
Resistance to energy metabolism - targeted therapy of AML cells residual in the bone marrow microenvironment

 ,
, Abstract
In response to the changing availability of nutrients and oxygen in the bone marrow microenvironment, acute myeloid leukemia (AML) cells continuously adjust their metabolic state. To meet the biochemical demands of their increased proliferation, AML cells strongly depend on mitochondrial oxidative phosphorylation (OXPHOS). Recent data indicate that a subset of AML cells remains quiescent and survives through metabolic activation of fatty acid oxidation (FAO), which causes uncoupling of mitochondrial OXPHOS and facilitates chemoresistance. For targeting these metabolic vulnerabilities of AML cells, inhibitors of OXPHOS and FAO have been developed and investigated for their therapeutic potential. Recent experimental and clinical evidence has revealed that drug-resistant AML cells and leukemic stem cells rewire metabolic pathways through interaction with BM stromal cells, enabling them to acquire resistance against OXPHOS and FAO inhibitors. These acquired resistance mechanisms compensate for the metabolic targeting by inhibitors. Several chemotherapy/targeted therapy regimens in combination with OXPHOS and FAO inhibitors are under development to target these compensatory pathways.
Keywords
INTRODUCTION
Acute myeloid leukemia (AML) comprises a highly aggressive, biologically heterogeneous group of hematopoietic disorders involving one or more cytogenetically or molecularly abnormal cell clones. It is primarily a disease of older adults. The standard of care for relapsed and refractory AML has progressed minimally in the past 30 years, with survival rates of less than 12% for patients over 65 years old[1]. Thus, novel therapeutic strategies that are more effective and carry a lower risk of organ damage than current treatments are urgently needed.
AML cells always face two major metabolic challenges: their high rate of proliferation imposes increased bioenergetic demands, and fluctuations in the availability of external nutrients and oxygen in the bone marrow (BM) microenvironment threaten cellular survival. In the BM, AML cells constantly modulate their metabolic state to adapt to this fluctuating microenvironment, shifting between quiescent, proliferative, and differentiated states[2-4]. Highly proliferative AML cells, drug-resistant AML cells, and leukemia stem cells (LSCs) that remain quiescent have been shown to depend on oxidative phosphorylation (OXPHOS). LSCs differ from bulk leukemia cells in that they possess stem cell characteristics including abnormal self-renewal capacity and drug resistance. Persistence of LSCs in the BM microenvironment after chemotherapy is considered an important factor in AML relapse[5]. Both quiescent AML cells and LSCs survive through metabolic activation of fatty acid oxidation (FAO) along with OXPHOS in their mitochondria. Hence, the reprogramming of energy metabolism processes in AML cells is recognized as a potential therapeutic target. Inhibition of OXPHOS and FAO can disrupt metabolic homeostasis, increase reactive oxygen species (ROS) production, and cause apoptosis in AML cells[2,6,7]. However, inhibition of this altered energy metabolism triggers various adaptive mechanisms in AML cells through their interaction with BM stromal cells. Thus, the BM microenvironment provides a setting in which secondary resistance to OXPHOS inhibition can develop, thereby contributing to the survival of AML cells. Therefore, strategies combining chemotherapy and specific molecular targeted therapy may have promise for eliminating BM-resident AML cells and LSCs.
In this review, we summarize the current state of knowledge about mitochondrial OXPHOS and fatty acid metabolism in residual AML cells in the BM microenvironment. We further describe the molecular mechanism by which AML cells acquire resistance to OXPHOS and FAO inhibitors. Finally, we evaluate potential therapeutic regimens combining OXPHOS and FAO inhibitors to target the metabolic vulnerabilities of BM-resident chemoresistant leukemia cells and LSCs.
MAIN TEXT
The BM microenvironment reprograms OXPHOS in AML cells
AML cells’ dependence on OXPHOS in the BM microenvironment
Whereas circulating AML cells are effectively eliminated by drug treatment, AML cells residing in the BM acquire resistance to chemotherapy. The BM microenvironment provides growth factors for leukemic cells, promotes immunosuppression, and supports leukemic cell survival. In response, leukemia cells adapt their metabolic state to this constantly changing environment[7-9].
Energy metabolism encompasses the molecular pathways whose products are involved in cellular energy production in the form of ATP. In leukemic cells, energy metabolism relies on OXPHOS and associated catabolic pathways, including glycolysis and fatty acid metabolism. The energy required for ATP production is produced by the mitochondrial potential, which causes protons to reenter the mitochondria through complex V. Fatty acid metabolism also supplies acetyl-CoA to the tricarboxylic acid (TCA) cycle through FAO [Figure 1].
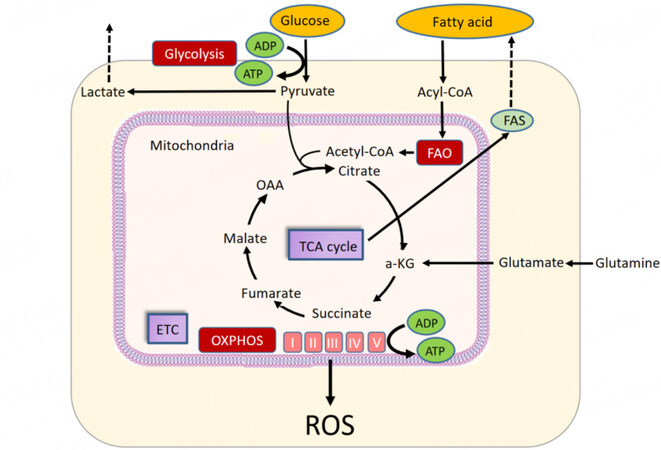
Figure 1. Energy metabolism in AML. Glucose is converted to pyruvate by glycolysis. Pyruvate is converted to acetyl-CoA for use in the tricarboxylic acid (TCA) cycle. ATP is produced by oxidative phosphorylation (OXPHOS) in the TCA cycle and electron transport chain (ETC). Fatty acid metabolism supplies acetyl-CoA to the TCA cycle via β-oxidation of fatty acids (FAO). Glutamine metabolism is the process of converting glutamine to glutamic acid. a-KG: Alpha-ketoglutarate; FAS: fatty acid synthase; OAA: oxaloacetic acid; ROS: reactive oxygen species.
Recently, the molecular mechanisms by which AML cells undergo metabolic reprogramming and those underlying the antileukemic efficacy of OXPHOS inhibitors have been demonstrated[7,10]. Actively proliferating AML cells respond to their increased energy and substrate demands via upregulation of OXPHOS, glycolysis, and related biochemical pathways. In turn, their bioenergetic efficacy strongly depends on extrinsic signals from the microenvironment[11].
The leukemic BM microenvironment is generally hypoxic; during disease progression, hypoxic areas in the BM expand[12-14]. Indeed, AML cells so strongly depend on OXPHOS for metabolism that they might cause hypoxia in the BM environment. These hypoxic niches are expanded, in part, through activation of the transcription factor hypoxia-inducible factor 1α (HIF-1α)[12,15]. Transcriptional complexes often include metabolic enzymes, which locally supply substrates and cofactors to these complexes[16]. For this reason, OXPHOS itself and the transcription factors that regulate it are attractive targets for novel therapeutic interventions.
The persistence of LSCs and treatment-resistant AML cells in the BM remains the major cause of failure to eradicate AML. Cancer stem cells were initially identified in AML[17,18] and subsequently validated in solid tumors. Across cancers, cancer stem cells share two important features: they can self-renew and produce differentiated progeny. The OXPHOS-dependent survival mechanism of LSCs is common to several solid tumor stem cells. For example, pancreatic cancer stem cells use OXPHOS for survival by accumulating the transcription coactivator peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), which enhances mitochondrial biogenesis and the oxygen consumption rate and is sensitive to inhibitors of mitochondrial respiration[19]. Reliance on OXPHOS has also been observed in other solid-tumor stem cells, including those in brain[10] and breast cancers[20].
Several recent studies have revealed how AML LSCs exploit OXPHOS[21]. The transcriptional and epigenetic signatures of leukemia-initiating LSCs are largely mutation-independent[22-25]. Instead, rewired cellular metabolism has been increasingly recognized to play a significant role in LSC maintenance and treatment resistance in AML[26]. In LSCs, several components of the electron transport chain (ETC) complexes I and V have been shown to be more abundant than in normal hematopoietic stem cells (HSCs)[27]. Notably, AML LSCs overexpress antiapoptotic BCL-2, which has been shown to regulate ATP/ADP exchange across the mitochondrial membrane by facilitating regulation of voltage-dependent anion channels and adenine nucleotides,[28,29] preventing the loss of coupled mitochondrial respiration during apoptosis[30].
Rewiring of mitochondrial function facilitates AML resistance to OXPHOS inhibition
Understanding the crosstalk between AML cells and their microenvironment is critical to targeting the pathways involved in the metabolic reprogramming of chemoresistant AML cells and LSCs. In preclinical studies, most AML models responded to inhibition of OXPHOS via targeting of ETC complex I[3]. However, several clinical trials have shown that the efficacy of these OXPHOS inhibitors is limited[31,32]. In trials of several types of solid tumors, one putative complex I inhibitor, carboxyamidotriazole, had no clinical benefit[31]. Mouse studies of BAY87-2243, a novel complex I inhibitor, demonstrated antitumor activity and no toxic effects, but the phase I trial was terminated because of unexpected toxic effects[32]. These findings indicate that OXPHOS inhibitors have a narrow therapeutic window and emphasize the need to better understand how the BM microenvironment enables AML cells to become resistant to the metabolic stress caused by OXPHOS inhibition.
One such mechanism occurring in the tumor microenvironment is the horizontal transfer of mitochondrial DNA from host to tumor cells. Studies using in vivo models have shown that this transfer reestablishes respiration and promotes tumorigenesis[33]. In OXPHOS-dependent AML cells, OXPHOS inhibition induced formation of tunneling nanotubes that enabled this mitochondrial DNA trafficking from BM stroma cells to AML cells[34]. In the formation of tunneling nanotubes, a filopodium-like protrusion is extended from one cell to another[35]. This process is positively regulated by activation of motor proteins such as Rho GTPases through actin polymerization[36,37] and filopodia formation through focal adhesion[38]. In addition, a recent study showed that a transmembrane complex gap junction channel opens under ROS-induced oxidative stress via PI3K-Akt activation to enable the transfer of mitochondrial DNA from stromal cells in the BM to HSCs [Figure 2][39].
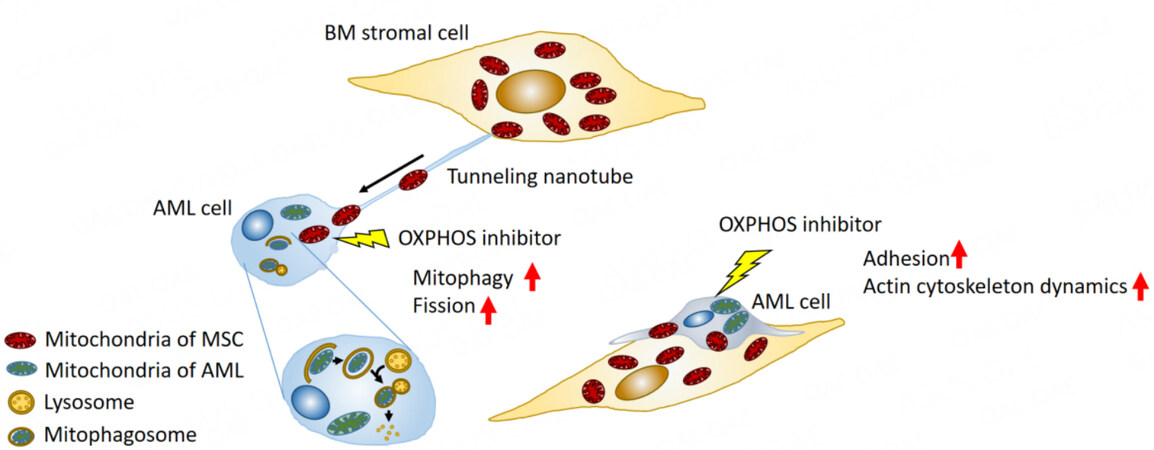
Figure 2. Secondary resistance to oxidative phosphorylation (OXPHOS) inhibition through bone marrow mesenchymal stem cells. OXPHOS inhibition stimulates cell adhesion and actin dynamics in AML cells. The bone marrow (BM) microenvironment facilitates the development of secondary resistance to OXPHOS inhibition by supporting direct mitochondrial trafficking via tunneling nanotubes from BM stromal cells to AML cells. This trafficking is accompanied by mitophagy and mitochondrial fission. MSC: Mesenchymal stem cell.
OXPHOS inhibition-induced horizontal transfer of mitochondria from BM stromal cells to AML cells is accompanied by endogenous mitochondrial fission and elimination of damaged mitochondria by mitophagy, both of which contribute to AML cell survival[34,40]. As the process by which damaged mitochondria are segregated for elimination by autophagy, mitochondrial fission is central to mitophagy[41-44]. Cellular metabolism and cell survival require efficient mitophagy[40,45]. Because of the centrality of these processes in the maintenance of mitochondrial function, both mitochondrial fission and crosstalk with BM stroma cells via tunneling nanotubes may be another mechanism by which AML cells develop resistance to OXPHOS inhibition[46].
LSCs have low rates of energy metabolism and cannot upregulate glycolysis after OXPHOS inhibition. Thus, they are particularly sensitive to OXPHOS blockade[47,48]. However, LSCs are able to maintain their stemness by mitochondrial fission and mitophagy, which balance mitochondrial functions such as energy production, ROS generation, and apoptosis regulation[44]. Given these competing pressures, LSCs have a fragile mitochondrial network. Thus, blocking both OXPHOS and other metabolic pathways is a promising strategy for overcoming OXPHOS resistance associated with the BM microenvironment. Two possible targets include the enzyme ASS1 and the lipid metabolism protein LRP1, both of which are overexpressed in OXPHOS inhibitor-treated AML cells in vivo. ASS1 is essential for the biosynthesis of arginine[49], and LRP1 contributes to hemin-induced autophagy in leukemia cells[50,51]. Further investigations will improve our understanding of how these enzymes shape the responses of LSCs to the metabolic and energetic effects of OXPHOS inhibition.
Several repurposed drugs have been shown to inhibit OXPHOS. Biguanides, including metformin, are routinely used for diabetes treatment and have been proposed for use in cancer because they inhibit complex I of the ETC in cancer cells[52]. However, metformin carries a risk of severe lactic acidosis[53,54], which is a safety concern for cancer patients. In addition, because high OXPHOS levels play a key role in cytarabine resistance, treatments combining cytarabine with OXPHOS inhibitors might be more effective than monotherapy with either type of agent[33]. Recently, a novel lipoate analog, devimistat, an inhibitor of two key TCA cycle enzymes, the pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes[55,56], showed a satisfactory safety profile in AML patients. In a phase I study, patients with relapsed or refractory AML who received a combination of devimistat with cytarabine and mitoxantrone had a complete remission rate of 50%[57].
In a phase II study of this combination, responses were observed in older patients but not in younger patients. In addition, RNA sequencing analysis showed a decrease in expression of mitochondria-related genes with aging, suggesting that age-related reduction in mitochondrial quality may be related to devimistat response[58]. These are encouraging findings that indicate that this approach would be particularly effective for older patients with the highest unmet medical needs. In sum, the judicious use of novel OXPHOS inhibitors in combination treatments may add to armamentarium of currently available therapeutics.
FAO metabolism of AML cells and LSCs in the fatty acid-abundant BM microenvironment
FAO of AML cells in the BM microenvironment
Adipocytes in the BM microenvironment support the survival of several types of tumor cells by stimulating FAO through fatty acid transfer[59]. While it was reported that BM adipocytes occupy approximately 60% of the BM in 65-year-old individuals[60], the development of BM adipocytes varies across different skeletal regions, and single-point iliac biopsy may not represent the BM environment of the skeletal system containing the red marrow and yellow marrow. In a previous study, leukemic cells have been shown to colonize in both red and yellow marrow regions, adhere to the cortical bone in the spine, and have enhanced activity in the proximal/distal femur[61]. In addition, radiation therapy accelerates the differentiation of mesenchymal stem cells into adipocytes in BM[62]. Such temporal and spatial changes in the BM microenvironment may play a key role in leukemia’s dynamic adaptation of FAO and in leukemia cells’ interactions with BM stromal cells.
AML cells generally obtain fatty acids for FAO from the extracellular microenvironment through lipolysis of stored triglycerides[63]. FAO is metabolically activated to promote leukemic cell survival by remodeling and lipolysis of BM adipocytes. FAO is an essential source of mitochondrial NADH and FADH2 for the ETC and provides acetyl-CoA to the TCA cycle to produce ATP[64]. BM adipocytes supply long-chain fatty acids, which are then taken up into the cytoplasm via the scavenger receptor CD36[65,66]. Fatty acids activation is a two-step reaction. In the first step, the fatty acids form acyl-CoA in the cytoplasm. Then, FAO breaks down acyl-CoA to form acetyl-CoA in the mitochondria. Carnitine O-palmitoyltransferase 1 (CPT1) catalyzes a rate-limiting step of FAO; this enzyme conjugates fatty acids to carnitine, which is required for fatty acids to translocate from the cytoplasm to the mitochondria[67]. The internalized fatty acids are further transferred to the AML cell nucleus by the lipid chaperone fatty acid-binding protein 4 (FABP4). In the nucleus, the fatty acids ligate to peroxisome proliferator-activated receptor γ (PPAR)[68]. Activated PPAR induces downstream target genes, including CD36, FABP4, and the antiapoptotic BCL2 [Figure 3][69].
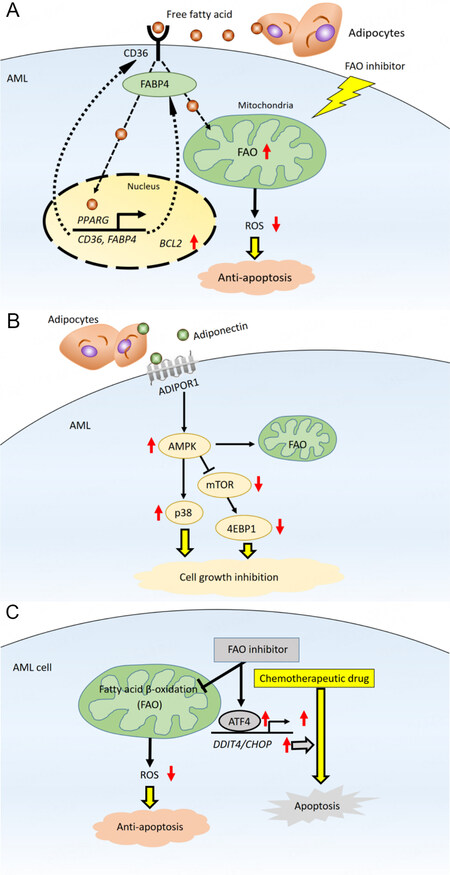
Figure 3. Bone marrow adipocytes promote fatty acid metabolism in AML. (A) Fatty acids derived through lipolysis of stored triglycerides in adipocytes induce upregulation of PPARG, CD36, and FABP4 gene transcription, which stimulates fatty acid endocytosis. In mitochondria, fatty acids are metabolized through fatty acid oxidation (FAO), decreasing mitochondrial reactive oxygen species (ROS) formation and intracellular oxidative stress, thereby reducing apoptosis; (B) transcriptional regulation and fatty acid metabolism pathways maintain AML cells in a quiescent state. Activation of AMPK, upregulation of p38 and associated induction of autophagy, and upregulation of antiapoptotic HSP chaperone proteins in this state lead to chemoresistance; (C) in mitochondria, fatty acids are consumed for FAO, resulting in diminished formation of mitochondrial ROS and decreased intracellular oxidative stress. Inhibition of FAO induces an integrated stress response that stimulates transcriptional activation of ATF4 and promotes apoptosis induced by chemotherapy. ADIPOR1: Adiponectin receptor 1; ATF4: activating transcription factor 4; AMPK: AMP-activated protein kinase; FABP4: fatty acid binding protein 4; p38: p38 mitogen-activated protein kinase.
As with AML cells, the specific BM microenvironment created by the interaction between LSCs and stromal adipocytes supports the metabolic demands of LSCs. LSCs induce adipocyte lipolysis, which drives FAO in LSCs and facilitates their survival[70,71]. Therefore, CD36 and CPT1 are potential targets for AML. A CD36 neutralizing antibody inhibited metastasis of human melanoma and breast cancer cells[72], and inhibition of CPT1 caused mitochondrial damage leading to cell death in primary AML cells[67].
FABP4 is important in FAO and cancer cell survival in both solid and hematologic cancers. Adipocytes are known to serve as fatty acid reservoirs in breast cancer and melanoma[73,74]. Ovarian cancer cells also survive and proliferate in an adipocyte-rich microenvironment[75]. When primary human omental adipocytes were co-cultured with ovarian cancer cells, the adipocytes underwent lipolysis, and FAO was induced in the cancer cells[63]. These processes are mediated by adipokines including interleukin-8 and by upregulation of FABP4 both in adipocytes and ovarian tumor cells. In studies of leukemia, AML cells co-cultured with BM adipocytes exhibited higher levels of FABP4[69], and knockdown of FABP4 prolonged survival in a mouse model of leukemia[71]. Thus, FABP4’s critical role in cancer cell survival involves its interactions with adipocytes.
Activation of β-adrenergic receptors, along with a G protein-coupled cascade that stimulates the lipolytic enzyme hormone-sensitive lipase (HSL), induces lipolysis of adipocytes[76,77]. Ovarian cancer cells have been found to upregulate HSL phosphorylation, thereby stimulating the release of free fatty acids from adipocytes[63]. AML blasts also induce HSL phosphorylation and, thus, activation of lipolysis in BM adipocytes[71].
BM adipocytes also increase AML cells’ expression of adiponectin and its downstream target, AMP-activated protein kinase (AMPK), a stress response kinase[69]. AMPK, an important modulator of energy metabolism, is activated upon ATP depletion. Its functions include upregulation of fatty acid uptake, FAO, and regulation of autophagy[78,79]. Levels of adiponectin, much of which is supplied by BM adipocytes, have been shown to increase during cancer therapy[80], and to promote chemotherapy resistance in myeloma cells via inducing adipokine secretion of adipokines and activating AMPK-dependent autophagy[81,82]. AMPK also positively regulates responses of an antiapoptotic chaperone heat shock protein that binds to denatured and unfolded proteins and promotes protein refolding or degradation to support AML cell survival[79]. In sum, leukemic cells often rely on fatty acids when they undergo metabolic stress, and the NADH and FADH2 generated by FAO support ATP production, redox homeostasis, biosynthesis, and cell survival.
FAO is involved in the interactions between LSCs and BM stromal cells[83]. LSCs rely on fatty acid uptake and consumption to shape their adaptation to the conditions of the BM microenvironment, their response to drugs, and their development of drug resistance[69]. Several such mechanisms have been identified. Mitochondrial uncoupling in AML cells negatively regulates Bak-dependent mitochondrial permeability transition[84]. In a study using samples from patients with relapsed AML, LSCs acquired the ability to counteract the loss of amino acid metabolism by upregulating FAO[85]. Specifically, this mechanism may underlie the development of resistance to treatment with azacitidine/venetoclax, a common induction regimen used mainly in older patients with AML[85,86]. In addition, a preclinical study demonstrated that cytarabine-resistant AML cells had enhanced FAO and OXPHOS[66]. Thus, targeting the metabolic vulnerabilities of chemoresistant LSCs, such as their dependence on FAO, may be a useful strategy for eradicating these cells.
FAO inhibitors and resistance acquired by compensatory metabolism in the BM microenvironment
FAO inhibition disrupts metabolic homeostasis, increases ROS levels, and induces expression of the integrated stress response mediator ATF4 in AML cells, all of which contribute to apoptosis[87]. Numerous studies have reported the anti-AML effect of inhibition of CPT1, the major rate-limiting enzyme in FAO[67,84,87,88]. CPT1 positively controls FAO by conjugating fatty acids with carnitine to transfer fatty acids into the mitochondrial matrix. Etomoxir is a pharmacological inhibitor of CPT1A, one of the isoforms of CPT1[89], frequently used to block free fatty acids from entering the mitochondria via CPT1. Although the clinical use of etomoxir has ceased because of adverse effects[90], the CPT1 inhibitor perhexiline can sensitize breast cancer cells to paclitaxel[91], and other CPT1 inhibitors[92] are currently being investigated for use in cancer therapy. The CPT1A inhibitor ST1326 has been shown to cause cell growth arrest, mitochondrial damage, and apoptosis in AML cells in a dose- and time-dependent manner[67]. Another novel FAO inhibitor, avocatin B, which is derived from avocados, decreased NADPH levels that were increased by FAO through acetyl-CoA and NADH production, inducing ROS-dependent cell death in AML cells[93,94]. Finally, the fatty acid synthase inhibitor orlistat has induced apoptosis in leukemic cells[2].
The intramitochondrial FAO enzyme very long-chain acyl-CoA dehydrogenase (VLCAD) is critical in supporting both FAO and OXPHOS in AML cells and LSCs. Recently, preclinical studies have demonstrated the antileukemia activity of a novel small-molecule VLCAD inhibitor, a polyhydroxylated fatty alcohol with a terminal alkyne (AYNE)[95]. AYNE reduced mitochondrial respiration by altering FAO, which led to reduced ATP production in AML cells, even though AYNE also moderately upregulated glycolysis. In a mouse model, pharmacological inhibition of VLCAD with AYNE significantly reduced the repopulation potential of leukemia cells and was well tolerated[95]. Notably, normal HSCs compensate for this reduced replicative capacity through glycolysis which maintains their ATP levels and thus their viability[95,96]. These findings demonstrate the importance of focusing on the specific metabolic vulnerabilities of residual AML cells and LSCs that survive chemotherapy-induced stress. Unfortunately, only a few FAO inhibitors have advanced from preclinical to clinical studies [Table 1].
FAO inhibitors in clinical trials on cancer treatment
| Compounds | Targeted enzyme | Clinical applications | Phase | ClinicalTrials.gov Identifier | Verified |
| Trimetazidine | 3-ketoacyl-C3-ketoacyl-CoA thiolase | Advanced Hepatocellular Carcinoma | Phase 3 | NCT03278444 | September 2017 |
| Trimetazidine | 3-ketoacyl-C3-ketoacyl-CoA thiolase | Intermediate-stage Hepatocellular Carcinoma | Phase 3 | NCT03274427 | September 2017 |
| Ranolazine | 3-ketoacyl-C3-ketoacyl-CoA thiolase | Prostate Cancers | N//A (Pilot Study) | NCT01992016 | December 2018 |
Because AML is heterogeneous and multiclonal, blocking only one part of this complex metabolic system may allow residual cells to adapt metabolically. For instance, it has been shown that BM-derived stromal cells, including adipocytes, diminish the antileukemia effects of FAO inhibitors in AML cells by increasing glycolysis and glucose and free fatty acid uptake[87]. This compensatory induction of glycolysis is a sustained source of ATP to AML cells and, in turn, induces substantial lactate production. Similarly, FAO-deficient Abcb11-knockout mice exhibited high FABP4 and CD36 expression and free fatty acid uptake[97]. In sum, it is clear that FAO inhibition initiates several different adaptive mechanisms that promote AML cell survival in the BM microenvironment.
For this reason, treatment options based on combination regimens have been tested. Although FAO inhibition alone can trigger compensatory activation of other metabolic pathways, FAO inhibitors can also synergize with conventional antitumor agents such as paclitaxel[91]. For example, FAO and OXPHOS are increased in cytarabine-resistant AML cells; FAO inhibition with etomoxir induced a metabolic shift from high to low OXPHOS, sensitizing the cells to cytarabine[66]. Similarly, the combination of avocatin B and cytarabine synergized to enhance ROS production and induce apoptosis in AML cells co-cultured with BM adipocytes[87]. The role of avocatin B in apoptosis induction was attributed to activation of endoplasmic reticulum stress-induced ATF4[87]. These findings suggest that AML cells treated with cytarabine exhibit increased dependence on FAO, which may account for the synergism of cytarabine and FAO inhibitors.
CONCLUSIONS
AML cells and LSCs both strongly depend on the production of mitochondrial biomass and on OXPHOS[66] and FAO[84] for survival. Compared to healthy HSCs, AML cells and LSCs are more susceptible to mitochondrial stress because their respiratory chain reserve capacity is lower[98]. These characteristic differences in the metabolism of AML cells and their normal hematopoietic-cell counterparts represent a specific vulnerability of leukemia cells and therefore are drawing a great deal of attention as targets for AML therapy. The results of studies in preclinical models using agents that target fatty acid metabolism have been encouraging.
Although they are vulnerable to the targeting of their metabolic pathways, AML cells, drawing on microenvironmental factors, can adapt to metabolic stress by activating metabolic bypass processes. Several in vivo studies and clinical trials have shown that the use of metabolic inhibitors alone is ineffective both because of their narrow therapeutic window and because of these adaptive mechanisms[87]. Alternatively, inhibition of FAO and other metabolic mechanisms along with conventional chemotherapy or targeted therapy may synergistically eradicate chemotherapy-resistant AML cells present in the BM.
In conclusion, understanding AML cell metabolism in the specific context of the BM microenvironment is crucial to improving therapies for AML. Because characteristics of the BM microenvironment enable the acquisition of resistance to OXPHOS and FAO inhibitors, drug combination strategies that interfere with these adaptations are needed. A more comprehensive understanding of the mechanisms of AML cell metabolism in future studies may reveal new treatment options targeting OXPHOS and FAO, enhance the efficacy of chemotherapeutic agents that target related pathways, reduce the toxicity of these agents, and enable the translation of new combinations of agents into clinically applicable treatment strategies.
DECLARATIONS
AcknowledgmentsWe thank Amy Ninetto, Scientific Editor, Research Medical Library, MD Anderson Cancer Center, for editing the manuscript.
Authors’ contributionsWrote manuscript: Tabe Y, Konopleva M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Hassan M, Abedi-Valugerdi M. Hematologic malignancies in elderly patients. Haematologica 2014;99:1124-7.
2. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009;23:537-48.
4. Raaijmakers MH. Niche contributions to oncogenesis: emerging concepts and implications for the hematopoietic system. Haematologica 2011;96:1041-8.
5. Ho TC, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016;128:1671-8.
6. Robinson AJ, Davies S, Darley RL, Tonks A. Reactive oxygen species rewires metabolic activity in acute myeloid leukemia. Front Oncol 2021;11:632623.
7. Romo-González M, Ijurko C, Hernández-Hernández Á. Reactive oxygen species and metabolism in leukemia: a dangerous liaison. Front Immunol 2022;13:889875.
8. Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol 2014;164:767-78.
9. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532.
10. Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036-46.
11. Hail N, Lotan R. Cancer chemoprevention and mitochondria: targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Mol Nutr Food Res 2009;53:49-67.
12. Benito J, Shi Y, Szymanska B, et al. Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One 2011;6:e23108.
13. Benito J, Zeng Z, Konopleva M, Wilson WR. Targeting hypoxia in the leukemia microenvironment. Int J Hematol Oncol 2013;2:279-88.
14. Rytelewski M, Harutyunyan K, Baran N, et al. Inhibition of oxidative phosphorylation reverses bone marrow hypoxia visualized in imageable syngeneic B-all mouse model. Front Oncol 2020;10:991.
15. Frolova O, Samudio I, Benito JM, et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther 2012;13:858-70.
16. Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013;502:489-98.
17. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730-7.
18. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645-8.
19. Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014;514:628-32.
20. Lee KM, Giltnane JM, Balko JM, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab 2017;26:633-47.e7.
21. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329-41.
22. Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011;17:1086-93.
23. Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 2015;6:8489.
24. George J, Uyar A, Young K, et al. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat Commun 2016;7:12166.
25. Ng SW, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016;540:433-7.
27. Raffel S, Klimmeck D, Falcone M, et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020;136:1507-19.
28. Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ 2000;7:1182-91.
29. Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell 1999;3:159-67.
30. Manfredi G, Kwong JQ, Oca-Cossio JA, et al. BCL-2 improves oxidative phosphorylation and modulates adenine nucleotide translocation in mitochondria of cells harboring mutant mtDNA. J Biol Chem 2003;278:5639-45.
31. Johnson EA, Marks RS, Mandrekar SJ, et al. Phase III randomized, double-blind study of maintenance CAI or placebo in patients with advanced non-small cell lung cancer (NSCLC) after completion of initial therapy (NCCTG 97-24-51). Lung Cancer 2008;60:200-7.
32. Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med 2013;2:611-24.
33. Tan AS, Baty JW, Dong LF, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015;21:81-94.
34. Saito K, Zhang Q, Yang H, et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv 2021;5:4233-55.
35. Abounit S, Delage E, Zurzolo C. Identification and characterization of tunneling nanotubes for intercellular trafficking. Curr Protoc Cell Biol 2015;67:12.0.1-.0.21.
36. Biran A, Perelmutter M, Gal H, et al. Senescent cells communicate via intercellular protein transfer. Genes Dev 2015;29:791-802.
37. Frei DM, Hodneland E, Rios-Mondragon I, et al. Novel microscopy-based screening method reveals regulators of contact-dependent intercellular transfer. Sci Rep 2015;5:12879.
39. Mistry JJ, Marlein CR, Moore JA, et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc Natl Acad Sci USA 2019;116:24610-9.
41. Kim DY, Jung SY, Kim YJ, et al. Hypoxia-dependent mitochondrial fission regulates endothelial progenitor cell migration, invasion, and tube formation. Korean J Physiol Pharmacol 2018;22:203-13.
42. Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016;351:275-81.
43. Cai J, Wang J, Huang Y, et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis 2016;7:e2459.
44. Pei S, Minhajuddin M, Adane B, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 2018;23:86-100.e6.
45. Wallace DC. Mitochondria and cancer: warburg addressed. Cold Spring Harb Symp Quant Biol 2005;70:363-74.
46. Marlein CR, Zaitseva L, Piddock RE, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017;130:1649-60.
47. Jones CL, Stevens BM, D'Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018;34:724-40.e4.
48. Schimmer AD. Novel mitochondrial mechanisms of cytarabine resistance in primary AML cells. Cancer Discov 2017;7:670-2.
49. Syed N, Langer J, Janczar K, et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis 2013;4:e458.
50. Xing P, Liao Z, Ren Z, et al. Roles of low-density lipoprotein receptor-related protein 1 in tumors. Chin J Cancer 2016;35:6.
51. Grosso RA, Caldarone PVS, Sanchez MC, Chiabrando GA, Colombo MI, Fader CM. Hemin induces autophagy in a leukemic erythroblast cell line through the LRP1 receptor. Biosci Rep 2019:39.
52. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304-5.
53. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2014;2:12.
54. Scotland S, Saland E, Skuli N, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013;27:2129-38.
55. Stuart SD, Schauble A, Gupta S, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab 2014;2:4.
56. Zachar Z, Marecek J, Maturo C, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med 2011;89:1137-48.
57. Pardee TS, Luther S, Buyse M, Powell BL, Cortes J. Devimistat in combination with high dose cytarabine and mitoxantrone compared with high dose cytarabine and mitoxantrone in older patients with relapsed/refractory acute myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol 2019;15:3197-208.
58. Anderson R, Miller LD, Isom S, et al. Phase II trial of cytarabine and mitoxantrone with devimistat in acute myeloid leukemia. Nat Commun 2022;13:1673.
59. Behan JW, Yun JP, Proektor MP, et al. Adipocytes impair leukemia treatment in mice. Cancer Res 2009;69:7867-74.
60. Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001;2:165-71.
61. Magome T, Froelich J, Holtan SG, et al. Whole-body distribution of leukemia and functional total marrow irradiation based on FLT-PET and dual-energy CT. Mol Imaging 2017;16:1536012117732203.
62. Islam MS, Stemig ME, Takahashi Y, Hui SK. Radiation response of mesenchymal stem cells derived from bone marrow and human pluripotent stem cells. J Radiat Res 2015;56:269-77.
63. Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17:1498-503.
64. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013;13:227-32.
65. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr 2014;34:281-303.
66. Farge T, Saland E, de Toni F, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 2017;7:716-35.
67. Ricciardi MR, Mirabilii S, Allegretti M, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 2015;126:1925-9.
68. Itoh T, Fairall L, Amin K, et al. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol 2008;15:924-31.
69. Tabe Y, Yamamoto S, Saitoh K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res 2017;77:1453-64.
70. Ye H, Adane B, Khan N, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 2016;19:23-37.
71. Shafat MS, Oellerich T, Mohr S, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017;129:1320-32.
72. Pascual G, Avgustinova A, Mejetta S, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41-5.
73. Balaban S, Shearer RF, Lee LS, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab 2017;5:1.
74. Zhang M, Di Martino JS, Bowman RL, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov 2018;8:1006-25.
75. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 2013;1831:1533-41.
77. Sengenes C, Bouloumie A, Hauner H, et al. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J Biol Chem 2003;278:48617-26.
78. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002;8:1288-95.
79. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012;32:2-11.
80. Cawthorn WP, Scheller EL, Learman BS, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab 2014;20:368-75.
81. Medina EA, Oberheu K, Polusani SR, Ortega V, Velagaleti GV, Oyajobi BO. PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia 2014;28:2080-9.
82. Ma R, Yu D, Peng Y, et al. Resveratrol induces AMPK and mTOR signaling inhibition-mediated autophagy and apoptosis in multiple myeloma cells. Acta Biochim Biophys Sin 2021;53:775-83.
83. Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA 2011;108:7950-5.
84. Samudio I, Harmancey R, Fiegl M, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 2010;120:142-56.
85. Jones CL, Stevens BM, D'Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2019;35:333-5.
86. Stevens BM, Jones CL, Pollyea DA, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer 2020;1:1176-87.
87. Tabe Y, Saitoh K, Yang H, et al. Inhibition of FAO in AML co-cultured with BM adipocytes: mechanisms of survival and chemosensitization to cytarabine. Sci Rep 2018;8:16837.
88. Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 2011;1807:726-34.
89. Schreurs M, Kuipers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev 2010;11:380-8.
90. Shim JK, Choi S, Yoon SJ, et al. Etomoxir, a carnitine palmitoyltransferase 1 inhibitor, combined with temozolomide reduces stemness and invasiveness in patient-derived glioblastoma tumorspheres. Cancer Cell Int 2022;22:309.
91. Wang T, Fahrmann JF, Lee H, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab 2018;27:1357.
92. Dhakal B, Li CMY, Li R, et al. The antianginal drug perhexiline displays cytotoxicity against colorectal cancer cells in vitro: a potential for drug repurposing. Cancers 2022;14:1043.
93. Lee EA, Angka L, Rota SG, et al. Targeting mitochondria with avocatin B induces selective leukemia cell death. Cancer Res 2015;75:2478-88.
94. Tcheng M, Samudio I, Lee EA, Minden MD, Spagnuolo PA. The mitochondria target drug avocatin B synergizes with induction chemotherapeutics to induce leukemia cell death. Leuk Lymphoma 2017;58:986-8.
95. Tcheng M, Roma A, Ahmed N, et al. Very long chain fatty acid metabolism is required in acute myeloid leukemia. Blood 2021;137:3518-32.
96. Mesbahi Y, Trahair TN, Lock RB, Connerty P. Exploring the metabolic landscape of AML: from haematopoietic stem cells to myeloblasts and leukaemic stem cells. Front Oncol 2022;12:807266.
97. Zhang Y, Li F, Patterson AD, et al. Abcb11 deficiency induces cholestasis coupled to impaired β-fatty acid oxidation in mice. J Biol Chem 2012;287:24784-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tabe Y, Konopleva M. Resistance to energy metabolism - targeted therapy of AML cells residual in the bone marrow microenvironment . Cancer Drug Resist 2023;6:138-50. http://dx.doi.org/10.20517/cdr.2022.133
AMA Style
Tabe Y, Konopleva M. Resistance to energy metabolism - targeted therapy of AML cells residual in the bone marrow microenvironment . Cancer Drug Resistance. 2023; 6(1): 138-50. http://dx.doi.org/10.20517/cdr.2022.133
Chicago/Turabian Style
Tabe, Yoko, Marina Konopleva. 2023. "Resistance to energy metabolism - targeted therapy of AML cells residual in the bone marrow microenvironment " Cancer Drug Resistance. 6, no.1: 138-50. http://dx.doi.org/10.20517/cdr.2022.133
ACS Style
Tabe, Y.; Konopleva M. Resistance to energy metabolism - targeted therapy of AML cells residual in the bone marrow microenvironment . Cancer Drug Resist. 2023, 6, 138-50. http://dx.doi.org/10.20517/cdr.2022.133
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 6
likes
Like This Article 6
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.