lipid peroxidation of plasma and intracellular membranes |
| Autophagy | Phagophore formation and fusion to the lysosome | Autophagy-related proteins (ATG) | Inhibition of lysosome acidification | Accumulation of toxic proteins or organelles leading to cellular stress |
Other cell death pathways
Pyroptosis is an inflammatory form of lytic RCD that frequently occurs in response to microbial infection by forming a multiprotein complex termed the inflammasome, which activates caspase 1 and forms Gasdermin D (GSDMD) dependent plasma-membrane pores[44]. Pyroptosis is mechanistically distinct from apoptosis and characterized by the absence of DNA fragmentation and the presence of nuclear condensation coupled with cell swelling. Large bubbles eventually form at the plasma membrane and rupture to expel cellular contents.
Mitotic catastrophe is a physiological mechanism by which the cells avoid aneuploidy and hence decrease tumorigenic potential. Accordingly, induction of mitotic catastrophe both precipitates oncogenesis and constitutes a therapeutic endpoint in cancer cells[45].
Inter-regulation and hierarchy between cell death pathways is complex and not completely understood. For instance, mitotic catastrophe is closely related to apoptosis by their common induction by cellular stress and DNA damage. For this reason, some authors suggest that mitotic catastrophe is not a distinct mechanism of death, but one that can occur through necrosis or apoptosis depending on the molecular profile of the cells[46]. Other examples of cross-talk between RCD include the link between ferroptosis and autophagy, in particular through ferritin degradation or the involvement of lysosomes in iron storage and redistribution[47,48]. Depletion of the ferroptosis trigger GPX4 also sensitizes cells to pyroptosis[49], necroptosis[50] and apoptosis[51]. Apoptosis and necroptosis are also linked through caspase 8 cleavage
RCD INDUCERS IN AML
Apoptotic inducers
Agents targeting intrinsic pathway
Most chemotherapies and radiotherapies are known to induce apoptosis of cancer cells in response to DNA damage or cellular stress[55]. As a consequence, in vitro and in vivo evidence indicates that TP53-mutated cells have impaired apoptosis signaling pathways, and these cells are typically less susceptible to cytotoxic chemotherapy[56].
Inhibitors of BCL2 family members
Besides VEN, several newly developed BCL-2 inhibitors are currently in various stages of investigation in AML and other leukemia models [Table 2][13,57-60]. In addition to BCL-2, other members of the BCL-2 family of antiapoptotic proteins (MCL-1, BCL-XL, BFL-1) are the target of small molecules with the goal of inducing BAK/BAX activation and promote intrinsic apoptosis. The antiapoptotic protein MCL-1 plays a critical role in inhibiting BAX/BAK activation. MCL1 dependency on leukemia blasts is associated with resistance to BCL2 inhibition by VEN. Several highly potent direct MCL-1 inhibitors have recently entered preclinical and clinical development [Table 2][61-64]. The therapeutic window of these inhibitors is narrow because of the high expression of MCL1 in cardiac and hepatic tissues[65]. Due to these safety concerns, indirect MCL1 inhibitors are also under evaluation. CDK9 inhibitors are in various stages of evaluation in AML [Table 2][66-73]. Addition of alvocidib to ICT improved response rates but not survival in a recently published phase 2 clinical trial[67]. Novel CDK9 inhibitors are currently in early phase trials [Table 2][74-78]. BCL-XL inhibition by navitoclax has been shown to be active in preclinical AML models[79,80]. Toxicity for platelets limited its clinical development; nevertheless, navitoclax in combination with ICT or targeted therapy is still under evaluation in acute lymphoblastic leukemia[81] or myelofibrosis (TRANSFORM-1, NCT04472598). Navitoclax in combination with VEN and decitabine may be a valuable option for VEN-refractory AML patients (NCT NCT05222984). Finally, BFL1 (BCL2A1) inhibition may also be an interesting option since the recent discovery of specific inhibitors, but the drug has not been specifically tested in leukemia models[82,83].
Drugs targeting main regulated cell death mechanisms in acute myeloid leukemia
| Therapeutic class | Mechanism of action | Drug | Trademark | Phase of development | References |
| BH3-mimetics | BCL2 inhibition by small molecule | BGB-11417 | - | Phase 1 (NCT04771130) | [57] |
| S65487 | - | Phase 1 (NCT03755154, NCT04742101) | [58] | ||
| APG-2575 | Lisaftoclax | Phase 1 (NCT03537482) | [59] | ||
| LP-108 | - | Phase 1 (NCT04139434) | [60] | ||
| Direct MCL1 inhibition by small molecule | S63845 | - | Phase 1 (NCT02979366, NCT03672695, NCT04629443) | [61] | |
| AZD5991 | - | Phase 1 (NCT03218683) | [62] | ||
| AMG176 | Tapotoclax | Phase 1 (NCT02675452) | [63] | ||
| AMG397 | Murizatoclax | Phase 1 (NCT03465540) | [64] | ||
| CDK9 kinase inhibitor | Indirect MCL1 inhibition by small molecule | Flavopiridol, HMR-1275 | Alvocidib | Phase 1 (NCT00407966, NCT03298984), phase 2 (NCT01349972) | [66,69] |
| SCH-727965 | Dinaciclib | Preclinical | [70,71] | ||
| P 1446A 05 | Voruciclib | Phase 1 (NCT03547115) | [72,73] | ||
| AZD4573 | - | Phase 1 (NCT03263637) | [74] | ||
| CYC065 | Fadraciclib | Phase 1 (NCT04017546) | [75,77] | ||
| TG02-101 | - | Preclinical | [78] | ||
| BH3 mimetics | BCL-XL inhibition | ABT-263 | Navitoclax | Phase 1 (NCT05222984) | - |
| BH3 mimetics | BFL1 inhibition | - | - | Preclinical | [82,83] |
| BAX/BAK activator | Direct BAX activation | BTSA1 | - | Preclinical | [84] |
| WEHI-9625 | - | Preclinical | [87,88] | ||
| Mitochondrial iron depletion | AM5 | Ironomycin | Preclinical | [53] | |
| TRAIL agonist | Death Receptor 5 (DR5) antibody | IGM-8444 | - | Phase 1 (NCT04553692) | [89] |
| TRAIL receptor agonist fusion protein | ABBV-621 | Eftozanermin alfa | Phase 1 (NCT03082209) | [79,80] | |
| FLIP inhibition | Direct FLIP inhibition | - | - | Preclinical | [92,93] |
| XIAP inhibition | Antisense oligonucleotide | LY2181308 | Gataparsen | Phase 2 (NCT00620321) | [98] |
| AEG35156 | Phase 1 NCT00363974, phase 2 NCT01018069 | [99,100] | |||
| SMAC/DIABLO mimetics | TL32711 | Birinapant | Phase 1 NCT01828346 phase 2 NCT01486784, NCT02147873 | [102,103] | |
| ASTX660 | Tolinapant | Phase 1 NCT02503423 | [105] | ||
| LCL161 | Phase 2 (NCT02098161) | [107] | |||
| SMAC/DIABLO mimetics + caspase 8 inhibition | TL32711 | Birinapant | Preclinical | [108] | |
| SMAC/DIABLO mimetics | BV6 | Preclinical | [109] | ||
| epigenetic therapies | Endogenous retroelements reactivation | - | Epigenetic therapies | Preclinical | [116] |
| Class 1 FIN | System xc- cystine/glutamate antiporter inhibition | - | Sulfasalazine | FDA approved in another indication | [118] |
| Class 2 FIN | GPX4 inhibition | ML162 | Altretamine | FDA-approved in another indication | [122] |
| Class 3 FIN | GSH metabolism inhibition | APR-246 | - | Phase 2 (NCT03931291) | [124] |
| Class 4 FIN | CoQ oxidoreductase FSP1 inhibition | - | - | Preclinical | [37,38] |
| Autophagy inhibitors | Lysosomal acidification blockade | - | Hydroxychloroquine | FDA-approved in another indication | [138,139] |
| PIK3C3/Vps34 inhibition | SAR405 | - | Preclinical | [140,141] | |
| Autophagy inducers | mTOR inhibition | - | Sirolimus | FDA-approved in another indication, phase 1 (NCT01869114) | [143] |
| RAD001 | Everolimus | FDA-approved in another indication, phase 1/2 (NCT00819546, NCT02638428, NCT01869114) | [144] |
BAX/BAK activators
BAK and BAX are crucial agents in promoting MOMP through protein oligomerization across the OMM. Recent findings showed direct activation of BAX by BTSA1, a pharmacological BAX activator that binds BAX with high affinity and specificity to the N-terminal activation site and induces conformational changes to BAX, leading to BAX-mediated apoptosis[84]. The histone deacetylase SAHA and its derivatives also have an affinity for BAX and induce its activation [Table 2] but have not been validated in AML models[85]. Other preclinical studies suggested a mitochondrial membrane-mediated spontaneous model of BAX activation. In this model, MOM plays a big role in orchestrating the turnover between cytosolic and membrane-bound BAX, its interaction through the α9 helix and the formation of macropore into the membrane. It is likely that lipids such as cardiolipin play a crucial role in this model[86]. Voltage Dependent Anion Channels (VDACs) are a family of membrane proteins that allow passage of both negatively and positively charged ions, NADH, ATP/ADP and other metabolites across the MOM. In particular, VDAC2 plays a role both in recruiting BAK to the MOM and in inhibiting its activation[17]. WEHI-9625 is a novel small molecule inducing BAK-mediated apoptosis in mice but is completely inactive against human BAK[87,88]. We found that ironomycin sequestered iron into lysosomes and subsequently reduced mitochondrial iron load, promoting the recruitment and non-canonical activation of BAX/BAK in AML in vitro and in vivo models. Crispr Cas9 screens uncovered the key metabolic and mitochondrial factors regulating this modality of non-canonical RCD[53].
Agents targeting extrinsic pathway
TRAIL Agonism
Agonists of the TNF-related apoptosis-inducing ligand (TRAIL) receptors (DR4/5) have been tested in AML, but response rates are low[13]. Novel antibodies against TRAILR1 and TRAILR2 have shown promising preclinical data along with synergy with VEN and are currently tested in phase 1 [Table 2][89]. Eftozanermin alfa (ABBV-621), a second-generation TRAIL receptor agonist binding to the death-inducing DR4 and DR5 receptors, is currently being tested in solid tumors and hematological malignancies
FLIP inhibition
FLIP (Fas-associated death domain (FADD)-like IL-1β-converting enzyme-inhibitory protein) is a multifunctional protein that plays a role in regulating the death-inducing signaling complex (DISC) and caspase 8 activation. CDK9 inhibitors and bromodomain inhibitors such as JQ1 have been shown to effectively decrease FLIP expression, leading to enhanced sensitivity to TRAIL-induced cell death in cancer[92]. Second-generation FLIP inhibitors have shown preclinical activity in multiple cancer cell lines including AML, and have a high potential for synergy with other apoptosis-targeting agents [Table 2][93,94].
XIAP inhibition
IAPs act as antiapoptotic proteins by inhibiting caspases and are promising therapeutic targets in AML. Inhibition of XIAP has been shown to sensitize AML cells to chemotherapy or BCL2 inhibitors[95,96]. Inhibition of XIAP by antisense strategies or peptides that bind and inhibit the BIR3 domain of XIAP has been tested in phase 1 studies[97-99] but failed in phase 2[100]. The SMAC/DIABLO mimetics (SM) birinapant is one of the most clinically advanced SM and is currently being tested in clinical trials for the treatment of certain solid and hematological cancers[101]. Birinapant showed limited efficacy as a single agent[102]. In a phase 2 randomized, double-blind study, birinapant plus AZA was not superior to AZA alone in advanced myelodysplastic syndromes (MDS) or chronic myelomonocytic leukemias[103]. The drug is also being tested in combination with chemotherapeutic agents and immune checkpoint inhibitors. Preclinical data also indicate that combination of Birinapant plus the multidrug resistance receptor 1 (MDR1) may circumvent birinapant resistance in AML[104]. ASTX660/Tolinapant is a dual antagonist of XIAP and cIAP is currently under investigation in phase 1/2 studies in solid tumors and in combination with hypomethylating agents in AML[105,106]. LCL161, an oral SMAC mimetic, has been tested in patients with myelofibrosis and showed a 30% objective response rate in a recently published phase 2 trial [Table 2][107].
Necroptosis inducers
SMAC mimetics combined with caspase 8 inhibition have been shown to trigger necroptosis in AML preclinical models[108-110]. RIPK1 inhibition enhanced the therapeutic efficacy of the HDAC inhibitor chidamide in FLT3-ITD positive AML, both in vitro and in vivo[111,112] and increased the efficacy of differentiating agents[113]. Other SMAC mimetics in combination with cytarabine or HMA also showed interesting preclinical results[114]. Therapeutic opportunities for cancer cell death induction through endogenous retroelements (EREs) reactivation have been recently described[115]. EREs are transcriptionally silent within mammalian genomes due to epigenetic mechanisms. Anticancer therapies targeting the epigenetic machinery reinduces ERE expression, inducing antiviral responses associated with consistent increased phosphorylation of RIPK1/3 and MLKL kinases associated with features of necroptosis in treated tumor cells [Table 2][116].
Ferroptosis inducers
Ferroptosis inducers (FINs) belong to four classes[117]. Class I FINs include pharmacological agents that limit intracellular glutathione (GSH) through the inhibition of the system xc- cystine/glutamate antiporter, as has been shown in non-leukemic models[28]. The main molecules of this class, which are active in AML models, are erastin and sulfasalazine[118,119]. Class II FINs directly inhibit the detoxifying enzyme GPX4. The lead compound in this class is RSL3 which covalently binds to GPX4. The antitumoral effect of GPX4 disruption has been shown in various models, including AML[120,121]. The FDA-approved alkylating agent altretamine has been shown to directly inhibit GPX4[122]. Class III FINs target GSH synthesis or cysteine synthetase such as buthionine sulfoximine (BSO), an irreversible inhibitor of rate-limiting enzyme in GSH synthesis, and also cisplatin[123]. Focusing on AML, APR-246, a drug known to restore the wild-type conformation of mutant TP53 protein, was shown to actually target GSH metabolism[124,125]. Class IV FINs disrupt the balance of iron metabolism and cellular reactive oxygen species (ROS). Dihydroartemisinin (DHA) belongs to this class[126-128], as well as drugs interfering with the antioxidant system CoQ10[37,38] or drugs modulating PUFA metabolism as they are highly sensitive to lipid oxidation [Table 2][129,130].
Iron chelation and overload play a crucial role in MDS and AML. Therapeutic interventions that modulate iron content and balance within blast cells are at least partially inducing ferroptosis[131]. Our group described in detail the mechanism of action of ironomycin that specifically sequesters ferrous iron into the lysosomes, and induces lipid peroxidation and cell death in several models of cancer stem cells as well as in AML through mitochondrial metabolism disruption[53,54,132].
Agent regulating autophagy
Autophagy deregulation in AML is well documented, but its impact on leukemogenesis remains unclear[133,134]. For example, in AML harboring an FLT3 internal tandem mutation (ITD), mTORC1 activation downstream the RET receptor tyrosine kinase suppresses FLT3 protein autophagic degradation[135]. In contrast, blocking autophagy in FLT3-ITD AML can increase the survival of mice with FLT3-ITD-driven AML[136]. These ambiguous results indicate the complexity of therapeutic intervention based on autophagy in AML. Another layer of complexity lies in the fact that autophagy is a dynamic process. For instance, blocking autophagy cargo can be done at earlier phases (autophagosome biogenesis) or at the later steps (endosome-lysosome fusion).
Autophagy inhibitors
Autophagy inhibition can be achieved by blocking the LC3 interacting regions (LIR) that orchestrate diverse stages of autophagy. This strategy was shown to sensitize cytotoxicity to cytarabine[137]. Late-stage autophagy inhibitors hydroxychloroquine and/or bafilomycin A1 (BafA1) block lysosomal acidification and are active on leukemia blasts and also sensitize cells to chemotherapy[138]. A randomized phase 2 study was recently published testing the addition of hydroxychloroquine to Imatinib in chronic myeloid leukemia and found no significant differences between the two arms[139]. SAR405, a highly potent small-molecule inhibitor of the phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3)/Vps34, induces a blockage at the late endosome-lysosome step autophagy flux and shows interesting preclinical efficacy in FLT3-ITD
Autophagy inducers
The mTORC1/S6K1 pathway is critical for the regulation of autophagy in AML initiation and progression, as reviewed by Ghosh & Kapur[142]. Therefore, the most studied drugs are the mTORC1 inhibitor sirolimus and everolimus. Despite promising data in preclinical models, clinical studies are ambiguous. Sirolimus combined with the chemotherapy regimen showed a high response rate in patients with baseline mTOR activation[143]. In a phase 1b study from the GOELAMS, everolimus plus chemotherapy improved the clinical outcomes of patients with AML[144]. But mTOR inhibition plus conventional chemotherapy did not show a clinical benefit in two other studies[145,146]. Therefore, mTOR inhibitors still need to be evaluated in AML, and several clinical trials are ongoing (NCT00819546, NCT02638428, NCT01869114).
Pharmacological induction of other cell death pathways
Small-molecule inhibitors of the serine dipeptidases DPP8 and DPP9 (DPP8/9) have been shown to induce pyroptosis in human myeloid cells, including cell lines, primary AML samples and mice models of human leukemia[147]. Constitutive innate immune activation is a pathogenetic driver of ineffective hematopoiesis and MDS, in particular through the NLRP3 inflammasome[148,149] Recent studies found that the inflammasome can be induced by gene mutations involving mRNA splicing by induction of cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING)[150]. As a consequence, the use of immunotherapies targeting inflammatory responses is the subject of many preclinical and clinical studies[151]. Mitotic catastrophe is induced by pharmacological agents targeting the mitotic machinery. Cell cycle inhibitors are currently in development in various hematological diseases, including AML[152,153].
UPCOMING CHALLENGES FOR TARGETING RCD IN AML
The key objectives of translating preclinical results into the clinic are to identify drugs that will treat resistant cells by inducing RCD, detect the population of patients who will respond to a given RCD and design new RCD inducers with high efficacy and low toxicity.
Target resistances with RCD
Cells resistant to treatment, called “persister cancer cells”, are largely responsible for relapse[154]. These cells develop antiapoptotic mechanisms as well as altered metabolism rendering them more susceptible to alternative RCD, in particular autophagy or ferroptosis that are directly related to the metabolic state of the cells[155,156]. For instance, preclinical data showed the xc- inhibitory activity of salazopyrine and its efficacy against primary AML samples in ex vivo cultures and in patient-derived AML models[118]. These preliminary results led to the hypothesis that the drug could be repositioned in AML. A clinical trial testing the addition of salazopyrine to ICT in older AML patients, named SALMA (EUDRACT no: 2022-001269-11), will be enrolling soon. Another example is the resistance to VEN, which is mediated through various mechanisms, including BCL2 family protein expression and occupation (MCL1 dependency), cellular differentiation state (monocytic versus stem cell-like), cellular metabolic state or sensitivity to mitochondrial machinery disruption[11]. One of the major limitations that emerged from both in vitro and clinical studies with the BH3-mimetics is the low sensitivity of TP53 mutated blasts to this class[157-159]. Strategies of treatment that overcome TP53-dependent apoptosis can be used, such as ironomycin that directly activates BAX/BAK in a BCL2-family protein-independent manner[53]. Optimized clinical-grade derivatives of ironomycin with better therapeutic windows are currently under development[160].
Identify new biomarkers
The second challenge is to identify the population of patients who will benefit from RCD inducers. Recent findings showed that ferroptosis-associated gene signatures can be assessed by transcriptomic methods such as RNA sequencing. These signatures predict survival and could possibly guide therapeutics by selecting patients who could be treated by the use of ferroptosis inducers[161-164]. Necroptosis transcriptomic signatures have also been found in MDS, indicating that such predictors could identify patients who would benefit from necroptosis inducers[165]. Functional assays such as BH3-profiling have been shown to be a highly efficient companion test that can predict response to the BH3-mimetics inhibiting BCL2 family proteins[20,166]. Our team published a translational proof-of-concept study in which relapsed or refractory AML patients were selected according to molecular and ex vivo drug sensitivity profiles[167]. Future clinical trials using companion tests based on molecular or functional approaches will confirm the feasibility and efficacy of this strategy.
Discovery of new RCD inducers
The use of targeted therapies has transformed AML treatment. Small molecule inhibitors of genes that underwent common somatic mutations, such as FLT3 or IDH inhibitors, have been approved recently for AML relapsed or refractory patients[168]. However, most of the patients cannot benefit from targeted therapies, because of a lack of targetable genetic alterations. On the contrary, BCL2 inhibition success story is based on the AML blast dependency to apoptosis independently of AML targetable mutations. Recent findings showed that AML cells are sensitive to ferroptosis induction, suggesting that efforts should be made to develop new ferroptosis inducers[169]. A validated approach is to use an unbiased strategy by performing high throughput screening of chemical libraries for their ability to induce ferroptosis (or other RCD) in vitro and in vivo[122]. In parallel, a better understanding of the ferroptotic molecular pathways can be obtained using CRISPR Cas-9 resistance screens of a given ferroptosis inducer that will identify crucial genes involved in cell death. We recently used this strategy to uncover the molecular mechanism of ironomycin[53]. This integrative strategy will help to design new tailored drugs and pave the way for future clinical trials based on RCD.
CONCLUSION
Treatment of AML has remained unchanged for more than 40 years with a one-size-fits-all intensive chemotherapy approach for eligible patients. In 2017, the FDA approved the utilization of two targeted therapies for patients with a molecular alteration in FLT3 or IDH genes[168]. Another significant advancement was made in 2020 with the publication of the VIALE-A study reporting the efficacy of VEN for the treatment of AML patients who are not eligible for intensive treatment[10]. Despite higher response and survival rates than before, there is still improvement to be made in the management of AML patients. A better understanding of molecular RCD pathways in collaboration with integrative translational studies will allow the design of new drugs or the repositioning of older ones to overcome resistance in AML.
Intrinsic apoptosis is initiated by the formation of pores permeabilizing the outer membrane of mitochondria (MOMP) induced by the oligomerization of BAX recruited from the cytosol and BAK inserted at the mitochondrial membrane. BAX and BAK interact with a series of antiapoptotic inhibitors proteins (BCL2, BCLXL, MCL1) and proapoptotic activator proteins (BIM, PUMA, NOXA, BID). MOMP induces a release of cytochrome c that cleaves caspase 9 and subsequently activates the effector caspases 3 and 7, leading to cell death. Extrinsic apoptosis is triggered by the ligation of cell death receptors (FAS, TRAIL and TNFR), which allow the assembly of the “death-inducing signaling complex” (DISC) and the subsequent activation of caspase 8. Caspase 8 induces the cleavage of BID, leading to the activation of BAX and intrinsic apoptosis and the direct activation of caspases 3 and 7, finally leading to cell death. Inhibitors of apoptosis (IAP) family, including XIAP, cIAP1 and cIAP2, are inhibitors of caspase 9 and the death receptor TNFR, whereas FLIP is the inhibitor of caspase 8. SMAC/DIABLO is a major inhibitor of IAPs. Necroptosis is induced by the ligation of cell death receptors and the activation of the receptor-interacting protein RIPK1 and RIPK3 that interacts with protein complex mixed lineage kinase domain-like pseudokinase (MLKL). Phosphorylation of MLKL results in the formation of pores on the cell membrane, followed by cell death. IAPs and Caspase 8 inhibit RIPK1 activation and block necroptosis. Ferroptosis is the result of lipid peroxidation triggered by the direct inhibition of the antioxidant enzyme GPX4 or by the blockage of the xc- cystine/glutamate antiporter (xCT-). Depletion of cysteine import and intracellular glutathione (GSH) increases lipid peroxides which is exacerbated by the Fenton reaction, where H2O2 and iron react to generate hydroxyl radicals. Autophagy is a process in which cytoplasmic components, including intracellular organelles, are degraded by the lysosomes. The first step of autophagy is the formation of phagophores, followed by the generation of double-membrane autophagosomes regulated by autophagy-regulated genes (ATG) and lysosomal fusion. Autophagy blockage or excess can induce cell death.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and design of the review: Garciaz S, Miller T, Collette Y, Vey N
Availability of data and materialsNot applicable.
Financial support and sponsorshipTM. work was supported by the National Cancer Institute (R37CA218259; TWM).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Armenta DA, Dixon SJ. Investigating nonapoptotic cell death using chemical biology approaches. Cell Chem Biol 2020;27:376-86.
2. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25:486-541.
3. Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology 1973;7:253-66.
4. Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135-47.
5. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424-47.
6. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022;140:1345-77.
8. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with > 30% blasts. Blood 2015;126:291-9.
9. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670-7.
10. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020;383:617-29.
11. Garciaz S, Saillard C, Hicheri Y, Hospital MA, Vey N. Venetoclax in acute myeloid leukemia: molecular basis, evidences for preclinical and clinical efficacy and strategies to target resistance. Cancers 2021;13:5608.
12. Maiti A, Rausch CR, Cortes JE, et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica 2021;106:894-8.
13. Maiti A, Carter BZ, Andreeff M, Konopleva MY. Beyond BCL-2 inhibition in acute myloid leukemia: other approaches to leverage the apoptotic pathway. Clin Lymphoma Myeloma Leuk 2022;22:652-8.
14. Lachowiez CA, Atluri H, DiNardo CD. Advancing the standard: venetoclax combined with intensive induction and consolidation therapy for acute myeloid leukemia. Ther Adv Hematol 2022;13:20406207221093964.
15. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014;15:49-63.
16. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010;11:621-32.
17. Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003;301:513-7.
18. Chen HC, Kanai M, Inoue-Yamauchi A, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 2015;17:1270-81.
19. Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 2013;5:a008722-a008722.
20. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002;2:183-92.
21. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol 2020;17:395-417.
22. Meier P, Vousden KH. Lucifer’s labyrinth--ten years of path finding in cell death. Mol Cell 2007;28:746-54.
23. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997;388:300-4.
24. Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther 2014;144:82-95.
25. Vercammen D, Beyaert R, Denecker G, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998;187:1477-85.
26. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112-9.
28. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060-72.
29. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med 2019;133:130-43.
30. Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014;136:4551-6.
31. Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 2017;3:232-43.
32. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273-85.
33. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015;59:298-308.
34. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 2016;113:E4966-75.
35. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91-8.
36. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81-90.
37. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688-92.
38. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693-8.
39. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 2020;6:41-53.
40. Soula M, Weber RA, Zilka O, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 2020;16:1351-60.
41. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med 2019;133:162-8.
42. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res 2019;29:347-64.
43. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov 2019;9:1167-81.
44. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009;7:99-109.
45. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011;12:385-92.
46. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008;15:1153-62.
47. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res 2016;26:1021-32.
48. Zhou Y, Shen Y, Chen C, et al. The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer? Cancer Biol Med 2019;16:630-46.
49. Lee BL, Stowe IB, Gupta A, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med 2018;215:2279-88.
50. Canli Ö, Alankuş YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016;127:139-48.
51. Seiler A, Schneider M, Förster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008;8:237-48.
52. Hughes SA, Lin M, Weir A, et al. Caspase-8-driven apoptotic and pyroptotic crosstalk causes cell death and IL-1β release in X-linked inhibitor of apoptosis (XIAP) deficiency. EMBO J 2023;42:e110468.
53. Garciaz S, Guirguis AA, Müller S, et al. Pharmacologic reduction of mitochondrial iron triggers a noncanonical BAX/BAK-dependent cell death. Cancer Discov 2022;12:774-91.
54. Mai TT, Hamaï A, Hienzsch A, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem 2017;9:1025-33.
55. Saxena K, DiNardo C, Daver N, Konopleva M. Harnessing apoptosis in AML. Clin Lymphoma Myeloma Leuk 2020;20 Suppl 1:S61-4.
56. Lowe SW, Bodis S, McClatchey A, et al. P53 status and the efficacy of cancer therapy in vivo. Science 1994;266:807-10.
57. Hu N, Guo Y, Xue H, et al. Abstract 3077: Preclinical characterization of BGB-11417, a potent and selective Bcl-2 inhibitor with superior antitumor activities in haematological tumor models. Cancer Res 2020;80:3077-3077.
58. Tiran AL, Claperon A, Davidson J, et al. Abstract 1276: identification of S65487/VOB560 as a potent and selective intravenous 2nd-generation BCL-2 inhibitor active in wild-type and clinical mutants resistant to Venetoclax. Cancer Res 2021;81:1276.
59. Ailawadhi S, Chanan-khan AAA, Chen Z, et al. First-in-human study of lisaftoclax (APG-2575), a novel BCL-2 inhibitor (BCL-2i), in patients (pts) with relapsed/refractory (R/R) CLL and other hematologic malignancies (HMs). J Clin Oncol 2021;39:7502.
60. Walker AR, Bergua Burgues JM, Montesinos P, et al. Phase 1 study of LP-108 as monotherapy and in combination with azacitidine in patients with relapsed or refractory myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML), or acute myeloid leukemia (AML). J Clin Oncol 2022;40:TPS7071-TPS7071.
61. Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538:477-82.
62. Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 2018;9:5341.
63. Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov 2018;8:1582-97.
64. Caenepeel S, Karen R, Belmontes B, et al. Abstract 6218: discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res 2020;80:6218-6218.
65. Wei AH, Roberts AW, Spencer A, et al. Targeting MCL-1 in hematologic malignancies: rationale and progress. Blood Rev 2020;44:100672.
66. Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res 2015;39:1312-8.
67. Zeidner JF, Foster MC, Blackford AL, et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7 + 3) in newly diagnosed acute myeloid leukemia. Haematologica 2015;100:1172-9.
68. Bogenberger J, Whatcott C, Hansen N, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017;8:107206-22.
69. Zeidner JF, Lee DJ, Frattini M, et al. Phase I study of alvocidib followed by 7 + 3 (cytarabine + daunorubicin) in newly diagnosed acute myeloid leukemia. Clin Cancer Res 2021;27:60-9.
70. Tibes R, Bogenberger JM. Transcriptional silencing of MCL-1 through cyclin-dependent kinase inhibition in acute myeloid leukemia. Front Oncol 2019;9:1205.
71. Baker A, Gregory GP, Verbrugge I, et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res 2016;76:1158-69.
72. Konopleva M, Brander DM, Patel K, et al. A phase 1 dose-escalation study of the oral CDK inhibitor voruciclib in patients with relapsed/refractory B-cell malignancies or acute myeloid leukemia (AML): preliminary results of the completed dose escalation stage in AML. Blood 2021;138:3423-3423.
73. Dey J, Deckwerth TL, Kerwin WS, et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci Rep 2017;7:18007.
74. Cidado J, Boiko S, Proia T, et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res 2020;26:922-34.
75. Frame S, Saladino C, MacKay C, et al. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS One 2020;15:e0234103.
76. Chantkran W, Hsieh YC, Zheleva D, Frame S, Wheadon H, Copland M. Interrogation of novel CDK2/9 inhibitor fadraciclib (CYC065) as a potential therapeutic approach for AML. Cell Death Discov 2021;7:137.
77. Borthakur GM, Kadia TM, Al Azzawi H, Zheleva D, Blake D, Chiao JH. Combining CDK2/9 inhibitor CYC065 with venetoclax, a BCL2 inhibitor, to treat patients with relapsed or refractory AML or MDS. Blood 2019;134:1379-1379.
78. Goh KC, Novotny-diermayr V, Hart S, et al. Abstract 2542: TG02, a novel multi-kinase inhibitor with potent anti-leukemic activity. Cancer Res 2010;70:2542-2542.
79. Kivioja JL, Thanasopoulou A, Kumar A, et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1+/FLT3-ITD+ acute myeloid leukemia. Leukemia 2019;33:1360-72.
80. Kontro M, Kumar A, Majumder MM, et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2017;31:301-9.
81. Pullarkat VA, Lacayo NJ, Jabbour E, et al. Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer Discov 2021;11:1440-53.
82. Harvey EP, Hauseman ZJ, Cohen DT, et al. Identification of a covalent molecular inhibitor of anti-apoptotic BFL-1 by disulfide tethering. Cell Chem Biol 2020;27:647-56.e6.
83. Liu N, Wang D, Lian C, et al. Selective covalent targeting of anti-apoptotic BFL-1 by a sulfonium-tethered peptide. Chembiochem 2021;22:340-4.
84. Reyna DE, Garner TP, Lopez A, et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 2017;32:490-505.e10.
85. Liang T, Zhou Y, Elhassan RM, Hou X, Yang X, Fang H. HDAC-Bax multiple ligands enhance Bax-dependent apoptosis in HeLa cells. J Med Chem 2020;63:12083-99.
86. Luo X, O'Neill KL, Huang K. The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved? F1000Res 2020;9:935.
87. van Delft MF, Chappaz S, Khakham Y, et al. A small molecule interacts with VDAC2 to block mouse BAK-driven apoptosis. Nat Chem Biol 2019;15:1057-66.
88. Yuan Z, Dewson G, Czabotar PE, Birkinshaw RW. VDAC2 and the BCL-2 family of proteins. Biochem Soc Trans 2021;49:2787-95.
89. Wang BT, Kothambawala T, Wang L, et al. Multimeric anti-DR5 IgM agonist antibody IGM-8444 is a potent inducer of cancer cell apoptosis and synergizes with chemotherapy and BCL-2 inhibitor ABT-199. Mol Cancer Ther 2021;20:2483-94.
90. Phillips DC, Buchanan FG, Cheng D, et al. Hexavalent TRAIL fusion protein eftozanermin alfa optimally clusters apoptosis-inducing TRAIL receptors to induce on-target antitumor activity in solid tumors. Cancer Res 2021;81:3402-14.
91. de Jonge MJ, Carneiro BA, Devriese L, et al. First-in-human study of abbv-621, a TRAIL receptor agonist fusion protein, in patients (Pts) with relapsed/refractory (RR) acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL). Blood 2019;134:3924.
92. Humphreys L, Espona-Fiedler M, Longley DB. FLIP as a therapeutic target in cancer. FEBS J 2018;285:4104-23.
93. Longley DB, Higgins C, Fox J, et al. Abstract 5220: development of first-in-class small molecule inhibitors of FLIP which activate caspase-8, the nodal regulator of apoptosis, necroptosis and pyroptosis. Cancer Res 2020;80:5220.
94. Higgins CA, Fox J, Roberts J, et al. Abstract 1342: development and preclinical evaluation of unique first-in-class small molecule inhibitors of the anti-apoptotic protein FLIP. Cancer Res 2021;81:1342.
95. Hashimoto M, Saito Y, Nakagawa R, et al. Combined inhibition of XIAP and BCL2 drives maximal therapeutic efficacy in genetically diverse aggressive acute myeloid leukemia. Nat Cancer 2021;2:340-56.
96. Zhou J, Lu X, Tan TZ, Chng WJ. X-linked inhibitor of apoptosis inhibition sensitizes acute myeloid leukemia cell response to TRAIL and chemotherapy through potentiated induction of proapoptotic machinery. Mol Oncol 2018;12:33-47.
97. Carter BZ, Gronda M, Wang Z, et al. Small-molecule XIAP inhibitors derepress downstream effector caspases and induce apoptosis of acute myeloid leukemia cells. Blood 2005;105:4043-50.
98. Erba HP, Sayar H, Juckett M, et al. Safety and pharmacokinetics of the antisense oligonucleotide (ASO) LY2181308 as a single-agent or in combination with idarubicin and cytarabine in patients with refractory or relapsed acute myeloid leukemia (AML). Invest New Drugs 2013;31:1023-34.
99. Schimmer AD, Estey EH, Borthakur G, et al. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol 2009;27:4741-6.
100. Schimmer AD, Herr W, Hänel M, et al. Addition of AEG35156 XIAP antisense oligonucleotide in reinduction chemotherapy does not improve remission rates in patients with primary refractory acute myeloid leukemia in a randomized phase II study. Clin Lymphoma Myeloma Leuk 2011;11:433-8.
101. Boddu P, Carter BZ, Verstovsek S, Pemmaraju N. SMAC mimetics as potential cancer therapeutics in myeloid malignancies. Br J Haematol 2019;185:219-31.
102. Frey NV, Luger S, Mangan J, et al. A phase I study using single agent birinapant in patients with relapsed myelodysplastic syndrome and acute myelogenous leukemia. Blood 2014;124:3758-3758.
103. Donnellan WB, Diez-campelo M, Heuser M, et al. A phase 2 study of azacitidine (5-AZA) with or without birinapant in subjects with higher risk myelodysplastic syndrome (MDS) or chronic myelomonocytic leukemia (CMML). J Clin Oncol 2016;34:7060.
104. Morrish E, Copeland A, Moujalled DM, et al. Clinical MDR1 inhibitors enhance Smac-mimetic bioavailability to kill murine LSCs and improve survival in AML models. Blood Adv 2020;4:5062-77.
105. Mita MM, LoRusso PM, Papadopoulos KP, et al. A phase I study of ASTX660, an antagonist of inhibitors of apoptosis proteins, in adults with advanced cancers or lymphoma. Clin Cancer Res 2020;26:2819-26.
106. Ward GA, Lewis EJ, Ahn JS, et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNFα-dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol Cancer Ther 2018;17:1381-91.
107. Pemmaraju N, Carter BZ, Bose P, et al. Final results of a phase 2 clinical trial of LCL161, an oral SMAC mimetic for patients with myelofibrosis. Blood Adv 2021;5:3163-73.
108. Brumatti G, Ma C, Lalaoui N, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med 2016;8:339ra69.
109. Safferthal C, Rohde K, Fulda S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 2017;36:1487-502.
110. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ 2016;23:748-56.
111. Hillert LK, Bettermann-Bethge K, Nimmagadda SC, Fischer T, Naumann M, Lavrik IN. Targeting RIPK1 in AML cells carrying FLT3-ITD. Int J Cancer 2019;145:1558-69.
112. Li J, Liao D, Wang F, et al. RIPK1 inhibition enhances the therapeutic efficacy of chidamide in FLT3-ITD positive AML, both in vitro and in vivo. Leuk Lymphoma 2022;63:1167-79.
113. Xin J, You D, Breslin P, et al. Sensitizing acute myeloid leukemia cells to induced differentiation by inhibiting the RIP1/RIP3 pathway. Leukemia 2017;31:1154-65.
114. Huang X, Xiao F, Li Y, Qian W, Ding W, Ye X. Bypassing drug resistance by triggering necroptosis: recent advances in mechanisms and its therapeutic exploitation in leukemia. J Exp Clin Cancer Res 2018;37:310.
115. Müller MD, Holst PJ, Nielsen KN. A systematic review of expression and immunogenicity of human endogenous retroviral proteins in cancer and discussion of therapeutic approaches. Int J Mol Sci 2022;23:1330.
116. Fresquet V, Garcia-Barchino MJ, Larrayoz M, et al. Endogenous retroelement activation by epigenetic therapy reverses the warburg effect and elicits mitochondrial-mediated cancer cell death. Cancer Discov 2021;11:1268-85.
117. Zhang J, Liu Y, Li Q, Xu A, Hu Y, Sun C. Ferroptosis in hematological malignancies and its potential network with abnormal tumor metabolism. Biomed Pharmacother 2022;148:112747.
118. Pardieu B, Pasanisi J, Ling F, et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 2022;36:1585-95.
119. Yu Y, Xie Y, Cao L, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2015;2:e1054549.
120. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317-31.
121. Yusuf RZ, Saez B, Sharda A, et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood 2020;136:1303-16.
122. Woo JH, Shimoni Y, Yang WS, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 2015;162:441-51.
123. Guo J, Xu B, Han Q, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat 2018;50:445-60.
124. Birsen R, Larrue C, Decroocq J, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022;107:403-16.
125. Hong Y, Ren T, Wang X, et al. APR-246 triggers ferritinophagy and ferroptosis of diffuse large B-cell lymphoma cells with distinct TP53 mutations. Leukemia 2022;36:2269-80.
126. Du J, Wang T, Li Y, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med 2019;131:356-69.
127. Smith KH, Budhraja A, Lynch J, et al. The heme-regulated inhibitor pathway modulates susceptibility of poor prognosis B-lineage acute leukemia to BH3-mimetics. Mol Cancer Res 2021;19:636-50.
128. Zhang X, Ai Z, Zhang Z, et al. Dihydroartemisinin triggers ferroptosis in multidrug-resistant leukemia cells. DNA Cell Biol 2022;41:705-15.
129. Beatty A, Singh T, Tyurina YY, et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat Commun 2021;12:2244.
130. Dierge E, Debock E, Guilbaud C, et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab 2021;33:1701-1715.e5.
131. Grignano E, Birsen R, Chapuis N, Bouscary D. From iron chelation to overload as a therapeutic strategy to induce ferroptosis in leukemic cells. Front Oncol 2020;10:586530.
132. Müller S, Sindikubwabo F, Cañeque T, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat Chem 2020;12:929-38.
133. Rafiq S, McKenna SL, Muller S, Tschan MP, Humbert M. Lysosomes in acute myeloid leukemia: potential therapeutic targets? Leukemia 2021;35:2759-70.
134. Seo W, Silwal P, Song IC, Jo EK. The dual role of autophagy in acute myeloid leukemia. J Hematol Oncol 2022;15:51.
135. Rudat S, Pfaus A, Cheng YY, et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018;32:2189-202.
136. Heydt Q, Larrue C, Saland E, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018;37:787-97.
137. Putyrski M, Vakhrusheva O, Bonn F, et al. Disrupting the LC3 interaction region (LIR) binding of selective autophagy receptors sensitizes AML cell lines to cytarabine. Front Cell Dev Biol 2020;8:208.
138. Dykstra KM, Fay HRS, Massey AC, et al. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv 2021;5:2087-100.
139. Horne GA, Stobo J, Kelly C, et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020;34:1775-86.
140. Dupont M, Huart M, Lauvinerie C, et al. Autophagy targeting and hematological mobilization in FLT3-ITD acute myeloid leukemia decrease repopulating capacity and relapse by inducing apoptosis of committed leukemic cells. Cancers 2022;14:453.
141. Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 2014;10:1013-9.
142. Ghosh J, Kapur R. Role of mTORC1-S6K1 signaling pathway in regulation of hematopoietic stem cell and acute myeloid leukemia. Exp Hematol 2017;50:13-21.
143. Kasner MT, Mick R, Jeschke GR, et al. Sirolimus enhances remission induction in patients with high risk acute myeloid leukemia and mTORC1 target inhibition. Invest New Drugs 2018;36:657-66.
144. Park S, Chapuis N, Saint Marcoux F, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013;27:1479-86.
145. Burnett AK, Das Gupta E, Knapper S, et al. Addition of the mammalian target of rapamycin inhibitor, everolimus, to consolidation therapy in acute myeloid leukemia: experience from the UK NCRI AML17 trial. Haematologica 2018;103:1654-61.
146. Liesveld JL, Baran A, Azadniv M, et al. A phase II study of sequential decitabine and rapamycin in acute myelogenous leukemia. Leuk Res 2022;112:106749.
147. Johnson DC, Taabazuing CY, Okondo MC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med 2018;24:1151-6.
148. Basiorka AA, McGraw KL, Eksioglu EA, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016;128:2960-75.
149. Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019;133:1039-48.
150. McLemore AF, Hou HA, Meyer BS, et al. Somatic gene mutations expose cytoplasmic DNA to co-opt the cGAS/STING/NLRP3 axis in myelodysplastic syndromes. JCI Insight 2022:7.
151. Chakraborty S, Shapiro LC, de Oliveira S, Rivera-Pena B, Verma A, Shastri A. Therapeutic targeting of the inflammasome in myeloid malignancies. Blood Cancer J 2021;11:152.
152. Zahr A, Borthakur G. Emerging cell cycle inhibitors for acute myeloid leukemia. Expert Opin Emerg Drugs 2017;22:137-48.
153. Sinha D, Duijf PHG, Khanna KK. Mitotic slippage: an old tale with a new twist. Cell Cycle 2019;18:7-15.
154. Conti G, Dias MH, Bernards R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021;13:1118.
155. Rodriguez R, Schreiber SL, Conrad M. Persister cancer cells: iron addiction and vulnerability to ferroptosis. Mol Cell 2022;82:728-40.
156. Poillet-Perez L, Sarry JE, Joffre C. Autophagy is a major metabolic regulator involved in cancer therapy resistance. Cell Rep 2021;36:109528.
157. Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021;137:2721-35.
158. Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov 2019;9:910-25.
159. Kim K, Maiti A, Loghavi S, et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021;127:3772-81.
160. Antoszczak M, Müller S, Cañeque T, et al. Iron-sensitive prodrugs that trigger active ferroptosis in drug-tolerant pancreatic cancer cells. J Am Chem Soc 2022;144:11536-45.
161. Cui Z, Fu Y, Yang Z, et al. Comprehensive analysis of a ferroptosis pattern and associated prognostic signature in acute myeloid leukemia. Front Pharmacol 2022;13:866325.
162. Shao R, Wang H, Liu W, et al. Establishment of a prognostic ferroptosis-related gene profile in acute myeloid leukaemia. J Cell Mol Med 2021;25:10950-60.
163. Song Y, Tian S, Zhang P, Zhang N, Shen Y, Deng J. Construction and validation of a novel ferroptosis-related prognostic model for acute myeloid leukemia. Front Genet 2021;12:708699.
164. Zhang X, Zhang X, Liu K, et al. HIVEP3 cooperates with ferroptosis gene signatures to confer adverse prognosis in acute myeloid leukemia. Cancer Med 2022;11:5050-65.
165. Montalban-Bravo G, Class CA, Ganan-Gomez I, et al. Transcriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia 2020;34:872-81.
166. Bhatt S, Pioso MS, Olesinski EA, et al. Reduced mitochondrial apoptotic priming drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer Cell 2020;38:872-890.e6.
167. Collignon A, Hospital MA, Montersino C, et al. A chemogenomic approach to identify personalized therapy for patients with relapse or refractory acute myeloid leukemia: results of a prospective feasibility study. Blood Cancer J 2020;10:64.
168. Kantarjian H, Kadia T, DiNardo C, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J 2021;11:41.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Garciaz S, Miller T, Collette Y, Vey N. Targeting regulated cell death pathways in acute myeloid leukemia. Cancer Drug Resist 2023;6:151-68. http://dx.doi.org/10.20517/cdr.2022.108
AMA Style
Garciaz S, Miller T, Collette Y, Vey N. Targeting regulated cell death pathways in acute myeloid leukemia. Cancer Drug Resistance. 2023; 6(1): 151-68. http://dx.doi.org/10.20517/cdr.2022.108
Chicago/Turabian Style
Garciaz, Sylvain, Thomas Miller, Yves Collette, Norbert Vey. 2023. "Targeting regulated cell death pathways in acute myeloid leukemia" Cancer Drug Resistance. 6, no.1: 151-68. http://dx.doi.org/10.20517/cdr.2022.108
ACS Style
Garciaz, S.; Miller T.; Collette Y.; Vey N. Targeting regulated cell death pathways in acute myeloid leukemia. Cancer Drug Resist. 2023, 6, 151-68. http://dx.doi.org/10.20517/cdr.2022.108
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 9
likes
Like This Article 9
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.