
Microvesicles: the functional mediators in sorafenib resistance

Abstract
Overcoming drug resistance in cancer therapies remains challenging, and the tumor microenvironment plays an important part in it. Microvesicles (MVs) are functional natural carriers of cellular information, participate in intercellular communication, and dynamically regulate the tumor microenvironment. They contribute to drug resistance by transferring functional molecules between cells. Conversely, due to their specific cell or tissue targeting ability, MVs are considered as carriers for therapeutic molecules to reverse drug resistance. Thus, in this mini-review, we aim to highlight the crucial role of MVs in cell-to-cell communication and therefore their diverse impact mainly on liver cancer progression and treatment. In addition, we summarize the possible mechanisms for sorafenib resistance (one of the main hurdles in hepatocellular carcinoma treatments) and recent advances in using MVs to reverse sorafenib resistance in liver cancer therapies. Identifying the functional role of MVs in cancer therapy might provide a new aspect for developing precise novel therapeutics in the future.
Keywords
INTRODUCTION
Liver cancer (LC), including hepatocellular carcinoma (HCC), hepatoblastoma, and cholangiocarcinoma, remains one of the malignant cancers worldwide with high mortality[1]. Surgery, radiotherapy and chemotherapy are considered as the main treatments for LCs at present, but their therapeutic effects are limited in advanced stages[2]. Sorafenib, an oral drug approved by the FDA for the treatment of advanced HCC, has shown excellent therapeutic effects by inhibiting tumor growth and angiogenesis in vitro[3,4]. However, reported resistance limits its therapeutic effects[5-9]. Microvesicles (MVs), functional mediators in cell-to-cell communication by transferring bioactive cargoes[10], play an important role in tumor progression and metastasis[11-14]. Interestingly, recent studies reported that HCC-derived MVs promoted sorafenib resistance in recipient liver cancer cells by transporting cancerous cargo compared to normal liver cell-derived MVs[15]. Thus, the role of these tiny functional vesicles needs to be cautiously studied for the in-depth understanding and successful treatment of HCC over sorafenib resistance.
Considering the necessity of overcoming sorafenib resistance while developing a successful treatment process for the challenging HCC, in the present review, we summarize the mechanism of action of sorafenib and sorafenib resistance. In addition, recent advances of MVs in intercellular communication and the intriguing contribution of MVs in cancer treatment are discussed. Especially, the review closely considers the possibilities for utilizing MVs as a potential therapeutic tool to alleviate sorafenib resistance in future cancer treatments.
SORAFENIB DRUG AND RESISTANCE
Mechanism of action of sorafenib
The revelation of the crucial involvement of Raf1 and vascular endothelial growth factor (VEGF) mediated signaling pathways in the molecular pathogenesis of liver cancer provided an interesting theoretic basis for applying sorafenib drugs to liver cancer treatment[16,17]. As a multikinase inhibitor, sorafenib strongly inhibits the tyrosine kinase Raf. Meanwhile, it has been shown to inhibit vascular endothelial growth factor receptor and platelet-derived growth factor receptor, which in turn inhibits the activation of other downstream multikinase that are normally essential for cell growth, angiogenesis, proliferation and metastasis of HCC cells [Figure 1]. Liu et al.[18] recorded that sorafenib inhibited the proliferation of HCC cells and reduced angiogenesis signal transduction in HCC tumor xenograft, promoting tumor cell apoptosis as well. In addition, the same therapeutic advances have also been revealed in clinical studies[19]. Nevertheless, several studies stated that the therapeutic effects of sorafenib varied among patients, some of whom experienced severe side effects[20-23].
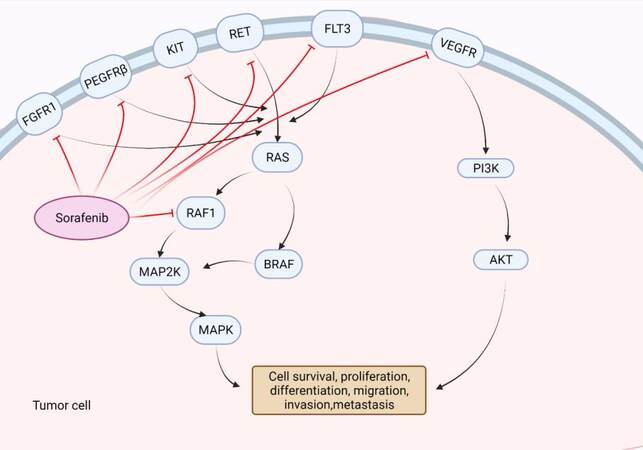
Figure 1. Mechanism of actions of sorafenib. Sorafenib inhibits tyrosine kinase receptor (VEGFR, PDGFRβ, Kit and RET) signaling and suppresses the activation of Raf, thus could suppress tumor progression by inhibiting angiogenesis and cell proliferation. Created with BioRender.com. VEGFR: Vascular endothelial growth factor receptor.
Mechanism of sorafenib resistance
Drug resistance limits the therapeutic effects of HCC treatments. Roughly 30% of patients were reported to respond to sorafenib well at the beginning, while the subsequently acquired resistance to sorafenib usually happens within six months[24], which is far from satisfactory. As an obvious contributor that hinders the effectiveness of cancer treatment, sorafenib resistance and the possible molecular mechanisms involved in sorafenib resistance become prominently important to be discussed here.
Due to the heterogenicity of liver cancer, some patients are resistant to sorafenib primarily, while others obtain sorafenib resistance during treatment, which further limits the application of the drug and leaves the treatment process questionable. Thus, the acquired resistance of sorafenib attracted the attention of a wide range of researchers[8,25]. To date, studies on the potential mechanism of sorafenib resistance have mainly focused on the activation of drug targets and downstream signaling, regulation of cell proliferation and apoptosis signaling[5-9]. In addition, stemness and mesenchymal states of sorafenib-resistant HCC cells provided a new aspect of this challenging problem[26]. Altogether, the mechanism of sorafenib resistance and its influence on the treatment process remain complicated and require more research for a better understanding.
The epidermal growth factor receptor (EGFR) has been found to be overexpressed or hyperactivated in the cancer cells of most liver cancer patients as well as be the reason for continuous activation of its downstream signaling of the Ras/Raf/MEK/ERK pathway. This contributes to the abnormal proliferation of cancer cells and therefore might promote sorafenib resistance[27]. For instance, an attenuated level of phosphorylated ERK was reported to be associated with sorafenib resistance in HCC[28]. In addition, hyperactivated EGFR/HER3 and its overexpressed ligands were reported to suppress the curative effect of sorafenib by interfering with the phosphorylation of EGFR/HER3, by which the enhanced anti-proliferative and pro-apoptotic abilities of sorafenib could be achieved during the treatment[29].
A body of evidence reveals that the PI3K/Akt pathway plays an important role in sorafenib resistance[5,30]. For instance, Chen et al.[5] found that exposure of Huh7 liver cancer cells to a high concentration of sorafenib could result in sorafenib resistance and an accelerated expression of Akt in the treated cells. Furthermore, the PI3K/Akt pathway has been identified to have a close relationship with cell apoptosis. In the pathway, the combination of pro-survival factor and tyrosine kinase receptor activates the kinase PI3K, which triggers the downstream cascade to endorse phosphorylation of Akt and thus contributes to the suppression of cell apoptosis. In turn, inhibition of Akt could make the tumor cell more responsive to sorafenib treatment[31]. Hence, silencing of PI3K/Akt signaling with Akt inhibitor alone or with other combination therapy has gained attention for reversing sorafenib resistance for better HCC treatment[32,33]. Furthermore, Src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP-1) has been reported to be activated by sorafenib, which in turn could negatively regulate pSTAT3 and suppress transduction of JAK/STAT signaling. Dysfunctional JAK/STAT has been observed in sorafenib-resistant liver cancer cells, including induced expression of pSTAT3 and its downstream anti-apoptotic protein Mcl-1 and reduced expression of SHP-1/pSHP-1[34].
Moreover, cancer stem cells (CSCs), which represent a subpopulation of cancer cells with a self-renewal nature, are considered to participate in tumorigenesis, drug resistance, tumor metastasis and recurrence, and they are innovative targets for cancer therapy[35-37]. Label-retaining cancer cells (LRCCs) can be used to label CSCs. Xin et al.[38] used this methodology to observe sorafenib treated HCC cells and found that LRCCs were highly enriched in the remaining HCC cells that escaped sorafenib treatment, which strongly evidenced the resistance to sorafenib-induced cytotoxicity and apoptosis. In addition, specific ATP binding box (ABC) transporters have been reported to be highly expressed on CSCs, which control the outflow of chemical agents to protect cells from toxic compound accumulation and damage, and hence could reduce the sensitivity of cells towards drug treatment[39,40]. ABCB1 has been attested to have a close relationship with multidrug resistance; thereby, knocking out ABCB1 in drug-resistant cancer cell lines made those cells more responsive to chemotherapies[41]. It was also revealed that CSCs isolated from HCC cell lines showed resistance to sorafenib both in vitro and in vivo with abnormal IL-6/STAT3 signaling[6]. Through VEGF, liver cancer stem cells could promote tumor angiogenesis to sustain their stemness as well as drug resistance features[42]. In addition, Wnt/β-catenin signaling, one of the classic pathways involved in stemness regulation[43,44], was proven to be hyperactivated in HCC cells, resulting in β-catenin accumulation in cytoplasm and nucleus, which finally led to enhanced self-renewal ability of CSCs[45,46]. Inhibition of Wnt signaling has been shown to be beneficial to CSC clearance and tumor development[46,47].
Dual biotransformation routes including oxidation and glucuronidation were witnessed in the sorafenib metabolism[48-50]. After hepatocellular uptake, sorafenib was N-oxidized by CYP3A4, one of the drug-metabolizing enzymes, to the pharmacologically active sorafenib-N-oxide metabolites[51,52]. However, CYP3A4 was identified as poorly expressed in liver cancer[53]. Well-studied oncomiRs (e.g., miR 21, miR-142 and miR-27b) overexpressed in HCC, which could be transferred by tumor-derived microvesicles (TMVs), were proved to be negatively associated with CYP3A4 mRNA in human liver[54], which might downregulate the expression of this main enzyme and thus could inhibit the active biotransformation of sorafenib drug. Apart from oxidation, sorafenib underwent glucuronidation, mainly mediated by UGT1A1 and UGT1A9, to inactive glucuronide metabolites[48,55]. A recent study revealed that sorafenib inhibits the above-mentioned UGT enzyme[56], which might be the blockage for sorafenib secretion to bile and later systemic circulation and clearance. Taken together, the insufficient oxidation and complex interaction between sorafenib and UGT enzymes need further investigation for a deep understanding of their role in resistance.
Anti-angiogenesis is one of the therapeutic effects of sorafenib, while the tumor vessel depletion along with pericyte could induce hypoxia and allow the maintenance and enhancement of CSCs in HCC[57,58]. Apart from specific niches, different stroma cells in the microenvironment render sorafenib sensitivity[59], where MVs play a significant role.
MVS: THE TINY MEDIATORS IN INTERCELLULAR COMMUNICATION
MVs, a subpopulation of extracellular vesicles (EVs), act as functional mediators by transferring bioactive molecules among various types of cells and thus have been considered as potential candidates in intercellular communication[10]. Through unconventional secretion mechanisms, eukaryotic cells were reported to release membrane-enclosed vesicles both in vivo and in vitro[60]. In general, MVs can be collected from cell culture media and blood from animals and patients via ultracentrifuge method, filtration, or commercial kits[61]. MVs are also identified as shedding microvesicles or microparticles, budding directly from the plasma membrane and are 100 nm-1000 nm in diameter. They participate in cell-to-cell communication by carrying the information from parental cells to others and orchestrate complicated physiological and pathological processes[14,62]. At the molecular level, symmetrical perturbation of membrane lipids leads to the surface expression of phospholipid serine acid pairs, which translocate from the inner lobule to the outer surface lobule of the membrane bilayer through special biological enzymes (floppases, flippases and scramblases), contributing to MVs formation[63]. However, the detailed mechanism of MVs formation and shedding remains to be further studied.
Evidence reveals that most MVs tend to be decomposed after shedding to release their cargo[64]. Especially, various cytoplasmic proteins, excluded from the classic signal-peptide secretion pathways, are discharged out regularly through MV decomposition[65]. For instance, fibroblast growth factor 2 (FGF2) was proved to be released in response to the breakdown of MVs derived from neurons, HCC and endothelial cells[66]. Similarly, MVs secreted by dendritic cells, macrophages and microglias have been demonstrated to release the pro-inflammatory cytokine interleukin 1B (IL-1B); IL-1B has also been found to aggregate in MVs along with proteases such as caspase 1[67]. A study on tumor-stromal interactions noted that when MVs were co-released with extracellular matrix metalloproteinase inducer (EMMPRIN/CD147), they prompted lung cancer cells to obtain increased mobility and invasion ability by promoting metalloproteinase to capture and digest extracellular matrix (ECM)[68]. In addition, Kornek et al.[69] demonstrated that EMMPRIN released by circulating T cell-derived MVs accelerated hepatic stellate cells to transform into a fibrolytic phenotype, which promotes ECM degradation.
Conversely, MVs are able to stimulate the receptor cells to complete signal transduction through their surface ligands. It has been confirmed that MVs derived from platelet arouse the intra-hemopoietic signal cascade by ligands such as CD40L/PF-4 and thus cause the proliferation and survival of hematopoietic cells[70]. Simultaneously, the adhesion molecules could be transferred to hematopoietic cells via MVs to enhance their adhesion to fibrinogen or endothelium[70]. Proteomic and transcriptomic studies have shown that MVs are natural vectors for transferring bioactive molecules, including protein, mRNA and miRNA, between cells[71], effectively facilitating intercellular communication[72,73]. In addition, MVs hold natural stability in the blood, low immunogenicity and special homing ability to specific organs or tissues[74]. Evidence shows that vesicle-associated integrins α6β4 and α6β1 are correlated with lung metastasis while αVβ5 with liver metastasis, which indicates the various origins of MVs encoded with different tags for the corresponding cells or organ targeting[74]. Moreover, it has also been demonstrated that MVs could possibly transport even complete organelles (e.g., mitochondria) to target cells[75], which declares its potential to act as a promising novel drug carrier system.
Several studies revealed that TMVs transport cancerous molecules to recipient cells, which eventually contribute to tumor progression and metastasis[11-14,76] [Figure 2]. Interestingly, the transport of anti-cancer drugs out of ovarian cancer cells through their secreted EVs supported the role of EVs in cancer progression and treatment. Conversely, the observation of more sorafenib resistance in HCC tumors that were treated with tumor cell-derived MVs added evidence to the noticeable link between MVs and sorafenib resistance[15,78].
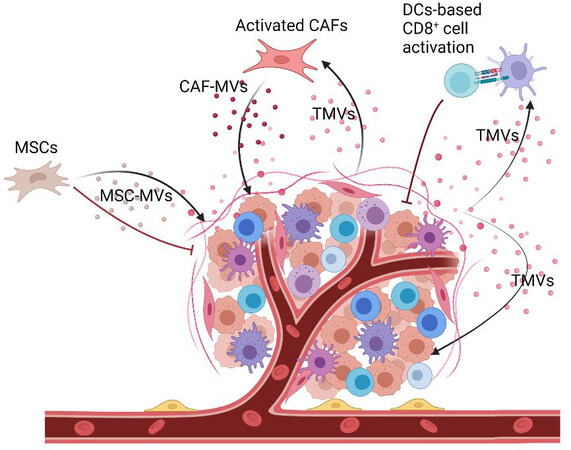
Figure 2. The bidirectional role of TMVs in tumor development. TMVs derived from tumor cells could promote tumor progression via transferring cancerous molecules[13,62]. Simultaneously, they act as a functional regulator to modulate DC cells based on CD8+ T cell activation, which provokes cytotoxic T cell infiltration and inhibits tumor development[77]. Created with BioRender.com. TMVs: Tumor-derived microvesicles.
THE DYNAMIC IMPACT OF MVS ON SORAFENIB RESISTANCE
MV-mediated sorafenib resistance
The physiological status of parental cells decides the composition of their secreted MVs, having direct or indirect effects on the uptake of MVs by recipient cells. Tumor cells that undergo hypoxic stress and obtain stemness or mesenchymal state for survival[57,58] promote tumor progression and therapy resistance. By inducing hypoxic stress, the hypoxia-inducible factor-1 α could stimulate the release of MVs along with the modulation of its packed cargoes[79]. Similarly, Wang et al.[80] reported that chemotherapeutic agents could enhance the secretion of ABCB-1-enriched EVs, which promote resistant phenotype transformation in recipient cells. Specifically, growth and pro-angiogenic factors transferred between CSCs and vascular niches via vesicles under hypoxia were witnessed[81] to limit the therapeutic effect of sorafenib.
Cancer-associated fibroblasts (CAFs), one of the main stroma cells, advance the self-renewal feature of CSCs in HCC and thus could induce sorafenib resistance by secretion of hepatocyte growth factor (HGF)[82,83]. A reduction in cancer cell stemness was recorded by inhibiting the paracrine behavior of CAFs[84]. Conversely, TMVs have been shown to activate CAFs to improve the mobility of tumor cells, while the activated fibroblasts could secret MVs and in turn facilitate tumor progression[12,85]. Moreover, small GTPases and RHO-associated protein kinase, key factors in the biogenesis of MVs[73], were found to be highly expressed in both CAFs and cancer cells[86], which indicates the possible role of CAFs in the alteration of MVs secretion.
As the messengers and mediators between cells, MVs could regulate the sorafenib sensitivity in the recipient cells via their cargoes [Table 1]. For example, miRNAs involved in the regulation of multiple mRNA targets in recipient cells were found to be enriched in TMVs[94-97]. In recent years, our group also found that HCC cell-derived MVs could increase sorafenib drug resistance by inducing FOXM1 expression via miR-25 transferred to the recipient liver cancer cells from the parent cells both in vitro and in vivo[15]. Similarly, long non-coding RNA (lnc-ROR and lnc-VLDLR)-enriched HCC cells-secreted vesicles (especially after sorafenib exposure) were proved to reduce apoptosis induced by chemotherapy[88,89]. However, MVs released from modified adipose tissue-derived MSCs have been shown to carry miR-199a/miR-122 and have the ability to improve chemosensitivity in HCC[90,91]. Elevated miR-214 in human cerebral endothelial cell-released vesicles was noted to enhance the anti-tumor efficacy of sorafenib[92] [Figure 3]. However, complete genomic and proteomic analyses of preclinical MVs cargoes need to be performed in the future.
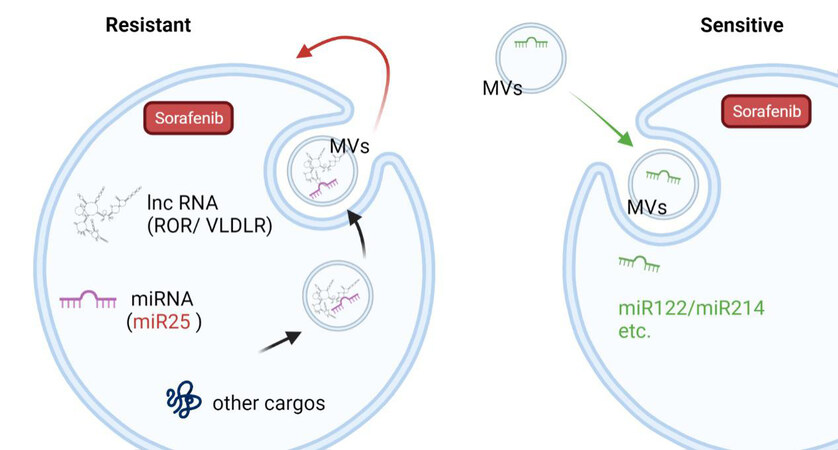
Figure 3. The dynamic role of MVs in sorafenib resistance modulation. MVs transfer biomolecules including miRNAs and lncRNAs to recipient cells and regulate their sensitivity to sorafenib. Created with BioRender.com. MVs: Microvesicles.
Key molecules transferred by MVs modulating sorafenib resistance
| Key molecules | MVs sources | Function | Ref. |
| miR-25 | HCC cells | Increase sorafenib resistance | [15] |
| miR-494-3p | GOLPH3 overexpressed HCC cells | Promote the angiogenesis ability of HUVECs and induce sorafenib resistance in HCC cells | [87] |
| lnc-ROR | HCC cells | Reduce chemotherapy-induced cell death | [88] |
| lnc-VLDLR | HCC cells | Reduce chemotherapy-induced cell death | [89] |
| miR-199a | Adipose tissue derived MSCs | Increase chemosensitivity in HCC | |
| miR-122 | Adipose tissue derived MSCs | Increase chemosensitivity in HCC | [91] |
| miR-214 | Human cerebral endothelial cells | Sensitize HCC cells to sorafenib treatment | [92] |
| siGRP78 | Modified bone-marrow-derived mesenchymal stem cells | Suppress sorafenib resistance | [93] |
Targeting MVs to reverse sorafenib resistance
Recently, the promising outcome of targeted gene therapy has motivated more researchers to meet sorafenib resistance in HCC. A lower expression of miR-34 has been found to have a direct relationship with patient survival, while the restoration of the miR-34a expression level in HCC cells has been noted to significantly downregulate the expression of BCL2 and enhance the cells’ sensitivity towards sorafenib-induced apoptosis and toxicity[7]. Dai et al.[98] found that BikDD gene therapy combined with low-dose sorafenib could enhance the anti-tumor efficacy of sorafenib and improve the survival rate of tumor-bearing mice. Furthermore, the simultaneous release of sorafenib and USP22 shRNA (shUSP22) by galactose-modified lipopolysaccharide revealed that the encapsulated sorafenib along with shUSP22 could obtain a synergistic anti-proliferative effect in HCC cells by inducing reactive oxygen cascade to promote the release of shUSP22 and inhibit the expression of USP22 in HCC cells, which promoted the accumulation of sorafenib by downregulating the expression level of multidrug resistance-related proteins[99].
Several emerging studies reported utilizing modified EVs to overcome chemoresistance during cancer treatments. As a naturally secreted nanoparticle, MVs exhibited excellent specific tissue homing ability and acted as good functional carriers for various therapeutic molecules[15,74,100]. To achieve bioactive and continuous drug containing MVs, the pre-loading strategy has been applied to modify donor cells. This strategy includes incorporating cargo into donor cells, so the donor cells could encapsulate the cargo during secretion production. Both biologically produced molecules (proteins and nucleic acids) and synthetic chemicals can be encapsulated into secreted vesicles[2]. By combined regulation of Akt/mTOR/PTEN, EVs secreted from human liver stem cells were proved to upgrade CSCs’ sensitivity to sorafenib[101]. Lou et al.[91] stated that treating HCCs with exosomes derived from miR-122 overexpressed AMSCs rendered HCCs more responsive to sorafenib. In the same way, miR-744-enriched exosomes have been demonstrated to be a potential tool for reducing sorafenib resistance[102]. Apart from utilizing the intrinsic homing ability, controllable targeting methods, including genetic engineering (pre-targeting)[103] and conjugation of ligands (post-targeting)[104], have also been developed for further clinical research. Engineering the donor cells by inserting sequences that encode desired targeting protein makes the affibody express on the surface of MVs[105]. In this way, our group also recently recorded that CRISPR system-carried engineered MVs derived from HEK293 cells could precisely disrupt IQGAP1, which is involved in PI3K/Akt signaling, and achieved an enhanced synergistic anti-cancer effect when combined with sorafenib treatment[106].
CONCLUSION
Sorafenib resistance remains challenging in HCC treatment, while targeting the suspicious genes involved in the resistance mechanism to shut off the pathological cycle provides a new chance for advancing the cancer treatment as well as the existing anti-cancer drug[107]. For instance, by delivering sorafenib and CRISPR system via nanoparticles, Zhang et al.[108] achieved effective modification of EGFR, following synergistic inhibition of angiogenesis and tumor cell proliferation. Even though emerging synthetic nanomaterials have shown great abilities as therapeutic vectors, their long-term safety remains uncertain.
MVs, cell-derived natural carriers, are advanced promising gene therapy vehicles that could specifically deliver therapeutic bioactive molecules[109]. Platelet-derived MVs have been reported to hold good biocompatibility to target leukemia cells naturally and thus have been utilized as targeted delivery vehicles for multiple drugs in leukemia treatment[110]. Macrophage-derived MVs have been noted to have the ability to transport cargoes specifically to hematopoietic stem and progenitor cells (HSPCs) both in vivo and in vitro. In addition, Kao et al.[100] showed that plasmids and small RNAs (miRNA and siRNA) that were encapsulated by macrophage-derived MVs exhibit successful modification of heat shock protein in the recipient HSPCs. In addition, MVs were used to transport engineered minicircle DNA by researchers to achieve good gene-mediated prodrug transformation and effectively promote tumor cell death in breast cancer cells as well as mouse models[111]. However, the unexpected combination, modification, and dissociation of the above-mentioned therapeutic nucleic acids are the main concerns in gene manipulation techniques, in which the off-target effect needs to be well controlled in future clinical trials.
Considering the challenges of sorafenib resistance and MVs as the functional regulator in cancer microenvironment, in this review, we discuss the potential role of MVs in sorafenib resistance. On the one hand, in cancer progression, MVs transport bioactive molecules to participate in cell-to-cell communication, making the microenvironment favor tumor growth, which could facilitate the tumor cells to be resistant to sorafenib treatment. On the other hand, therapeutic molecules could be transferred specifically to tumor cells to alleviate the sorafenib resistance via MVs. Thus, a better understanding of these tiny players’ role in tumor microenvironment and cancer progression is necessary to appropriately use this double-edged sword for precise anti-cancer therapies in the future.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: He C, Jaffar Ali D
Provided overview: Sun B, Sun BC, Xiao ZD
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the National Natural Science Foundation of China (No. 81671807), the Key Research & Development Program of Jiangsu Province (BE2020777) and Fundamental Research Funds for the Central Universities (2242018K3DN05) to Z.D.X.; Fundamental Research Funds for the Central Universities (2242021K10004) to B.S.; and the Jiangsu Postdoctoral Research Foundation(1601001C) and Fundamental Research Funds for the Central Universities (2242016R20017) to J.A.D.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835-44.
3. Chang YS, Adnane J, Trail PA, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol 2007;59:561-74.
4. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64:7099-109.
5. Chen KF, Chen HL, Tai WT, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther 2011;337:155-61.
6. Li Y, Chen G, Han Z, Cheng H, Qiao L, Li Y. IL-6/STAT3 Signaling contributes to sorafenib resistance in hepatocellular carcinoma through targeting cancer stem cells. Onco Targets Ther 2020;13:9721-30.
7. Yang F, Li QJ, Gong ZB, et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol Cancer Res Treat 2014;13:77-86.
8. Zhu YJ, Zheng B, Wang HY, et al. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin 2017;38:614-22.
9. Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett 2015;367:1-11.
10. Lemoinne S, Thabut D, Housset C, et al. The emerging roles of microvesicles in liver diseases. Nat Rev Gastroenterol Hepatol 2014;11:350-61.
11. Camussi G, C. Deregibus M, Tetta C. Tumor-derived microvesicles and the cancer microenvironment. Curr Mol Med 2012;13:58-67.
12. Clancy JW, Tricarico CJ, D’Souza-Schorey C. Tumor-derived microvesicles in the tumor microenvironment: how vesicle heterogeneity can shape the future of a rapidly expanding field. Bioessays 2015;37:1309-16.
13. Balaj L, Lessard R, Dai L, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun 2011;2:180.
14. D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev 2012;26:1287-99.
15. Jaffar Ali D, He C, Xu H, et al. Microvesicles mediate sorafenib resistance in liver cancer cells through attenuating p53 and enhancing FOXM1 expression. Life Sci 2021;271:119149.
16. Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis 2007;27:55-76.
17. Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006;130:1117-28.
18. Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006;66:11851-8.
19. Llovet JM, Ricci S, Mazzaferro V, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
20. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006;24:4293-300.
21. Awada A, Hendlisz A, Gil T, et al. Phase I safety and pharmacokinetics of BAY 43-9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br J Cancer 2005;92:1855-61.
22. Ratain MJ, Eisen T, Stadler WM, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:2505-12.
23. Ziogas IA, Tsoulfas G. Ziogas IA, Tsoulfas G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J Clin Oncol 2017;8:203-13.
24. Tang DP, Lin RJ, Wang XJ. Sorafenib plus capecitabine of patients with advanced hepatocellular carcinoma. China Pharm 2008;19:848-9.
25. Tang W, Chen Z, Zhang W, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther 2020;5:87.
26. Xia S, Pan Y, Liang Y, Xu J, Cai X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2020;51:102610.
27. Ito Y, Takeda T, Sakon M, et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer 2001;84:1377-83.
28. Zhang Z, Zhou X, Shen H, Wang D, Wang Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: evidence from an in vitro study. BMC Med 2009;7:41.
29. Blivet-Van Eggelpoël MJ, Chettouh H, Fartoux L, et al. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol 2012;57:108-15.
30. Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014;13:1589-98.
31. Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005;16:797-803.
32. Wu B, Li A, Zhang Y, et al. Resistance of hepatocellular carcinoma to sorafenib can be overcome with co-delivery of PI3K/mTOR inhibitor BEZ235 and sorafenib in nanoparticles. Expert Opin Drug Deliv 2020;17:573-87.
33. Zhang H, Wang Q, Liu J, Cao H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol Lett 2018;15:9377-84.
34. Chen KF, Tai WT, Hsu CY, et al. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur J Med Chem 2012;55:220-7.
35. Miranda A, Hamilton PT, Zhang AW, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci USA 2019;116:9020-9.
36. Lo RC, Leung CO, Chan KK, et al. Cripto-1 contributes to stemness in hepatocellular carcinoma by stabilizing Dishevelled-3 and activating Wnt/β-catenin pathway. Cell Death Differ 2018;25:1426-41.
37. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008;27:1749-58.
38. Xin HW, Ambe CM, Hari DM, et al. Label-retaining liver cancer cells are relatively resistant to sorafenib. Gut 2013;62:1777-86.
39. Sukowati CH, Rosso N, Crocè LS, Tiribelli C. Hepatic cancer stem cells and drug resistance: Relevance in targeted therapies for hepatocellular carcinoma. World J Hepatol 2010;2:114-26.
40. Fung SW, Cheung PF, Yip CW, et al. The ATP-binding cassette transporter ABCF1 is a hepatic oncofetal protein that promotes chemoresistance, EMT and cancer stemness in hepatocellular carcinoma. Cancer Lett 2019;457:98-109.
41. Yang Y, Qiu JG, Wei MN, et al. Targeting ABCB1-mediated tumor multidrug resistance by CRISPR/Cas9-based genome editing. Am J Transl Res 2016;8:3986-94.
42. Cheng CC, Chao WT, Liao CC, et al. The roles of angiogenesis and cancer stem cells in sorafenib drug resistance in hepatocellular carcinoma. Onco Targets Ther 2019;12:8217-27.
43. Nio K, Yamashita T, Kaneko S. The evolving concept of liver cancer stem cells. Mol Cancer 2017;16:4.
44. Wang N, Wang S, Li MY, et al. Cancer stem cells in hepatocellular carcinoma: an overview and promising therapeutic strategies. Ther Adv Med Oncol 2018;10:1758835918816287.
45. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445-64.
46. Tang Y, Berlind J, Mavila N. Inhibition of CREB binding protein-beta-catenin signaling down regulates CD133 expression and activates PP2A-PTEN signaling in tumor initiating liver cancer cells. Cell Commun Signal 2018;16:9.
47. Tang J, Tao ZH, Wen D, et al. MiR-612 suppresses the stemness of liver cancer via Wnt/β-catenin signaling. Biochem Biophys Res Commun 2014;447:210-5.
48. Tlemsani C, Huillard O, Arrondeau J, et al. Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin Drug Metab Toxicol 2015;11:785-94.
49. Zimmerman EI, Roberts JL, Li L, et al. Ontogeny and sorafenib metabolism. Clin Cancer Res 2012;18:5788-95.
50. Edginton AN, Zimmerman EI, Vasilyeva A, Baker SD, Panetta JC. Sorafenib metabolism, transport, and enterohepatic recycling: physiologically based modeling and simulation in mice. Cancer Chemother Pharmacol 2016;77:1039-52.
51. Gerth K, Kodidela S, Mahon M, Haque S, Verma N, Kumar S. Circulating extracellular vesicles containing xenobiotic metabolizing CYP enzymes and their potential roles in extrahepatic cells via cell-cell interactions. Int J Mol Sci 2019;20:6178.
52. Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis 2017;21:1-20.
53. Alonso-peña M, Sanchez-martin A, Sanchon-sanchez P, Soto-muñiz M, Espinosa-escudero R, Marin JJ. Pharmacogenetics of hepatocellular carcinoma and cholangiocarcinoma. Cancer Drug Resist 2019;2:680-709.
54. Liu JE, Ren B, Tang L, et al. The independent contribution of miRNAs to the missing heritability in CYP3A4/5 functionality and the metabolism of atorvastatin. Sci Rep 2016;6:26544.
55. Karbownik A, Szkutnik-Fiedler D, Grabowski T, et al. Pharmacokinetic drug interaction study of Sorafenib and morphine in rats. Pharmaceutics 2021;13:2172.
56. Miners JO, Chau N, Rowland A, et al. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: Implications for hyperbilirubinemia. Biochem Pharmacol 2017;129:85-95.
57. Cui CP, Wong CC, Kai AK, et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017;66:2149-59.
58. Won C, Kim BH, Yi EH, et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015;62:1160-73.
59. Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500-4.
60. Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 2010;123:1603-11.
61. Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. Biomed Res Int 2018;2018:8545347.
62. Bian X, Xiao YT, Wu T, et al. Microvesicles and chemokines in tumor microenvironment: mediators of intercellular communications in tumor progression. Mol Cancer 2019;18:50.
63. Hugel B, Martínez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005;20:22-7.
64. Sedgwick AE, D’Souza-Schorey C. The biology of extracellular microvesicles. Traffic 2018;19:319-27.
65. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol 2009;19:43-51.
66. Schiera G, Proia P, Alberti C, Mineo M, Savettieri G, Di Liegro I. Neurons produce FGF2 and VEGF and secrete them at least in part by shedding extracellular vesicles. J Cell Mol Med 2007;11:1384-94.
67. Pizzirani C, Ferrari D, Chiozzi P, et al. Stimulation of P2 receptors causes release of IL-1beta-loaded microvesicles from human dendritic cells. Blood 2007;109:3856-64.
68. Sidhu SS, Mengistab AT, Tauscher AN, LaVail J, Basbaum C. The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions. Oncogene 2004;23:956-63.
69. Kornek M, Popov Y, Libermann TA, Afdhal NH, Schuppan D. Human T cell microparticles circulate in blood of hepatitis patients and induce fibrolytic activation of hepatic stellate cells. Hepatology 2011;53:230-42.
70. Baj-krzyworzeka M, Majka M, Pratico D, et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp Hematol 2002;30:450-9.
71. van Dommelen SM, Vader P, Lakhal S, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release 2012;161:635-44.
72. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013;200:373-83.
73. Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018;19:213-28.
74. Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329-35.
75. Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006;20:1487-95.
76. Wu S, Luo M, To KKW, et al. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol Cancer 2021;20:17.
77. Dionisi M, De Archangelis C, Battisti F, et al. Tumor-derived microvesicles enhance cross-processing ability of clinical grade dendritic cells. Front Immunol 2018;9:2481.
78. Qu Z, Wu J, Wu J, Luo D, Jiang C, Ding Y. Exosomes derived from HCC cells induce sorafenib resistance in hepatocellular carcinoma both in vivo and in vitro. J Exp Clin Cancer Res 2016;35:159.
79. Patton MC, Zubair H, Khan MA, Singh S, Singh AP. Hypoxia alters the release and size distribution of extracellular vesicles in pancreatic cancer cells to support their adaptive survival. J Cell Biochem 2020;121:828-39.
80. Wang X, Qiao D, Chen L, et al. Chemotherapeutic drugs stimulate the release and recycling of extracellular vesicles to assist cancer cells in developing an urgent chemoresistance. Mol Cancer 2019;18:182.
81. Yao H, Liu N, Lin MC, Zheng J. Positive feedback loop between cancer stem cells and angiogenesis in hepatocellular carcinoma. Cancer Lett 2016;379:213-9.
82. Lau EY, Lo J, Cheng BY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep 2016;15:1175-89.
83. Li Y, Li S, Wang J, Liu G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol 2019;37:730-43.
84. Sun L, Wang Y, Wang L, et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J Exp Clin Cancer Res 2019;38:170.
85. Zhang W, Zhao P, Xu XL, et al. Annexin A2 promotes the migration and invasion of human hepatocellular carcinoma cells in vitro by regulating the shedding of CD147-harboring microvesicles from tumor cells. PLoS One 2013;8:e67268.
86. Whatcott CJ, Ng S, Barrett MT, Hostetter G, Von Hoff DD, Han H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS One 2017;12:e0183871.
87. Gao Y, Yin Z, Qi Y, et al. Golgi phosphoprotein 3 promotes angiogenesis and sorafenib resistance in hepatocellular carcinoma via upregulating exosomal miR-494-3p. Cancer Cell Int 2022;22:35.
88. Takahashi K, Yan IK, Kogure T, Haga H, Patel T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio 2014;4:458-67.
89. Takahashi K, Yan IK, Wood J, Haga H, Patel T. Involvement of extracellular vesicle long noncoding RNA (linc-VLDLR) in tumor cell responses to chemotherapy. Mol Cancer Res 2014;12:1377-87.
90. Lou G, Chen L, Xia C, et al. MiR-199a-modified exosomes from adipose tissue-derived mesenchymal stem cells improve hepatocellular carcinoma chemosensitivity through mTOR pathway. J Exp Clin Cancer Res 2020;39:4.
91. Lou G, Song X, Yang F, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol 2015;8:122.
92. Semaan L, Zeng Q, Lu Y, et al. MicroRNA-214 enriched exosomes from human cerebral endothelial cells (hCEC) sensitize hepatocellular carcinoma to anti-cancer drugs. Oncotarget 2021;12:185-98.
93. Li H, Yang C, Shi Y, Zhao L. Exosomes derived from siRNA against GRP78 modified bone-marrow-derived mesenchymal stem cells suppress Sorafenib resistance in hepatocellular carcinoma. J Nanobiotechnology 2018;16:103.
94. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009;10:704-14.
95. Rabinowits G, Gerçel-Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung Cancer 2009;10:42-6.
96. Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008;10:1470-6.
97. Ohshima K, Inoue K, Fujiwara A, et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS One 2010;5:e13247.
98. Dai HY, Chen HY, Lai WC, Hung MC, Li LY. Targeted expression of BikDD combined with metronomic doxorubicin induces synergistic antitumor effect through Bax activation in hepatocellular carcinoma. Oncotarget 2015;6:23807-19.
99. Xu S, Ling S, Shan Q, et al. Self-activated cascade-responsive Sorafenib and USP22 shRNA co-delivery system for synergetic hepatocellular carcinoma therapy. Adv Sci (Weinh) 2021;8:2003042.
100. Kao CY, Papoutsakis ET. Engineering human megakaryocytic microparticles for targeted delivery of nucleic acids to hematopoietic stem and progenitor cells. Sci Adv 2018;4:eaau6762.
101. Fonsato V, De Lena M, Tritta S, et al. Human liver stem cell-derived extracellular vesicles enhance cancer stem cell sensitivity to tyrosine kinase inhibitors through Akt/mTOR/PTEN combined modulation. Oncotarget 2018;9:36151-65.
102. Wang G, Zhao W, Wang H, et al. Exosomal MiR-744 Inhibits proliferation and Sorafenib chemoresistance in hepatocellular carcinoma by targeting PAX2. Med Sci Monit 2019;25:7209-17.
103. Cooper JM, Wiklander PB, Nordin JZ, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord 2014;29:1476-85.
104. Kooijmans SA, Aleza CG, Roffler SR, van Solinge WW, Vader P, Schiffelers RM. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J Extracell Vesicles 2016;5:31053.
105. Yoshioka Y, Ochiya T. Extracellular vesicles as novel nanocarriers for therapeutic delivery. Nucleic Acid Nanotheranostics 2019:391-407.
106. He C, Jaffar Ali D, Xu H, et al. Epithelial cell -derived microvesicles: A safe delivery platform of CRISPR/Cas9 conferring synergistic anti-tumor effect with sorafenib. Exp Cell Res 2020;392:112040.
107. Wang X, Tai Z, Zhang W, Gao S. Current status of gene therapy for hepatocellular carcinoma, with a focus on gene delivery approaches. Curr Gene Ther 2015;15:120-41.
108. Zhang BC, Luo BY, Zou JJ, et al. Co-delivery of Sorafenib and CRISPR/Cas9 based on targeted core-shell hollow mesoporous organosilica nanoparticles for synergistic HCC therapy. ACS Appl Mater Interfaces 2020;12:57362-72.
109. Burnouf T, Agrahari V, Agrahari V. Extracellular vesicles as nanomedicine: hopes and hurdles in clinical translation. Int J Nanomedicine 2019;14:8847-59.
110. Kailashiya J, Gupta V, Dash D. Engineered human platelet-derived microparticles as natural vectors for targeted drug delivery. Oncotarget 2019;10:5835-46.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
He C, Jaffar Ali D, Sun B, Sun BC, Xiao ZD. Microvesicles: the functional mediators in sorafenib resistance. Cancer Drug Resist 2022;5:749-61. http://dx.doi.org/10.20517/cdr.2021.137
AMA Style
He C, Jaffar Ali D, Sun B, Sun BC, Xiao ZD. Microvesicles: the functional mediators in sorafenib resistance. Cancer Drug Resistance. 2022; 5(3): 749-61. http://dx.doi.org/10.20517/cdr.2021.137
Chicago/Turabian Style
He, Cong, Doulathunnisa Jaffar Ali, Bo Sun, Bei-Cheng Sun, Zhong-Dang Xiao. 2022. "Microvesicles: the functional mediators in sorafenib resistance" Cancer Drug Resistance. 5, no.3: 749-61. http://dx.doi.org/10.20517/cdr.2021.137
ACS Style
He, C.; Jaffar Ali D.; Sun B.; Sun B.C.; Xiao Z.D. Microvesicles: the functional mediators in sorafenib resistance. Cancer Drug Resist. 2022, 5, 749-61. http://dx.doi.org/10.20517/cdr.2021.137
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 22
likes
Like This Article 22
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.