
Insight into the molecular mechanisms of gastric cancer stem cell in drug resistance of gastric cancer

 , ...
, ... Abstract
Gastric cancer (GC) is one of the most common causes of cancer-related death worldwide, and gastric cancer stem cells (GCSCs) are considered as the major factor for resistance to conventional radio- and chemotherapy. Accumulating evidence in recent years implies that GCSCs regulate the drug resistance in GC through multiple mechanisms, including dormancy, drug trafficking, drug metabolism and targeting, apoptosis, DNA damage, epithelial-mesenchymal transition, and tumor microenvironment. In this review, we summarize current advancements regarding the relationship between GCSCs and drug resistance and evaluate the molecular bases of GCSCs in drug resistance.
Keywords
INTRODUCTION
According to GLOBOCAN estimates in 2020, gastric cancer (GC) is the fifth cause of global cancer incidence and the fourth leading cause of cancer mortality[1]. The incidence rates of GC vary widely across the world, with the highest rates in East Asia and Eastern Europe[1]. GC, most cases of which are gastric adenocarcinoma (GAC), is histologically divided into two subtypes [intestinal and diffuse (Lauren classification)][2] or four subtypes [papillary, tubular, mucinous, and poorly cohesive (WHO classification)][3]. Based on genomic and epigenomic alterations, the most well-defined molecular-based classification systems include The Cancer Genome Atlas (TCGA) classification [EBV positive (EBV), microsatellite instable (MSI), genomically stable (GS), and chromosomal instable (CIN)][4] and the Asian Cancer Research Group (ACRG) classification [microsatellite instable (MSI), microsatellite stable TP53 inactive (MSS/TP53 inactive), MSS TP53 active (MSS/TP53 active), and MSS with epithelial–mesenchymal transition (EMT) features (MSS/EMT)][5]. Despite advances in the field of early diagnosis in GC, most cases are still diagnosed at an advanced stage[6] with unresectable or metastatic disease. Although current systemic treatments, including surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy [Table 1] for advanced GC patients, have been considerably improved during recent decades, most patients with advanced GC die from tumor relapse and metastasis. The prognosis of advanced and metastatic GC remains poor, and the 5-year survival rate is < 10%[7].
A summary of systemic treatments in gastric cancer
| Treatment approaches | Mechanisms | Regimen | Efficacy | Adverse effects |
| Surgery | Surgical resection | Open surgery, laparoscopic surgery, endoscopic resection, robotic surgery | Primary choice for early-stage GC | Low |
| Chemotherapy | Target and kill fast-dividing cells | Fluoropyrimidines, platinums, taxanes, irinotecan, etc. | The standard-of-care treatment for advanced GC | Damage to normal and healthy tissues |
| Radiotherapy | Ionizing radiation to target and kill tumor tissue | Ionizing radiation | Curative and palliative treatment | Damage to normal and healthy tissues |
| Targeted therapy | Target the specific molecules (HER2, VEGF, VEGFR, etc.) | Monoclonal antibodies and small molecule inhibitors | Use in combination with chemotherapy in first- and second-line settings | Relatively low |
| Immunotherapy | Block the binding of ligands to checkpoint receptors and re-activate the human cellular immune response | Immune checkpoint inhibitors (PD-1, PD-L1, and CTLA-4) | Use in second- and third-line settings | Relatively low |
Early GC patients can be cured with surgery alone. For advanced unresectable patients, chemotherapy represents the backbone of systemic therapy, and chemotherapy with or without radiotherapy has been integrated into standard-of-care therapies. Cytotoxic chemotherapy has been demonstrated to be effective for advanced GC patients, and the common cytotoxic chemotherapy drugs include fluoropyrimidines (e.g., fluorouracil, capecitabine, and S-1), platinums (e.g., cisplatin and oxaliplatin), taxanes (e.g., paclitaxel and docetaxel), topoisomerase inhibitors (e.g., irinotecan), and anthracyclines (e.g., doxorubicin and epirubicin). In patients with human epidermal growth factor 2 (HER2)-negative, advanced gastric adenocarcinoma, the current first-line treatment consists of two- or three-drug regimens. Doublet therapies are the combination of platinum derivatives (cisplatin and oxaliplatin) and fluoropyrimidine analogs (5-fluorouracil, capecitabine, and S-1). Three-drug regimens are the triplet combinations adding taxanes or anthracyclines (doxorubicin and epirubicin) to the doublet regimen. Table 2 summarizes the landmark trials for first-line treatment of advanced gastric cancer. However, the clinical benefit from these treatments is limited due to the toxicity of chemotherapeutic drugs and the development of drug resistance.
Landmark trials in first-line treatment of advanced gastric cancer
| Studies | Treatment regimen | ORR | mPFS (mo), P-value | mOS (mo), P-value | Reference |
| MacDonald, et al. 1980 | Fluorouracil, doxorubicin, mitomycin (FAM) | 42% | NR | 5.5 | [8] |
| Wils, et al. 1991 | Fluorouracil , doxorubicin, methotrexate (FAMTX) vs. FAM | 41%/9% | NR | 9.7/6.7 | [9] |
| Webb, et al. 1997 | Epirubicin, cisplatin, fluorouracil (ECF) vs. FAMTX | 45%/21% | 7.4/3.4, P = 0.00006 | 8.9/5.7, P = 0.0009 | [10] |
| Van Cutsem, et al. 2006 | Cisplatin, fluorouracil (CF) vs. Docetaxel,cisplatin, fluorouracil (DCF) | 37%/25% | 5.6/3.7, P < 0.001 | 8.2/9.6, P = 0.02 | [11] |
| Cunningham, et al. 2008 | Epirubicin, cisplatin, fluorouracil (ECF) vs. Epirubicin, cisplatin, capecitabine (ECX) vs. Epirubicin, oxaliplatin, fluorouracil (EOF) vs. Epirubicin, oxaliplatin, capecitabine (EOX) | 41%/46%/42%/48% | 6.2/6.7/6.5/7.0 | 9.9/9.9/9.3/11.2 | [12] |
| Kang, et al. 2009 | Cisplatin, capecitabine (XP) vs. Cisplatin, fluorouracil (FP) | 41%/29% | 5.6/5.0, P < 0.001 | 10.5/9.3, P = 0.008 | [13] |
| Shah, et al. 2010 | DCF, granulocyte stimulating factor (G-CSF) vs. modified DCF (mDCF) | 33%/49% | 6.5/9.7, P = 0.2 | 12.6/18.8, P = 0.007 | [14] |
| Koizumi, et al. 2014 | S-1, Docetaxel vs. S-1 | 38.8%/26.8% | 5.3/4.2, P < 0.001 | 12.5/10.8, P = 0.032 | [15] |
| Guimbaud, et al. 2014 | epirubicin, cisplatin, capecitabine (ECX) vs. Folinic acid, Fluorouracil, Irinotecan (FOLFIRI) | 39.2%/37.8% | 5.3/5.8, P = 0.96 | 9.5/9.7, P = 0.95 | [16] |
More recently, targeted therapies have been developed for gastric cancer patients. These include trastuzumab, lapatinib, and margetuximab for epidermal growth factor receptor-2 (HER-2); bevacizumab for vascular endothelial growth factor (VEGF); ramucirumab, apatinib, and regorafenib for vascular endothelial growth factor receptor (VEGFR); cetuximab and panitumumab for endothelial growth factor receptor (EGFR); bemarituzumab for fibroblast growth factor receptor (FGFR); everolimus for mTOR; and zolbetuximab for Claudin 18.2. Various targeted therapy approaches have been investigated; however, several clinical trials in gastric cancer have failed due to the tumor heterogeneity and the difficulty of screening the beneficiary population for targeted therapeutic drugs. Furthermore, immunotherapy is being developed to block the binding of ligands to checkpoint receptors and re-activate the human cellular immune response. These are immune checkpoint inhibitors (ICIs) including nivolumab and pembrolizumab as PD-1 inhibitors, durvalumab and avelumab as PD-L1 inhibitors, and ipilimumab and tremelimumab as CTLA-4 inhibitors. Several trials have demonstrated that the benefits of immunotherapy only or with cytotoxic chemotherapy are relatively limited[17]. Thus, with relatively low response rates, the use of immunotherapy has only led to limited approval in the second-line treatment setting for GC[18]. More and more promising targeted therapies and immunotherapies are being investigated, and these will likely further improve outcomes for patients. Table 3 summarizes the landmark trials for targeted therapy and immunotherapy of advanced gastric cancer.
Landmark trials in targeted therapy and immunotherapy [immune checkpoint inhibitors (ICIs)] for the treatment of advanced GC
| Items | Drugs | Treatment regimen | ORR, P-value | mPFS (mo), P-value | mOS (mo), P-value | Reference |
| Targeted therapies | HER-2 | |||||
| Trastuzumab | Capecitabine or 5- FU plus cisplatin with Trastuzumab vs. Capecitabine or 5- FU plus cisplatin | 47%/35%, P = 0.0017 | 6.7/5.5, P = 0.0002 | 13.8/11.1, P = 0.0046 | [19] | |
| Trastuzumab | TrastuzumabDeruxtecan vs. Irinotecan or Paclitaxel | 51%/14%, P < 0.001 | 5.6/3.5, P = 0.01 | 12.5/8.4 | [20] | |
| Lapatinib | Paclitaxel with Lapatinib vs. Paclitaxel | 27%/9%, P < 0.001 | 5.4/4.4, NS | 11.0/8.9, NS | [21] | |
| Lapatinib | Capecitabine plus oxaliplatin with Lapatinib vs. Capecitabine plus oxaliplatin | 53%/39%, P = 0.0031 | 6.0/5.4, P = 0.0381 | 12.2/10.5, NS | [22] | |
| Margetuximab | Margetuximab vs. Trastuzumab | 25%/14%, P < 0.001 | 5.8/4.9, P = 0.03 | 21.6/19.8, NS | [23] | |
| VEGF | ||||||
| Bevacizumab | Capecitabine plus cisplatin with Bevacizumab vs. Capecitabine plus cisplatin | 46%/37.4%, P = 0.0315 | 6.7/5.3, P = 0.0037 | 12.1/10.1, NS | [24] | |
| VEGFR | ||||||
| Ramucirumab | Ramucirumab vs. placebo | 3%/3%, NS | 2.1/1.3, P < 0.0001 | 5.3/3.8, P = 0.047 | [25] | |
| Ramucirumab | Paclitaxel with Ramucirumab vs. Paclitaxel | 28%/16%, P = 0.0001 | 4.4/2.9, P = 0.0001 | 9.6/7.4, P = 0.017 | [26] | |
| Apatinib | Apatinib vs. placebo | 2.84%/0%, NS | 2.6/1.8, P < 0.001 | 6.5/4.7, P = 0.0149 | [27] | |
| Regorafenib | Regorafenib vs. placebo | NR | 2.6/0.9, P < 0.001 | 5.8/4.5, NS | [28] | |
| EGFR | ||||||
| Cetuximab | Capecitabine plus cisplatin with Cetuximab vs. Capecitabine plus cisplatin | 30%/29%, NS | 4.4/5.6, NS | 9.4/10.7, NS | [29] | |
| Panitumumab | Epirubicin, oxaliplatin, and capecitabine with Panitumumab vs. Epirubicin, oxaliplatin, and capecitabine | 46%/42%, NS | 6.0/7.4, NS | 8.8/11.3, P = 0.013 | [30] | |
| FGFR | ||||||
| Bemarituzumab | Modified FOLFOX6 with Bemarituzumab vs. modified FOLFOX6 | 47%/33% | 9.5/7.4, P = 0.073 | Not reached/12.9, P = 0.027 | [31] | |
| mTOR | ||||||
| Everolimus | Everolimus vs. placebo | 4.5%/2.1% | 1.7/1.4, P < 0.001 | 5.4/4.3, NS | [32] | |
| Claudin 18.2 | ||||||
| Zolbetuximab | EOX with Zolbetuximab vs. EOX | 39.0%/25.0%, P = 0.034 | 7.5/5.3, P < 0.0005 | 13.0/8.3, P < 0.0005 | [33] | |
| Immunotherapy-Immune Checkpoint Inhibitors (ICIs) | PD-1 | |||||
| Nivolumab | Nivolumab vs. placebo | 11.2%/0%, P = 0.0088 | 6.1/1.61, P < 0.0001 | 11.6/5.26, P < 0.0001 | [34] | |
| Nivolumab | CTx (S-1 or capecitabine plus oxaliplatin) with Nivolumab vs. CTx (S-1 or capecitabine plus oxaliplatin) | 57.5%/47.8%, P = 0.0088 | 10.45/8.34, P = 0.0007 | 17.45/17.15, NS | [35] | |
| Pembrolizumab | Pembrolizumab vs. paclitaxel | 16%/14% | 1.5/4.1, P = 0.0007 | 9.1/8.3, P = 0.0421 | [36] | |
| PD-L1 | ||||||
| Durvalumab | Durvalumab plus tremelimumab, 2 L vs. Durvalumab, 2 L vs. Tremelimumab, 2 L vs. Durvalumab, tremelimumab, 3 L vs. Durvalumab, tremelimumab, 2 L/3 L | 7.4%/0%/8.3%/4.0% | 1.8/1.6/1.7/1.8 | 3.4/3.2/7.7/10.6, | [37] | |
| Avelumab | Avelumab vs. chemotherapy (paclitaxel or irinotecan) | 2.2%/4.3% | 1.4/2.7, NS | 4.6/5.0, NS | [38] | |
| CTLA-4 | ||||||
| Ipilimumab | Ipilimumab vs. first-line chemotherapy | 1.8%/7.0% | 2.72/4.90, P = 0.034 | 12.7/12.1 | [39] | |
| Tremelimumab | Durvalumab plus tremelimumab, 2 L vs. Durvalumab, 2 L vs. Tremelimumab, 2 L & Durvalumab, tremelimumab, 3 L vs. Durvalumab, tremelimumab, 2 L/3 L | 7.4%/0%/8.3%/4.0% | 1.8/1.6/1.7/1.8 | 3.4/3.2/7.7/10.6, | [37] | |
Drug resistance leads to pharmacological treatment failure and poor outcomes for advanced GC patients. The mechanisms of drug resistance of GC are divided into seven groups, according to the previously proposed classification[40]: change in drug intracellular concentration (MOC-1), change in drug metabolism (MOC-2), change in drug targets (MOC-3), change in DNA repair (MOC-4), change in apoptosis and survival (MOC-5), change in tumor cell microenvironment (MOC-6), and phenotypic transformation (MOC-7) [Figure 1]. We summarize the updated knowledge of the molecular mechanisms attributed to drug resistance in GC in Figure 2.
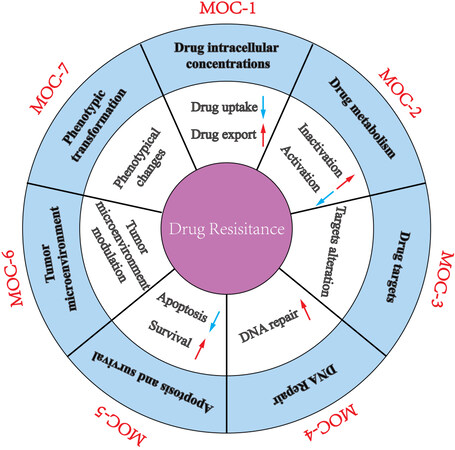
Figure 1. A schematic diagram depicting the molecular mechanisms accounting for drug resistance in gastric cancer.
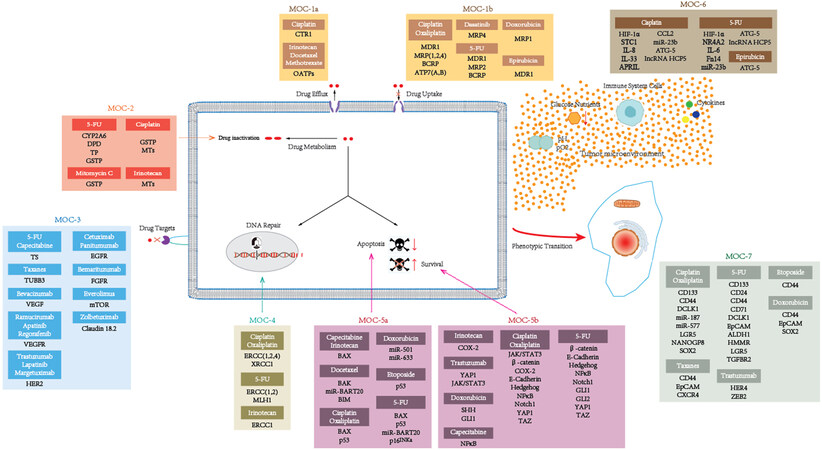
Figure 2. Proteins and non-coding RNAs accounting for drug resistance in gastric cancer. This figure is based on the work of Marin et al.[40].
Cancer stem cells (CSCs) are a subpopulation of stem cell-like cancer cells that are responsible for cancer pathogenesis including initiation, development, drug resistance, metastasis, and cancer recurrence[41-43]. In recent years, accumulating evidence indicates the presence of CSCs in various types of cancers, including brain[44], breast cancer[45], head and neck cancer[46], renal cancer[47], colon cancer [48-50], pancreatic cancer[51-52], liver cancer[53], lung cancer[54], prostate cancer[55], and melanoma[56], and targeting CSCs may be essential to prevent tumor relapse and spread[57]. Moreover, growing evidence suggests that there are several signaling pathways preferentially associated with CSCs[58-60], including Hedgehog, Notch, WNT/β-catenin, JAK/STAT, PI3K/PTEN, and NF-κB pathways, which contribute to the survival, self-renewal, and differentiation properties of CSCs[61].
CSC-targeting therapies are currently being investigated to reverse chemoresistance, including chemotherapeutic and biological agents that target stemness pathways including Hedgehog, Notch, Hippo/YAP1, JAK/STAT, and Wnt/β-catenin pathways; cancer stem cell surface markers including CD24, CD44, CD54, CD71, CD90, CD133, ALDH, CXCR4, EpCAM, LGR5, Sox2, and Oct4; the CSC microenvironment; and others[62-64]. However, these current strategies to target CSCs are not specific to CSCs, leading to toxic effects on normal tissues.
CSCs in GC were first identified from a panel of human GC cell lines[65]. Cancer stem cells from either human GC cell lines or tumor tissues were isolated using cell surface markers such as CD24, CD44, CD54, CD71, CD90, CD133, Lgr5, ALDH1, EpCAM, and CXCR4[63, 66-67]. Although studies suggest the presence of gastric cancer stem cells (GCSCs), the origin of GCSCs is currently unclear and controversial. Two major hypotheses propose that GCSCs are derived from normal gastric stem cells (GSCs) or from bone marrow-derived mesenchymal stem cells (BM-MSCs)[68, 69].
In recent years, growing evidence shows that GCSCs play important roles in drug resistance in GC. Thus, understanding GCSC functions and their roles in drug resistance, as well as defining the molecular mechanisms of drug resistance, will help identify potential anticancer drug targets and develop new chemotherapeutic drugs and effective therapeutic strategies to improve the clinical outcomes of GC patients. In this review, we summarize our current understanding of the roles of GCSCs in GC drug resistance, as well as provide a comprehensive analysis of the potential molecular mechanisms by which CSCs contribute to drug resistance in GC.
GCSCs AND DRUG RESISTANCE
Substantial studies have demonstrated that GCSCs are resistant to conventional radio-/chemotherapy. Aldehyde dehydrogenase (ALDH) is generally highly expressed in stem cells and considered as a CSC marker[70]. Gastric cancer cells with high expression of ALDH showed strong resistance to 5-fluorouracil (FU) and cisplatin; thus, high expression of ALDH in GC cell lines is believed to play a key role in resistance to chemotherapeutic drugs in GC[71,72]. Similarly, upregulation of LGR5, another GCSC marker, significantly enhanced cell stemness and drug resistance in MGC803 cells[73]. Further studies have shown that LGR5+ GCSCs are resistant to cisplatin treatment[74]. We recently showed that CD44+/CD54+ GCSCs isolated from cancer tissues can survive and expand after treatment with 5-FU and cisplatin[75]. Consistent with our results, another study showed that KHDRBS3 plays an important role in the acquisition of 5-FU resistance by regulating CD44 variant expression[76]. These results show that GCSCs play a key role in the acquisition of drug resistance in GC.
Accumulating evidence suggests the drug resistance capability of GCSCs is significantly higher than that of GC cells, and GCSCs can be enriched in GC after chemotherapy. Compared with GC cells, GCSCs showed stronger resistance to chemotherapeutic drugs 5-FU and oxaliplatin[77]. CSCs can be isolated or enriched by CSC-specific surface markers or through stem cell side population (SP) analysis[78]. Similarly, GCSCs isolated from GC cell lines by the SP method showed more resistance to chemotherapy[79]. Further study showed that CD44+ GCSCs isolated from tumor tissues were significantly enriched after treatment with 5-FU[80]. Another study demonstrated that ALDH+ CSCs in GC cell cultures can be enriched after treatment with cisplatin and 5-fluorouracil[81]. Meanwhile, clinical studies have revealed that resistance to anticancer drugs of GC is mainly associated with GCSCs. Patients with high CD133 expression exhibited stronger drug resistance, higher relapse rate, and lower five-year survival rate compared with patients with low CD133 expression[82]. Similarly, patients with high CD44 and CD133 expression showed worse survival[83]. Furthermore, expression of LGR5 and CD133 was identified to be significantly associated with poor clinical outcomes, and patients who are LGR5+ and CD133+ showed a lower overall survival rate than those who are LGR5- and CD133-[84]. The results from a phase II clinical trial show that GC patients with high expression CD44 who received chemotherapy with vismodegib, a hedgehog inhibitor, held a survival advantage[85]. Therefore, GCSCs are a major factor in GC resistance to radiation and chemotherapy.
THE UNDERLYING MECHANISMS FOR GCSCS REGULATING THE DRUG RESISTANCE
Drug resistance is a multifactorial phenomenon involving various components and multiple interrelated pathways, which work together to contribute to the development of this phenomenon. Various CSC-associated signaling pathways and molecular mechanisms have been described as implicated in CSC drug resistance[86]. To our knowledge, the underlying molecular mechanisms by which GCSCs contribute to chemoresistance include dormancy, drug trafficking, drug metabolism and targeting, apoptosis and cell death, DNA damage, epithelial-mesenchymal transition (EMT), and tumor microenvironment. The molecular mechanisms attributed to drug resistance in GCSCs are described below based on the previously proposed classification (MOC-1-7)[40], and a schematic outline is summarized in Figure 3.
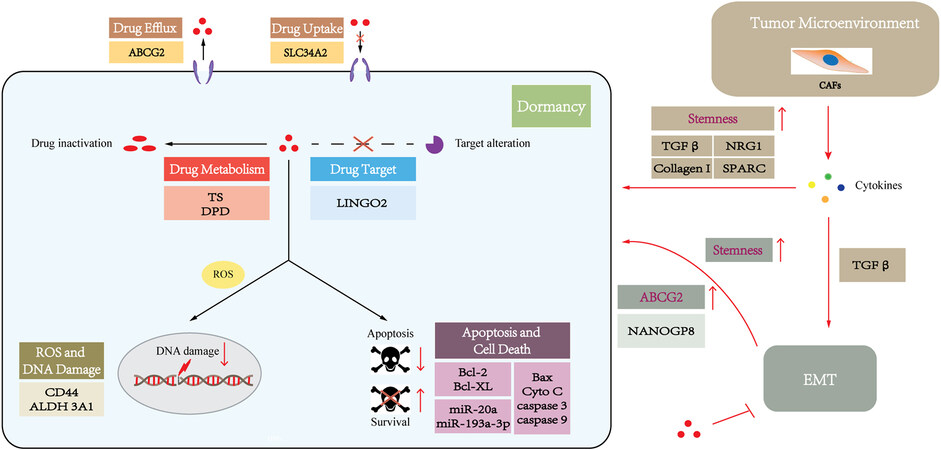
Figure 3. A schematic diagram depicting the molecular mechanisms accounting for chemoresistance in GCSCs. This figure is based on the work of Marin et al.[40].
Dormancy
Tumor dormancy contributes to the development of chemoresistance, metastasis, and cancer recurrence. CSCs are frequently in a quiescent state in which CSCs can remain in the G0/G1 stage with a low proliferation rate[87,88]. As most conventional chemotherapeutic drugs target proliferating cells, quiescence properties support CSCs to become resistant to radio- and chemotherapy[72,89-90]. Accordingly, 5-FU-resistant GC cells with CSC features were found to be mainly quiescent cells, which remained in the G0/1 phase[91]. Similarly, IL-17 enhances the proliferative capacity of quiescent gastric stem cells[92], potentially promoting these transformed GCSCs to be sensitive to chemotherapy.
Changes in drug uptake, efflux, metabolism, and targeting
Reduced drug uptake
One of the most studied mechanisms of cancer drug resistance is reducing the uptake of drugs. The uptake of drugs into tumor cells can be active transportation mediated by the membrane transporters. Most of these membrane transporters are solute carrier (SLC) proteins, which play an essential role in drug uptake. The SLC protein family contains more than 300 proteins that mediate the absorption of multiple types of substrates, including amino acids, sugars, organic cations, anions, etc., as well as chemotherapeutic drugs[93]. Unexpectedly, SLC34A2 was found to be increased in CD44+ GCSCs, and suppression of SLC34A2 in GCSCs reduced the effects of chemoresistance[94]. This result is consistent with SLC34A2 potentially having an oncogenic role in GC cells[95]. However, the detailed molecular mechanism remains unclear, which requires further investigation.
Increased drug efflux
Another mechanism of drug resistance is associated with the increased efflux of cytotoxic drugs by active ATP-binding cassette (ABC) transporter proteins, which is known as “drug efflux”. Forty-eight ABC transporter members have been identified in humans and are divided into seven distinct subfamilies (ABCA-ABCG) with different functions. Only 13 ABC transporters (ABCA2/3, ABCB1/2/5, ABCC1/2/3/4/5/6/10, and ABCG2) have been directly associated with chemoresistance[96]. Three major ABC transporters, including P-glycoprotein (P-gp/MDR1/ABCB1), multidrug resistance-associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2), have been found in various drug-resistant cancer cell lines and tissues and studied extensively for their correlation to multidrug resistance (MDR)[97]. These ABC transporters can lower the intracellular drug concentration by pumping out chemotherapy-based agents, including vinblastine, vincristine, doxorubicin, daunorubicin, actinomycin-D, and taxanes[98,99].
CSCs express a high number of ABC transporter proteins on their cell surface[64,86], and ABC transporters in CSCs have been shown to play an important role in drug resistance[72,100-101]. Therefore, ABC transporters are widely used as surface markers for CSC identification and isolation[57].
ABCG2 is a major multidrug resistance pump, which is a downstream target of the sonic hedgehog (SHH)-glioma-associated oncogene homolog (GLI) signaling pathway[102]. Recent studies have indicated ABCG2 plays a pivotal role in drug resistance in GCSCs. CD44+/Musashi-1+ GCSCs with increased expression of ABCG2 exhibited resistant to doxorubicin[103]. Moreover, the inactivation of SHH-GLI signaling pathways decreased ABCG2 expression, rendering GCSCs more chemosensitive to doxorubicin[103]. This result implies that ABCG2 is a potential therapeutic target against CSCs to overcome drug resistance. Moreover, inhibition of ABCG2 expression by genistein, which is the predominant isoflavone in soy products, could inhibit gastric cancer stem cell-like features and reduce the chemoresistance of GCSCs[104]. Another study also demonstrated miR-132 could enhance cisplatin resistance in LGR5+ GCSCs via the SIRT1/CREB/ABCG2 signaling pathway[74]. However, the roles of other ABC transporter proteins of GCSCs in drug resistance are not clearly defined, and further investigations are needed to explore the roles of ABCs in cancer therapies against GCSCs.
Altered drug metabolism
Thymidylate synthase (TS) and dihydropyrimidine dehydrogenase (DPD) are both key 5-FU metabolic enzymes. TS is a target enzyme of 5-FU, and 5-FU exerts an anticancer effect by its conversion into fluorodeoxyuridine monophosphate (FdUMP) which can form a ternary complex with TS to cause suppression of the de novo synthesis of dTMP. DPD is the initial and rate-limiting enzyme that translates 5-FU into metabolites without cytotoxicity. TS and DPD, which are representative markers of 5-FU resistance, were shown to be significantly upregulated in a 5-FU-resistant CSC-like cell population in GC[76]. Hence, metabolic inactivation or alteration of the anticancer drugs in GCSCs enable CSCs to resist therapy and strengthen their stemness.
Changed drug targeting
Drug targeting altering by changing the expression and function of drug targets is also one of the major causes of drug resistance. Receptors of tyrosine kinase and its downstream signaling pathway play a pivotal role in carcinogenesis and tumor development and constitute the targets for tyrosine kinase inhibitors (TKIs). For instance, the EGFR signaling pathway is involved in the pathogenesis and progression of cancers by activation of either RAS/RAF/MEK/ERK or PI3K/AKT/mTOR cascade[105]. The activation of MEK, a component of the EGFR/Ras/RAF/MEK/ERK signaling pathway, caused drug resistance to MEK inhibitors[106]. Correspondingly, silencing LINGO2, a GCSC-related marker, reduced AKT, ERK, and MEK phosphorylation[107], suggesting that activation of AKT, ERK, and MEK in GCSCs is responsible for chemoresistance to the inhibitors targeting these kinases.
Inhibition of apoptosis and cell death
One of the primary goals of most anticancer agents is to cause tumor-selective cell death. The resistance to apoptosis, one of the key regulatory events leading to cell death, is the hallmark of cancer. Apoptosis occurs through extrinsic and intrinsic pathways that are dependent on caspase activation and mitochondrial outer membrane permeabilization (MOMP), respectively. The extrinsic apoptotic pathway is often related to ligands such as TNF-α, TNF-α-related apoptosis-inducing ligand (TRAIL), and Fas-ligand (FasL) and cell death receptors such as TNFR, TRAILR, FasR, linker proteins, and caspases 3, 6, 7, and 8. The intrinsic pathway is triggered by mitochondrial membrane disturbance following various stimuli including DNA damage and radio-/chemotherapy. Pro-apoptotic proteins such as Bax and Bak, as well as anti-apoptotic proteins such as Bcl2 and Bcl-XL are involved. Both intrinsic and extrinsic pathways activate caspases and ultimately lead to cell apoptosis. Increasing evidence suggests that disruption of the apoptotic pathway impacts resistance to anticancer drugs in GCSCs. Pro-apoptotic proteins including Bax, cytochrome C, caspase 9a, cleaved caspase 3, and cleaved caspase 9 were observed to be downregulated, while anti-apoptotic proteins Bcl-2 and Bcl-XL were upregulated in CD44+ GC cells compared with CD44- GC cells[108]. Moreover, in CD44+ GC cells, inhibition of miR-193a-3p can induce apoptosis by activating the mitochondrial apoptotic pathway and enhance the chemotherapeutic response of cisplatin[108]. Thus, GCSCs can induce resistance to drug-mediated apoptosis by upregulation or activation of anti-apoptotic proteins or downregulation or mutation of pro-apoptotic proteins. Similarly, miR-20a could increase cisplatin resistance in GC cells via modulating the anti-apoptotic factors livin and survivin[109,110], whereas miRNA-19b, -20a, and -92a are proven to promote GCSCs properties[111]. miR-20a may also be involved in the development of chemoresistance in GCSCs by modulating apoptosis through livin and survivin.
Repair and prevention of DNA damage
The dynamic balance between DNA damage and repair depends on the type of injury and the activity of a variety of repair mechanisms: nucleotide-excision repair (NER), base-excision repair (BER), mismatch repair (MMR), non-homologous end-joining (NHEJ), and homologous recombination (HR) systems. DNA damage-inducing agents are among the most effective treatment regimens in clinical chemotherapy. However, GCSCs can be resistant to DNA damage by drug treatment-induced reactive oxygen species (ROS) scavenging. Gastrointestinal cancer cells with high CD44 expression exhibited an enhanced capacity for GSH synthesis, resulting in defense against ROS[112]. CSC marker ALDH can facilitate detoxification by scavenging of ROS, as well as by producing antioxidant compounds such as NADP[113]. Aldehyde dehydrogenase 3A1 was found to be upregulated in gastric cancer stem-like cells[114]. Moreover, in multiple GC cell lines and hematopoietic malignancies, ALDH is highly expressed in ROS-low cells, and ALDH-high/ROS-low cells may be cancer-initiating cells (CISs)[115-117], which are also called CSCs. These data indicate that ALDH+ GCSCs can enhance the resistance to chemotherapy or radiochemotherapy by reducing the level of ROS and avoiding DNA damage.
Epithelial-mesenchymal transition
Epithelial-mesenchymal transition (EMT) is a process of lineage transition whereby epithelial cells lose their epithelial traits and acquire mesenchymal cell phenotypes, with corresponding changes in cell morphology and expression of surface markers[118]. EMT facilitates tumor cell migration, invasion, metastasis, and drug resistance[119]. Several cytokines, chemokines, and growth factors can trigger EMT by activation of a group of EMT-inducing transcription factors (EMT-TFs) such as SNAIL, SLUG, ZEB1/2, and TWIST[120]. EMT is regulated by a wide, complex, interactive molecular network including exogenous inducers, intracellular regulatory miRNA, epigenetic modulators, and cellular signaling pathways including MAPK, ERK, PI3K, SMADs, and Wnt/β-catenin[121].
EMT has been shown to regulate the acquisition of stemness in multiple cancer cells[122] and promote CSC stemness and quiescence that increase drug resistance[123]. EMT could induce CSC characteristics that increase drug resistance through different mechanisms including the hedgehog, Wnt, Notch, and Musashi signaling pathways, as well as the epigenetic regulator Bmi1[124,125].
EMT activation confers drug resistance in CSCs through other mechanisms, including promoting drug efflux by increased levels of ABC pumps or inhibition of cell apoptosis by elevated expression of anti-apoptotic proteins such as Bcl-XL[123-124,126]. Correspondingly, NANOGP8, one of the pseudogenes in the NANOG gene family, is identified to be the main regulator of GCSCs, which can promote EMT/stemness and enhance chemoresistance[127]. NANOGP8 may confer gastric cancer cells with chemoresistance by upregulation of ABCG2[127]. However, the exact molecular mechanisms responsible for EMT and the resulting drug resistance in GCSCs remain uncertain.
Adaptation to tumor microenvironment
CSCs are found in a specialized tumor microenvironment (TME), known as the niche, which is mainly composed of extracellular matrix (ECM), cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs), and endothelial, mesenchymal, and immune cells, and those conditions promote CSC adaptation[128-129]. Reciprocal interactions between CSCs and the niche are critical for CSCs to maintain their stemness properties and promote tumor initiation, metastasis, and drug resistance[130].
Increasing evidence highlights that the TME takes part in therapeutic resistance in GCSCs, largely involving CAFs, which remarkably influence the TME via the secretion of various growth factors, cytokines, and chemokines[131]. The main component secreted by CAFs is TGFβ, which induces EMT[132] and promotes the acquisition of GCSC features[133], ultimately leading to drug resistance[134]. Another study showed that CAFs can also promote stemness by the secretion of NRG1, which activates the NF-κB signaling in GC[135]. Moreover, CAFs can induce drug resistance not only by promoting stem-related signaling pathways in GCSCs but also by secreting type I collagen, which contributes to decreasing drug uptake[136]. Additionally, a recent study demonstrated that low expression of gastric CAF-derived SPARC (secreted protein acidic and rich in cysteine) can promote GCSC transformation and 5-FU resistance[137], suggesting that CAF-secreted SPARC may be involved in the regulation of drug resistance of GCSCs. Collectively, this evidence implicates an important role of the TME in the development of drug resistance of GC.
Exosomes
Exosomes (about 30-200 nm) are small extracellular vesicles (EVs) that originate from endosomes and are secreted by live cells into the extracellular space through the fusion of multivesicular bodies (MVBs) with the plasma membrane[138]. They are composed of a transmembrane protein-containing lipid bilayer and cell-state-specific molecules including DNAs, mRNAs, ncRNAs, and proteins in the vesicle lumen. Exosomes, as carriers, mediate cell-to-cell communication and substance exchange via the transfer of donor cell-derived contents to recipient cells[139]. Increasing evidence suggests that tumor-derived exosomes play critical roles in many aspects of cancer, including tumor growth, metastasis, angiogenesis, immunity, and other processes, and can be used as potential diagnostic biomarkers or therapeutic targets for cancer patients[140-142]. Recent studies showed exosomes are associated with the transfer of the drug resistance phenotype, and cancer cells could develop drug resistance after the incorporation of exosomes from drug-resistant cancer cells. Studies indicated exosomal PD-L1 promotes chemoresistance via inducing T cell exhaustion, by which the T cells cannot be reinvigorated by anti-PD-1 treatment[143]. Inhibition of exosomal PD-L1 has also been reported to enhance the efficacy of anti-PD-L1 treatment[143,144]. Furthermore, another study showed chemotherapeutic agents stimulated the secretion of ABCB1-enriched exosomes from drug-resistant cells and increased the transfer of ABCB1 to the recipient cancer cells, thus assisting these sensitive cancer cells in developing the resistant phenotype[145]. More recently, it has been shown that exosomal transference of wild-type EGFR to EGFR-mutated sensitive cancer cells promotes resistance to the mutant-selective EGFR inhibitor osimertinib by activating the MAPK and PI3K/AKT signaling pathways[146]. Thus, exosomes could be novel therapeutic targets, which could overcome resistance to chemotherapeutic drugs[105, 106] or antibody-based approaches[143, 144], and they also might serve as a predictive biomarker for clinical responses to anti-PD-1 therapy[144].
Increasing evidence also highlights that exosomes are involved in the drug resistance of CSCs. The underlying mechanisms are complex, including cell cycle blockage, increased drug efflux, upregulation of detoxifying enzymes, enhanced anti-apoptotic capacity and DNA repair efficiency, inducing EMT process, and immunosuppression[147,148]. However, the physiological and functional properties of exosomes in GCSCs are still unknown and need further investigation.
Extrachromosomal circular DNA
Extrachromosomal circular DNA (eccDNA) refers to a type of double-stranded circular DNA that originates from but is independent of chromosomes, which is widely present in various eukaryotic cells and can be derived from anywhere in a genome with sizes ranging from hundreds of base pairs (bp) to several megabases (Mb)[149]. According to the size and origin, eccDNAs can be categorized into organelle eccDNAs such as mitochondrial DNAs (mtDNAs) or non-organelle eccDNA such as telomeric circle (t-circles), microDNA (100-400 bp), small polydispersed circular DNA (spcDNA) (100 bp-10 kb), episomes, and double minutes (DMs) (100 kb-3 Mb)[150]. eccDNAs play important roles in gene regulation, sponging of transcription factors, environmental adaptation and evolution, aging, immune response, cell-to-cell communications, and tumor development[151,152].
eccDNAs have been shown to help cancer cells develop drug resistance via various mechanisms [Figure 4]. (A) Amplification of drug target genes: For instance, DMs, which contain the gene coding for dihydrofolate reductase (DHFR), were identified to be amplified and associated with the development of methotrexate (MTX) resistance[153]. (B) Amplification of multidrug resistance (MDR) genes: DMs, bearing the multidrug resistance 1 (MDR1) gene, were amplified in human epidermoid carcinoma cells and caused resistance to various anticancer drugs by upregulation of MDR1[154]. (C) “Hide and seek” mechanism: EGFRvIII, an oncogenic variant, can induce tumor cells to be more sensitive to EGFR tyrosine kinase inhibitor (TKI). Previous studies have demonstrated that erlotinib resistance in glioblastoma is caused by the elimination of DMs containing EGFRvIII[155,156]. However, after erlotinib withdrawal, the mutant EGFR re-emerged on DMs, which induced GBM cells to be re-sensitive to erlotinib treatment[156]. Through this “hide and seek” mechanism, cancer cells can evade drug therapy by dynamic modulation of drug-targeted oncogenes residing on eccDNAs. (D) Increasing intratumoral heterogeneity: eccDNAs can drive heterogeneity among daughter tumor cells, thus inducing these cells to obtain survival advantage under drug pressure[150]. (E) Increasing homologous recombination activity: Homologous recombination is associated with eccDNA biogenesis. Recent studies have shown that homologous recombination activity was increased in DM-carrying MTX-resistant colon cancer cells, whereas inhibition of homologous recombination activity decreased the expression of DM-containing genes and enhanced drug sensitivity in MTX-resistant cells[157].
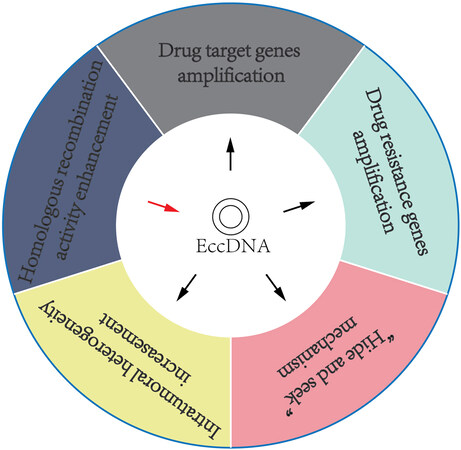
Figure 4. Overview of our current understanding of cancer drug resistance mechanisms induced by eccDNAs.
eccDNAs contribute to a variety of features in cancers and may serve as novel, promising molecular markers to shed new insights into the diagnosis, prognosis, and treatment of cancer patients. However, the functions and underlying mechanisms of eccDNA in CSCs are still unclear and require further exploration.
Helicobacter pylori infection
Helicobacter pylori (H. pylori) infection remains a main risk factor in the development of GC. In 1994, H. pylori was diagnosed as a Group I carcinogen by the WHO (World Health Organization)[158]. The stem cell hypothesis of cancer formation is that stem or progenitor cells can acquire CSC characteristics, evade homeostatic control, and lead to carcinogenesis. H. pylori has been shown to induce EMT and cancer stem cell (CSC)-like properties in gastric epithelial cells[159,160] and gastric cancer cells[161,162]. Data from multiple studies show that H. pylori may directly interact with gastric stem/progenitor cells[163-164] or bone marrow-derived cells (BMDCs)[165] to impact the status and properties of these cells, which could be responsible for generating GCSCs. Moreover, H. pylori infection can induce inflammation, impact the local microenvironment, and affect gastric stem/progenitor cells and their differentiation by inducing genetic or epigenetic alterations[166-168]. H. pylori infection can mediate oncogenic transformation by inducing GCSCs generation or affecting gastric stem/progenitor cells. However, the underlying mechanisms leading to GCSC emergence and the resulting drug resistance in GCSCs in response to H. pylori infection are awaiting further investigation.
CONCLUSION
Conventional radio-/chemotherapy provides a limited effect on prolonging the survival of advanced GC patients, and, recently, accumulating evidence shows that GCSCs are resistant to conventional chemotherapy and play a direct role in tumor metastasis and relapse. Based on the extensive evidence presented in this review, it is obvious that GCSCs regulate tumor radio-/chemoresistance via multiple intrinsic and extrinsic mechanisms. This review aims to provide an understanding of the precise mechanisms underlying GCSC resistance to chemotherapeutic drugs. Identifying the molecules and revealing insight into their interaction networks through further investigations may help to discover novel targets of anticancer therapy, develop new therapeutic approaches for the prevention of tumor recurrence and resistance, and increase the lifespan of GC patients.
However, to date, molecular mechanisms of drug resistance in GCSC remain largely unclear. Many aspects are still in need of further clarification: (1) to find more key components or molecules for regulating GCSCs resistance to the anticancer agents; (2) to define the precise molecular mechanisms and clarify how GCSCs coordinate these different, complex molecular pathways to response the chemotherapeutic drugs; and (3) to find the specific GCSC markers related to its response to the anticancer agents, so as to evaluate the effectiveness of different drugs and therapeutic strategies. More importantly, many more need to be proven to be effective in the clinic. We are at the beginning of understanding drug resistance from gastric cells to GCSCs. More basic and clinical studies should be done to increase the knowledge about the mechanisms of drug resistance to improve the outcome of advanced GC patients.
DECLARATIONS
Authors’ contributionsWrote the manuscript: Xiong J, Fu L
Literature review and analysis: Zhang T, Lan P, Zhang S
Planed and designed figures: Xiong J, Fu L
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from the National Natural Science Foundation of China (No. 82173003), the National Key R&D Program of China (No. 2017YFA0503900), the Science and Technology Program of Guangdong Province in China (No. 2019B030301009), the Industry and Information Technology Foundation of Shenzhen (No. 20180309100135860), the SZU Top Ranking Project (No. 86000000210), the Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515010989), and the Medical Scientific Research Foundation of Guangdong Province (A2019434)
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209-49.
2. Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965;64:31-49.
3. Fléjou JF. WHO Classification of digestive tumors: the fourth edition. Ann Pathol 2011;31:S27-31.
4. Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202-9.
5. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015;21:449-56.
7. Jim MA, Pinheiro PS, Carreira H, Espey DK, Wiggins CL, Weir HK. Stomach cancer survival in the United States by race and stage (2001-2009): findings from the CONCORD-2 study. Cancer 2017;123 Suppl 24:4994-5013.
8. MacDonald JS, Schein PS, Woolley PV, et al. 5-Fluorouracil, doxorubicin, and mitomycin (FAM) combination chemotherapy for advanced gastric cancer. Ann Intern Med 1980;93:533-6.
9. Wils JA, Klein HO, Wagener DJ, et al. Sequential high-dose methotrexate and fluorouracil combined with doxorubicin-a step ahead in the treatment of advanced gastric cancer: a trial of the European Organization for Research and Treatment of Cancer Gastrointestinal Tract Cooperative Group. J Clin Oncol 1991;9:827-31.
10. Webb A, Cunningham D, Scarffe JH, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol 1997;15:261-7.
11. Cutsem E, Moiseyenko VM, Tjulandin S, et al; V325 Study Group. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006;24:4991-7.
12. Cunningham D, Starling N, Rao S, et al. Upper gastrointestinal clinical studies group of the national cancer research institute of the united kingdom. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36-46.
13. Kang YK, Kang WK, Shin DB, et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 2009;20:666-73.
14. Shah MA, Janjigian YY, Stoller R, et al. Randomized multicenter phase II study of modified docetaxel, cisplatin, and fluorouracil (DCF) versus DCF plus growth factor support in patients with metastatic gastric adenocarcinoma: a study of the US gastric cancer consortium. J Clin Oncol 2015;33:3874-9.
15. Koizumi W, Kim YH, Fujii M, et al. JACCRO and KCSG Study Group. Addition of docetaxel to S-1 without platinum prolongs survival of patients with advanced gastric cancer: a randomized study (START). J Cancer Res Clin Oncol 2014;140:319-28.
16. Guimbaud R, Louvet C, Ries P, et al. Prospective, randomized, multicenter, phase III study of fluorouracil, leucovorin, and irinotecan versus epirubicin, cisplatin, and capecitabine in advanced gastric adenocarcinoma: a French intergroup (Fédération Francophone de Cancérologie Digestive, Fédération Nationale des Centres de Lutte Contre le Cancer, and Groupe Coopérateur Multidisciplinaire en Oncologie) study. J Clin Oncol 2014;32:3520-6.
17. Li K, Zhang A, Li X, Zhang H, Zhao L. Advances in clinical immunotherapy for gastric cancer. Biochim Biophys Acta Rev Cancer 2021;1876:188615.
18. Hsu A, Raufi AG. Advances in Systemic Therapy for gastric cancer. Gastrointest Endosc Clin N Am 2021;31:607-23.
19. Bang Y, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687-97.
20. Shitara K, Bang YJ, Iwasa S, et al. DESTINY-Gastric01 Investigators. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med 2020;382:2419-30.
21. Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN-a randomized, phase III study. J Clin Oncol 2014;32:2039-49.
22. Hecht JR, Bang YJ, Qin SK, et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO-013/LOGiC--a randomized phase III trial. J Clin Oncol 2016;34:443-51.
23. Rugo HS, Im SA, Cardoso F, et al. SOPHIA Study Group. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol 2021;7:573-84.
24. Ohtsu A, Shah MA, Van Cutsem E, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29:3968-76.
25. Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31-9.
26. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224-35.
27. Li J, Qin S, Xu J, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol 2016;34:1448-54.
28. Pavlakis N, Sjoquist KM, Martin AJ, et al. Regorafenib for the treatment of advanced gastric cancer (INTEGRATE): a multinational placebo-controlled phase II trial. J Clin Oncol 2016;34:2728-35.
29. Lordick F, Kang Y, Chung H, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:490-9.
30. Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481-9.
31. Catenacci DVT, Rasco D, Lee J, et al. Phase I escalation and expansion study of bemarituzumab (FPA144) in patients with advanced solid tumors and FGFR2b-selected gastroesophageal adenocarcinoma. J Clin Oncol 2020;38:2418-26.
32. Ohtsu A, Ajani JA, Bai YX, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol 2013;31:3935-43.
33. Sahin U, Koslowski M, Dhaene K, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res 2008;14:7624-34.
34. Kang Y, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461-71.
35. Shitara K, Van Cutsem E, Bang YJ, et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: the KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol 2020;6:1571-80.
36. Shitara K, Özgüroğlu M, Bang Y, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018;392:123-33.
37. Kelly RJ, Lee J, Bang YJ, et al. Safety and Efficacy of durvalumab and tremelimumab alone or in combination in patients with advanced gastric and gastroesophageal junction adenocarcinoma. Clin Cancer Res 2020;26:846-54.
38. Bang YJ, Ruiz EY, Van Cutsem E, et al. Phase III, randomised trial of avelumab versus physician's choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol 2018;29:2052-60.
39. Bang YJ, Cho JY, Kim YH, et al. Efficacy of sequential ipilimumab monotherapy versus best supportive care for unresectable locally advanced/metastatic gastric or gastroesophageal junction cancer. Clin Cancer Res 2017;23:5671-8.
40. Marin JJG, Perez-Silva L, Macias RIR, et al. Molecular bases of mechanisms accounting for drug resistance in gastric adenocarcinoma. Cancers (Basel) 2020;12:2116.
41. Pützer BM, Solanki M, Herchenröder O. Advances in cancer stem cell targeting: how to strike the evil at its root. Adv Drug Deliv Rev 2017;120:89-107.
42. Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer 2012;12:767-75.
44. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396-401.
45. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983-8.
46. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007;104:973-8.
47. Bussolati B, Dekel B, Azzarone B, Camussi G. Human renal cancer stem cells. Cancer Lett 2013;338:141-6.
48. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106-10.
49. Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007;445:111-5.
50. Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA 2007;104:10158-63.
51. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007;67:1030-7.
52. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313-23.
53. Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007;132:2542-56.
54. Lundin A, Driscoll B. Lung cancer stem cells: progress and prospects. Cancer Lett 2013;338:89-93.
55. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005;65:10946-51.
56. Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 2005;65:9328-37.
57. Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020;5:8.
58. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res 2017;36:41.
59. Mohammed MK, Shao C, Wang J, et al. Wnt/β-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis 2016;3:11-40.
60. Bekaii-Saab T, El-Rayes B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017;123:1303-12.
62. Nunes T, Hamdan D, Leboeuf C, et al. Targeting cancer stem cells to overcome chemoresistance. Int J Mol Sci 2018;19:4036.
63. Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol 2016;51:313-26.
64. Li Y, Wang Z, Ajani JA, Song S. Drug resistance and cancer stem cells. Cell Commun Signal 2021;19:19.
65. Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009;27:1006-20.
66. Qiao XT, Gumucio DL. Current molecular markers for gastric progenitor cells and gastric cancer stem cells. J Gastroenterol 2011;46:855-65.
67. Fu L, Bu L, Yasuda T, et al. Gastric cancer stem cells: current insights into the immune microenvironment and therapeutic targets. Biomedicines 2020;8:7.
68. Ray K. Stem cells: Settling the stomach - tracing gastric stem cells. Nat Rev Gastroenterol Hepatol 2016;13:626.
70. Toledo-Guzmán ME, Hernández MI, Gómez-Gallegos ÁA, Ortiz-Sánchez E. ALDH as a stem cell marker in solid tumors. Curr Stem Cell Res Ther 2019;14:375-88.
71. Zhi QM, Chen XH, Ji J, et al. Salinomycin can effectively kill ALDH(high) stem-like cells on gastric cancer. Biomed Pharmacother 2011;65:509-15.
72. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015;31:16-27.
73. Wang B, Chen Q, Cao Y, et al. LGR5 is a gastric cancer stem cell marker associated with stemness and the EMT SIgnature genes NANOG, NANOGP8, PRRX1, TWIST1, and BMI1. PLoS One 2016;11:e0168904.
74. Zhang L, Guo X, Zhang D, et al. Upregulated miR-132 in Lgr5+ gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol Carcinog 2017;56:2022-34.
75. Xiong J, Wang S, Chen T, et al. Verteporfin blocks Clusterin which is required for survival of gastric cancer stem cell by modulating HSP90 function. Int J Biol Sci 2019;15:312-24.
76. Ukai S, Honma R, Sakamoto N, et al. Molecular biological analysis of 5-FU-resistant gastric cancer organoids; KHDRBS3 contributes to the attainment of features of cancer stem cell. Oncogene 2020;39:7265-78.
77. Zhu Z, Xu J, Li L, et al. Effect of gastric cancer stem cell on gastric cancer invasion, migration and angiogenesis. Int J Med Sci 2020;17:2040-51.
78. Shimoda M, Ota M, Okada Y. Isolation of cancer stem cells by side population method. In: Papaccio G, Desiderio V, editors. Cancer stem cells. New York: Springer; 2018. pp. 49-59.
80. Chen W, Zhang X, Chu C, et al. Identification of CD44+ cancer stem cells in human gastric cancer. Hepatogastroenterology 2013;60:949-54.
81. Nishikawa S, Konno M, Hamabe A, et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int J Oncol 2013;42:1437-42.
82. Ishigami S, Ueno S, Arigami T, et al. Prognostic impact of CD133 expression in gastric carcinoma. Anticancer Res 2010;30:2453-2457. [PMID: 20651407].
83. Wang T, Ong CW, Shi J, et al. Sequential expression of putative stem cell markers in gastric carcinogenesis. Br J Cancer 2011;105:658-65.
84. Wattanawongdon W, Bathpho TS, Tongtawee T. Co-expression of LGR5 and CD133 cancer stem cell predicts a poor prognosis in patients with gastric cancer. Turk J Gastroenterol 2021;32:261-8.
85. Katoh M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin Sci (Lond) 2019;133:953-70.
86. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol 2020;60:166-80.
87. Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and cancer stem cells: an enigma for cancer therapeutic targeting. Cancer stem cells. Elsevier; 2019. pp. 43-84.
88. Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis 2021;12:835.
90. Takeishi S, Nakayama KI. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci 2016;107:875-81.
91. Xu ZY, Tang JN, Xie HX, et al. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci 2015;11:284-94.
92. Jiang YX, Yang SW, Li PA, et al. The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the underlying mechanisms. Oncogene 2017;36:1256-64.
93. Muley H, Fadó R, Rodríguez-Rodríguez R, Casals N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem Pharmacol 2020;177:113959.
94. Zhang L, Guo X, Zhang L, et al. SLC34A2 regulates miR-25-Gsk3β signaling pathway to affect tumor progression in gastric cancer stem cell-like cells. Mol Carcinog 2018;57:440-50.
95. Zhang JX, Xu Y, Gao Y, et al. Decreased expression of miR-939 contributes to chemoresistance and metastasis of gastric cancer via dysregulation of SLC34A2 and Raf/MEK/ERK pathway. Mol Cancer 2017;16:18.
96. Wang JQ, Wu ZX, Yang Y, et al. ATP-binding cassette (ABC) transporters in cancer: a review of recent updates. J Evid Based Med 2021;14:232-56.
97. Li W, Zhang H, Assaraf YG, et al. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 2016;27:14-29.
98. Gottesman MM, Pastan IH. The role of multidrug resistance efflux pumps in cancer: revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene. J Natl Cancer Inst 2015;107:djv222.
99. Duan H, Liu Y, Gao Z, Huang W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm Sin B 2021;11:55-70.
100. Kim JK, Jeon HY, Kim H. The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch Pharm Res 2015;38:389-401.
101. Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther 2011;89:491-502.
102. Pasello M, Giudice AM, Scotlandi K. The ABC subfamily A transporters: multifaceted players with incipient potentialities in cancer. Semin Cancer Biol 2020;60:57-71.
103. Xu M, Gong A, Yang H, et al. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett 2015;369:124-33.
104. Huang W, Wan C, Luo Q, Huang Z, Luo Q. Genistein-inhibited cancer stem cell-like properties and reduced chemoresistance of gastric cancer. Int J Mol Sci 2014;15:3432-43.
105. Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006;366:2-16.
106. Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev 2017;53:61-9.
107. Jo JH, Park SB, Park S, et al. Novel gastric cancer stem cell-related marker LINGO2 is associated with cancer cell phenotype and patient outcome. Int J Mol Sci 2019;20:555.
108. Lee SD, Yu D, Lee DY, Shin HS, Jo JH, Lee YC. Upregulated microRNA-193a-3p is responsible for cisplatin resistance in CD44(+) gastric cancer cells. Cancer Sci 2019;110:662-73.
109. Zhu M, Zhou X, Du Y, et al. miR-20a induces cisplatin resistance of a human gastric cancer cell line via targeting CYLD. Mol Med Rep 2016;14:1742-50.
110. Du Y, Zhu M, Zhou X, et al. miR-20a enhances cisplatin resistance of human gastric cancer cell line by targeting NFKBIB. Tumour Biol 2016;37:1261-9.
111. Shao Q, Xu J, Guan X, et al. In vitro and in vivo effects of miRNA-19b/20a/92a on gastric cancer stem cells and the related mechanism. Int J Med Sci 2018;15:86-94.
112. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011;19:387-400.
113. Muzio G, Maggiora M, Paiuzzi E, Oraldi M, Canuto RA. Aldehyde dehydrogenases and cell proliferation. Free Radic Biol Med 2012;52:735-46.
114. Wu D, Mou YP, Chen K, et al. Aldehyde dehydrogenase 3A1 is robustly upregulated in gastric cancer stem-like cells and associated with tumorigenesis. Int J Oncol 2016;49:611-22.
115. Ikeda J, Mamat S, Tian T, et al. Reactive oxygen species and aldehyde dehydrogenase activity in Hodgkin lymphoma cells. Lab Invest 2012;92:606-14.
116. Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int 2017;2017:5619472.
117. Gasparetto M, Smith CA. ALDHs in normal and malignant hematopoietic cells: potential new avenues for treatment of AML and other blood cancers. Chem Biol Interact 2017;276:46-51.
119. Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): a biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol 2017;232:3261-72.
120. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol 2019;21:102-12.
121. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420-8.
122. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep 2017;50:117-25.
123. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20:69-84.
124. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611-29.
125. Wilson MM, Weinberg RA, Lees JA, Guen VJ. Emerging mechanisms by which EMT programs control stemness. Trends Cancer 2020;6:775-80.
126. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741-51.
127. Ma X, Wang B, Wang X, Luo Y, Fan W. NANOGP8 is the key regulator of stemness, EMT, Wnt pathway, chemoresistance, and other malignant phenotypes in gastric cancer cells. PLoS One 2018;13:e0192436.
128. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 2016;11:47-76.
129. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 2019;24:25-40.
130. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell 2019;24:41-53.
131. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer 2019;18:70.
132. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer 2014;110:724-32.
133. Choi YJ, Kim N, Chang H, et al. Helicobacter pylori-induced epithelial-mesenchymal transition, a potential role of gastric cancer initiation and an emergence of stem cells. Carcinogenesis 2015;36:553-63.
134. Huang L, Wu RL, Xu AM. Epithelial-mesenchymal transition in gastric cancer. Am J Transl Res 2015;7:2141-58. [PMID: 26807164].
135. Han ME, Kim HJ, Shin DH, Hwang SH, Kang CD, Oh SO. Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J Gastroenterol 2015;50:645-56.
136. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest 2006;116:1955-62.
137. Ma Y, Zhu J, Chen S, et al. Low expression of SPARC in gastric cancer-associated fibroblasts leads to stemness transformation and 5-fluorouracil resistance in gastric cancer. Cancer Cell Int 2019;19:137.
139. Kalluri R, LeBleu VS. function
141. Tai YL, Chen KC, Hsieh JT, Shen TL. Exosomes in cancer development and clinical applications. Cancer Sci 2018;109:2364-74.
142. Dai J, Su Y, Zhong S, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther 2020;5:145.
143. Chen G, Huang AC, Zhang W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018;560:382-6.
144. Poggio M, Hu T, Pai CC, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019;177:414-427.e13.
145. Wang X, Qiao D, Chen L, et al. Chemotherapeutic drugs stimulate the release and recycling of extracellular vesicles to assist cancer cells in developing an urgent chemoresistance. Mol Cancer 2019;18:182.
146. Wu S, Luo M, To KKW, et al. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol Cancer 2021;20:17.
147. Wang J, Zheng Y, Zhao M. Exosome-based cancer therapy: implication for targeting cancer stem cells. Front Pharmacol 2016;7:533.
148. Lee NK, Kothandan VK, Kothandan S, Byun Y, Hwang SR. Exosomes and cancer stem cells in cancer immunity: current reports and future directions. Vaccines (Basel) 2021;9:441.
149. Zuo S, Yi Y, Wang C, et al. Extrachromosomal circular DNA (eccDNA): from chaos to function. Front Cell Dev Biol 2021;9:792555.
150. Wang M, Chen X, Yu F, Ding H, Zhang Y, Wang K. Extrachromosomal circular DNAs: origin, formation and emerging function in cancer. Int J Biol Sci 2021;17:1010-25.
151. Paulsen T, Kumar P, Koseoglu MM, Dutta A. Discoveries of extrachromosomal circles of DNA in normal and tumor cells. Trends Genet 2018;34:270-8.
152. Ling X, Han Y, Meng J, et al. Small extrachromosomal circular DNA (eccDNA): major functions in evolution and cancer. Mol Cancer 2021;20:113.
153. Curt GA, Carney DN, Cowan KH, et al. Unstable methotrexate resistance in human small-cell carcinoma associated with double minute chromosomes. N Engl J Med 1983;308:199-202.
154. Ruiz JC, Choi KH, von Hoff DD, Roninson IB, Wahl GM. Autonomously replicating episomes contain mdr1 genes in a multidrug-resistant human cell line. Mol Cell Biol 1989;9:109-15.
155. Nathanson DA, Gini B, Mottahedeh J, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014;343:72-6.
156. Nikolaev S, Santoni F, Garieri M, et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 2014;5:5690.
157. Cai M, Zhang H, Hou L, et al. Inhibiting homologous recombination decreases extrachromosomal amplification but has no effect on intrachromosomal amplification in methotrexate-resistant colon cancer cells. Int J Cancer 2019;144:1037-48.
159. Lin L, Wei H, Yi J, et al. Chronic CagA-positive Helicobacter pylori infection with MNNG stimulation synergistically induces mesenchymal and cancer stem cell-like properties in gastric mucosal epithelial cells. J Cell Biochem 2019;120:17635-49.
160. Song X, Xin N, Wang W, Zhao C. Wnt/β-catenin, an oncogenic pathway targeted by H. pylori in gastric carcinogenesis. Oncotarget 2015;6:35579-88.
161. Courtois S, Haykal M, Bodineau C, et al. Autophagy induced by Helicobacter pylori infection is necessary for gastric cancer stem cell emergence. Gastric Cancer 2021;24:133-44.
162. Tiffon C, Giraud J, Molina-Castro SE, et al. TAZ controls helicobacter pylori-induced epithelial-mesenchymal transition and cancer stem cell-like invasive and tumorigenic properties. Cells 2020;9:1462.
163. Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci USA 2005;102:5186-91.
164. Giannakis M, Chen SL, Karam SM, Engstrand L, Gordon JI. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci USA 2008;105:4358-63.
165. Varon C, Dubus P, Mazurier F, et al. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 2012;142:281-91.
166. Korkaya H, Paulson A, Charafe-Jauffret E, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol 2009;7:e1000121.
167. Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006;66:6063-71.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Xiong J, Zhang T, Lan P, Zhang S, Fu L. Insight into the molecular mechanisms of gastric cancer stem cell in drug resistance of gastric cancer. Cancer Drug Resist 2022;5:794-813. http://dx.doi.org/10.20517/cdr.2022.11
AMA Style
Xiong J, Zhang T, Lan P, Zhang S, Fu L. Insight into the molecular mechanisms of gastric cancer stem cell in drug resistance of gastric cancer. Cancer Drug Resistance. 2022; 5(3): 794-813. http://dx.doi.org/10.20517/cdr.2022.11
Chicago/Turabian Style
Xiong, Jixian, Tiantian Zhang, Penglin Lan, Shuhong Zhang, Li Fu. 2022. "Insight into the molecular mechanisms of gastric cancer stem cell in drug resistance of gastric cancer" Cancer Drug Resistance. 5, no.3: 794-813. http://dx.doi.org/10.20517/cdr.2022.11
ACS Style
Xiong, J.; Zhang T.; Lan P.; Zhang S.; Fu L. Insight into the molecular mechanisms of gastric cancer stem cell in drug resistance of gastric cancer. Cancer Drug Resist. 2022, 5, 794-813. http://dx.doi.org/10.20517/cdr.2022.11
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 26
likes
Like This Article 26
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.