
Association between genome-wide epigenetic and genetic alterations in breast cancer tissue and response to HER2-targeted therapies in HER2-positive breast cancer patients: new findings and a systematic review
 0
0Abstract
Recent evidence suggests that genetic and epigenetic mechanisms might be associated with acquired resistance to cancer therapies. The aim of this study was to assess the association of genome-wide genetic and epigenetic alterations with the response to anti-HER2 agents in HER2-positive breast cancer patients. PubMed was screened for articles published until March 2021 on observational studies investigating the association of genome-wide genetic and epigenetic alterations, measured in breast cancer tissues or blood, with the response to targeted treatment in HER2-positive breast cancer patients. Sixteen studies were included in the review along with ours, in which we compared the genome-wide DNA methylation pattern in breast tumor tissues of patients who acquired resistance to treatment (case group, n = 6) to that of patients who did not develop resistance (control group, n = 6). Among genes identified as differentially methylated between the breast cancer tissue of cases and controls, one of them, PRKACA, was also reported as differentially expressed in two studies included in the review. Although included studies were heterogeneous in terms of methodology and study population, our review suggests that genes of the PI3K pathway may play an important role in developing resistance to anti-HER2 agents in breast cancer patients. Genome-wide genetic and epigenetic alterations measured in breast cancer tissue or blood might be promising markers of resistance to anti-HER2 agents in HER2-positive breast cancer patients. Further studies are needed to confirm these data.
Keywords
INTRODUCTION
The human epidermal growth factor receptor 2 (HER2) is a transmembrane tyrosine kinase receptor and belongs to the epidermal growth factor receptor (EGFR) family[1]. It comprises an extracellular domain (ECD), a transmembrane segment, and an intracellular region[2]. HER2 gene amplification and receptor overexpression, which occur in approximately 15%-20% of breast cancer patients, are important markers for poor prognosis, including a more aggressive disease and shorter survival[3]. In addition, HER2-positive status is considered a predictive marker of response to HER2-targeted drugs[4]. Detection of receptor overexpression via immunohistochemistry (IHC) and/or HER2 gene amplification using in situ hybridization (ISH) techniques in breast cancer tissue, including fluorescent ISH (FISH), determines patients’ eligibility to receive anti-HER2 therapies[5]. Food and Drug Administration (FDA)-approved anti-HER2 agents currently used in clinical settings in combination with chemotherapy comprises recombinant monoclonal antibodies that bind the ECD of HER2, such as trastuzumab (Herceptin®), pertuzumab (Perjeta®) and trastuzumab emtansine (T-DM1, Kadcyla®), and small molecule tyrosine kinase inhibitors, like lapatinib (TYKERB®), that inhibit enzyme function of the intracellular catalytic domain of HER2 and other EGFR members[6,7].
Although targeted treatment with anti-HER2 agents has significantly improved the disease-free and overall survival rates of metastatic and early-stage HER2-positive breast cancer patients[8-11], resistance to anti-HER2 therapy, both primary and acquired, has emerged as a major clinical problem in the treatment of HER2-positive breast cancer patients[12-14]. Even though several molecular mechanisms of resistance to anti-HER2 agents have been proposed in preclinical models, no clinically applicable strategy to overcome resistance to these targeted treatments has been identified yet[15]. Therefore, there is an urgent need to identify reliable predictive molecular markers of treatment failure with the ultimate goal of developing targeted drugs that can overcome resistance.
Studies indicate that genetic factors, including single nucleotide polymorphisms (SNPs), copy number variations (CNVs), HER2 mutations, and HER2 splice variants, might influence treatment effectiveness toward targeted therapies in HER2-positive breast cancer patients or HER2-positive breast cancer cell lines[16-28]. Recent evidence suggests that epigenetic regulatory mechanisms, including DNA methylation and microRNAs (miRNAs), might play a role in acquiring resistance to cancer therapies[29-32]. DNA methylation occurs through the covalent attachment of a methyl group on cytosine residues in CpG dinucleotides and contributes to transcriptional regulations[33]. While DNA methylation in the immediate vicinity of the transcriptional start site (TSS) generally represses gene expression, methylation in the gene body (far from annotated TSS) may stimulate elongation and is, therefore, positively associated with gene expression[34,35]. miRNA are approximately 22 nucleotides long non-coding RNAs that regulate gene expression in a sequence-specific manner[36].
The aim of this pilot study was to analyze the association between DNA methylation patterns in breast cancer specimens and response to trastuzumab in a cohort of 12 trastuzumab-treated, non-metastatic HER2-positive breast cancer patients. Additionally, a systematic review that combines these findings with all available published results on the association of genome-wide genetic and epigenetic alterations in breast cancer tissue or blood with response to anti-HER2 treatment in HER2-positive breast cancer patients is also reported.
MATERIAL AND METHODS
Pilot study of new findings
Study population and data collection
The study population consisted of 12 women (six cases and six controls) selected among 106 trastuzumab-treated patients with non-metastatic, HER2-positive breast cancer diagnosed between July 1, 2005 and December 31, 2010 at the Centre des Maladies du Sein, a specialized breast center in Quebec City, Canada. Information on tumor characteristics and prognostic factors at the time of diagnosis (baseline) and follow-up information were collected from medical records. The clinical endpoint in this study was disease-free survival (DFS). All breast cancer recurrences (locoregional, contralateral breast, and distant) were considered as events, whereas death (from any cause) before recurrence and loss to follow-up were considered as censoring events.
Over a mean follow-up period of 6.22 years, 22 patients out of 106 experienced recurrence. Eight cases were randomly selected among all patients who developed recurrence during follow-up and six had a sufficient amount of primary breast cancer tissue available for DNA extraction (see below). Of note, baseline characteristics of the six selected cases were comparable to the total population of cases (n = 22) for all characteristics except tumor grade (the proportion of grade III tumors among the selected cases was 67% vs. 41% for the total population of cases) [Supplementary Table 1]. For each case, one control was selected from the 84 patients who had not developed recurrence and were alive at the date of the case’s recurrence. Controls were matched to cases for the following factors: age at diagnosis (with 5-year age categories), estrogen receptor (ER) status, year of diagnosis (with 2-year categories), and menopausal status. The number of samples used was determined upon the availability of samples and not evaluated using a statistical sample size calculation. All patients provided written informed consent. Ethical approval of the study was obtained from the Research Ethics Committee of the Centre de Recherche du CHU de Québec
Gene methylation assessment
To ensure that DNA methylation was analyzed to the greatest possible extent in breast cancer tissue and to reduce contamination with other cell types (lymphocytes, adipocytes, fibroblasts), tissue microarray (TMA) blocks containing formalin-fixed, paraffin-embedded (FFPE) breast cancer tissue cores (1 mm in diameter) were constructed for each patient, as previously described[37]. From each TMA block, one section was stained with hematoxylin and eosin (H&E) to verify the cellular composition of the cores. Cores were removed from TMA blocks if they contained abnormal tissue or if epithelial tumor tissue occupied < 70% of the core area before proceeding to DNA extraction. H&E sections were prepared from different levels of the TMA blocks: at the beginning, at regular intervals (every tenth 10-µm-thick serial section), and after the last section. DNA was extracted from tissue cores using GeneJET FFPE DNA Purification kit (ThermoScientific, Ottawa, Canada) with minor modifications to the manufacturer’s instructions in which samples were incubated with Digestion buffer for six minutes and incubated with Proteinase K solution for 180 minutes.
DNA samples were sent to Génome Québec Innovation Center (Montreal, Canada). Methylation was measured with the Illumina HumanMethylation450 BeadChip array (Illumina Inc., San Diego, CA, USA) following the manufacturer’s instructions for the bisulfite treatment, Infinium FFPE quality control (Illumina FFPE QC kit, Illumina, Inc., CA, USA), and DNA restoration. This BeadChip interrogates 482,421 CpG sites, 3091 non-CpG sites, 65 random SNPs, and covers 21,231 RefSeq genes. It uses two distinct oligonucleotide probes (Infinium I and Infinium II) to assess methylation levels[38].
Statistical analysis
Raw β-values, defined as the ratio of the methylated probe intensity to the overall intensity (sum of the methylated and unmethylated probe intensities)[39], were imported into the R statistical programming environment (version 3.2.2). Since M-values [logit transformed β-values, calculated as 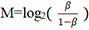 ] are considered more reliable in the detection rate and true positive rate for both highly methylated and unmethylated CpG sites compared to β-values[39], M-values were used for statistical analyses.
] are considered more reliable in the detection rate and true positive rate for both highly methylated and unmethylated CpG sites compared to β-values[39], M-values were used for statistical analyses.
Quality control was performed with the qcReport function from the minfi package, and none of the 12 samples were excluded due to bad quality control. The Dasen method from the WateRmelon package, also known as data-driven separate normalization, was used to background correct and quantile normalize data based on methylated and unmethylated intensities, separately, by probe types (Infinium I and II)[40]. A probe filtration step was performed to remove CpG sites corresponding to probes that could affect our analysis, including probes with bad detection (detection P-value > 0.01); unique probes having a common single nucleotide polymorphism (SNP) in European individuals at the interrogated CpG loci or the single-base extension according to the list published by Chen et al.[41]; probes that can hybridize to multiple loci also listed by Chen et al.[41]; and probes located on X and Y chromosomes. A total of 76,161 unique probes were removed, leaving 406,260 autosomal probes for the analysis. Data were verified for confounding batch effects due to separate chips[42], and none were observed. All samples passed quality-control tests and were therefore retained in the analysis.
Baseline characteristics between cases and controls were compared using Fisher’s exact test for categorical variables, Student’s t-test for follow-up time and Wilcoxon-Mann-Whitney test for the other continuous variables. The difference in global methylation levels between median M-values of cases and controls was assessed using a Wilcoxon signed-rank test for paired samples. Differentially methylated probes (DMPs) were identified using LIMMA (robust linear regression method), taking into account the matching factors between cases and controls (i.e., age at diagnosis, ER status, year of diagnosis, and menopausal status). Multiple testing correction was performed using false discovery rate (FDR) estimation (cut-off < 5%). In addition, we used a log2-fold change |log2FC| (i.e., the difference between mean M-values measured in breast cancer tissues of resistant patients and controls) > 2.0 as a cut-off to identify probes that were strongly differentially methylated between cases and controls.
Systematic review of published findings
A systematic review was conducted and reported according to the 2020 Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines[43].
Eligibility criteria
Population: We included studies of HER2-positive breast cancer patients treated with any type of anti-HER2 agent (trastuzumab [Herceptin®], lapatinib [TYKERB®], pertuzumab [Perjeta®], trastuzumab emtansine [trastuzumab-DM1, Kadcyla®], erlotinib [Traceva®], gefitinib [Iressa®]) regardless of age, stage, and menopausal status.
Exposure: To be included, a study had to measure response-specific survival using including pathologic complete response (pCR), disease-free survival (DFS), progression-free survival (PFS), or event-free survival (EFS).
Outcome: We considered all assessments of genetic and epigenetic alterations at the genome-wide level in breast cancer tissue or blood, whatever the measurement method.
Types of studies: Any observational or randomized controlled study that assessed the association between genome-wide genetic and epigenetic alterations in breast cancer specimens or blood and the response to anti-HER2 agents in HER2-positive breast cancer patients was eligible. Case reports were excluded.
Only full available articles in English were included.
Information sources
Dragic D searched the PubMed biomedical database from inception to the search date of March 25, 2021, to identify eligible studies.
Search strategy
The search strategy was developed by Dragic D and Furrer D, approved by Diorio C, using controlled vocabulary search terms and free-text words related to HER2-positive breast cancer, genome-wide genetic and epigenetic alterations and treatment outcome [Supplementary Table 2]. No restrictions regarding language were applied.
Selection process
The references identified by the search strategy were selected according to the predefined eligibility criteria in a two-step process: titles and abstracts were screened by one author (Dragic D), and full texts of retained articles were examined by two authors (Dragic D and Furrer D). Disagreements between the two authors were discussed until a consensus was reached, and whenever required, a third review author (Diorio C) was consulted.
Data collection process
We designed a data extraction form for this review. Data were extracted by two authors (Furrer D and Dragic D) for half of the included studies. A third author (Diorio C) was consulted when discrepancies between both authors could not be resolved. For the remaining studies, the extraction was done by one author (Dragic D), and when needed, a second author (Diorio C) was involved. Additionally, the authors of studies of interest that lacked data to evaluate eligibility (n = 1) or other measures needed for this review
Data items
For all selected articles, study characteristics (study design and sample size), patient’s characteristics (age, stage, ER and PR status, menopausal status, and treatment received), assessment of genetic and epigenetic alterations (tissue processing, DNA, RNA or miRNA extraction method, assessment method, and parameters used), as well as statistical methods and study results, were collected. The study’s definition of response to targeted treatment was recorded.
Risk of bias assessment
Studies included in the review were assessed for risk of bias using the “Risk Of Bias in Non-randomized Studies of Interventions (ROBINS-I) tool[44]. The following domains were assessed: selection of participants in the study, exposure measurement, outcome measurement, potential confounding factors, missing data, and selective reporting.
Assessment of the risk of bias was done by two authors (Furrer D and Dragic D) for half of the included studies. Inconsistencies were discussed to reach a consensus. For the remaining studies, the assessment was done by one author (Dragic D), and when required, a second reviewer (Furrer D) was consulted.
Assessment of heterogeneity
Differences between studies, including study design, patient characteristics (age, menopausal status, ethnicity, and treatment received), tumor characteristics (stage, ER status), assessment of genome-wide genetic and epigenetic alterations (tissue processing, extraction method, and measurement method), and different levels of risk of bias were considered for exploring possible sources of heterogeneity.
Synthesis methods
Considering that high heterogeneity between studies was expected, quantitative data synthesis was not considered appropriate. Instead, we adopted a qualitative systematic review approach to investigate the relationship between epigenetic and genetic alterations and response to HER2-targeted therapies in HER2-positive breast cancer patients. The selection process was detailed using the PRISMA 2020 flow diagram. Extracted data were first reported in a table gathering summary characteristics of all included studies. Additional information specific to the epigenetic or genetic method used was detailed in several tables. Results and genes identified by several studies were also highlighted in a table. If pathway analysis was not presented, we performed pathway analysis using the list of the differentially expressed genes reported by the study authors and the PANTHER online software (Protein Analysis Through Evolutionary Relationships).
RESULTS
Pilot study of new findings
Genome-wide DNA methylation data in 12 breast cancer specimens were obtained from six trastuzumab-treated HER2-positive breast cancer patients who experienced recurrence during follow-up (cases) and six individually matched patients who had not developed recurrence and were alive at the date of the case’s recurrence (controls). Baseline characteristics of cases and controls are summarized in Table 1. Baseline characteristics for both groups were comparable in clinicopathological characteristics (tumor grade, lymph node status, and tumor size) and treatment received. Compared to controls, a higher proportion of cases (50%) had a body mass index > 25 kg/m2, although not statistically significant.
Baseline characteristics of the whole study population, case group and control group
| Factor | Whole study (n = 12) | Cases (n = 6) | Controls (n = 6) | P-value |
| Mean ± SD | Mean ± SD | Mean ± SD | ||
| Age (years) | 51.3 ± 5.6 | 51.3 ± 6.1 | 51.2 ± 5.6 | 0.87 |
| Body mass index (kg/m2) | 23.8 ± 2.2 | 23.8 ± 2.6 | 23.9 ± 1.6 | 0.94 |
| Follow-up time (years) | 5.5 ± 2.9 | 3.8 ± 2.8 | 7.2 ± 2.1 | 0.04 |
| n (%) | n (%) | n (%) | ||
| Grade I/II III | 3 (23%) 9 (77%) | 2 (33%) 4 (67%) | 1 (17%) 5 (83%) | 1.00 |
| Lymph node status Negative Positive | 1 (8%) 11 (92%) | 0 (0%) 6 (100%) | 1 (17%) 5 (83%) | 1.00 |
| Tumor size (cm) ≤ 5 > 5 | 11 (92%) 1 (8%) | 5 (83%) 1 (17%) | 6 (100%) 0 (0%) | 1.00 |
| Estrogen receptor status Negative Positive | 4 (33%) 8 (67%) | 2 (33%) 4 (67%) | 2 (33%) 4 (67%) | 1.00 |
| Progesterone receptor status Negative Positive | 6 (50%) 6 (50%) | 3 (50%) 3 (50%) | 3 (50%) 3 (50%) | 1.00 |
| Menopausal status Pre Post | 4 (33%) 8 (67%) | 2 (33%) 4 (67%) | 2 (33%) 4 (67%) | 1.00 |
| Radiotherapy No Yes | 1 (8%) 11 (92%) | 1 (17%) 5 (83%) | 0 (0%) 6 (100%) | 1.00 |
| Endocrine therapy No Yes | 4 (33%) 8 (67%) | 2 (33%) 4 (67%) | 2 (33%) 4 (67%) | 1.00 |
| Chemotherapy No Yes | 0 (0%) 12 (100%) | 0 (0%) 6 (100%) | 0 (0%) 6 (100%) | 1.00 |
| Trastuzumab treatment completed No Yes | 0 (0%) 12 (100%) | 0 (0%) 6 (100%) | 0 (0%) 6 (100%) | 1.00 |
Global methylation levels between cases and controls were not statistically different: the median M-values of cases were 0.487, and the median M-values of controls were 0.504 (P-value: 0.844). At probe methylation levels, we identified 2,009 CpGs (1,382 genes) that were differentially methylated between cases and controls: 1,200 DMPs (885 genes) were significantly hypermethylated and 809 DMPs (497 genes) were significantly hypomethylated in tumor tissues of cases compared to those of controls after multiple testing correction (FDR < 0.05).
Fifteen genes had a |log2FC| > 2.0: ten genes (SIX2, PLEC1, ZNF833, RAI1, ZNF598, USP4, DOCK1, UNC84A, KLF16, PRKACA) were significantly hypermethylated, and five genes [STK33, TBXT (alias T), KCNH7, ADAMTS2, FAM19A5] were significantly hypomethylated in breast cancer tissues of cases compared to controls. Results are reported in Table 2.
Genes differentially methylated in breast cancer tissues of cases compared to controls
| CpG sites | Chr | Gene | Gene region | CpG island region | LogFC | q-value |
| cg08788717 | chr11 | STK33 | TSS200 | Island | -2.835 | 0.036 |
| cg02149708 | chr6 | TBXT (T) | TSS200 | Island | -2.384 | 0.038 |
| cg26974327 | chr2 | KCNH7 | TSS200 | OpenSea | -2.150 | 0.037 |
| cg05214690 | chr5 | ADAMTS2 | 1stExon | Island | -2.127 | 0.015 |
| cg22643811 | chr22 | FAM19A5 (TAFA5) | 1stExon | Island | -2.024 | 0.015 |
| cg26391832 | chr2 | SIX2 | TSS1500 | S_Shore | 2.042 | 0.028 |
| cg21672292 | chr8 | PLEC1 | Body | Island | 2.097 | 0.039 |
| cg26590664 | chr19 | ZNF833 | TSS200 | N_Shore | 2.114 | 0.030 |
| cg21771200 | chr19 | ZNF833 | TSS200 | N_Shore | 2.270 | 0.028 |
| cg02147681 | chr17 | RAI1 | 5'UTR | Island | 2.235 | 0.035 |
| cg03654304 | chr16 | ZNF598 | Body | Island | 2.287 | 0.038 |
| cg18886444 | chr3 | USP4 | TSS1500 | S_Shore | 2.366 | 0.038 |
| cg26353296 | chr3 | USP4 | TSS1500 | S_Shore | 2.381 | 0.034 |
| cg06406458 | chr10 | DOCK1 | Body | OpenSea | 2.028 | 0.044 |
| cg26987690 | chr7 | UNC84A | Body | S_Shore | 2.037 | 0.037 |
| cg08287334 | chr19 | KLF16 | Body | Island | 2.230 | 0.049 |
| cg19586199 | chr19 | PRKACA | TSS200; body | N_Shelf | 2.582 | 0.030 |
Systematic review of published findings
Study selection
Of the 758 references retrieved by electronic search in PubMed, we reviewed 52 full-text documents, and fifteen met the eligibility criteria [Figure 1]. This review also included our pilot study that assessed the association between DNA methylation patterns in breast cancer specimens and response to trastuzumab in a cohort of 12 HER2-positive breast cancer patients.
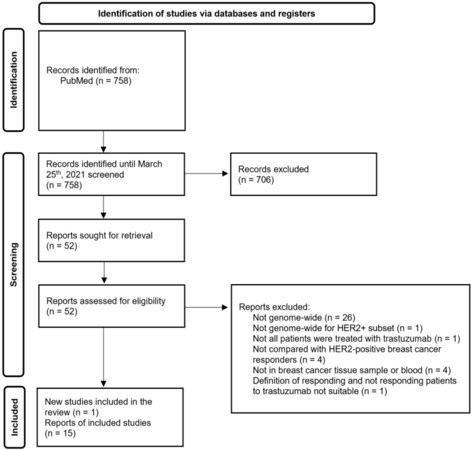
Figure 1. Process flow for article selection (PRISMA 2020 flow diagram).
Study characteristics
In all selected studies, epigenetic and genetic patterns were measured genome-wide in breast cancer tissues or blood. DNA methylation patterns were evaluated in one study (our pilot study), gene expression profile in nine studies[45-53], miRNA expression in two studies[54,55], long intergenic non-coding RNA (lincRNA) profile in one study[50], copy number alteration (CNA) profile in two studies[48,56], protein expression in one study[57] and mutations in two studies[58,59]. One study reported both genome-wide gene expression and genome-wide CNA patterns[48], and one reported both genome-wide gene and genome-wide lincRNA expression profiles[50]. The majority of studies were retrospective (n = 10)[46,47,49-51,54-57] including ours, and six studies were prospective[45,48,52,53,58,59] [Table 3].
Summary characteristics of studies reporting on the association between genome-wide epigenetic and genetic modifications and response to HER2-targeted therapies in HER2-positive breast cancer patients (n = 16)
| Study design | Prospective studies: n = 6 (Harris[45], Guarneri[48], Shi[59], Sorokin[52], Perez[53], Lesurf[58]) Retrospective studies: n = 10 (Khoury[46], Gámez-Pozo[47], Triulzi[49], Du[54], Merry[50], Ohzawa[55], Zhao[51], Yang[57], Walsh[56], Furrer [2022]) |
| HER2-positive breast cancer patients | Number of patients: 9 to 849 Age: median - 50 to 59 y (n = 6) (Gámez-Pozo[47], Triulzi[49], Ohzawa[55], Walsh[56], Yang[57], Lesurf[58]); mean - 51.3 to 55.2 y (n = 2) (Furrer [2022], Sorokin[52]); < 45 y in 61% (n = 1) (Harris[45]); < 50 y in 48.3% (n = 1) (Perez[53]); < 65 y in 90.3% (n = 1) (Shi[59]); NR (n = 5) (Khoury[46], Merry[50], Guarneri[48], Zhao[51], Du[54]) Stage: Non metastatic I-III: n = 1 (Du[54]) Non metastatic II-III: n = 4 (Harris[45], Ohzawa[55], Sorokin[52], Furrer [2022]) Non metastatic NR: n = 3 (Guarneri[48], Triulzi[49], Perez[53]) Metastatic IV: n = 2 (Gámez-Pozo[47], Walsh[56]) Non metastatic and metastatic: n = 1 (Khoury[46]) NR: n = 5 (Merry[50], Shi[59], Zhao[51], Yang[57], Lesurf[58]) Treatment received: Neoadjuvant NVB and T: n = 1 (Harris[45]) Adjuvant T: n = 1 (Khoury[46]) Adjuvant or neoadjuvant CT (ATC-based, TAX-based, ATC + TAX, Other CT) and T: n = 1 (Gámez-Pozo[47]) Neoadjuvant CT (PTX and FEC) plus either T (arm A), L (arm B), or T + L (arm C): n = 1 (Guarneri[48]) Neoadjuvant PTX plus either T, L, or T + L: n = 1 (Shi[59]) Adjuvant CT and T: n = 4 (Triulzi[49], Du[54], Merry[50], Ohzawa[55]) Neoadjuvant CT and T: n = 2 (Zhao[51], Lesurf[58]) Adjuvant T only or T plus DTX/PTX + CBDCA/PTX/DTX + CBDCA/Cap/NVB/Gem: n = 1 (Sorokin[52]) Neoadjuvant therapy or surgical treatment and T: n = 1 (Yang[57]) T: n = 1 (Walsh[56]) Adjuvant ATC + CTX, PTX, T, or ATC + CTX, PTX + T, T: n = 1 (Perez[53]) |
| Genome-wide profiling method | Methylation: n = 1 (Furrer [2022]) miRNA expression: n = 2 (Du[54], Ohzawa[55]) Gene expression: n = 9 (Harris[45], Khoury[46], Gámez-Pozo[47], Guarneri[48], Triulzi[49], Merry[50], Zhao[51], Sorokin[52], Perez[53]) CNA: n = 2 (Guarneri[48], Walsh[56]) lincRNA expression: n = 1 (Merry[50]) Protein expression: n = 1 (Yang[57]) Mutations: n = 2 (Lesurf[58], Shi[59]) |
| Outcome | Disease-free survival (DFS): n = 4 (Khoury[46], Du[54], Sorokin[52], Furrer [2022]) Relapse-free survival (RFS): n = 2 (Perez[53], Triulzi[49]) Pathological complete response (pCR): n = 10 (Harris[45], Gámez-Pozo[47], Guarneri[48], Merry[50], Shi[59], Ohzawa[55], Zhao[51], Yang[57], Walsh[56], Lesurf[58]) |
DFS was reported in four studies including ours[46,52,54], RFS in two studies[49,53], and pCR in ten studies[45,47,48,50,51,55-59]. DFS was defined as the time between the diagnosis and recurrence (locoregional recurrence, recurrence in the contralateral breast, and distant breast cancer recurrence) or death; RFS was defined as the time from the start of trastuzumab treatment to the first local, regional or distant recurrence event; and pCR as the absence of invasive breast cancer in the breast and axillary lymph nodes at the time of surgery. Besides ours, the other fifteen included studies were published between 2007 and 2020 and involved between 9 and 849 HER2-positive, anti-HER2 therapy-treated breast cancer patients [Table 3].
Studies of genome-wide DNA methylation and response to targeted treatment
Characteristics of the study (our new findings) that evaluated the association between genome-wide DNA methylation patterns measured in breast cancer tissues and response to targeted treatment in HER2-positive, trastuzumab-treated breast cancer patients are presented in Supplementary Table 3. The study was retrospective. The mean age of included HER2-positive breast cancer patients was 51.3 years. The proportion of ER-positive breast cancer patients was 67%, and the proportion of positive lymph node status was 92%. Sixty-seven percent of HER2-positive breast cancer patients were postmenopausal. DNA methylation was assessed using Infinium HumanMethylation450 BeadChip array. Breast cancer patients were treated with adjuvant chemotherapy and trastuzumab. The study reported DFS. HER2-positive breast cancer patients were of Caucasian ethnicity. Tumor cell fraction was ≥ 70%.
Studies of genome-wide gene expression profile and response to targeted treatment
Characteristics of the nine studies that examined the association between genome-wide gene expression profiles measured in breast cancer tissues and response to HER2-targeted therapies in HER2-positive breast cancer patients are presented in Supplementary Table 4. Five studies were retrospective[46,47,49-51], and four studies were prospective[45,48,52,53]. The median age of HER2-positive breast cancer patients was not reported in four studies[46,48,50,51] and varied from 52 to 58 years in two studies[47,49]. The mean age was 55.2 years in one study[52], 60.9% of participants were < 45 years in another[45], and 48.3% of participants were < 50 years in one other study[53]. The proportion of ER-positive breast cancer patients was reported in three studies and varied between 43.4% and 69.6%[45,47,49]. In eight studies, patients received trastuzumab[45-47,49-53], and in the remaining study, patients were treated with trastuzumab, lapatinib or both[48]. pCR was reported in five studies[45,47,48,50,51], RFS in two studies[49,53], and DFS in two studies[46,52]. Gene expression profile was evaluated in fresh frozen tissue in five studies[45,46,48,50,51] and in FFPE tissues in four studies[47,49,52,53]. Gene expression profile was assessed using HumanHT-12 v3 BeadChip[46], HumanHT-12 v4 BeadChip[49], Affymetrix Human Genome U219 array[47], Affymetrix GeneChip 3’ IVT Express kit[48], Illumina HiSeq 2500 platform[50], Illumina HiSeq 3000[52], GeneChip U133 Plus 2.0 Gene Array[45], DASL technology[53] and Agilent UNC Perou Lab Homo sapiens 1X44K Custom Array/Illumina HumanHT-12 WG-DASL V4.0 R2 expression BeadChip/Affymetrix technology using the HG-U133 Plus 2.0 GeneChip array[51]. Tumor cell fraction was not reported in five studies[47,48,50,51,53], and was at least 70% in three studies[46,49,52]. In the remaining study, breast cancer samples of each patient contained at least 500 breast cancer cells[45] [Supplementary Table 4].
Study of genome-wide miRNA expression profile and response to targeted treatment
Characteristics of the two studies that examined the association between genome-wide miRNA expression profile and response to targeted treatment in HER2-positive trastuzumab-treated breast cancer patients are presented in Supplementary Table 5. Both studies had a retrospective design[54,55]. miRNA expression was measured genome-wide in FFPE breast cancer tissues using Agilent miRNA assay[54] or Agilent SurePrint G3 Human miRNA microarray[55] in cohorts of 14 and 40 HER2-positive breast cancer patients. Patients were treated with trastuzumab. DFS[54] and pCR[55] were reported. Tumor cell fraction was not reported in one study[54] and was at least 80% in the other[55].
Study of genome-wide lincRNA expression profile and response to targeted treatment
Characteristics of the single study that examined the association between genome-wide lincRNA expression profile and response to targeted treatment in HER2-positive trastuzumab-treated breast cancer patients are presented in Supplementary Table 6. The study had a retrospective design. lincRNA expression was measured genome-wide in fresh frozen breast cancer tissues using the Illumina HiSeq 2500 platform in a cohort of 13 HER2-positive breast cancer patients. Patients were treated with trastuzumab and pCR was reported. Tumor cell fraction was not reported.
Study of genome-wide CNA profile and response to targeted treatment
Characteristics of the two studies that examined the association between genome-wide CNA profile and response to HER2-targeted therapies in HER2-positive breast cancer patients are presented in Supplementary Table 7. The studies had a prospective design[48,56]. CNA profile was assessed genome-wide in fresh frozen breast cancer tissues using Affymetrix Genome-wide Human SNP array[48] or Illumina HiSeq[56] in cohorts of 68 and 11 HER2-positive breast cancer patients. Patients were treated with trastuzumab, lapatinib or both in one study[48] and trastuzumab in the other[56]. pCR was reported. Tumor cell fraction was not reported.
Study of genome-wide protein expression profile and response to targeted treatment
Characteristics of the only study that examined the association between genome-wide protein expression profile and response to targeted treatment in HER2-positive trastuzumab-treated breast cancer patients are presented in Supplementary Table 8. The study had a retrospective design[57]. The protein expression profile was assessed genome-widely in blood using TMT-6 plex Isobaric Label Reagent Set in a cohort of 6 HER2-positive breast cancer patients. Patients were treated with trastuzumab. pCR was reported.
Study of genome-wide mutation profile and response to targeted treatment
Characteristics of the two studies that examined the association between genome-wide somatic or germline mutation profile and response to targeted treatment in HER2-positive breast cancer patients are presented in Supplementary Table 9. The studies had a prospective design[58,59]. Somatic mutation profile was assessed genome-wide in fresh frozen breast cancer tissues for the two studies. Germline mutation profile was assessed genome-widely in blood using Illumina HiSeq 2000[59], and whole genome sequencing (WGS) and whole exome sequencing (WES) + Illumina HiSeq 2000 platform[58] in cohorts of 48 and 203 HER2-positive breast cancer patients. Patients were treated with trastuzumab. pCR was reported. Tumor cell fraction was not reported in one study[58] and was at least 10% in the other study[59].
Risk of bias in studies
Overall, studies ranged on average from low to critical risk of bias [low (n = 2)[53,59], moderate (n = 4, including our study)[47,49,57], serious (n = 1)[54], and critical (n = 9)][45,46,48,50-52,55,56,58], most commonly due to confounding. The bias evaluation of each included study is presented in Supplementary Table 10.
Results of individual studies
Genome-wide gene expression profile in breast cancer tissues and response to anti-HER2 agents in HER2-positive breast cancer patients
The genes reported to be differentially expressed in breast cancer tissues of cases compared to controls for each identified study are presented in Supplementary Table 11. Higher expression of the following genes was consistently observed in two different studies: ESR1[49,50], RBP1, SLP1[46,50], EPS8L1[50,52], MEP1B, UTP15, RRAS2, GRB10, FAM98A, RBMS2, RPL9, RPAIN, MORF4L1, LLPH, MTMR1, FRYL, FLCN, CLINT1, ERICH1[47,52], ZNF281, ANKRD52, PIAS3, BRD1, RNF146, CLDN12, PROM2, COMMD5, VMP1, DEDD, ZNF439, CRIP2, PRSS16, GUK1, RRN3, PLEKHG3, JUN, BCL9, SLC25A37, CRYZ, RNF24, PSMG3, PAQR7, ABT1, WNT7B, SLC35B2, SYTL4, NUPR1, DPY19L1, DAZAP1, EEF1D, SGPP1, GALNT2, SPA17, RAD51, MBD6, KIF1C, C1QTNF3, BLOC1S2, SLC2A10, ZNF740, ADCK2, SLC41A1, RAB4A, CRIP1, ZNF552, CARHSP1, POFUT1, EMC10, BAX, HOXC4, DDR1, CTSD, FEN1, SULT1A1, DUSP14, IRF9, TMC4, MUC1, LMAN2, LASP1, SHROOM1[50,52], NSL1, ENAH, UBE2Q2, GNPAT, THBS2, TBCEL, FAM46A, ZNF678, TSEN15, ZNF674, CNIH4, ASAH1, SELT, ARFGAP3, TATDN3, FBLN1, MOSPD1, PPCS, NUCKS1, PGBD2, ACBD3, ORMDL2, AMMECR1, TNFRSF19, FOSL2, PYCR2, WSB1, TROVE2, RWDD3[47,50], LYSMD3[47,50], APOB, SLC3A2, CST3[52,57], BOC[45,52], DDX27, IL17RC, PKP3, WNK2[52,53] AGRN, ATRN, NACC1, PCSK6, PIR, PLAUR, QSOX1, RHBDL2, SEC22B, SERPINH1, TCEAL1[50,53], CSDE1, SCUBE3, TMEM167A[47,53]. Lower expression of the following genes was consistently reported in two different studies: ORMDL3[49,50], NME1[46,50], SLC35B1[46,50], UTP18[46,50], PHB[46,50], S100B, TOM1L1[50,52], HIST1H2BG, PRR13[47,50], NXPH1, CKS2, NETO2, SLC12A2[46,52], ACSL5, CD3E, GIMAP7, GZMK, PPARG, SELL, VCAM1[50,53], CLEC10A, CTLA4, FGL2, TSPAN7[52,53], DENND3, TIAM1[47,53]. The following genes were reported to be higher expressed in breast cancer samples of cases compared to controls in two different studies but were reported to be lower expressed in another study: SFRP1[45,46,50], ACTR1B, FASTK, TMEM219, NDUFA3[47,50,52], ATP5I[47,50,52]. The following genes were reported to be lower expressed in breast cancer samples of cases compared to controls in two different studies but were reported to be higher expressed in another study: PABPC1[47,50,52], ARPC1A, TXN[47,50,52], TOB1[46,50,52]. Higher expression of the GOLGA2[47,50,52] and PHF21A[47,50,52] genes was consistently observed in three different studies.
Gámez-Pozo et al. and Sorokin et al. identified several pathways associated with response to trastuzumab, including those involved in EGF receptor signaling, PI3K, apoptosis signaling, and p53[47,52].
In our study, we observed overlap between the identified differentially expressed genes and the strongly differentially methylated (i.e., |log2FC| > 2.0) genes. PRKACA was hypermethylated in our study (within the TSS region as well as the gene body), upregulated in one study[50], and downregulated in another study[47].
Genome-wide miRNA expression profile in breast cancer tissues and response to trastuzumab in HER2-positive breast cancer patients treated with trastuzumab
Du et al. identified seven upregulated and two downregulated miRNAs in breast cancer tissues of cases compared to controls[54]. Ohzawa et al. identified four upregulated and ten downregulated miRNAs in breast cancer tissues of cases compared to controls[55] [Supplementary Table 13]. Regarding the miRNAs identified by Du et al., 902 genes were predicted to be targeted by miR-150-5p, 47 genes by miR-4734, 570 genes by miR-361-5p, 1,134 genes by miR-26a-5p, 416 genes by miR-365a-3p, 701 genes by miR-155-5p, 737 genes by miR-205-5p, 1,384 genes by miR-106b-5p, and 187 genes by miR-424-3p (as illustrated in a database for miRNA target prediction and functional annotations available online at www.mirdb.org)[54]. For the miRNAs reported by Ohzawa et al., 484 genes were predicted to be targeted by miR-210, 242 genes by miR-31-3p, 891 genes by miR-449a, 801 genes by miR-449b-5p, 21 genes by miR-106b-3p, 263 genes by miR-1180, 242 genes by miR-1238-5p, 1,133 genes by miR-142-5p, 902 genes by miR-150-5p, 1,409 genes by miR-181c-5p, 1,266 genes by miR-182-5p, 438 genes by miR-20a-5p, 1,084 genes by miR-218-5p, 1,249 genes by miR-3609, 270 genes by miR-362-5p, 420 genes by miR-3620-3p, 676 genes by miR-4418, 272 genes by miR-4506, 410 genes by miR-4657, 406 genes by miR-505-3p, and 392 genes by miR-505-5p[55].
We observed that among the 344 genes that were reported to be differentially expressed (at the mRNA level) between breast cancer tissues of cases and controls in at least two studies, 170 were predicted to be targeted by miRNA identified as differentially expressed between breast cancer tissues of cases and controls in Ohzawa et al.[55] or Du et al.[54] [Supplementary Table 14]. We observed that among the 15 genes identified as differentially methylated in our study, five (KCNH7, ADAMTS2, SIX2, DOCK1 and ZNF598) were predicted to be targeted by miRNA identified as differentially expressed in breast cancer tissues of cases compared to controls in the study of Ohzawa et al.[55] or Du et al.[54] [Supplementary Table 14].
Genome-wide lincRNA expression profile in breast cancer tissues and response to targeted treatment in HER2-positive breast cancer patients treated with trastuzumab
Merry and collaborators observed that 371 lincRNAs were differentially expressed in non-pCR samples compared to pCR samples, where 33 lincRNAs showed decreased expression and 338 increased expression[50] [Supplementary Table 15]. Among these 371 genes, 97 were reported to be differentially expressed at the mRNA level in breast cancer tissues of cases compared to controls in at least two studies: FAM84B, PHF21A, NDUFV3, COMMD6, SRP9, S100B, WDR26, LYSMD3, CSTB, C5orf39, FOXA1, GALM, ITGB2, SP140, UTRN, SAA2, SHB, ZFP64, ZC3H12B, NADSYN1, B4GALNT4, AP2B1, USP16, ARL4D, SYNPO2, FAIM3, CBR1, EFR3B, IL18, LOC389493, FBXO16, FAM114A1, MAP3K9, TOX3, ZNF681, IDH3B, TPBG, PRKACB, UBE2A, ID2, IRX3, CILP, COL5A2, WRB, MBOAT1, GCA, SATB2, HERC6, RALGPS2, NUF2, MIA3, FAM91A1, FAM5C, C1orf227, RPP4, MYOT, PRKAA2, MAP1LC3B, PEX3, MEST, F13A1, CREB1, LRCH2, PCF11, POLR3G, RORA, USP3, TSHZ3, CXCR4, CCNH, CCM2, ZNF814, RAPH1, ZPBP2, FBXW4, ODF3B, CROCC, SH3RF2, HEATR6, CDK13, ATF3-1, ATF3, DTL, IARS2, RC3H1, RC3H1, URB2, RHOU, RC3H1, RC3H1, SUPT3H, ABI1, OTUD7B, GPATCH2, RNF2, IRF2BP2.
Among the differentially expressed lincRNAs reported by Merry et al.[50], we observed 44 genes that were predicted to be target genes of miRNA identified as differentially expressed in the study of Ohzawa et al.[55] or Du et al.[54] [Supplementary Table 14].
None of the genes whose lincRNAs were reported to be differentially expressed between patients showing pCR and those showing non-pCR in Merry et al. was reported as differentially methylated with a
Genome-wide copy number alterations in breast cancer tissue and response to targeted treatment in HER2-positive breast cancer patients treated with anti-HER2 agents
In their analysis of the association of genome-wide CNA in breast cancer tissues of HER2-positive breast cancer patients who received trastuzumab, lapatinib, or both, with response to anti-HER2 treatment, Guarneri et al. reported that, unlike pCR patients, non-pCR patients showed a CNA signature[48]. Overall, the authors observed CN alterations, mainly CN gains, in 557 genes located on chromosomes 1, 8, 17, 20
We observed that among the 557 genes that were reported to show CN alteration in non-pCR samples compared to pCR-samples in the study of Guarneri et al.[48], 279 were predicted to be targeted by miRNA identified as differentially expressed in breast cancer tissues of cases compared to controls in the study of Du et al.[54] or Ohzawa et al.[55] [Supplementary Table 14].
Among the genes showing CN alterations in non-pCR samples compared to pCR-samples in the study of Guarneri et al.[48], 18 genes (FAM84B, SRP9, WDR26, MIA3, FAM91A1, FAM5C, C1orf227, ATF3, DTL, IARS2, RC3H1, URB2, RHOU, RC3H1, OTUD7B, GPATCH2, RNF2, IRF2BP2) showed higher lincRNA expression and one (FAIM3) showed lower lincRNA expression in Merry et al.[50].
No overlap was observed between genes showing CN alterations in non-pCR samples compared to pCR-samples in Guarneri et al. and genes identified as differentially methylated with a |log2FC| > 2.0 in our study[48]. However, when we consider the whole list of differentially methylated genes, we identified 27 genes that were differentially methylated in breast cancer tissues of cases compared to controls in our study among the 557 genes showing CNV variations in the study of Guarneri et al.[48]. Of these 27 differentially methylated genes, nine were hypomethylated (PLD5, GJD2, C20orf85, APCDD1L, VASH2, PSEN2, FMN2, EDN3, ACTN2) and 18 were hypermethylated (TRAF5, TSEN15, TRIB1, TMEM206, SNX31, SMG7, RGS1, PRG4, PGBD5, NID1, KCNV1, GPATCH2, EXT1, CDK18, CAPN9, ADSS, ABR, C1orf55).
Genome-wide protein expression profile in blood of breast cancer cases compared to controls
Out of the 18 genes that were reported as differentially expressed (five downregulated and 13 upregulated) in the blood of breast cancer cases compared to controls by Yang et al.[57] [Supplementary Table 17], three
None of the differentially expressed genes in the study of Yang et al. were differentially methylated in our study[57].
Genome-wide somatic and germline mutations profile in breast cancer tissues of cases compared to controls
Whereas Shi and collaborators observed that higher somatic mutation frequency in the PIK3CA gene in the breast tissues of cases was associated with trastuzumab resistance[59], Lesurf et al. reported that no somatic or germline mutations were associated with response to trastuzumab in breast cancer tissues of cases compared to controls[58] [Supplementary Table 18].
DISCUSSION
In a cohort of 12 HER2-positive breast cancer patients treated with trastuzumab, interrogation of DNA methylation using the Infinium HumanMethylation450 BeadChip allowed identifying genes that were differentially methylated between trastuzumab-resistant and trastuzumab-sensitive HER2-positive breast cancer patients. Interestingly, among the strongly differentially methylated genes, we observed genes associated with human cancer, including DOCK1, ADAMTS2, PLEC1, USP4, and PRKACA[60-69].
The guanine nucleotide exchange factor DOCK1 (Dedicator of cytokinesis protein 1) is involved in cytoskeletal rearrangements required for phagocytosis of apoptotic cells and cell mobility[70]. A recent study reported that DOCK1 inhibition leads to suppressed migration of the triple-negative breast cancer cell lines MDA-MB-157 and MDA-MB-231[68,71]. ADMATS2 (ADAM metallopeptidase with thrombospondin type 1 motif 2) belongs to the ADAM metallopeptidase with thrombospondin type 1 motif and processes collagen precursors into mature collagen molecules[72]. It has been proposed that ADAMTS2 exerts an anti-tumor effect by inhibiting intratumoral vascularization[73]. The PLEC gene encodes the pectin protein, which plays a role in maintaining tissue integrity[74]. A recent study suggests that a PLEC gene polymorphism (rs138924815) might increase the risk for familial testicular cancer[75]. USP4 (Ubiquitin carboxyl-terminal hydrolase 4) is a deubiquitinating enzyme that removes conjugated ubiquitin from target proteins[76]. It has been reported that USP4 expression was decreased in breast cancer tissue samples compared to paired normal breast tissues[63]. Moreover, USP4 expression was associated with decreased proliferation in two HER2-negative breast cancer cell lines (MCF7 and BT549)[63]. The PRKACA gene encodes for a protein kinase that plays a role in controlling cellular processes such as glucose metabolism and cellular division[72]. Of note, one recent study suggests that PRKACA expression might be associated with the development of trastuzumab resistance in HER2-positive breast cancer patients[64]. The authors observed that in a subgroup of HER2-positive breast cancer patients who developed trastuzumab resistance (three out of five patients), PRKACA expression was highly increased in the breast cancer sample obtained after the onset of trastuzumab resistance compared to the pre-treatment sample. Considering that in our study, the PRKACA gene was hypermethylated within the gene body and that hypermethylation within this gene region often promotes gene elongation and, therefore, gene expression, we can postulate that our results might be concordant with those reported by Moody et al.[64]. Although in our study, DNA methylation was exclusively measured in pre-treatment samples and Moody et al. observed increased PRKACA expression only in breast cancer samples obtained after the onset of recurrence[64].
Among all genes that we identified as strongly differentially methylated between trastuzumab-resistant and trastuzumab-sensitive HER2-positive breast cancer patients, one of them, PRKACA, was reported as being higher expressed in breast cancer tissues of cases compared to controls in the study conducted by Merry and collaborators[50] and as being lower expressed in breast cancer tissues of cases compared to controls in the study of Gámez-Pozo et al.[47]. In our study, the PRKACA gene was hypermethylated within the TSS region and the gene body. Our observation could be partly concordant with the results reported by
In one of the studies retained in our systematic review, the authors created a predictive model to differentiate HER2-positive trastuzumab-treated breast cancer patients with a higher risk of relapse from those with a lower risk in a cohort of 53 patients[49]. The validity of this predictive model was then confirmed in an independent and bigger data set. The authors observed that differentially expressed genes in the breast cancer tissues of patients identified as at low risk for relapse in the independent data set using this model were associated with the immune system. When we performed gene enrichment analysis of genes showing CN alteration in the study of Guarneri and collaborators, we observed that pathways associated with the immune response (inflammation mediated by chemokine and cytokine signaling pathway) were overrepresented. The immune system's involvement in response to anti-HER2 agents in HER2-positive breast cancer patients has also been reported in other studies[77-82].
To our knowledge, we conducted the first systematic review on the association of epigenetic and genetic alterations in breast cancer tissues or blood with the response to anti-HER2 agents in HER2-positive breast cancer patients. Sixteen studies were included in this review, and very few overlaps between studies were found. The most consistent results were the higher expression of GOLGA2 and PHF21A genes and the higher expression and CN gain of MIA3, WDR26 and C1orf133 genes observed in three different studies. Among these five genes, WDR26 has been shown to promote breast cancer growth and metastasis via the PI3K/AKT signaling pathway[83]. Gámez-Pozo and collaborators also observed that genes identified as differentially expressed in breast tumor tissues of cases compared to controls were overrepresented in the PI3K pathway[47]. Interestingly, the study by Shi and collaborators[59] also reported that PI3K mutations were associated with response to anti-HER2 agents in HER2-positive breast cancer patients and other previously published studies[84-86]. Taken together, these results suggest that genes of the PI3K pathway might play a relevant role in the development of resistance to anti-HER2 agents in breast cancer patients. Exploring the ramifications of these and other findings in larger cohorts or datasets like TCGA should be considered in future studies.
Several factors might explain why we only observed a few overlaps in our systematic review. Tumor cell content varied from > 40% to > 80% between studies, and in several publications (n = 4), this information was not provided. Contamination with cell types other than breast cancer cells can modify the observed pattern of genetic and epigenetic markers, as DNA methylation and other epigenetic or genetic markers widely vary across tissues and cellular types[87]. Moreover, the type of outcome evaluated in the study might also play a role, as the assessment of pCR in the neoadjuvant setting might mainly identify patients who did not primarily respond to targeted treatment (primary resistance), whereas the evaluation of DFS in the adjuvant setting might allow identifying patients who initially respond to targeted treatment but who develop resistance over time (acquired resistance). Moreover, epigenetic and genetic markers can be influenced by clinicopathological data, including age, stage, ER status, menopausal status, and ethnicity[88-96]. Unfortunately, it was difficult to evaluate this aspect, as patients’ clinicopathological data were not extensively reported in the majority of the included studies. The risk of bias in most studies was due to confounding.
CONCLUSION
In conclusion, although the sample size of the present pilot study was small and despite the lack of validation cohort, using a high-throughput analysis, we identified genes that were differentially methylated in breast cancer tissues of HER2-positive trastuzumab-treated breast cancer patients who developed resistance toward this drug compared to those who responded to targeted therapy. One of the most differentially methylated genes, PRKACA, has been reported to be differentially expressed in breast cancer tissues of trastuzumab-resistant compared to trastuzumab-sensitive HER2-positive breast cancer patients in two studies included in our systematic review. Although we identified very few genes that overlap between studies, our review suggests that some of the genes acting in the PI3K pathway, such as PRKACA[97], might play an important role in developing resistance to anti-HER2 agents in breast cancer patients. Although the associations between PI3KCA mutations and PI3K dysfunctions and anti-HER2 treatment resistance are well documented in the literature[86,98-100], further studies on this topic are needed, which may help to unveil carcinogenic mechanisms involved in this pathway.
Although the observations reported in the retained studies were only marginally concordant, our work and the studies presented in this article suggest that knowledge gathered from these high-throughput studies could be useful for the identification of novel biomarkers of trastuzumab resistance. This might promote the development of new targeted drugs that could be administered to trastuzumab-resistant HER2-positive breast cancer patients.
DECLARATIONS
Authors’ contributionsConceived, acquired, analyzed and interpreted the data, wrote the original draft: Furrer D, Dragic D
Analyzed the data, edited the manuscript: Fournier F, Droit A, Jacob S
Reviewed and edited the manuscript: Chang SL
Conceived and designed the study, acquired, analyzed and interpreted the data, wrote and edited the manuscript, supervised and acquired funding for the study: Diorio C
All authors read and approved the final manuscript.
Availability of data and materialsAll data generated or analysed during this study are included in this published article [and its supplementary information files].
Financial support and sponsorshipThis work was supported by the Canadian Breast Cancer Foundation-Canadian Cancer Society Development award (No.703003) and the Fondation du cancer du sein du Québec and the Banque de tissus et de données of the Réseau de recherche sur le cancer of the Fonds de recherche du Québec - Santé (FRQS), which is affiliated with the Canadian Tumour Repository Network and a FRQS Senior Investigator Award to Diorio C. Furrer D was supported by doctoral fellowships from FRQS and the Laval University Cancer Research Center. Dragic D is supported by doctoral fellowships from the University Laval Cancer Research Center and the University Paris-Saclay Doctoral School of Public Health EDSP.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateEthical approval of the study was obtained from the Research Ethics Committee of the Centre de Recherche du CHU de Québec (# 2016-2802).
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127-37.
2. Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu Rev Biochem 1987;56:881-914.
3. Soerjomataram I, Louwman MW, Ribot JG, Roukema JA, Coebergh JW. An overview of prognostic factors for long-term survivors of breast cancer. Breast Cancer Res Treat 2008;107:309-30.
4. Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol 2010;7:98-107.
5. Wolff AC, Hammond ME, Hicks DG, et al. American Society of Clinical Oncology. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997-4013.
6. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 2011;9:16-32.
7. Poon KA, Flagella K, Beyer J, et al. Preclinical safety profile of trastuzumab emtansine (T-DM1): mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol Appl Pharmacol 2013;273:298-313.
8. Balduzzi S, Mantarro S, Guarneri V, et al. Trastuzumab-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev 2014:CD006242.
9. Moja L, Tagliabue L, Balduzzi S, et al. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst Rev 2012:CD006243.
10. Swain SM, Baselga J, Kim SB, et al. CLEOPATRA Study Group. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724-34.
11. Xu ZQ, Zhang Y, Li N, et al. Efficacy and safety of lapatinib and trastuzumab for HER2-positive breast cancer: a systematic review and meta-analysis of randomised controlled trials. BMJ Open 2017;7:e013053.
12. D’Amato V, Raimondo L, Formisano L, et al. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev 2015;41:877-83.
13. de Melo Gagliato D, Jardim DL, Marchesi MS, Hortobagyi GN. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016;7:64431-46.
14. Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 2006;3:269-80.
15. Zhang S, Huang WC, Li P, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 2011;17:461-9.
16. Beauclair S, Formento P, Fischel JL, et al. Role of the HER2 [Ile655Val] genetic polymorphism in tumorogenesis and in the risk of trastuzumab-related cardiotoxicity. Ann Oncol 2007;18:1335-41.
17. Borley A, Mercer T, Morgan M, et al. Impact of HER2 copy number in IHC2+/FISH-amplified breast cancer on outcome of adjuvant trastuzumab treatment in a large UK cancer network. Br J Cancer 2014;110:2139-43.
18. Boulbes DR, Arold ST, Chauhan GB, et al. HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol Oncol 2015;9:586-600.
19. Furrer D, Jacob S, Michaud A, Provencher L, Lemieux J, Diorio C. Association of tobacco use, alcohol consumption and HER2 polymorphisms with response to trastuzumab in HER2-positive breast cancer patients. Clin Breast Cancer 2018;18:e687-94.
20. Gavin PG, Song N, Kim SR, et al. Association of polymorphisms in FCGR2A and FCGR3A with degree of trastuzumab benefit in the adjuvant treatment of ERBB2/HER2-positive breast cancer: analysis of the NSABP B-31 trial. JAMA Oncol 2017;3:335-41.
21. Han X, Diao L, Xu Y, et al. Association between the HER2 Ile655Val polymorphism and response to trastuzumab in women with operable primary breast cancer. Ann Oncol 2014;25:1158-64.
22. Hurvitz SA, Betting DJ, Stern HM, et al. Analysis of Fcγ receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin Cancer Res 2012;18:3478-86.
23. Jackson C, Browell D, Gautrey H, Tyson-Capper A. Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol 2013;2013:973584.
24. Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008;26:1789-96.
25. Norton N, Olson RM, Pegram M, et al. Association studies of Fcγ receptor polymorphisms with outcome in HER2+ breast cancer patients treated with trastuzumab in NCCTG (Alliance) trial N9831. Cancer Immunol Res 2014;2:962-9.
26. Scaltriti M, Rojo F, Ocaña A, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst 2007;99:628-38.
27. Tamura K, Shimizu C, Hojo T, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol 2011;22:1302-7.
28. Toomey S, Madden SF, Furney SJ, et al. The impact of ERBB-family germline single nucleotide polymorphisms on survival response to adjuvant trastuzumab treatment in HER2-positive breast cancer. Oncotarget 2016;7:75518-25.
29. He DX, Gu F, Gao F, et al. Genome-wide profiles of methylation, microRNAs, and gene expression in chemoresistant breast cancer. Sci Rep 2016;6:24706.
30. Klajic J, Busato F, Edvardsen H, et al. DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors. Clin Cancer Res 2014;20:6357-66.
31. Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol 2012;8:999-1014.
32. Ye XM, Zhu HY, Bai WD, et al. Epigenetic silencing of miR-375 induces trastuzumab resistance in HER2-positive breast cancer by targeting IGF1R. BMC Cancer 2014;14:134.
33. Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011;2:607-17.
34. Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget 2012;3:462-74.
35. Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010;466:253-7.
36. Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta 2010;1803:1231-43.
37. Furrer D, Jacob S, Caron C, Sanschagrin F, Provencher L, Diorio C. Tissue microarray is a reliable tool for the evaluation of HER2 amplification in breast cancer. Anticancer Res 2016;36:4661-6.
38. Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011;98:288-95.
39. Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010;11:587.
40. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 2013;14:293.
41. Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013;8:203-9.
42. Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 2010;11:733-9.
43. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. PLoS Med 2021;18:e1003583.
44. Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016;355:i4919.
45. Harris LN, You F, Schnitt SJ, et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res 2007;13:1198-207.
46. Khoury T, Kanehira K, Wang D, et al. Breast carcinoma with amplified HER2: a gene expression signature specific for trastuzumab resistance and poor prognosis. Mod Pathol 2010;23:1364-78.
47. Gámez-Pozo A, Pérez Carrión RM, Manso L, et al. The Long-HER study: clinical and molecular analysis of patients with HER2+ advanced breast cancer who become long-term survivors with trastuzumab-based therapy. PLoS One 2014;9:e109611.
48. Guarneri V, Dieci MV, Frassoldati A, et al. Prospective biomarker analysis of the randomized CHER-LOB study evaluating the dual anti-HER2 treatment with trastuzumab and lapatinib plus chemotherapy as neoadjuvant therapy for HER2-positive breast cancer. Oncologist 2015;20:1001-10.
49. Triulzi T, De Cecco L, Sandri M, et al. Whole-transcriptome analysis links trastuzumab sensitivity of breast tumors to both HER2 dependence and immune cell infiltration. Oncotarget 2015;6:28173-82.
50. Merry CR, McMahon S, Forrest ME, et al. Transcriptome-wide identification of mRNAs and lincRNAs associated with trastuzumab-resistance in HER2-positive breast cancer. Oncotarget 2016;7:53230-44.
51. Zhao B, Zhao Y, Sun Y, et al. Alterations in mRNA profiles of trastuzumab‑resistant Her‑2‑positive breast cancer. Mol Med Rep 2018;18:139-46.
52. Sorokin M, Ignatev K, Barbara V, et al. Molecular pathway activation markers are associated with efficacy of trastuzumab therapy in metastatic HER2-positive breast cancer better than individual gene expression levels. Biochemistry (Mosc) 2020;85:758-72.
53. Perez EA, Thompson EA, Ballman KV, et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J Clin Oncol 2015;33:701-8.
54. Du F, Yuan P, Zhao ZT, et al. A miRNA-based signature predicts development of disease recurrence in HER2 positive breast cancer after adjuvant trastuzumab-based treatment. Sci Rep 2016;6:33825.
55. Ohzawa H, Miki A, Teratani T, et al. Usefulness of miRNA profiles for predicting pathological responses to neoadjuvant chemotherapy in patients with human epidermal growth factor receptor 2-positive breast cancer. Oncol Lett 2017;13:1731-40.
56. Walsh N, Andrieu C, O’Donovan P, et al. Whole-exome sequencing of long-term, never relapse exceptional responders of trastuzumab-treated HER2+ metastatic breast cancer. Br J Cancer 2020;123:1219-22.
57. Yang T, Fu Z, Zhang Y, Wang M, Mao C, Ge W. Serum proteomics analysis of candidate predictive biomarker panel for the diagnosis of trastuzumab-based therapy resistant breast cancer. Biomed Pharmacother 2020;129:110465.
58. Lesurf R, Griffith OL, Griffith M, et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. Ann Oncol 2017;28:1070-7.
59. Shi W, Jiang T, Nuciforo P, et al. Pathway level alterations rather than mutations in single genes predict response to HER2-targeted therapies in the neo-ALTTO trial. Ann Oncol 2019;30:1018.
60. Berthon AS, Szarek E, Stratakis CA. PRKACA: the catalytic subunit of protein kinase A and adrenocortical tumors. Front Cell Dev Biol 2015;3:26.
61. Kordowski F, Kolarova J, Schafmayer C, et al. Aberrant DNA methylation of ADAMTS16 in colorectal and other epithelial cancers. BMC Cancer 2018;18:796.
62. Kumar S, Rao N, Ge R. Emerging roles of ADAMTSs in angiogenesis and cancer. Cancers (Basel) 2012;4:1252-99.
63. Li Y, Jiang D, Zhang Q, Liu X, Cai Z. Ubiquitin-specific protease 4 inhibits breast cancer cell growth through the upregulation of PDCD4. Int J Mol Med 2016;38:803-11.
64. Moody SE, Schinzel AC, Singh S, et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene 2015;34:2061-71.
65. Nguyen HH, Kim T, Nguyen T, Hahn MJ, Yun SI, Kim KK. A selective inhibitor of ubiquitin-specific protease 4 suppresses colorectal cancer progression by regulating β-catenin signaling. Cell Physiol Biochem 2019;53:157-71.
66. Qin N, Han F, Li L, et al. Deubiquitinating enzyme 4 facilitates chemoresistance in glioblastoma by inhibiting P53 activity. Oncol Lett 2019;17:958-64.
67. Tomasini MD, Wang Y, Karamafrooz A, et al. Conformational landscape of the PRKACA-DNAJB1 chimeric kinase, the driver for fibrolamellar hepatocellular carcinoma. Sci Rep 2018;8:720.
68. Tomino T, Tajiri H, Tatsuguchi T, et al. DOCK1 inhibition suppresses cancer cell invasion and macropinocytosis induced by self-activating Rac1P29S mutation. Biochem Biophys Res Commun 2018;497:298-304.
69. Zhang B, Li H, Yin C, et al. Dock1 promotes the mesenchymal transition of glioma and is modulated by MiR-31. Neuropathol Appl Neurobiol 2017;43:419-32.
70. Laurin M, Côté JF. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev 2014;28:533-47.
71. Liang Y, Wang S, Zhang Y. Downregulation of Dock1 and Elmo1 suppresses the migration and invasion of triple-negative breast cancer epithelial cells through the RhoA/Rac1 pathway. Oncol Lett 2018;16:3481-8.
72. Kesteloot F, Desmoulière A, Leclercq I, et al. ADAM metallopeptidase with thrombospondin type 1 motif 2 inactivation reduces the extent and stability of carbon tetrachloride-induced hepatic fibrosis in mice. Hepatology 2007;46:1620-31.
73. Dubail J, Kesteloot F, Deroanne C, et al. ADAMTS-2 functions as anti-angiogenic and anti-tumoral molecule independently of its catalytic activity. Cell Mol Life Sci 2010;67:4213-32.
74. Thorolfsdottir RB, Sveinbjornsson G, Sulem P, et al. A missense variant in PLEC increases risk of atrial fibrillation. J Am Coll Cardiol 2017;70:2157-68.
75. Paumard-Hernández B, Calvete O, Inglada Pérez L, et al. Whole exome sequencing identifies PLEC, EXO5 and DNAH7 as novel susceptibility genes in testicular cancer. Int J Cancer 2018;143:1954-62.
76. Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 2009;78:363-97.
77. Fumagalli D, Venet D, Ignatiadis M, et al. RNA sequencing to predict response to neoadjuvant anti-HER2 therapy: a secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol 2017;3:227-34.
78. Triulzi T, Regondi V, De Cecco L, et al. Early immune modulation by single-agent trastuzumab as a marker of trastuzumab benefit. Br J Cancer 2018;119:1487-94.
79. Brodsky AS, Xiong J, Yang D, et al. Identification of stromal ColXα1 and tumor-infiltrating lymphocytes as putative predictive markers of neoadjuvant therapy in estrogen receptor-positive/HER2-positive breast cancer. BMC Cancer 2016;16:274.
80. Hendricks WPD, Briones N, Halperin RF, et al. PD-1-associated gene expression signature of neoadjuvant trastuzumab-treated tumors correlates with patient survival in HER2-positive breast cancer. Cancers (Basel) 2019;11:1566.
81. Powles RL, Redmond D, Sotiriou C, et al. Association of T-Cell receptor repertoire use with response to combined trastuzumab-lapatinib treatment of HER2-positive breast cancer: secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol 2018;4:e181564.
82. Varadan V, Gilmore H, Miskimen KL, et al. Immune signatures following single dose trastuzumab predict pathologic response to preoperativetrastuzumab and chemotherapy in HER2-positive early breast cancer. Clin Cancer Res 2016;22:3249-59.
83. Ye Y, Tang X, Sun Z, Chen S. Upregulated WDR26 serves as a scaffold to coordinate PI3K/AKT pathway-driven breast cancer cell growth, migration, and invasion. Oncotarget 2016;7:17854-69.
84. Loibl S, Majewski I, Guarneri V, et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann Oncol 2019;30:1180.
85. Takada M, Higuchi T, Tozuka K, et al. Alterations of the genes involved in the PI3K and estrogen-receptor pathways influence outcome in human epidermal growth factor receptor 2-positive and hormone receptor-positive breast cancer patients treated with trastuzumab-containing neoadjuvant chemotherapy. BMC Cancer 2013;13:241.
86. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007;12:395-402.
87. Zhang B, Zhou Y, Lin N, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res 2013;23:1522-40.
88. Galanter JM, Gignoux CR, Oh SS, et al. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. Elife 2017;6:e20532.
89. Benevolenskaya EV, Islam AB, Ahsan H, et al. DNA methylation and hormone receptor status in breast cancer. Clin Epigenetics 2016;8:17.
90. Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev 2018;174:3-17.
91. Abba MC, Hu Y, Sun H, et al. Gene expression signature of estrogen receptor alpha status in breast cancer. BMC Genomics 2005;6:37.
92. Adkins RM, Krushkal J, Tylavsky FA, Thomas F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res A Clin Mol Teratol 2011;91:728-36.
93. Sempere LF, Christensen M, Silahtaroglu A, et al. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 2007;67:11612-20.
94. Van Grembergen O, Bizet M, de Bony EJ, et al. Portraying breast cancers with long noncoding RNAs. Sci Adv 2016;2:e1600220.
96. Yerukala Sathipati S, Ho SY. Identifying a miRNA signature for predicting the stage of breast cancer. Sci Rep 2018;8:16138.
97. Le X, Antony R, Razavi P, et al. Systematic functional characterization of resistance to pi3k inhibition in breast cancer. Cancer Discov 2016;6:1134-47.
99. Rimawi MF, De Angelis C, Contreras A, et al. Low PTEN levels and PIK3CA mutations predict resistance to neoadjuvant lapatinib and trastuzumab without chemotherapy in patients with HER2 over-expressing breast cancer. Breast Cancer Res Treat 2018;167:731-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Furrer D, Dragic D, Chang SL, Fournier F, Droit A, Jacob S, Diorio C. Association between genome-wide epigenetic and genetic alterations in breast cancer tissue and response to HER2-targeted therapies in HER2-positive breast cancer patients: new findings and a systematic review. Cancer Drug Resist 2022;5:995-1015. http://dx.doi.org/10.20517/cdr.2022.63
AMA Style
Furrer D, Dragic D, Chang SL, Fournier F, Droit A, Jacob S, Diorio C. Association between genome-wide epigenetic and genetic alterations in breast cancer tissue and response to HER2-targeted therapies in HER2-positive breast cancer patients: new findings and a systematic review. Cancer Drug Resistance. 2022; 5(4): 995-1015. http://dx.doi.org/10.20517/cdr.2022.63
Chicago/Turabian Style
Furrer, Daniela, Dzevka Dragic, Sue-Ling Chang, Frédéric Fournier, Arnaud Droit, Simon Jacob, Caroline Diorio. 2022. "Association between genome-wide epigenetic and genetic alterations in breast cancer tissue and response to HER2-targeted therapies in HER2-positive breast cancer patients: new findings and a systematic review" Cancer Drug Resistance. 5, no.4: 995-1015. http://dx.doi.org/10.20517/cdr.2022.63
ACS Style
Furrer, D.; Dragic D.; Chang S.L.; Fournier F.; Droit A.; Jacob S.; Diorio C. Association between genome-wide epigenetic and genetic alterations in breast cancer tissue and response to HER2-targeted therapies in HER2-positive breast cancer patients: new findings and a systematic review. Cancer Drug Resist. 2022, 5, 995-1015. http://dx.doi.org/10.20517/cdr.2022.63
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 29
likes
Like This Article 29
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.