
Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer

Abstract
Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors combined with endocrine therapy have transformed the treatment of estrogen receptor-positive (ER+) and human epidermal growth factor receptor 2 negative (HER2-) metastatic breast cancer. However, some patients do not respond to this treatment, and patients inevitably develop resistance, such that novel biomarkers are needed to predict primary resistance, monitor treatment response for acquired resistance, and personalize treatment strategies. Circumventing the spatial and temporal limitations of tissue biopsy, newly developed liquid biopsy approaches have the potential to uncover biomarkers that can predict CDK4/6 inhibitor efficacy and resistance in breast cancer patients through a simple blood test. Studies on circulating tumor DNA (ctDNA)-based liquid biopsy biomarkers of CDK4/6 inhibitor resistance have focused primarily on genomic alterations and have failed thus far to identify clear and clinically validated predictive biomarkers, but emerging epigenetic ctDNA methodologies hold promise for further discovery. The present review outlines recent advances and future directions in ctDNA-based biomarkers of CDK4/6 inhibitor treatment response.
Keywords
INTRODUCTION
Breast cancer is the most diagnosed cancer in women globally, with approximately 70% of diagnoses being tumors that express estrogen receptors (ER+) but not human epidermal growth factor receptor 2
In the past decade, novel therapeutic strategies for metastatic breast cancer patients have been implemented in the clinic. Among these, the new standard treatment for ER+/HER2- locally advanced and metastatic breast cancer consists of cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors combined with endocrine therapy[4]. Three CDK4/6 inhibitors have been approved (i.e., abemaciclib, palbociclib, and ribociclib) after displaying significant clinical benefit in pivotal phase III clinical trials[5-12]. CDK4/6 inhibitors have been shown to improve response rate, median progression-free survival (PFS), health-related quality of life, and overall survival (OS) of metastatic breast cancer patients[13-19]. However, CDK4/6 inhibitor resistance remains a significant obstacle. A minority of patients have intrinsic resistance, defined as progression (without response) within six months of starting treatment. Even for patients who experience initial response and clinical benefit from these agents, acquired resistance inevitably develops over subsequent months (median PFS ranges from 23.8-28.2 months in the first-line metastatic setting)[11,17,20]. Therefore, biomarkers are urgently needed to predict CDK4/6 inhibitor efficacy or resistance in metastatic breast cancer patients, allowing clinicians to tailor treatment and potentially add additional therapies for patients at high risk of early progression.
Biomarkers for CDK4/6 inhibitors have been thoroughly investigated through molecular profiling of tumor material, but to date, the only clinically available biomarker remains breast cancer subtype as defined by traditional tissue markers (i.e., ER+/HER2-)[21,22]. Despite significant research efforts, tumor heterogeneity and difficulties distinguishing endocrine resistance from CDK4/6 inhibitor resistance have impeded predictive biomarker discovery[23]. Given practical challenges to obtaining and repeating metastatic tissue biopsies, blood-based profiling of tumor-derived material (i.e., “liquid biopsy”) has significant potential to facilitate biomarker explorations. For instance, circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) are promising liquid biopsy analytes because they harbor cancer-specific molecular aberrations[24].
In this review, we discuss biomarker-directed treatment for breast cancer, mechanisms of resistance to CDK4/6 inhibitors, and attempts to uncover ctDNA liquid biopsy biomarkers of efficacy and resistance to CDK4/6 inhibitors. Lastly, we highlight undeveloped areas for future advances, namely epigenetic-based liquid biopsy biomarkers for patients treated with CDK4/6 inhibitors.
BIOMARKER-DIRECTED PRECISION ONCOLOGY AND BREAST CANCER
Precision oncology relies on molecular information from each patient’s cancer to optimize and individualize treatment regimens. Leveraging these patient-specific molecular biomarkers allows clinicians to select the best treatment for individual patients to improve therapeutic efficacy and reduce adverse effects on healthy cells[25]. Molecular biomarkers include prognostic biomarkers, which may provide information on the expected disease course independent of treatment, and predictive biomarkers, which provide insight into the effect of a specific therapy. Both prognostic and predictive biomarkers may be used to personalize treatment via risk-stratification or directing effective treatments.
Current standard-of-care breast cancer treatments provide several archetypal examples of biomarker-directed precision oncology. For instance, OncotypeDx is a commercial 21-gene assay for ER+ early-stage breast cancer that returns a recurrence score indicating the probability of relapse without adjuvant chemotherapy, with higher scores associated with a poorer prognosis. OncotypeDx serves a prognostic role; its clinical utility stems from identifying patients with a higher absolute recurrence risk and, therefore, a higher likelihood of benefiting from adjuvant chemotherapy[26,27]. Similarly, the MammaPrint microarray assay is a prognostic biomarker that uses the expression levels of 70 genes to classify patients according to recurrence risk[28,29].
Other breast cancer biomarkers highlight the impact of predictive biomarkers in precision oncology. Intrinsic molecular subtypes and associated hormone receptors (ER, PR) and HER2 expression levels are critical for drug selection in breast cancer patients[30]. For instance, HER2+ breast cancer preferentially responds to HER2-targeted agents, such as trastuzumab and trastuzumab emtansine (T-DM1)[31]. Likewise, hormone receptor expression denotes tumors that preferentially respond to endocrine therapy. Resistance to endocrine therapy can occur over time through a variety of mechanisms (e.g., genetic alterations of ESR1, increased activity of cyclin-dependent kinases (CDKs), and mitogen-signaling pathways such as PI3K and RAS, or a decrease in proteins that inhibit CDKs such as p16, p21, and p27), several of which converge on the cyclin D-CDK4/6 axis[32]. Therefore, simultaneous treatment with endocrine therapy and CDK4/6 inhibitors has emerged as a highly successful treatment paradigm for ER+/HER2- metastatic breast cancer.
MOLECULAR MECHANISMS OF CDK4/6 INHIBITOR EFFICACY AND RESISTANCE
The cyclin D-CDK4/6-Retinoblastoma protein (Rb) axis regulates cell cycle progression from G1 to the S phase [Figure 1]. Before entering the cell cycle, Rb is hypophosphorylated and bound to the E2F transcription factors (TFs), causing their inhibition[33]. When the appropriate mitogenic signals are present, quiescent cells may enter the cell cycle at the G1 phase. These mitogenic signals lead to the expression of cyclin D, which competes with CDKN2 family proteins to bind CDK4/6, forming the cyclin D-CDK4/6 complex[34]. This active complex can then phosphorylate Rb, causing a conformational change and subsequent release of the E2F TFs, which drive S phase entry and further cell cycle progression via downstream transcriptional activation[33]. Furthermore, the cyclin D-CDK4/6 complex triggers the forkhead box protein M1 (FOXM1) TF, promoting the advancement of later cell cycle phases (G2/M)[35]. ER+ breast cancer is highly reliant on an intact cyclin D-CDK4/6-Rb axis, as estrogen drives cyclin D1 expression leading to the formation of the cyclin D-CDK4/6 complex, ultimately inducing cell proliferation through the CDK4/6 pathway[36]. CDK4/6 inhibitors leverage this by binding the ATP domain of CDK4/6 and halting progression from the G1 to S phase of the cell cycle[37].
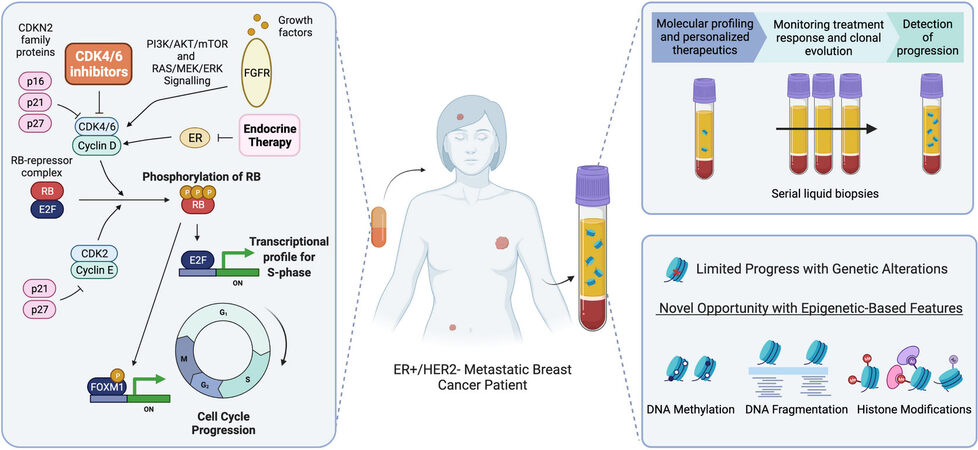
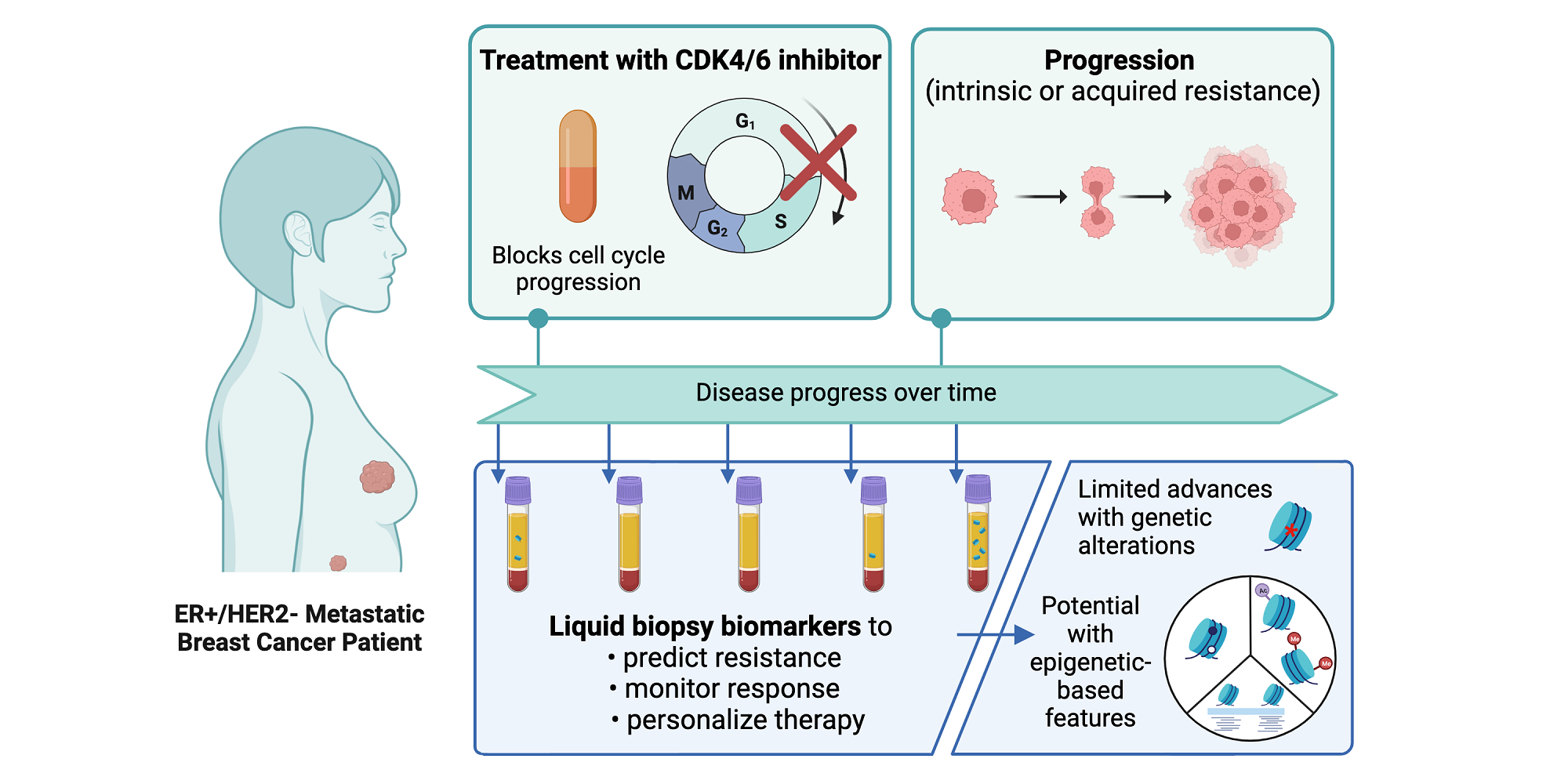
Figure 1. Overview of CDK4/6 inhibitor treatment and liquid biopsy in ER+/HER2- metastatic breast cancer patients. CDK4/6 inhibitors prevent the phosphorylation of Rb and downstream activation of the transcriptional profile required for progression to the S-phase of the cell cycle. In the anti-CDK4/6 therapy clinical setting, a liquid biopsy may be used to direct therapy, monitor patient response over time, and detect progression. Currently, research efforts focused on monitoring genetic alterations in cfDNA through liquid biopsy have made limited progress. Epigenetic profiling of cfDNA may reveal new biomarkers of CDK4/6 inhibitor efficacy or resistance.
The mechanisms of resistance to CDK4/6 inhibitors have yet to be fully elucidated, and in many cases, the clinical relevance of putative mechanisms discovered in preclinical models remains unconfirmed[38,39]. Recognized resistance mechanisms include amplification of members of the cyclin D-CDK4/6 axis or downregulation of CDK4/6 repressor proteins, such as p21 and p27, which may thwart the direct effects of these drugs[38,40-42]. Additionally, alterations in RB1, FAT1, or the PI3K/AKT/mTOR and KRAS signaling pathways may act to circumvent the G1/S checkpoint in the presence of CDK4/6 inhibitors, inducing cell cycle progression independent of the cyclin D-CDK4/6 axis[38,39,43-46].
CIRCULATING CELL-FREE DNA FOR LIQUID BIOPSY AND COMPANION DIAGNOSTICS
In recent years, liquid biopsy approaches have emerged, intending to provide molecular information for biomarker-directed precision oncology from a fluid sample[24]. Traditionally, accessing this information has required invasive procedures, like tissue biopsy, which are not always feasible depending on the nature and location of the tumor, as well as the health of the patient. These limitations are particularly relevant for metastatic breast cancer patients, where ER+/HER2- breast cancer recurrences often occur past 5 years. In this clinical setting, the primary tissue sample may not represent metastatic disease, and the metastatic sites are often in areas difficult to biopsy (e.g., bone and lung)[47,48]. Here, a liquid biopsy may bypass the procedural, spatial, and temporal obstacles of tissue biopsy by encompassing disease heterogeneity and new metastases while being easily repeated throughout tumor progression and different lines of treatment[49]. Therefore, liquid biopsy is advancing as a valuable approach to enable contemporaneous and minimally invasive testing of tumor-specific analytes in cancer patients[50].
For most liquid biopsy applications, the body fluid of choice is peripheral blood plasma. This has previously presented challenges in detecting brain tumors or metastatic sites, likely due to the blood-brain barrier; however, newer liquid biopsy technologies have led to improvements in sensitivity[51,52]. Regardless, other accessible fluids may be more informative depending on the tumor type (e.g., saliva for oral cancer, urine for bladder cancer, and cerebrospinal fluid for glioma)[53]. There are also a variety of components from the tumor which may be assessed through liquid biopsy, including intact tumor cells (e.g., CTCs), cell-free DNA (cfDNA), extracellular vesicles, cell-free RNA, and more[24]. Herein, we focus our discussion on ctDNA as a liquid biopsy biomarker of response to anti-CDK4/6 therapy due to the wealth of recent studies on this topic and the precedent of ctDNA in other settings of precision oncology (e.g., for EGFR tyrosine kinase inhibitors in lung cancer)[49,54]. Other liquid biopsy analytes and their role as biomarkers for CDK4/6 inhibitor treatment have been reviewed elsewhere[23,55,56].
ctDNA molecules containing tumor-derived genetic and epigenetic features are released into the bloodstream as tumor cells die[49]. ctDNA usually represents a small portion of the total circulating cfDNA pool, where most other cfDNA molecules are derived from cells of hematopoietic origin and alternative tissues[57-59]. The release of ctDNA is related to cellular turnover (i.e., many apoptotic and necrotic cells), during which a subset of double-stranded DNA fragments associated with chromatin components enters the extracellular space[60-62]. ctDNA levels can be as low as < 0.01% of the entire cfDNA pool, with variations depending on tumor size, stage, anatomical location, treatment, and the biological propensity of tumor cells to release their DNA into the circulation[51,58,63-65]. Once in the circulation, clearance of ctDNA by multiple mechanisms (e.g., nuclease-mediated degradation, phagocytosis, and renal excretion) occurs quickly with a half-life between 16 minutes and two hours[66-68]. This property enables cfDNA to portray a “real-time” snapshot of the patient’s disease state[49,69].
Analysis of ctDNA through liquid biopsy may be relevant in many clinical stages of CDK4/6 inhibitor treatment, such as prognostication, personalizing therapeutics (e.g., determining which patients should receive anti-CDK4/6 therapy or adding other agents), monitoring treatment response, and identifying resistance [Figure 1]. ctDNA can be analyzed by polymerase chain reaction (PCR)[70,71], which targets a single gene locus, or next-generation sequencing (NGS), which simultaneously profiles dozens or hundreds of genes[64,72-74]. Of relevance in ER+/HER2- advanced breast cancer, the therascreen PIK3CA RGQ PCR kit detects PIK3CA mutations to help direct PI3Ka inhibitor treatment (alpelisib)[75,76]. NGS-based liquid biopsy assays often include PIK3CA in addition to genes of potential relevance to resistance mechanisms to endocrine therapy, such as ESR1 and PTEN[77]. Thus, although not yet established in the CDK4/6 setting, ctDNA shows considerable promise as means of biomarker detection for genotype-guided precision oncology.
GENETIC-BASED LIQUID BIOPSY BIOMARKERS OF CDK4/6 INHIBITOR EFFICACY AND RESISTANCE
Currently, there are no clinically validated liquid biopsy biomarkers to distinguish patients with differential benefits to anti-CDK4/6 therapy. Here we report the main ctDNA liquid biopsy biomarkers for CDK4/6 inhibitor treatment investigated at baseline or time of progression. The main focus of the studies outlined in this section are alterations of genes related to cell cycle regulation using ctDNA and their relationship to outcomes [Table 1].
Summary of the main genomic alterations interrogated in ctDNA as biomarkers for CDK4/6 inhibitor resistance
| Genomic alteration | ctDNA sample | Cohort | Prevalence | Technique | Main findings | Reference |
| RB1 | Baseline | PALOMA-3: Palbociclib plus fulvestrant | 27 of 156 patients (17.3%) | Target panel NGS (17 genes) | Patients in the palbociclib treatment arm with loss of RB1 had worse median PFS compared to wild-type (exact PFS not reported) | O’Leary |
| Baseline | MONALEESA 2,3,7: Ribociclib plus endocrine therapy | 26 of 1,534 patients (1.7%) | Target panel NGS (~600 genes) | Patients with RB1 mutated tumors did not have significantly different median PFS with ribociclib treatment compared to placebo (mutant: 9.2 vs. 3.7 months placebo vs. ribociclib arm) | Bertucci | |
| Progression | Three case reports: Palbociclib plus fulvestrant or ribociclib plus letrozole | NA | Custom library for RB1 and TP53 coding sequence/ Guardant 360 assay (73 genes) | Patients had five different loss-of-function genetic alterations of RB1 after exposure to CDK4/6 inhibitors and coinciding with resistance | Condorelli | |
| Baseline and End-of-treatment | PALOMA-3: Palbociclib plus fulvestrant | Acquired in 6 of 127 patients (4.7%) | Exome sequencing/ Target panel NGS/ ddPCR | Patients exclusively acquired RB1 alterations in the palbociclib treatment arm | O’Leary | |
| ESR1 | Baseline | PALOMA-3: Palbociclib plus fulvestrant | 91 of 360 patients (25.3%) | ddPCR | Patients in the palbociclib treatment arm had similarly improved median PFS regardless of ESR1 status, although patients in the placebo arm with ESR1 mutations had worse PFS compared to wild-type (palbociclib arm: 9.4 vs. 9.5 months mutant vs. wild-type; placebo arm: 3.6 vs. 5.4 months mutant vs. wild-type) | Fribbens |
| Baseline and End-of-treatment | PALOMA-3: Palbociclib plus fulvestrant | Acquired in 25 of 195 patients (12.8%) | Exome sequencing/ Target panel NGS/ ddPCR | Patients acquired ESR1 Y537S mutations in both treatment arms and were associated with improved median PFS compared to patients who did not acquire the mutation (13.7 vs. 7.4 months acquired vs. not acquired) | O’Leary | |
| Baseline | PALOMA-3: Palbociclib plus fulvestrant | 72 of 331 patients (21.8%) | Target panel NGS (17 genes) | Patients in the placebo arm with ESR1 mutations had worse PFS compared to wild-type (exact PFS not reported) | O’Leary | |
| Baseline | MONARCH-2: abemaciclib and fulvestrant | 147 of 248 patients (59.3%) | ddPCR | Patients in the abemaciclib treatment arm had improved PFS regardless of ESR1 status but observed a higher numerical median PFS in patients with ESR1 mutant tumors compared to wild-type (abemaciclib arm: 20.7 vs. 15.3 months mutant vs. wild-type; placebo arm: 13.1 vs. 11.3 months mutant vs. wild-type). Patients with ESR1 mutations also had improved OS (abemaciclib arm: not reached vs. 52.2 months mutants vs. wild-type; placebo arm: 42.2 vs. 29.4 months mutant vs. wild-type) | Tolaney | |
| PIK3CA | Baseline | PALOMA-3: Palbociclib plus fulvestrant | 129 of 395 patients (33%) | BEAMing assay | Patients in the palbociclib treatment arm had similarly improved PFS regardless of PIK3CA status (palbociclib arm: 9.5 vs. 9.9 months mutated vs. wild-type; placebo arm: 3.6 vs. 4.6 months mutated vs. wild-type) | Critstofanilli |
| Baseline | MONARCH-2: abemaciclib and fulvestrant | 96 of 219 patients (43.8%) | ddPCR | Patients in the abemaciclib treatment arm had similarly improved PFS regardless of PIK3CA status, although patients in the placebo treatment arm with PIK3CA mutations had worse median PFS compared to wild-type (abemaciclib arm: 17.1 vs. 16.9 months mutant vs. wild-type; placebo arm: 5.7 vs. 12.3 months mutant vs. wild-type) | Tolaney | |
| Baseline | Palbociclib or ribociclib plus fulvestrant or letrozole | 12 of 30 patients (40%) | ddPCR | Patients treated with palbociclib or ribociclib plus endocrine therapy with PIK3CA mutations had a worse median PFS compared to wild-type (7.44 vs. 12.9 months mutant vs. wild-type) | Del Re | |
| Baseline | PALOMA-3: Palbociclib plus fulvestrant | 55 of 331 patients (16.6%) | Target panel NGS (17 genes) | PIK3CA mutations were not identified as predictive (exact PFS not reported) | O’Leary | |
| Baseline and End-of-treatment | PALOMA-3: Palbociclib plus fulvestrant | Acquired in 15 of 195 patients (7.6%) | Exome sequencing/Target panel NGS/ddPCR | Patients acquired PIK3CA mutations in both treatment arms and were associated with improved median PFS compared to patients who did not acquire a PIK3CA mutation (12.7 vs. 9.2 months acquired vs. not acquired) | O’Leary | |
| Baseline | MONALEESA-7: Ribociclib plus endocrine therapy | 139 of 489 patients (28%) | Target panel NGS (~600 genes) | Patients in the ribociclib treatment arm had improved median PFS compared to the placebo arm, and this was more prominent in patients with wild-type PIK3CA compared to PIK3CA mutations (ribociclib arm: 14.8 vs. 24.7 months mutant vs. wild-type; placebo arm 12.9 vs. 12.2 months mutant vs. wild-type) | Bardia | |
| FGFR | Progression | MONALEESA-2: Ribociclib plus letrozole | 20 of 427 patients (5%) | Guardant360 assay (73 genes) | Patients in the ribociclib treatment arm with FGFR1 alterations had a worse median PFS (10.61 vs. 24.84 months mutant vs. wild-type), although significance was not achieved due to the small sample size | Formisano |
| Baseline | PALOMA-3: Palbociclib plus fulvestrant | 20 of 401 patients (4.9%) | Target panel NGS (17 genes) | Patients with FGFR1 amplifications had a worse median PFS in both treatment arms (palbociclib arm: 3.9 vs. 12 months mutant vs. wild-type; placebo arm: 1.8 vs. 4.8 months mutant vs. wild-type) | O’Leary | |
| Baseline and End-of-treatment | PALOMA-3: Palbociclib plus fulvestrant | Acquired in 2 of 195 patients (1%) | Exome sequencing/Target panel NGS/ddPCR | Patients acquired FGFR2 alterations with no apparent difference between treatment arms. | O’Leary | |
| TP53 | Baseline | PALOMA-3: Palbociclib plus fulvestrant | 52 of 331patients (15.7%) | Target panel NGS (17 genes) | Patients with TP53 mutations had a worse median PFS in both treatment arms, and no interaction with treatment was observed (palbociclib arm: 3.7 vs. 12.7 months mutant vs. wild-type; placebo arm: 1.8 vs. 5.4 months mutant vs. wild-type) | O’Leary |
| Baseline | MONALEESA-7: Ribociclib plus endocrine therapy | 92 of 489 patients (19%) | Target panel NGS (~600 genes) | Patients with TP53 mutations had a worse median PFS in both treatment groups (ribociclib arm: 9.2 vs. 24.7 months mutant vs. wild-type; placebo arm: 7.2 vs. 13.0 months mutant vs. wild-type) | Bardia | |
| KRAS | Baseline and on treatment | Palbociclib plus fulvestrant | 66 of 106 patients (62.2%) | ddPCR | Patients treated with palbociclib and fulvestrant with baseline KRAS mutations had a worse median PFS compared to wild-type (3 vs. 17.8 months mutant vs. wild-type) | Raimondi |
| CCND1 | Baseline | MONALEESA-7: Ribociclib plus endocrine therapy | 51 of 489 patients (10%) | Target panel NGS (~600 genes) | Patients with CCND1 alterations had a worse median PFS in both treatment arms and patients with altered CCND1 also had a significant treatment interaction effect (ribociclib arm: 12.9 vs. 22.1 months mutant vs. wild-type; placebo arm: 5.5 vs. 11.3 months mutant vs. wild-type) | Bardia |
| MYC | Baseline | MONALEESA-7: Ribociclib plus endocrine therapy | 35 of 489 patients (7.1%) | Target panel NGS (~600 genes) | Patients with MYC alterations had a worse median PFS in both treatment arms (ribociclib arm: 7.3 vs. 24.7 months mutant vs. wild-type; placebo arm: 7.2 vs. 12.9 months mutant vs. wild-type) | Bardia |
Most studied resistance mechanisms converge on Rb modulation since it is the central target of CDK4/6 to control cell cycle progression[38]. Genetic alterations of RB1 may cause its inactivation and confer resistance to CDK4/6 inhibitors. For example, in the PALOMA-3 trial, loss of RB1 detected via baseline ctDNA was associated with worse PFS for patients in the palbociclib plus fulvestrant treatment group[78]. These results suggest that RB1 alterations may be prognostic and potentially predictive; however, this study could not determine the treatment interaction effect due to the small sample size. This was further supported by an analysis of ctDNA from patients in the MONALEESA 2, 3, and 7 trials, which found that for patients with RB1 mutations, ribociclib plus endocrine therapy did not significantly improve median PFS[79]. Furthermore, a small clinical report of three patients was the first to identify loss-of-function RB1 mutations in ctDNA sampled at the time of acquired CDK4/6 inhibitor resistance[44]. This was later supported by an analysis of PALOMA-3 matched baseline and end-of-treatment ctDNA samples, which found loss-of-function RB1 mutations were exclusively acquired in 4.7% of patients treated with palbociclib plus fulvestrant, suggesting they were selected for in treatment resistance. However, due to the small number of acquired RB1 mutations, no associations were made to PFS[39]. Altogether, these results and biological support of RB1 loss-of-function as a resistance mechanism suggest that genomic alterations of RB1 detected in ctDNA may be predictive of CDK4/6 inhibitor resistance. However, the low prevalence of these alterations suggests other resistance mechanisms are involved, and RB1 mutations may serve as a predictive biomarker of resistance for a limited portion of patients on CDK4/6 inhibitors.
In addition to RB1 mutations, genomic alterations in ESR1 have been investigated due to the ER mitogenic pathway being critical to cyclin D-CDK4/6 dependence and the inactivation having a potential role in resistance to CDK4/6 inhibitors[46]. First, multiple analyses of the PALOMA-3 trial revealed that patients with baseline ctDNA ESR1 mutations had a worse median PFS solely in the placebo treatment arm[78,80]. These findings support the role of ESR1 mutations as a biomarker of endocrine resistance, and no treatment interaction effect was observed, indicating the lack of predictive potential of ESR1 mutations for CDK4/6 inhibitor resistance. A later study of PALOMA-3 found that 13 of 195 patients lost ESR1 mutations between baseline and end-of-treatment, whereas 25 patients in both treatment groups acquired the alteration, suggesting ESR1 mutations promote resistance to fulvestrant and do not predict CDK4/6 inhibitor treatment benefit[39]. Lastly, a recent analysis of ctDNA from the MONARCH-2 trial found that abemaciclib plus fulvestrant improved PFS regardless of ESR1 status, but patients with ESR1 mutations had a higher numerical median PFS in both treatment arms[81]. This study also observed an unexpectedly high prevalence of ESR1 mutations and increased OS for patients with ESR1 mutations. The contrast of these findings is unclear but may be explained by differences in sample size, patient criteria, and analytical techniques. Altogether, ESR1 mutations are not a promising candidate biomarker for CDK4/6 inhibitor resistance and may be more informative for endocrine resistance.
PIK3CA mutations have also been interrogated by ctDNA due to their upstream role in cell cycle regulation through the PI3K/AKT/mTOR pathway, which interacts with estrogen receptors and potentially impacts CDK4/6 inhibitor resistance[38]. For instance, analyses of the PALOMA-3 trial found that palbociclib plus fulvestrant treatment similarly improved PFS in patients with mutated or wild-type PIK3CA in baseline ctDNA, indicating that PIK3CA genomic alterations are not predictive of CDK4/6 inhibitor benefit[13,78]. In support of this finding, a study of baseline ctDNA from the MONARCH-2 trial found patients in the abemaciclib treatment arm had similarly improved PFS regardless of PIK3CA mutation status[81]. In addition, patients in the placebo arm with mutant PIK3CA had a worse median PFS, suggesting the role of PIK3CA mutations in endocrine therapy resistance instead of as a biomarker of CDK4/6 inhibitor treatment. In contrast, another study of baseline ctDNA of advanced breast cancer patients treated with palbociclib or ribociclib found that patients with PIK3CA mutations had a shorter median PFS than wild-type[82]. Due to a lack of a control treatment arm, this study could not assess the treatment effect with PIK3CA. Still, these findings suggest PIK3CA mutations may act as a prognostic biomarker for advanced breast cancer patients receiving treatment with CDK4/6 inhibitors.
Furthermore, the lack of predictive potential of PIK3CA has been supported in multiple studies. An additional PALOMA-3 analysis of matched baseline and end-of-treatment ctDNA samples found that 7.6% of patients acquired PIK3CA mutations across both treatment arms, suggesting that PIK3CA mutations emerge due to fulvestrant resistance[39]. Interestingly, patients with acquired PIK3CA mutations had an improved PFS than those who did not; however, this trend was seen with the acquisition of any new mutation (including ESR1), suggesting that novel alterations are more likely to emerge in patients with a longer duration of treatment. Moreover, a recent study analyzing ctDNA from the MONALEESA-7 trial found that patients in the ribociclib treatment arm had an improved median PFS, but this was more pronounced in patients with wild-type PIK3CA[83]. However, this difference was not statistically significant, reinforcing the limited potential of PIK3CA as a predictive biomarker of CDK4/6 inhibitor resistance.
In addition, FGFR genetic alterations have been investigated in plasma ctDNA of patients treated with CDK4/6 inhibitors due to evidence of abnormal FGFR signaling, driving CCND1 overexpression and MAPK activation, contributing to resistance[38]. One analysis of baseline ctDNA from the MONALEESA-2 trial found that for patients in the ribociclib treatment arm, FGFR1 alterations were related to worse PFS, although significance was not achieved due to the small sample size[41]. In further support, a study of ctDNA from the PALOMA-3 trial found that FGFR1 amplifications were associated with worse PFS in both treatment groups, suggesting that the alteration may be related to endocrine resistance[78]. Furthermore, in an assessment of pre- and post-treatment ctDNA samples from PALOMA-3, FGFR2 was acquired in 2 of 195 patients, with no apparent difference between treatment groups[39]. Altogether these findings support FGFR alterations, specifically FGFR1 amplifications, as a possible prognostic biomarker for patients treated with CDK4/6 inhibitors and endocrine therapy.
TP53 is a tumor suppressor gene whose product induces antiproliferative factors, such as CDKN1A, and its modification in ctDNA has also been investigated in many studies. For example, genomic alterations of TP53 in ctDNA from the PALOMA-3 trial were assessed, revealing that patients with TP53 mutations had significantly worse PFS in both treatment arms, and no interaction with treatment was observed[78]. This work also found that TP53 mutations were connected to a distinct aggressive phenotype with more metastases but could not rule out the presence of these mutations due to clonal hematopoiesis. Moreover, a MONALEESA-7 biomarker analysis found that TP53 mutations in baseline ctDNA were associated with progression independently from treatment with ribociclib and did not predict response to therapy[83]. Overall, these studies support TP53 alterations as prognostic biomarkers and suggest they are not candidate predictive biomarkers of anti-CDK4/6 therapy.
In addition, genomic alterations of KRAS in ctDNA have recently been interrogated due to its role upstream of CDK4/6, transducing mitogenic signaling and affecting cyclin D1[38]. One study investigated KRAS mutations in ctDNA of 106 HR+/HER2- metastatic breast cancer patients treated with palbociclib plus fulvestrant and found that after 18 months, all patients with KRAS alterations had progressive disease[45]. In contrast, only one KRAS wild-type patient had progressed. Accordingly, patients with mutated KRAS had a worse median PFS compared to wild-type, supporting the potential of KRAS mutations as a prognostic biomarker. Further study with a control treatment group is required to determine the treatment interaction effect before conclusions can be made. For instance, previous studies on ctDNA from the PALOMA-3 trial have investigated KRAS mutations, but likely due to the low frequency of aberrations, have not made associations with PFS[39,78].
Many other genomic alterations have been investigated in plasma ctDNA of patients treated with CDK4/6 inhibitors due to their role around the cyclin D-CDK4/6-Rb axis or in breast cancer in general, namely CCND1, CDK4, CDK6, CDKN1, CDKN2, NF1, ERBB2, AKT1, NRAS, HRAS, GATA3, and MYC[39,78,83]. In particular, a recent study of the MONALEESA-7 samples found that patients with CCND1 alterations in baseline ctDNA had a worse median PFS for both treatment arms, indicating the role of CCND1 as a prognostic biomarker[83]. The benefit of ribociclib was also greater in patients with CCND1 altered ctDNA, supporting CCND1 as a candidate predictive biomarker. Alterations in MYC have also been reported as a potential prognostic biomarker for patients treated with CDK4/6 inhibitors in combination with or solo endocrine therapy[83]. Otherwise, associations with PFS and treatment have not been reported for many of the above alterations.
In summary, current work suggests ESR1 and PIK3CA mutations in ctDNA have limited CDK4/6 inhibitor biomarker potential, which may be clouded due to implications with endocrine resistance. Alterations in FGFR1, TP53, and MYC may also be prognostic biomarkers of resistance to anti-CDK4/6 therapy, whereas RB1, KRAS, and CCND1 may be prognostic and putative predictive biomarkers of CDK4/6 inhibitor resistance. Although numerous potential biomarkers have been identified, many studies have not confirmed the effect of interaction with treatment, and further validation is needed. While considering this, the evidence so far indicates that no singular genetic alteration will serve as an ideal prognostic or significantly predictive biomarker for CDK4/6 inhibitor efficacy or resistance. While some potential predictive biomarkers, such as RB1 alterations, seem promising, their low prevalence indicates multifactorial resistance mechanisms. As such, analyses that only evaluate one genomic alteration are limited in assessing whether other alterations add predictive value. To assess the vast landscape of CDK4/6 inhibitor resistance mechanisms, future studies should consider a broader range of genomic alterations in ctDNA or, as discussed in later sections, expand to longitudinal monitoring to assess ctDNA dynamics or investigate epigenetic-based features.
DYNAMIC ctDNA BIOMARKERS OF CDK4/6 INHIBITOR EFFICACY AND RESISTANCE
Monitoring changes in ctDNA levels throughout treatment may reveal more prognostic and predictive information than a single time point measurement. Serial monitoring of ctDNA levels could provide a dynamic biomarker that allows personalized modifications to treatment, such as adding or switching to a more effective therapy for non-responsive patients[84]. In investigations of the utility of dynamic liquid biopsy biomarkers, questions remain concerning sampling time points, assays used, and change thresholds. This section discusses research on dynamic ctDNA biomarkers relevant to CDK4/6 inhibitor treatment [Table 2].
Summary of dynamic ctDNA biomarkers investigated in the CDK4/6 inhibitor treatment setting
| Technique | ctDNA sample time points | Genetic alteration | Cohort | Metric | Main findings | Reference |
| ddPCR | Baseline, cycle 1 day 15, and progression | ESR1 and PIK3CA | PALOMA-3: Palbociclib plus fulvestrant | High and low CDR15 based on a threshold determined by Harrell’s c-index and Benjamini-Hochberg p-value corrections | All patients in the palbociclib treatment arm had a CDR15 less than one. For PIK3CA, patients with a high CDR15 had a worse median PFS than those with a low CDR15 (4.1 vs. 11.2 months high vs. low) | O’Leary |
| ddPCR | Baseline, day 15, day 30, and progression | ESR1 | ALCINA: Palbociclib plus fulvestrant | Ratio relative to baseline (mutant copies/mL) | All patients experienced a decrease in ctDNA on day 15 relative to baseline. Patients with early progression had increased ctDNA on day 30, and patients with longer PFS had lower or consistent ctDNA levels relative to baseline. ESR1 mutations ctDNA on day 30, as opposed to ctDNA clearance, was correlated with worse PFS | Jeannot |
| ddPCR monitoring one tumor-specific mutation per patient | Baseline, day 15, day 30 and progression | PIK3CA (n = 21), TP53 (n = 2), or AKT1 (n = 2) | ALCINA: Palbociclib plus fulvestrant | Ratio relative to baseline (mutant copies/mL) | All patients had a decrease in ctDNA on day 15, but this was not associated with PFS. Patients with undetectable ctDNA on day 30 had an improved PFS compared to those with detectable ctDNA (25 vs. 3 months undetectable vs. detectable, respectively). ctDNA ratios (day 30/baseline) greater than or less than one were significantly associated with PFS | Darrigues |
| Guardant360 assay | Baseline, four weeks | 73 genes | Palbociclib or ribociclib plus endocrine therapy | mVAFR for the 79 mutations found between baseline and week 4 assess in groups of high, medium, and low mVAFR groups and as a continuous variable | mVAFR was significantly associated with PFS, whereas single timepoint mean VAFs or absolute changes in mean VAF were not. Patients with high mVAFR had a worse median PFS than those with low mVAFR (4.2 months vs. not reached high vs. low) | Martinez-Saez |
| mFAST-seq | Various | Aneuploidy | CDK4/6 inhibitor plus endocrine therapy | z-score trajectories | Raised z-score trajectories were significantly related to worse PFS, whereas baseline z-scores were not predictive of progression. A single z-score increased in a consecutive blood sample at any follow-up point was not associated with PFS | Dandachi |
Dynamic ctDNA levels in response to CDK4/6 inhibitor treatment were first investigated by monitoring PIK3CA and ESR1 mutant ctDNA from the PALOMA-3 trial collected at baseline, cycle one day 15, and progression[85]. PIK3CA and ESR1 mutations were detected in 100 and 114 of 455 baseline samples (22% and 25.6%), with 73 and 65 matched day 15 samples, respectively. For both PIK3CA and ESR1, a significant decline in ctDNA occurred on day 15, with a more apparent decrease in mutant PIK3CA ctDNA in the palbociclib arm and mutant ESR1 ctDNA in the placebo arm. This group also defined a circulating DNA ratio (CDR15) as the concentration of ctDNA on day 15 compared to baseline. They found that all patients on palbociclib had a CDR15 less than one, possibly due to the cytostatic effect of CDK4/6 inhibitors. For PIK3CA, patients with CDR15 above the median had a worse PFS than those below the median; however, this was not seen for ESR1. Using an optimal threshold determined by Harrell’s c-index and Benjamini-Hochberg p-value corrections, they found that patients with a high PIK3CA CDR15 had an inferior median PFS than those with a low CDR15. Ultimately, this study determined that relative change in ctDNA based on commonly truncal PIK3CA mutations was predictive of PFS for patients treated with palbociclib and fulvestrant. Alternatively, ctDNA dynamics based on ESR1 mutations, which are generally subclonal due to selection of prior endocrine therapy, were not predictive of clinical outcome. Altogether, these results indicate that early evaluation of ctDNA dynamics with truncal mutations may be a predictive biomarker of PFS for patients on CDK4/6 inhibitor treatment.
In addition, a subsequent study also assessed ESR1 mutations longitudinally in the ACLINA cohort of 59 ER+/HER2- metastatic breast cancer patients treated with palbociclib plus fulvestrant[86]. They found ESR1 mutations in 28.8% of the baseline samples, but these were not associated with PFS. In addition, they found that all patients experienced a decrease in ctDNA on day 15 relative to baseline. In contrast, on day 30, patients with early progression had increased ctDNA, and patients with longer PFS had lower or consistent ctDNA levels. They found that the presence of ESR1 mutant ctDNA on day 30, as opposed to ctDNA clearance, was correlated with worse PFS, suggesting the potential of ctDNA detection on day 30 of treatment as a prognostic biomarker.
Further support for ctDNA ratios on day 30 as a biomarker comes from additional analysis of ctDNA from patients in the ALCINA cohort[87]. First, this study used archived tumor tissue to identify trackable mutations based on a panel of 15 driver genes. Next, they assessed serial plasma samples for 25 patients with either PIK3CA, TP53, or AKT1 mutations and found that baseline ctDNA levels had no association with PFS. In addition, they found that all patients had a decrease in ctDNA on day 15, which was not associated with PFS. In contrast, three kinetic patterns appeared on day 30, with nine patients displaying a continuous decreased or undetected ctDNA, one patient with consistent ctDNA levels, and five patients with an increase in ctDNA. Patients with undetectable ctDNA on day 30 had an improved PFS compared to those with detectable ctDNA. Furthermore, the radiological response had high concordance with ctDNA detection, with general decreases or undetectable ctDNA for non-progressive disease compared to rising ctDNA between days 15 and 30 for progressive disease. However, this study assessed concentrations of ctDNA with respect to disease progression and found overlap in progressive versus non-progressive disease at all time points, highlighting the challenge of absolute abundance as a biomarker. Therefore, they assessed ctDNA ratios relative to baseline and found low ratios on day 15, with no significant difference in decline between patients with and without disease progression. Instead, they found that ctDNA ratios on day 30 relative to baseline distinguished patients with non-progressive disease (ratio < 1) and progressive disease (ratio > 1) and that high ctDNA ratios were associated with worse PFS. This study supports that monitoring of ctDNA is related to radiological progression and demonstrates the potential of ctDNA dynamics on day 30 relative to baseline as a prognostic biomarker for patients treated with CDK4/6 inhibitors.
In contrast to the studies highlighted above, ctDNA dynamics can be monitored using the combined signal from profiling changes in many mutated genes instead of specific mutations. For instance, a recent study assessed ctDNA levels at baseline and four weeks for 45 patients treated with CDK4/6 inhibitors and endocrine therapy using the 73-gene Guardant360 assay[88]. This work defined a mean variant allele fraction ratio (mVAFR) as an average of mutations found between baseline and week 4 for each patient. They found that mVAFR was significantly associated with PFS, whereas single timepoint mean VAFs or absolute changes in mean VAF were not. One consideration of this study is that they assessed PFS with respect to three mVAFR groups (high, medium, and low) with cutoffs based on their cohort before assessing mVAFR as a continuous variable. Another limitation of this study was that they could not distinguish mutations from clonal hematopoiesis. Altogether, this study illustrates that early ctDNA dynamics in multiple genes may act as a biomarker to identify patients who are likely to progress on CDK4/6 inhibitors and endocrine therapy, which may provide the opportunity to modify or add treatments early on.
An additional study evaluated ctDNA using an untargeted sequencing technique, modified Fast Aneuploidy Screening Test-Sequencing System (mFAST-seq) in longitudinal samples from 49 HR+/HER2- metastatic breast cancer patients treated with CDK4/6 inhibitors[89]. In this work, associations between z-score measurements, which are surrogate measurements to ctDNA fraction, were made to clinical outcomes using joint models, which link data over a protracted period of time and time-to-event. They found that raised z-score trajectories were significantly related to worse PFS, whereas baseline z-scores were not predictive of progression. Interestingly, they found that a single rise z-score in a consecutive blood sample at any follow-up point was not associated with PFS. This study highlights the use of different assays in dynamic monitoring and reinforces that trajectories, as opposed to single time points, may be useful biomarkers of progression on anti-CDK4/6 therapy. A limitation of this approach is that mutations were not assessed, which may be relevant in the future for potential interventions or modifications to treatments. Also, this study evaluated z-score measurements at 181 time points for 49 patients, but these were not standardized and did not address the optimal time to sample. They also observed that some patients with progressive disease did not have raised z-scores over time and may be due to long intervals in sampling failing to detect an increase. This may be elucidated by shorter and more consistent sampling in future studies.
Overall, the current literature on monitoring ctDNA dynamics for patients treated with CDK4/6 inhibitors is limited. Differences in methods, patient populations, sampling time points, assays, and change thresholds lead to discordance across studies. Determining ideal change thresholds and timepoints will be essential for downstream clinical decisions. Future research should evaluate a more comprehensive range of consistent time points. Another important consideration is the way mutations are interrogated in ctDNA. Methods can vary dramatically by the number of tumor-specific markers assessed. ddPCR and related single-locus approaches can have high analytical sensitivity and specificity for a specific mutation, but other mutations and subclones that may be relevant to treatment resistance are ignored. Broader targeted panel sequencing approaches can be more robust by simultaneously interrogating multiple mutations and accounting for potential mechanisms of resistance[64,72]. Bespoke assays designed for each patient based on tumor tissue sequencing results [e.g., TARgeted DIgital Sequencing (TARDIS) and Signatera] show promise for sensitive and specific ctDNA detection[63,73,90,91], although emergent subclones that are not present in the tissue specimen can be missed[54]. Future studies may benefit from combining these approaches to achieve high analytical performance while enabling broad discovery.
EPIGENETIC-BASED LIQUID BIOPSY BIOMARKERS OF CDK4/6 INHIBITOR EFFICACY AND RESISTANCE
As outlined in the previous sections, most CDK4/6 inhibitor ctDNA studies have focused on genetic alterations (e.g., small nucleotide variants, copy number aberrations, etc.). In contrast, epigenetic alterations have been relatively understudied in this context. Emerging methodologies now make it easier to profile epigenetic aberrations in ctDNA, including DNA methylation, fragmentation, and histone modifications[92]. This new generation of liquid biopsy investigations has expanded the potential diagnostic use of cfDNA compared to genetic alterations on their own (e.g., providing information on the tissue of origin). Investigating epigenetic-based features has also expanded the number of cfDNA fragments of interest beyond solely mutated tumor-derived fragments. Supporting these new methods is a maturing research infrastructure (e.g., bioinformatics infrastructure, machine learning tools) for handling increasingly complex cancer liquid biopsy data[93]. Since genetic alterations in ctDNA have failed to identify clear predictive biomarkers of CDK4/6 inhibitor efficacy and resistance, there is interest in exploring the potential value of epigenetic-based biomarkers in this setting.
Phenotype of CDK4/6 inhibitor resistance and gene expression profiling
Support for epigenetic liquid biopsy approaches comes from previous work highlighting that phenotypic biomarkers that reflect transcriptomic programs are likely to predict response to CDK4/6 inhibitors. As summarized below, multiple gene expression analyses have demonstrated the effect of CDK4/6 inhibitors on proliferation and cell cycle genes, and patterns associated with resistance have been proposed.
An analysis of ER+/HER2- breast cancer patients in the NeoPalAna trial explored gene expression changes through serial tissue biopsies at baseline, cycle one day 1, cycle one day 15, and surgery. High expression of CCNE1, CCND3, and CDKN2D at cycle one day 15 was associated with resistance to neoadjuvant palbociclib plus anastrozole but not anastrozole alone[94]. These gene expression changes suggest that resistant tumors have continual E2F1 activity. Similarly, a gene expression analysis on tissue samples from baseline and surgery from patients treated with preoperative palbociclib found large-scale changes in genes related to proliferation and cell cycle after treatment, including a significant decrease in CCNE2 expression in antiproliferative responders compared to nonresponders[95].
A substudy of the PALOMA-3 trial yielded partially confirmatory findings[96]. Gene expression of 2534 cancer-related genes from 302 patient tumors revealed that high expression of CCNE1 - but not other genes related to cell cycle regulation (CDK4, CDK6, CCND1, and RB1) - was associated with resistance to palbociclib (median PFS palbociclib arm: 7.6 vs. 14.1 months high vs. low; placebo arm: 4.0 vs. 4.8 months high vs. low). The increased predictive power of CCNE1 mRNA in metastatic biopsies suggests that sampling closer to treatment allows improved identification of predictive biomarkers, which may be facilitated by liquid biopsy. The authors also confirmed the potential role of CCNE1 as a predictive biomarker in an independent validation cohort of breast cancer patients from the preoperative palbociclib study, where high CCNE1 levels were associated with a decreased antiproliferative effect with palbociclib.
Further support for CCNE1 mRNA as a predictive biomarker stems from a series of studies examining expression levels relative to those of RB1. One study of ER+/HER2- preclinical models found that joint decreased expression of RB1 and increased expression of CCNE1 commonly occurred at resistance[97]. The ratio of CCNE1 to RB1 was then confirmed to be associated with palbociclib resistance among patients in the NeoPalAna trial. Another study of patients treated with abemaciclib and anastrozole alone or combined within the neoMONARCH trial found that resistant tumors had higher expression levels of CCNE1 and lower levels of RB1[98]. High tumor CCNE1 expression was again associated with poor PFS among 391 patients treated with letrozole plus ribociclib in the MONALEESA-2 trial[41]; interestingly, this study also identified FGFR1 expression as a putative biomarker for CDK4/6 inhibitors (PFS of 22 months vs. not reached for patients with high vs. low FGFR1 expression, respectively).
One limitation of many studies correlating gene expression and PFS is that high and low expression thresholds are often determined above and below the median expression level in the cohort. Thresholds that are biased to the study cohort make it challenging to assess the prognostic or predictive potential, make comparisons across studies, and translate biomarker development to the clinic. Regardless, the current evidence supports that gene expression may predict CDK4/6 inhibitor efficacy or resistance. Considering gene signature assays can become routine in the clinical management of ER+ breast cancer patients, such as MammaPrint and OncotypeDx, specific gene signatures that are predictive of therapy with CDK4/6 inhibitors may be defined and developed[27,29]. However, these assays require tissue samples, which have barriers to accessibility and are often limited by the quality and quantity of RNA after FFPE chemical degradation[99]. This becomes especially difficult to derive from archival tissue specimens collected years before metastatic relapse. Many of these obstacles may be overcome by assessing transcriptional and epigenetic profiles with liquid biopsy.
Opportunities of non-mutational signatures of cfDNA
The potential of epigenetic mechanisms and biomarkers for CDK4/6 inhibitor resistance has so far not been examined in detail, and investigating these avenues may provide novel insight. For instance, one recent study showed that treatment with CDK4/6 inhibitors in ER+ breast cancer causes extensive enhancer activation through activator protein-1 (AP-1) transcriptional changes. They found that the widespread chromatin remodeling with CDK4/6 inhibitor treatment may explain the effects of these drugs beyond cell cycle arrest and may be involved in early adaptations leading to resistance[100]. These epigenomic changes may also be inferred from various features of ctDNA, such as methylation, fragmentation patterns, and histone modifications [Figure 1].
DNA methylation, namely 5-methylcytosines at CpG sites, is a vital part of cell-type-specific transcriptional regulation, and methylation profiles differ between tumor and normal tissues[101]. These differential methylation patterns are maintained in plasma cfDNA and can classify cancer types with high sensitivity in both early and late-stage disease[102,103]. Differential plasma cfDNA methylation patterns can also be leveraged to delineate the contribution of various tissues to the cfDNA pool and infer the expression of genes implicated in cancer[57,103,104]. One study investigated methylation-based biomarkers in the context of CDK4/6 inhibitors by assessing the methylation status of ESR1 in plasma cfDNA from a cohort of 49 HR+/HER2- metastatic breast cancer patients treated mostly with endocrine therapy and CDK4/6 inhibitors. Using samples from baseline and at three months, they assessed methylation levels at two main promoters with methylation-specific ddPCR. They found that a greater than 2-fold increase in promoter B or both promoters was associated with a worse prognosis[105]. While this study has various limitations, such as a small heterogeneous cohort, inability to dissect endocrine and CDK4/6 inhibitor effects, and analysis of limited loci, it paves the way for both epigenetic and dynamic cfDNA biomarker discovery approaches.
Future studies of DNA methylation in the CDK4/6 inhibitor treatment setting should consider other technical approaches. For instance, while the analysis of limited CpG sites is practical in many clinical settings for simplicity of interpretation and lower costs, expanding to genome-wide explorations may uncover novel candidate resistance biomarkers. Furthermore, bisulfite conversion is necessary for most methylation techniques but can result in excessive loss of DNA due to degradation, which is a substantial challenge for low-input cfDNA samples[106]. Forthcoming research should leverage other methods that surpass these limitations by enriching specifically for methylated fragments of cfDNA before sequencing[104,107,108]. Moreover, hydroxymethylation is a related epigenetic modification produced by TET enzymes during cytosine demethylation and acts as a marker of active promoters[109]. Though less studied in cfDNA, similar enrichment techniques have been developed to assess regions with 5-hydroxymethyl cytosines[110,111]. These approaches may be especially useful for revealing surrogates of gene expression that could confer sensitivity or resistance to CDK4/6 inhibitors.
Phenotypic information can also be inferred from distinct fragmentation patterns between ctDNA and other sources of cfDNA. The fragmentation of cfDNA is a non-random process associated with chromatin structure, gene expression, and nuclease content. Differences in nucleosome occupancy patterns at open versus closed chromatin and across varying gene expression levels affect where nucleases can access and fragment the DNA[92]. This, in turn, is reflected in the physical characteristics of the cfDNA fragments and their distribution over the genome (i.e., fragmentation features), revealing information about cell and tissue of origin. Early work on cfDNA fragments revealed differences in fragment length, a phenomenon that multiple studies have leveraged to enrich ctDNA and improve the accuracy of cancer detection[112-117].
Beyond fragment length, many other fragmentation features have also been investigated, including relative sequence coverage[118-124], end motifs[125-129], and more[130-132]. There is substantial evidence that these features convey information about DNA protection from digestion, which multiple studies have used to create cfDNA deduced nucleosome maps[58,118,120,133]. The fragmentation profiles from this work have been correlated with gene expression profiles and permitted identification of cancer type[118,120,123].
The association of fragmentation profiles with transcriptional activity may present opportunities to infer existing candidate gene expression biomarkers of CDK4/6 inhibitor resistance (e.g., CCNE1 expression) with cfDNA in blood plasma while simultaneously permitting assessment of clinically actionable mutations to direct subsequent therapies. While promising, there remain several practical hurdles to implementing such biomarkers in the clinic. Pre-analytical variables could influence fragmentation features, and their extraction from sequencing data requires complex bioinformatics analysis. However, if these hurdles can be overcome, fragmentation-based biomarkers could greatly extend the utility of ctDNA analysis in the CDK4/6 inhibitor biomarker setting and beyond.
Another class of epigenetic modification has recently been proposed for liquid biopsy applications: post-translational modifications of nucleosomal histones in circulation. These modifications differ in euchromatin compared to heterochromatin and may signal transcriptionally active (e.g., H3K4me3, H3K36me3) or repressed (e.g., H3K27me3, H3K9me3) regions of the genome[134]. Furthermore, histone modifications may signal chromatin remodeling and transcriptional changes between cancer and healthy cells. Initial work found a global decrease in repressive histone markers across multiple cancer types[135,136]. In a recent study, cell-free chromatin immunoprecipitation and sequencing (ChIP-seq) was conducted to identify regions associated with transcriptionally active histone modifications[137]. This approach revealed signals reflective of distinct tissues and cancer types, potentially expanding the toolbox for biomarker discovery in the context of CDK4/6 inhibitors for breast cancer patients.
Overall, epigenetic profiling of cfDNA has many potential benefits that have gone mostly unexplored in the context of breast cancer resistance to CDK4/6 inhibitors. More investigation is needed to elucidate epigenetic signatures and determine which features are the most informative from the vast list growing in the literature. A combination of approaches may increase the predictive power compared to any method alone and increase the ability to direct subsequent therapies[138-140].
CONCLUSION
Biomarkers are essential for precision oncology, and despite widespread research efforts, no clinically validated biomarkers beyond breast cancer subtype have been established to guide the use of CDK4/6 inhibitors. Liquid biopsy presents many benefits for biomarker development due to being minimally invasive and encompassing the heterogeneity of metastatic sites. Accordingly, putative predictive biomarkers of resistance to treatment with CDK4/6 inhibitors have been found in ctDNA such as RB1, KRAS, and CCND1 genomic alterations; dynamic changes in ctDNA, such as changes in truncal mutations between baseline and cycle one day 15 or 30, or ctDNA clearance on day 30. Expression of E2F target genes such as CCNE1 is also associated with resistance and could someday be reflected through emerging epigenetic ctDNA analysis methodologies. In addition, prognostic biomarkers such as ESR1, PIK3CA, FGFR1, TP53, and MYC alterations have also been identified.
To advance further biomarker discovery, future studies of trials or cohorts that included a control arm (e.g., endocrine therapy alone) will be especially valuable to permit the assessment of treatment interaction effects. In addition, it would be beneficial to investigate a broader range of alterations within ctDNA, given the low likelihood that a single alteration will be an ideal biomarker to the vast mechanisms of resistance. Thresholds for expression or dynamic changes in ctDNA should also be determined independently from the patient cohort in which they are studied to increase generalizability and reproducibility. Moreover, future work on ctDNA dynamics should sample a larger range of time points. There is also enormous potential for phenotype-based biomarkers, which may reflect widespread epigenetic changes and inform changes in tumor biology associated with resistance. There is a lack of established mutation-based liquid biopsy biomarkers, and features of cfDNA related to methylation, fragmentation, and histone modifications are uncharted in the context of CDK4/6 inhibitor resistance. These ctDNA epigenomic profiles should be leveraged for new biomarker discovery. Although this review has focused on the ER+/HER2- subtype of breast cancer, it is conceivable that novel predictive biomarkers could identify other patients likely to respond. Lastly, there have been substantial investigations into novel targeted agents for treatment after CDK4/6 inhibitor resistance. For instance, preclinical and clinical data indicate that treatment with novel endocrine therapies, PI3K/MAPK pathway inhibitors, downstream mitotic kinase inhibitors, and DNA-damage related inhibitors may each have a role in the treatment of CDK4/6 inhibitor-resistant disease[46]. With further development, liquid biopsy biomarkers may help direct these subsequent therapies in the future, increasing the likelihood of successful clinical development and the potential impact on patient outcomes.
DECLARATIONS
AcknowledgementsFigures were created with BioRender.com.
Authors’ contributionsConceptualized the manuscript: Main SC, Cescon DW, Bratman SV
Performed the literature review and wrote the original manuscript: Main SC
Supervised and edited the manuscript: Cescon DW, Bratman SV
Availability of data and materialsNot applicable.
Financial support and sponsorshipS.C.M. is supported by the Canadian Institutes of Health Research (CIHR) Frederick Banting and Charles Best Canadian Graduate Scholarship. S.V.B. is supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at the Princess Margaret Cancer Centre and the Dr. Mariano Elia Chair in Head & Neck Cancer Research at University Health Network and the University of Toronto.
Conflicts of interestMain SC has no conflicts of interest to declare. Cescon DW reports consultancy and advisory fees from AstraZeneca, Exact Sciences, Eisai, Gilead, GlaxoSmithKline, Merck, Novartis, Pfizer and Roche; research funding to their institution from GlaxoSmithKline, Inivata, Merck, Pfizer and Roche; is a member of a trial steering committee for AstraZeneca, Merck and GlaxoSmithKline; and holds a holds a patent (US62/675,228) for methods of treating cancers characterized by a high expression level of spindle and kinetochore associated complex subunit 3 (ska3) gene. Bratman SV is inventor on patents related to cell-free DNA mutation and methylation analysis technologies that are unrelated to this work and have been licensed to Roche Molecular Diagnostics and Adela, respectively. Bratman SV is a co-founder of, has ownership in, and serves in a leadership role at Adela.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Hart CD, Migliaccio I, Malorni L, Guarducci C, Biganzoli L, Di Leo A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat Rev Clin Oncol 2015;12:541-52.
2. Pan H, Gray R, Braybrooke J, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med 2017;377:1836-46.
3. Mehta RS, Barlow WE, Albain KS, et al. Overall survival with fulvestrant plus anastrozole in metastatic breast cancer. N Engl J Med 2019;380:1226-34.
4. Burstein HJ, Somerfield MR, Barton DL, et al. Endocrine treatment and targeted therapy for hormone receptor–positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: ASCO Guideline update. J Clin Oncol 2021;39:3959-77.
5. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016;375:1925-36.
6. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 2015;373:209-19.
7. Sledge GW, Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol 2017;35:2875-84.
8. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 2017;35:3638-46.
9. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for hr-positive, advanced breast cancer. N Engl J Med 2016;375:1738-48.
10. Slamon DJ, Neven P, Chia S, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol 2018;36:2465-72.
11. Tripathy D, Im SA, Colleoni M, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol 2018;19:904-15.
12. Portman N, Alexandrou S, Carson E, Wang S, Lim E, Caldon CE. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr Relat Cancer 2019;26:R15-30.
13. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phas. Lancet Oncol 2016;17:425-39.
14. Im SA, Lu YS, Bardia A, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med 2019;381:307-16.
15. Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med 2018;379:1926-36.
16. Slamon DJ, Neven P, Chia S, et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl J Med 2020;382:514-24.
17. Johnston S, Martin M, Di Leo A, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. npj Breast Cancer 2019;5:5.
18. Schettini F, Giudici F, Giuliano M, et al. Overall survival of CDK4/6-inhibitor-based treatments in clinically relevant subgroups of metastatic breast cancer: systematic review and meta-analysis. J Natl Cancer Inst 2020;112:1089-97.
19. Rugo HS, Cristofanilli M, Loibl S, et al. Prognostic factors for overall survival in patients with hormone receptor-positive advanced breast cancer: analyses from PALOMA-3. Oncologist 2021;26:e1339-46.
20. Rugo HS, Finn RS, Diéras V, et al. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat 2019;174:719.
21. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009;11:R77.
22. Finn RS, Liu Y, Zhu Z, et al. Biomarker analyses of response to cyclin-dependent kinase 4/6 inhibition and endocrine therapy in women with treatment-naïve metastatic breast cancer. Clin Cancer Res 2020;26:110-21.
23. Asghar US, Kanani R, Roylance R, Mittnacht S. Systematic review of molecular biomarkers predictive of resistance to CDK4/6 inhibition in metastatic breast cancer. JCO Precis Oncol 2022;6:e2100002.
24. Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 2019;20:71-88.
25. Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med 2020;12:1-19.
26. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817-26.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 2006;24:3726-34.
28. van’t Veer LJ, Dai H, Van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530-6.
29. Cardoso F, van’t Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717-29.
30. Prat A, Pineda E, Adamo B, et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. The Breast 2015;24:S26-35.
31. von Minckwitz G, Huang CS, Mano MS, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med 2019;380:617-28.
32. Rugo HS, Vidula N, Ma C. Improving response to hormone therapy in breast cancer: new targets, new therapeutic options. Am Soc Clin Oncol Educ B 2016;35:e40-54.
33. Bertoli C, Skotheim JM, De Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518-28.
34. Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci 2008;121:3853-7.
35. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011;20:620-34.
36. Sabbah M, Courilleau D, Mester J, Redeuilh G. Estrogen induction of the cyclin D1 promoter: involvement of a cAMP response-like element. Proc Natl Acad Sci USA 1999;96:11217-22.
37. Thu K, Soria-Bretones I, Mak T, Cescon D. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle 2018;17:1871-85.
38. Álvarez-Fernández M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 2020;37:514-29.
39. O’Leary B, Cutts RJ, Liu Y, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov 2018;8:1390-403.
40. Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017;36:2255-64.
41. Formisano L, Lu Y, Servetto A, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun 2019;10:1373.
42. Herrera-Abreu MT, Palafox M, Asghar U, et al. Early Adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res 2016;76:2301-13.
43. Li Z, Razavi P, Li Q, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell 2018;34:893-905.e8.
44. Condorelli R, Spring L, O’Shaughnessy J, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018;29:640-5.
45. Raimondi L, Raimondi FM, Pietranera M, et al. Assessment of resistance mechanisms and clinical implications in patients with KRAS mutated-metastatic breast cancer and resistance to CDK4/6 inhibitors. Cancers (Basel) 2021;13:1928.
46. Wander SA, Cohen O, Gong X, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov 2020;10:1174-93.
47. Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010;28:3271-7.
48. Gerratana L, Fanotto V, Bonotto M, et al. Pattern of metastasis and outcome in patients with breast cancer. Clin Exp Metastasis 2015;32:125-33.
49. Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223-38.
50. Cescon DW, Bratman SV, Chan SM, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer 2020;1:276-90.
51. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24.
52. Nassiri F, Chakravarthy A, Feng S, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med 2020;26:1044-7.
53. Andersson D, Kubista M, Ståhlberg A. Liquid biopsy analysis in cancer diagnostics. Mol Aspects Med 2020;72:100839.
54. Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol 2021;18:297-312.
55. Migliaccio I, Leo A, Galardi F, et al. Circulating biomarkers of CDK4/6 inhibitors response in hormone receptor positive and HER2 negative breast cancer. Cancers (Basel) 2021;13:2640.
56. González-Conde M, Yañez-Gómez C, López-López R, Costa C. Liquid biopsy: a new tool for overcoming CDKi resistance mechanisms in luminal metastatic breast cancer. J Pers Med 2021;11:407.
57. Sun K, Jiang P, Chan KCA, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci 2015;112:E5503-12.
58. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016;164:57-68.
59. Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:1-12.
60. Rostami A, Lambie M, Yu CW, Stambolic V, Waldron JN, Bratman S V. Senescence, necrosis, and apoptosis govern circulating cell-free DNA release kinetics. Cell Rep 2020;31:107830.
61. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001;61:1659-65.
62. Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA: apoptosis and active DNA release. Clin Chim Acta 2001;313:139-42.
63. Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446-51.
64. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547-55.
65. Kurtz DM, Soo J, Co Ting Keh L, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol 2021;39:1537-47.
66. Yao W, Mei C, Nan X, Hui L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: a qualitative study. Gene 2016;590:142-8.
67. Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 1999;64:218.
68. To EWH, Chan KCA, Leung SF, et al. Rapid clearance of plasma epstein-barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin Cancer Res 2003;9:3254-9.
69. Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985-90.
70. Li M, Diehl F, Dressman D, Vogelstein B, Kinzler KW. BEAMing up for detection and quantification of rare sequence variants. Nat Methods 2006;3:95-7.
71. Postel M, Roosen A, Laurent-Puig P, Taly V, Wang-Renault SF. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective. Expert Rev Mol Diagn 2018;18:7-17.
72. Newman AM, Bratman S V. , To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548-54.
73. McDonald BR, Contente-Cuomo T, Sammut SJ, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med 2019;11:1-14.
74. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4:136ra68.
75. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA -mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 2019;380:1929-40.
76. US Food and Drug Administration. therascreen PIK3CA RGQ PCR kit FDA . Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190004A.pdf2019 [Last accessed on 1 Jun 2021].
77. Razavi P, Dickler MN, Shah PD, et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat Cancer 2020;1:382-93.
78. O’Leary B, Cutts RJ, Huang X, et al. Circulating tumor DNA markers for early progression on fulvestrant with or without palbociclib in ER+ advanced breast cancer. JNCI J Natl Cancer Inst 2021;113:309-17.
79. Bertucci F, Ng CKY, Patsouris A, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569:560-4.
80. Fribbens C, O’Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 2016;34:2961-8.
81. Tolaney SM, Toi M, Neven P, et al. Clinical significance of PIK3CA and ESR1 mutations in circulating tumor DNA: analysis from the MONARCH 2 study of abemaciclib plus fulvestrant. Clin Cancer Res 2022;28:1500-6.
82. Del Re M, Crucitta S, Lorenzini G, et al. PI3K mutations detected in liquid biopsy are associated to reduced sensitivity to CDK4/6 inhibitors in metastatic breast cancer patients. Pharmacol Res 2021;163:105241.
83. Bardia A, Su F, Solovieff N, et al. Genomic profiling of premenopausal HR+ and HER2- metastatic breast cancer by circulating tumor DNA and association of genetic alterations with therapeutic response to endocrine therapy and ribociclib. JCO Precis Oncol 2021;5:PO.20.00445.
84. Sanz-Garcia E, Zhao E, Bratman S V, Siu LL. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: current research, opportunities, and challenges. Sci Adv 2022;8:1-15.
85. O’Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun 2018;9:1-10.
86. Jeannot E, Darrigues L, Michel M, et al. A single droplet digital PCR for ESR1 activating mutations detection in plasma. Oncogene 2020;39:2987-95.
87. Darrigues L, Pierga JY, Bernard-Tessier A, et al. Circulating tumor DNA as a dynamic biomarker of response to palbociclib and fulvestrant in metastatic breast cancer patients. Breast Cancer Res 2021;23:1-10.
88. Martínez-Sáez O, Pascual T, Brasó-Maristany F, et al. Circulating tumor DNA dynamics in advanced breast cancer treated with CDK4/6 inhibition and endocrine therapy. npj Breast Cancer 2021;7:8.
89. Dandachi N, Posch F, Graf R, et al. Longitudinal tumor fraction trajectories predict risk of progression in metastatic HR + breast cancer patients undergoing CDK4/6 treatment. Mol Oncol 2021;15:2390-400.
90. Henriksen TV, Tarazona N, Frydendahl A, et al. Circulating tumor DNA in stage III colorectal cancer, beyond minimal residual disease detection, toward assessment of adjuvant therapy efficacy and clinical behavior of recurrences. Clin Cancer Res 2022;28:507-17.
91. Coombes RC, Page K, Salari R, et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res 2019;25:4255-63.
92. Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021;372:eaaw3616.
93. Im YR, Tsui DWY, Diaz LA, Wan JCM. Next-generation liquid biopsies: embracing data science in oncology. Trends in Cancer 2021;7:283-92.
94. Ma CX, Gao F, Luo J, et al. NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor-positive breast cancer. Clin Cancer Res 2017;23:4055-65.
95. Arnedos M, Bayar MA, Cheaib B, et al. Modulation of Rb phosphorylation and antiproliferative response to palbociclib: the preoperative-palbociclib (POP) randomized clinical trial. Ann Oncol 2018;29:1755-62.
96. Turner NC, Liu Y, Zhu Z, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol 2019;37:1169-78.
97. Guarducci C, Bonechi M, Benelli M, et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. npj Breast Cancer 2018;4:38.
98. Hurvitz SA, Martin M, Press MF, et al. Potent cell-cycle inhibition and upregulation of immune response with abemaciclib and anastrozole in neoMONARCH, Phase II neoadjuvant study in HR+/HER2- breast cancer. Clin Cancer Res 2020;26:566-80.
99. Evers DL, He J, Kim YH, Mason JT, O’Leary TJ. Paraffin embedding contributes to RNA aggregation, reduced RNA yield, and low RNA quality. J Mol Diagnostics 2011;13:687-94.
100. Watt AC, Cejas P, DeCristo MJ, et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat Cancer 2021;2:34-48.
102. Nuzzo PV, Berchuck JE, Korthauer K, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med 2020;26:1041-3.
103. Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31:745-59.
104. Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563:579-83.
105. Gerratana L, Basile D, Franzoni A, et al. Plasma-based longitudinal evaluation of ESR1 epigenetic status in hormone receptor-positive HER2-negative metastatic breast cancer. Front Oncol 2020;10:1-12.
106. Ørntoft MB, Jensen SØ, Hansen TB, Bramsen JB, Andersen CL. Comparative analysis of 12 different kits for bisulfite conversion of circulating cell-free DNA. Epigenetics 2017;12:626-36.
107. Shen SY, Burgener JM, Bratman S V. , De Carvalho DD. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat Protoc 2019;14:2749-80.
108. Huang J, Soupir AC, Wang L. Cell-free DNA methylome profiling by MBD-seq with ultra-low input. Epigenetics 2022;17:239-52.
109. Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet 2012;13:7-13.
110. Song CX, Yin S, Ma L, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res 2017;27:1231-42.
111. Li W, Zhang X, Lu X, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res 2017;27:1243-57.
112. Underhill HR, Kitzman JO, Hellwig S, et al. Fragment length of circulating tumor DNA. Kwiatkowski DJ, ed. PLoS Genet 2016;12:e1006162.
113. Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10:1-14.
114. Hellwig S, Nix DA, Gligorich KM, et al. Automated size selection for short cell-free DNA fragments enriches for circulating tumor DNA and improves error correction during next generation sequencing. PLoS One 2018;13:1-24.
115. Liu X, Liu L, Ji Y, et al. Enrichment of short mutant cell-free DNA fragments enhanced detection of pancreatic cancer. EBioMedicine 2019;41:345-56.
116. Underhill HR. Leveraging the fragment length of circulating tumour DNA to improve molecular profiling of solid tumour malignancies with next-generation sequencing: a pathway to advanced non-invasive diagnostics in precision oncology? Mol Diagn Ther 2021;25:389-408.
117. Heitzer E, Speicher MR. One size does not fit all: size-based plasma DNA diagnostics. Sci Transl Med 2018;10:1-4.
118. Ulz P, Thallinger GG, Auer M, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 2016;48:1273-8.
119. Zhu G, Guo YA, Ho D, et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat Commun 2021;12:2229.
120. Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385-9.
121. Ivanov M, Baranova A, Butler T, Spellman P, Mileyko V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 2015;16:1-12.
122. Ulz P, Perakis S, Zhou Q, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 2019;10:4666.
123. Peneder P, Stütz AM, Surdez D, et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat Commun 2021;12:3230.
124. Mathios D, Johansen JS, Cristiano S, et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun 2021;12:5060.
125. Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genomics 2015;8:29.
126. Jin C, Liu X, Zheng W, et al. Characterization of fragment sizes, copy number aberrations and 4-mer end motifs in cell-free DNA of hepatocellular carcinoma for enhanced liquid biopsy-based cancer detection. Mol Oncol 2021;15:2377-89.
127. Serpas L, Chan RWY, Jiang P, et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci 2019;116:641-9.
128. Jiang P, Sun K, Peng W, et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov 2020;10:664-73.
129. Zhitnyuk Y V, Koval AP, Alferov AA, et al. Deep cfDNA fragment end profiling enables cancer detection. Mol Cancer 2022;21:26.
130. Jiang P, Xie T, Ding SC, et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res 2020;30:1144-53.
131. Sun K, Jiang P, Cheng SH, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res 2019;29:418-27.
132. Jiang P, Sun K, Tong YK, et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci USA 2018;115:E10925-33.
133. Murtaza M, Caldas C. Nucleosome mapping in plasma DNA predicts cancer gene expression. Nat Genet 2016;48:1105-6.
134. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21:381-95.
135. Deligezer U, Akisik EE, Erten N, Dalay N. Sequence-specific histone methylation is detectable on circulating nucleosomes in plasma. Clin Chem 2008;54:1125-31.
136. Deligezer U, Yaman F, Darendeliler E, et al. Post-treatment circulating plasma BMP6 mRNA and H3K27 methylation levels discriminate metastatic prostate cancer from localized disease. Clin Chim Acta 2010;411:1452-6.
137. Sadeh R, Sharkia I, Fialkoff G, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol 2021;39:586-98.
138. Lasseter K, Nassar AH, Hamieh L, et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet Med 2020;22:1366-73.
139. Hu X, Luo K, Shi H, et al. Integrated 5-hydroxymethylcytosine and fragmentation signatures as enhanced biomarkers in lung cancer. Clin Epigenetics 2022;14:15.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Main SC, Cescon DW, Bratman SV. Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer. Cancer Drug Resist 2022;5:727-48. http://dx.doi.org/10.20517/cdr.2022.37
AMA Style
Main SC, Cescon DW, Bratman SV. Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer. Cancer Drug Resistance. 2022; 5(3): 727-48. http://dx.doi.org/10.20517/cdr.2022.37
Chicago/Turabian Style
Main, Sasha C., David W. Cescon, Scott V. Bratman. 2022. "Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer" Cancer Drug Resistance. 5, no.3: 727-48. http://dx.doi.org/10.20517/cdr.2022.37
ACS Style
Main, SC.; Cescon DW.; Bratman SV. Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer. Cancer Drug Resist. 2022, 5, 727-48. http://dx.doi.org/10.20517/cdr.2022.37
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 40 clicks
Cite This Article 40 clicks

 Like This Article 15
likes
Like This Article 15
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.