
Mechanisms of neratinib resistance in HER2-mutant metastatic breast cancer

 ,
, Abstract
Human epidermal growth factor receptor 2 (HER2) is a major drug target and clinical biomarker in breast cancer treatment. Targeting HER2 gene amplification is one of the greatest successes in oncology, resulting in the use of a wide array of HER2-directed agents in the clinic. The discovery of HER2-activating mutations as novel therapeutic targets in breast and other cancers marked a significant advance in the field, which led to the metastatic breast and other solid tumor trials MutHER (NCT01670877), SUMMIT (NCT01953926), and one arm of plasmaMATCH (NCT03182634). These trials reported initial clinical benefit followed by eventual relapse ascribed to either primary or acquired resistance. These resistance mechanisms are mediated by additional secondary genomic alterations within HER2 itself and via hyperactivation of oncogenic signaling within the downstream signaling axis.
Keywords
INTRODUCTION
Human epidermal growth factor receptor 2 (HER2)-positive breast cancers have long been treated with targeted therapy, comprising either monoclonal antibodies, such as trastuzumab or pertuzumab, which bind to the extracellular domain of HER2, or tyrosine kinase inhibitors (TKIs), such as the reversible inhibitors lapatinib and tucatinib and the irreversible inhibitor neratinib[1]. Genome sequencing efforts have recently identified recurrent somatic mutations in the HER2 (ERBB2) gene in HER2-negative (non-amplified) breast cancer. Recurrent HER2 mutations have been proven to be oncogenic drivers in both preclinical experiments and clinical trials[2-9]. Activating HER2 mutations typically fall into four categories, with distribution dependent on tumor type: single nucleotide variants (SNVs) in the extracellular domain, particularly S310F/Y; SNVs in the transmembrane domain; SNVs in the kinase domain; and small insertions in exon 20[4,10,11]. HER2 mutations constitutively activate the tyrosine kinase receptor activity, leading to upregulation of downstream phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling[6,12]. HER2 mutations are rare in primary cancers, occurring in 2%-12% of solid tumors depending on tumor type and disease stage. In breast cancer, HER2 mutations vary in frequency from ~2% to 8% depending on disease stage and histology (higher in lobular)[7,10,11,13,14] and have been associated with poor prognosis[15,16]. The prevalence of HER2 mutations is higher in patients with metastatic breast cancer (MBC) that has progressed after primary endocrine therapy (~6%), and these mutations have been causally associated with antiestrogen resistance[12,13,17]. Furthermore, HER2 and estrogen receptor 1 gene (ESR1) mutations are mutually exclusive in primary breast cancer, suggesting that HER2 mutations are independent predictive and prognostic markers in estrogen receptor (ER)-positive MBC[11,12,16].
Neratinib is an orally available, second-generation, pan-HER TKI that irreversibly binds to cysteine residues Cys773 and Cys805 in the ATP pocket of the tyrosine kinase domain of epidermal growth factor receptor (EGFR), HER2, and HER4[18]. Neratinib inhibits autophosphorylation, downstream signaling, and growth of EGFR- or HER2-dependent cell lines, with cellular half-maximal inhibitory concentration
Clinically, the utility of neratinib, alone or in combination with other agents, in patients with heavily pretreated HER2-mutant breast and other cancers was explored in the phase II SUMMIT and MutHER trials[4,7]. The SUMMIT trial demonstrated clinical benefit from single-agent neratinib in patients with several solid tumor types, including HER2-mutant breast cancers[4]. For patients with HER2-mutant, hormone receptor (HR)-positive MBC, both the SUMMIT and MutHER trials were amended to combine neratinib with fulvestrant to suppress both HER2 and HR signaling simultaneously. This dual combination was clinically active in heavily pretreated patients with HER2-mutant, HR-positive MBC, including those who had received prior fulvestrant and cyclin-dependent kinase (CDK)4/6 inhibitor therapy. In SUMMIT, the overall response rate (ORR) for neratinib monotherapy in patients with HER2-mutant, HR-positive MBC
Unfortunately, patients who initially derived benefit from neratinib or neratinib plus fulvestrant in these studies eventually relapsed with metastatic disease, and a comparison of the genomic landscape of tumor tissue or ctDNA before treatment and upon progression revealed the acquisition of additional genomic aberrations[8,9,22]. Mechanisms of acquired resistance appeared to occur primarily via the development of secondary HER2 genomic alterations (mutations or amplification), whereas intrinsic resistance was observed not only in patients whose baseline tumors had more than one HER2 alteration, but also via alterations in the HER3/PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) and MAPK signaling axis[8,9,22] [Figure 1].
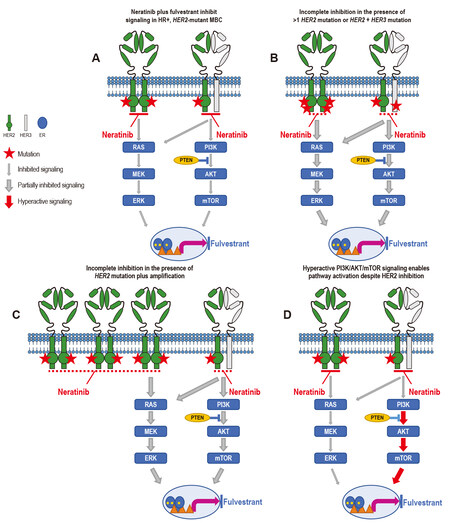
Figure 1. Mechanisms of resistance to neratinib. (A) Neratinib plus fulvestrant inhibit signaling in HR+, HER2-mutant MBC. In patients whose tumors harbor a single somatic activating mutation (red star) in HER2, neratinib strongly inhibits (thin gray arrow) HER2 pathway signaling, whereas fulvestrant inhibits ER signaling, leading to tumor growth inhibition. (B) Neratinib is less effective against/partially inhibits signaling in (thick gray arrow) tumors with more than one HER2 mutation or HER2 mutation plus HER3 mutation or (C) HER2 mutation plus amplification, whether these dual alterations are intrinsic or acquired. (D) Hyperactivation (thick red arrow) of downstream signaling can also preclude the effect of neratinib on mutant HER2. ER: Estrogen receptor; HR: hormone receptor; MBC: metastatic breast cancer.
MECHANISMS OF RESISTANCE
Accumulation of additional HER2 genomic events
Among patients with heavily pretreated MBC, the presence of more than one de novo HER2-activating event before treatment trended with a lack of benefit from neratinib alone or in combination with fulvestrant in trials to date. In the SUMMIT trial, six of the seven patients whose pre-treatment tumors harbored more than one HER2-activating event (second HER2 mutation, n = 2; copy-number amplification, n = 3; or both, n = 2) did not derive clinical benefit[9]. In MutHER, three out of 48 patients had dual HER2 mutations at enrollment; two of those three patients did not experience clinical benefit[7,8].
In patients whose treatment-naive tumors harbored one HER2 mutation and who initially derived clinical benefit from neratinib-containing treatments, the one consistently observed mechanism of acquired resistance was the accumulation of a second (or further) additional HER2 alteration[8,9]. In SUMMIT, three of nine patients with HR-positive, HER2-mutant MBC, who were treated with neratinib plus fulvestrant and who had both pre- and post-treatment tumors available for central sequencing, had additional HER2-activating events in the post-treatment tumor[9]. One patient had amplification of the mutant allele, one acquired a gatekeeper mutation, and one had amplification plus two acquired HER2 hotspot mutations. Secondary HER2 mutations were also detected in seven of 16 patients with paired pre-treatment and progression ctDNA who derived clinical benefit, including two of the three described above. Among patients in MutHER who had paired ctDNA samples, acquired HER2 mutations were detected upon progression in three of six patients with clinical benefit following neratinib monotherapy, in four of seven patients with benefit following neratinib plus fulvestrant, and in one who experienced short-term stable disease[8]. Although several of the tumors acquired gatekeeper mutations (T798I and L785F)[23,24], the acquisition of additional sensitizing mutations or variants of unknown significance was also reported [Table 1]. Beyond HER2, no other acquired genetic event was consistently observed. These findings suggest that HER2-mutant MBCs are dependent on HER2 signaling even upon disease progression.
HER2 alterations detected following neratinib-containing regimens in clinical trials of HER2-mutant MBC (compiled from the works of Ma et al.[7], Ma et al.[8], and Smyth et al.[9])
| Trial | Regimen | HER2 mutations detected at baseline | Best response | Acquired HER2 alterations |
| MutHER | ||||
| N | L755S, P761del | PR | R678Q, V697L | |
| N + F | G778_P780dup | PR | D808Ha, T798Ib, I767M | |
| N + F | S310F | PR | L755S, D769Y, G776V, T798Ib, L841V | |
| N + F | G778_P780dup | PR | S310Y, S310F, I767M, T798Ib | |
| N | L869R; amplification | SD | D1011Dc | |
| N | L869R, D769Y | SD | S310F, I767M, T862A, T798Ib | |
| N + F | V777L | SD | S310F | |
| N + F | L755S | SD* | S310F | |
| SUMMIT | ||||
| N + F | S310F | CR | L785Fb,d | |
| N + F | G778_P780dup | PR | I767M, S310Y, amplificationd | |
| N + F | L869R | PR | S310Y, D769Y, L755S, T798Ib | |
| N + F | V697L | PR | Amplified mutant alleled | |
| N + F | V777L | PR | T798Ib | |
| N + F | L755S, L755P | SD | T862A, S310F | |
| N + F | G776V | SD | I767M | |
| N + F | L755S | PD* | D769H, D962Ha, K1171Na, D1016Ya, D1089Ya | |
Aberrant HER3/PI3K/mTOR signaling
In preclinical models of HR-positive breast cancer, the recurrent HER2 L755S and V777L mutations constitutively upregulated HER3 phosphorylation, particularly upon treatment with fulvestrant, resulting in hyperactivation of the HER3/PI3K/AKT/mTOR signaling axis and leading to antiestrogen resistance[12,22]. Structural modeling of the HER2 L755S mutation revealed a loss of flexibility in the active state, allowing for increased HER2/HER3 heterodimerization and upregulation of PI3K/AKT/mTOR signaling[12]. HER3 mutations have been modeled to stabilize HER2/HER3 dimerization and increase HER2 signaling[25,26], and preclinical models showed that dual HER2/HER3 mutations further enhanced oncogenicity and promoted resistance to HER2-targeted therapies, including neratinib[26]. In SUMMIT, pre-existing concurrent activating HER3 mutations were associated with poor treatment outcomes in patients with HER2-mutant MBC[9]. Further analysis of data from SUMMIT patients with HER2-mutant tumors across multiple tumor types revealed that mTOR pathway alterations were associated with a lack of clinical benefit with single-agent neratinib. Preclinically, hyperactivation of mTOR signaling was an actionable acquired mechanism of resistance to neratinib in HER2-mutant cell lines and patient-derived xenograft (PDX) models[22]. Interestingly, however, PIK3CA mutations per se were not associated with a lack of clinical benefit with neratinib plus fulvestrant in HER2-mutant MBC[8,9], and no other single gene or mutation in the HER3/PI3K/AKT/mTOR signaling axis was clearly associated with acquired neratinib resistance.
Acquisition of somatic HER2 mutations in HER2-positive breast cancer
Although this review focuses specifically on the acquisition of resistance to neratinib in HER2-negative, HER2-mutant MBC, the acquisition of HER2 mutations in HER2-positive breast cancer also merits consideration. The co-occurrence of HER2 mutations and amplification has been associated with poor response to trastuzumab and lapatinib, although neratinib has been shown to be effective against HER2-positive, HER2-mutant preclinical models and in patients whose breast tumors had coincident HER2 amplification and mutation, suggesting neratinib as monotherapy may be effective in this setting[27]. In preclinical models of HER2-positive breast cancer, HER2 reactivation in lapatinib-resistant derivatives was driven by the acquisition of a HER2 L755S mutation, which could be overcome by neratinib or afatinib[28]. Finally, recent findings from plasmaMATCH demonstrate that the incidence of HER2 mutations in HER2-positive cancers increases with the number of lines of HER2-directed therapy[29]. Regardless of initial HER2-positive or HER2-mutant status, accumulation of genomic events within HER2 itself is prevalent upon exposure to HER2-targeted agents. Whether or not there is a clinical difference in response depending on which type of alteration is the initial driver remains to be seen.
Taken together, these findings provide a rationale for the combination of multiple HER2 inhibitors or inhibitors of the downstream signaling axis in patients with HER2-mutant breast cancer, a therapeutic strategy that has already proven highly effective in HER2-positive breast cancer.
OVERCOMING NERATINIB RESISTANCE IN HER2–MUTANT MBC
The following are three possible approaches to overcoming neratinib resistance in HER2-mutant MBC:
Dual HER2 targeting: neratinib plus monoclonal antibody or antibody–drug conjugate
The dual targeting of HER2 either upfront or at disease progression has proven to be effective in patients with HER2-mutant MBC. In MutHER, adding trastuzumab after disease progression on neratinib plus fulvestrant led to re-response in four of five patients, with a concomitant decrease in ctDNA[8]. In SUMMIT, the HER2-mutant breast cohorts were recently amended to treat patients upfront with the triple combination of neratinib, fulvestrant, and trastuzumab. This combination, in fact, demonstrated encouraging clinical activity in SUMMIT patients with heavily pretreated HR-positive, HER2-negative, HER2-mutant MBC who had previously received a CDK4/6 inhibitor (n = 33; ORR of 42.4%, CBR of 51.5%, median DOR of 14.4 months, median PFS of 7.0 months)[30,31]. Preclinically, neratinib combined with trastuzumab in HER2-mutant cancer models yielded more robust inhibition of HER2 signaling and growth than either agent alone[5,32].
Neratinib induces HER2 receptor ubiquitination and endocytosis[33]; combining neratinib with a HER2-directed antibody–drug conjugate may therefore enable increased payload internalization. In HER2-mutant PDX models, the combination of neratinib and trastuzumab emtansine (T-DM1) or trastuzumab deruxtecan (T-DXd) did, in fact, show synergistic tumor growth inhibition[34]. Safety and preliminary efficacy of neratinib plus T-DM1 have been demonstrated in patients with HER2-positive breast cancer[35]; clinical trials of neratinib plus antibody–drug conjugates are similarly warranted in the HER2-mutant MBC setting.
Combination with PI3K, mTOR, MEK, or CDK4/6 inhibitors
Combining neratinib with inhibitors of the downstream signaling axis or with CDK4/6 inhibitors may be a second approach to prolonging response to neratinib in patients with HER2-mutant MBC. First, preclinical data in HER2/HER3 double mutant cell lines show that the combination of a PI3K inhibitor (alpelisib) with neratinib overcame neratinib resistance[26]. Second, the combination of the mTOR inhibitor everolimus with neratinib arrested the growth of neratinib-resistant, ER-positive, HER2-mutant organoids and xenografts[22]. Third, in two HER2-positive breast and colorectal PDX models harboring activating HER2 mutations (V777L and R678Q) derived from patients who had been treated with HER2-targeted therapies, the combination of neratinib with the MEK inhibitor trametinib, the mTOR inhibitors everolimus or sapanisertib, or the CDK4/6 inhibitor palbociclib synergistically decreased tumor volume to a greater extent than any of the agents alone. These combinations were well tolerated in HER2-positive preclinical PDX models[36]. A clinical trial to study the safety and tolerability of neratinib combined with trametinib, everolimus, or palbociclib in metastatic solid tumors with HER family alteration or KRAS mutation is currently underway (NCT03065387)[37]. Given the promising efficacy of neratinib-containing regimens post CDK4/6 inhibitor in the SUMMIT trial[31], first-line treatment with neratinib plus a CDK4/6 inhibitor and endocrine therapy could warrant investigation if the combination is deemed tolerable.
Sequential treatment of neratinib with a second TKI
Sequential TKI treatment has long been standard in EGFR-mutant non-small cell lung cancer, and a similar approach could be investigated for HER2-mutant MBC. The HER2 gatekeeper mutation T798I is recurrent in HER2-mutant MBC upon clinical progression following neratinib. Preclinically, another second-generation TKI, afatinib, and AZ5104, the metabolite of the third-generation TKI osimertinib, blocked HER2 T798I mutation-induced cell growth and signaling[23]. These findings support the clinical investigation of sequencing TKI therapy in HER2-mutant cancers that develop gatekeeper mutations.
CONCLUSION
HER2-activating mutations are a targetable alteration in MBC and can be inhibited by neratinib. In heavily pretreated patients with MBC, more than one alteration in the HER2 signaling pathway, whether in the HER2 gene itself or downstream in the signaling cascade, may preclude initial response. Furthermore, patients with a single HER2 mutation who derive initial clinical benefit appear to become resistant via the acquisition of additional HER2 mutations and/or amplification. Dual HER2-targeting via the addition of trastuzumab to neratinib, in combination with fulvestrant for patients with HR-positive MBC, has exhibited strong clinical activity against HER2-mutant MBC[8,30,31]; targeting both HER2 and CDK4/6 together may warrant exploration as part of a front-line approach in this setting. Future analysis of plasma samples from patients receiving dual HER2-targeting will elucidate whether acquired resistance occurs via the same mechanisms. Combination and/or sequencing of neratinib plus additional agents targeting either HER2 or downstream or alternative pathway members may be required for more durable clinical benefit. Any combination approach will require diligent clinical management given the gastrointestinal toxicity profile of neratinib, although neratinib dose escalation may help to mitigate adverse events[38].
Future studies may consider molecularly guided approaches beyond genomics, including but not limited to evaluation of changes in gene or protein expression or protein phosphorylation status, to inform the design of rational drug combinations and lead to improved outcomes for patients with HER2-mutant MBC.
DECLARATIONS
AcknowledgmentsThe authors would like to acknowledge Ron Bose, MD, PhD, and Cynthia Ma, MD, PhD, for providing data from MutHER and for critical review of the manuscript, and Miller Medical Communications for editorial assistance, funding for which was provided by Puma Biotechnology, Inc.
Authors’ contributionsConceived of this review article and compiled data from SUMMIT and MutHER publications: Eli LD
Made substantial contributions to data interpretation and manuscript writing: Eli LD, Kavuri SM
Availability of data and materialsNot applicable.
Financial support and sponsorshipSMK’s research is funded by Susan G. Komen (CCR16380599) and the Department of Defense (W81XWH-18-1-0040 and W81XWH-18-1-0084).
Conflicts of interestLDE is an employee and shareholder of Puma Biotechnology, Inc. SMK is a stakeholder in NeoZenome Therapeutics Inc.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer 2020;126:4278-88.
2. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 2013;3:224-37.
3. Shimamura T, Ji H, Minami Y, et al. Non-small-cell lung cancer and Ba/F3 transformed cells harboring the ERBB2 G776insV_G/C mutation are sensitive to the dual-specific epidermal growth factor receptor and ERBB2 inhibitor HKI-272. Cancer Res 2006;66:6487-91.
4. Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189-94.
5. Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov 2015;5:832-41.
6. Zabransky DJ, Yankaskas CL, Cochran RL, et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci USA 2015;112:E6205-14.
7. Ma CX, Bose R, Gao F, et al. Neratinib efficacy and circulating tumor DNA detection of HER2 mutations in HER2 nonamplified metastatic breast cancer. Clin Cancer Res 2017;23:5687-95.
8. Ma CX, Luo J, Freedman RA, et al. The phase II MutHER study of neratinib alone and in combination with fulvestrant in HER2-mutated, non-amplified metastatic breast cancer. Clin Cancer Res 2022;28:1258-67.
9. Smyth LM, Piha-Paul SA, Won HH, et al. Efficacy and determinants of response to HER kinase inhibition in HER2-mutant metastatic breast cancer. Cancer Discov 2020;10:198-213.
10. Schram A, Won HH, Andre F, et al. Abstract LB-103: landscape of somatic ERBB2 mutations: findings from AACR GENIE and comparison to ongoing ERBB2 mutant basket study. Cancer Res 2017;77:LB103.
11. Connell CM, Doherty GJ. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open 2017;2:e000279.
12. Croessmann S, Formisano L, Kinch LN, et al. Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER2 mutant breast cancer. Clin Cancer Res 2019;25:277-89.
13. Razavi P, Chang MT, Xu G, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018;34:427-438.e6.
14. Desmedt C, Zoppoli G, Gundem G, et al. Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol 2016;34:1872-81.
15. Wang T, Xu Y, Sheng S, et al. HER2 somatic mutations are associated with poor survival in HER2-negative breast cancers. Cancer Sci 2017;108:671-7.
16. Kurozumi S, Alsaleem M, Monteiro CJ, et al. Targetable ERBB2 mutation status is an independent marker of adverse prognosis in estrogen receptor positive, ERBB2 non-amplified primary lobular breast carcinoma: a retrospective in silico analysis of public datasets. Breast Cancer Res 2020;22:85.
17. Nayar U, Cohen O, Kapstad C, et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet 2019;51:207-16.
18. Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res 2004;64:3958-65.
19. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688-700.
20. Saura C, Oliveira M, Feng YH, et al. NALA Investigators. Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2-positive metastatic breast cancer previously treated with ≥ 2 HER2-directed regimens: phase III NALA trial. J Clin Oncol 2020;38:3138-49.
21. Turner NC, Kingston B, Kilburn LS, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol 2020;21:1296-308.
22. Sudhan DR, Guerrero-Zotano A, Won H, et al. Hyperactivation of TORC1 drives resistance to the pan-HER tyrosine kinase inhibitor neratinib in HER2-mutant cancers. Cancer Cell 2020;37:183-199.e5.
23. Hanker AB, Brewer MR, Sheehan JH, et al. An acquired HER2T798I gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discov 2017;7:575-85.
24. Smyth L, Piha-Paul S, Saura C, et al. Abstract PD3-06: neratinib + fulvestrant for HER2-mutant, HR-positive, metastatic breast cancer: updated results from the phase 2 SUMMIT trial. Cancer Res 2019;79(4 Suppl):PD3-06.
25. Collier TS, Diraviyam K, Monsey J, Shen W, Sept D, Bose R. Carboxyl group footprinting mass spectrometry and molecular dynamics identify key interactions in the HER2-HER3 receptor tyrosine kinase interface. J Biol Chem 2013;288:25254-64.
26. Hanker AB, Brown BP, Meiler J, et al. Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell 2021;39:1099-1114.e8.
27. Cocco E, Javier Carmona F, Razavi P, et al. Neratinib is effective in breast tumors bearing both amplification and mutation of ERBB2 (HER2). Sci Signal 2018;11:eaat9773.
28. Xu X, De Angelis C, Burke KA, et al. HER2 reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2+ breast cancer. Clin Cancer Res 2017;23:5123-34.
29. Kingston B, Cutts RJ, Bye H, et al. Genomic profile of advanced breast cancer in circulating tumour DNA. Nat Commun 2021;12:4479.
30. Jhaveri K, Saura C, Guerrero-Zotano A, et al. Abstract PD1-05: latest findings from the breast cancer cohort in SUMMIT - a phase 2 ‘basket’ trial of neratinib + trastuzumab + fulvestrant for HER2 -mutant, hormone receptor-positive, metastatic breast cancer. Cancer Res 2021;81:PD1-05.
31. Jhaveri K, Park H, Waisman J, et al. Abstract GS4-10: neratinib + fulvestrant + trastuzumab for hormone receptor-positive, HER2-mutant metastatic breast cancer and neratinib + trastuzumab for triple-negative disease: Latest updates from the SUMMIT trial. Cancer Res 2022;82:GS4-10.
32. Ivanova E, Kuraguchi M, Xu M, et al. Use of ex vivo patient-derived tumor organotypic spheroids to identify combination therapies for HER2 mutant non-small cell lung cancer. Clin Cancer Res 2020;26:2393-403.
33. Zhang Y, Zhang J, Liu C, et al. Neratinib induces ErbB2 ubiquitylation and endocytic degradation via HSP90 dissociation in breast cancer cells. Cancer Lett 2016;382:176-85.
34. Bose R, Li S, Primeau TM, et al. Abstract PS4-13: irreversible inhibition of HER2 activating mutations with neratinib enhances the pre-clinical efficacy of trastuzumab emtansine and trastuzumab deruxtecan. Cancer Res 2021;81:PS4-13.
35. Abraham J, Montero AJ, Jankowitz RC, et al. Safety and efficacy of T-DM1 plus neratinib in patients with metastatic HER2-positive breast cancer: NSABP foundation trial FB-10. J Clin Oncol 2019;37:2601-9.
36. Zhao M, Scott S, Evans KW, et al. Combining neratinib with CDK4/6, mTOR, and MEK inhibitors in models of HER2-positive cancer. Clin Cancer Res 2021;27:1681-94.
37. Piha-Paul SA, Fu S, Hong DS, et al. Phase I study of the pan-HER inhibitor neratinib given in combination with everolimus, palbociclib or trametinib in advanced cancer subjects with EGFR mutation/amplification, HER2 mutation/amplification or HER3/4 mutation. J Clin Oncol 2018;36(15 Suppl):TPS2611.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Eli LD, Kavuri SM. Mechanisms of neratinib resistance in HER2-mutant metastatic breast cancer. Cancer Drug Resist 2022;5:873-81. http://dx.doi.org/10.20517/cdr.2022.48
AMA Style
Eli LD, Kavuri SM. Mechanisms of neratinib resistance in HER2-mutant metastatic breast cancer. Cancer Drug Resistance. 2022; 5(4): 873-81. http://dx.doi.org/10.20517/cdr.2022.48
Chicago/Turabian Style
Eli, Lisa D., Shyam M. Kavuri. 2022. "Mechanisms of neratinib resistance in HER2-mutant metastatic breast cancer" Cancer Drug Resistance. 5, no.4: 873-81. http://dx.doi.org/10.20517/cdr.2022.48
ACS Style
Eli, LD.; Kavuri SM. Mechanisms of neratinib resistance in HER2-mutant metastatic breast cancer. Cancer Drug Resist. 2022, 5, 873-81. http://dx.doi.org/10.20517/cdr.2022.48
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 6
likes
Like This Article 6
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.