
Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo

Abstract
Prostate cancer (PCa) is a leading cause of cancer-related morbidity and mortality in men globally. Despite improvements in the diagnosis and treatment of PCa, a significant proportion of patients with high-risk localized disease and all patients with advanced disease at diagnosis will experience progression to metastatic castration-resistant prostate cancer (mCRPC). Multiple drugs are now approved as the standard of care treatments for patients with mCRPC that have been shown to prolong survival. Although the majority of patients will respond initially, primary and secondary resistance to these therapies make mCRPC an incurable disease. Several molecular mechanisms underlie the development of mCRPC, with the androgen receptor (AR) axis being the main driver as well as the key drug target. Understanding resistance mechanisms is crucial for discovering novel therapeutic strategies to delay or reverse the progression of the disease. In this review, we address the diverse mechanisms of drug resistance in mCRPC. In addition, we shed light on emerging targeted therapies currently being tested in clinical trials with promising potential to overcome mCRPC-drug resistance.
Keywords
INTRODUCTION
Prostate cancer (PCa) is a leading cause of mortality and morbidity among men worldwide, where 12.5% of men are prone to be diagnosed with PCa during their lifetime[1]. According to the Globocan data collected from 185 countries in 2020, an estimated 1.4 million new PCa cases were diagnosed, with an incidence rate of 37.5 per 100,000 males, accounting for 14.1% of all male cancer diagnoses. In addition, 375,000 deaths due to PCa were recorded, comprising 6.8% of cancer deaths in men[2]. PCa was discovered to be androgen-sensitive by Huggins and Hodges in 1941[3]. The activation of androgen receptor (AR)-mediated signaling is an essential route for regulating PCa cell growth and proliferation; thereby, alterations in this cascade hold a great influence on cancer progression[4]. This relation between androgen and PCa leads to androgen deprivation treatment (ADT), which includes surgical castration through orchiectomy to remove the source of androgen or medical castration through antiandrogen and gonadotropin-releasing hormone analogs, thus reducing androgens to castrate levels[3]. Interventions such as prostatectomy or radiotherapy along with ADT for localized PCa are the standard of care for localized disease, yet ADT remains the backbone of first-line treatment for locally advanced and metastatic PCa[5]. For patients treated with long-term gonadal androgen suppression, PCa cells become resistant to ADT within 2-3 years and the malignancy progresses even when the serum testosterone level is below the castrate level. This stage of prostate cancer, known as castration-resistant prostate cancer (CRPC), can progress to the lethal form of metastatic CRPC (mCRPC), resulting in rapid mortality with mean overall survival historically of 16-18 months[6].
Although curative treatment has not yet been reached, multiple therapeutic modalities for mCRPC patients have been accomplished[7-16] [Table 1].
Summary table of the drugs and agents indicated for mCRPC
| Class | Agent | MOA | Studies enrolled with indications | Trial result |
| Hormonal therapy | Abiraterone Acetate | Irreversible selective inhibitor of CYP17-alpha-hydroxylase and C17, 20-lyase coupled with a modest AR antagonist activity[7] | COU-AA-301; post-docetaxel[8] | ABI increased median overall survival when compared to the placebo group by 3.9 months |
| COU-AA-302; pre-docetaxel[9] | ABI significantly increased median overall survival when compared to the placebo group by 8.2 months | |||
| AR antagonists | Enzalutamide | AR antagonist impedes nuclear receptor translocation and DNA binding, induces apoptosis | AFFIRM; post-docetaxel[10] | Overall survival increased by 4.8 months in comparison to the control group |
| Darolutamide | ARCADES[11] | 86% of patients treated with 1400 mg dose of darolutamide had a 50% or greater decrease in PSA | ||
| Cytotoxic chemotherapy | Docetaxel | Inhibits microtubular depolymerization arresting their function | TAX 327; first-line chemotherapy[12] | DOC combined with prednisone increased overall survival by 2.9 months when compared to the control group treated with mitoxantrone plus PDN |
| Cabazitaxel | TROPIC, Phase III, randomized, open-label[13] | Overall survival increased by 2.4 months in the treatment group (CBZ+ PDN) compared to patients treated with a combination of PDN and mitoxantrone | ||
| Calcium-mimetic | Ra-223 | Emits high energy alpha particles after adhering selectively to regions of elevated bone turnover | ALSYMPCA, Phase III, randomized[14] | Patients placed on Ra-223 treatment had 3.6 months increase in survival compared to the placebo group |
| PARP inhibitor | Olaparib | Impede PARP causing cumulative DNA and cell damage | PROfound Phase III, randomized[15] | Patients with mutations in BRCA1, BRCA2, or ATM genes assigned to receive olaparib experienced a 4.4-month increase in survival compared to the control arm receiving ENZ or ABI plus prednisone |
| Dendritic-cell vaccine | Sipuleucel-T | Infused within APCs to present PAP peptides in vivo activating CD4+ and CD8+ T cells | IMPACT trial, Phase III[16] | Utilizing sipuleucel-T extended overall survival among men with mCRPC by 10 months |
Inhibitors of CYP17
The emerging speculative conveyance regarding the defiance of castration resistance classes to androgen manipulating therapies has been fully debunked recently, thus reemphasizing the chief role of the androgen signaling axis as a subject to therapy development[17]. Cytochrome P450 enzyme 17 (CYP17) has been recognized as a vital mediator in steroidal biosynthesis, especially androgen. To impede this vicious pathway, several drugs, such as antifungal ketoconazole, were depicted as potential options; however, prohibitive toxicities paired with minute survival benefits raised the need for better alternatives[18]. Abiraterone (ABI) acetate functions as an irreversible selective steroidal agent for the microsomal complex CYP17, thus restraining two functionalities: cortisol biosynthesis in the adrenal gland (via C21 17-α-hydroxylation) and steroid production in testis and adrenal gland (lyase property)[19]. As a result, both testosterone supply and cortisol will be inhibited, causing a flux of augmented steroid levels upstream of the target halt. Therefore, prednisone (PDN), a corticosteroid that binds to the glucocorticoid receptor (GR), is often co-administered with ABI, seeking in return a diminished side effect for mineralocorticoid excess syndrome[20]. Significantly ameliorated radiographic progression-free survival (rPFS) and overall survival were reported in a large Phase III trial for asymptomatic CRPC patients treated with ABI (Abiraterone) and PDN compared to PDN alone (16.5 vs. 8.2 months; HR: 0.52; 95%CI: 0.45-0.61; P < 0.0001)[9]. Based on the data presented by such trials, the FDA approved ABI as a management therapy for CRPC patients. Subsequently, the drug has also been shown to improve survival in patients with hormone-sensitive metastatic disease and is also approved as first-line therapy in combination with ADT[21].
AR antagonists
The oral AR antagonist enzalutamide (ENZ) lashes with high affinity, impeding nuclear receptor translocation and DNA binding, thereby inducing apoptosis[22]. Based on the results of two Phase III placebo-controlled studies where a 4.8-month median overall survival benefit was reported in chemotherapy-naïve mCRPC patients, both the FDA and EMA ratified ENZ usage for mCRPC previously treated with docetaxel[23]. Notably, the steroidal biosynthesis impact was nullified with ENZ utilization, thus shifting away from PDN prescription during the treatment regimen[24].
Cytotoxic chemotherapy
By reversibly stabilizing the microtubules and promoting their assembly, docetaxel (DOC) fosters a mitotic block on cancerous cells. After augmentation of these bundles, the natural dynamics of mitosis would be impaired, consequently leading to apoptosis, which is further induced by phosphorylation of the Bcl-2 oncoprotein[25]. Furthermore, impaired microtubules functionality surges p53 buildup in the nucleus, thus driving its downstream signaling forward[26]. Notably, due to their impact on microtubules, it has been shown that taxanes also possess antiangiogenic capabilities paired with impeding AR trafficking and accumulation[27]. This drug was granted FDA and EMA approval based on the results of the TAX 327 study conducted on patients diagnosed with CRPC and subjected to DOC treatment paired with PDN compared to the standard approach of mitoxantrone and PDN in the control arm. The results demonstrate an overall increased survival benefit (19.2 months vs. 16.3 months; P < 0.004) reinforced with significant amelioration in pain relief[27]. The incorporation of PDN augments the efficacy of DOC and results in superior outcomes as a result of the additive benefit from the two drugs, each known to be effective in PCa through targeting different signaling, AR and GR. In addition, prednisone also has anti-inflammatory properties and can treat pain, nausea, and edema[28-30]. Moreover, the cytotoxic tubulin-binding drug cabazitaxel (CBZ) has demonstrated a prevailing advantage in enhancing the survival of patients with mCRPC after being assigned to a DOC-based treating regimen, causing a 30% decline in risk of death when compared to those treated with mitoxantrone (panel). These results granted CBZ US Food and Drug Administration approval for being prescribed as a second-line treatment in mCRPC[13].
DRUG RESISTANCE IN mCRPC
Despite the significant survival benefit of the currently approved therapies, which can alleviate symptoms and prolong overall survival, mCRPC remains incurable as primary and secondary resistance develops rapidly[31]. Drug resistance can develop due to mechanisms intrinsic to the biology of PCa or by more general mechanisms shared with diverse tumor types, as can be seen in Figure 1.
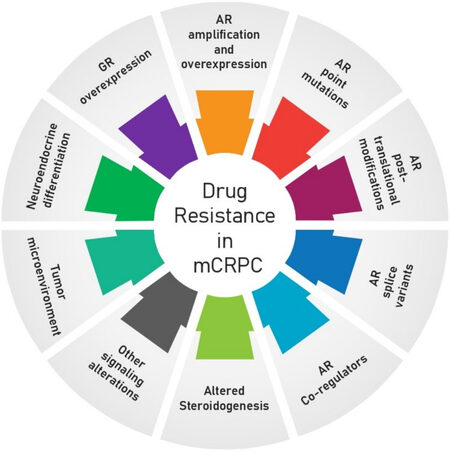
Figure 1. Mechanisms of drug resistance in mCRPC. Several mechanisms of drug resistance are well defined in CRPC, including AR amplification and overexpression, AR point mutations, AR post-translational modifications, AR splice variants, AR co-regulators, altered steroidogenesis, GR overexpression, neuroendocrine differentiation, tumor microenvironment, and other signaling alterations. AR: Androgen receptor; GR: glucocorticoid receptor; mCRPC: metastatic castration-resistance prostate cancer.
AR amplification and overexpression
AR gene amplification has been recognized in clinical studies as the most common genetic alteration deriving AR reactivation and progression to CRPC[32]. AR overexpression can result from different mechanisms such as gene amplification, increased histone acetylation/phosphorylation at enhancers sites, overexpression of co-regulators, or enhanced protein stability[33,34]. The increase in AR expression was found to be consistently linked with the development of resistance to ADT, resulting in PCa progression from a castration-sensitive phenotype to a castration-resistant one. This increase is capable of sensitizing PCa cells to castrate concentrations of androgen and converting the action of AR antagonists to agonists[24,35].
Among patients with CRPC, up to 80% exhibit a significant upregulation in AR transcripts and protein levels[36], in comparison to < 1% of the primary androgen-dependent PCa cases[37]. This aberration has been detected, at high frequency, in circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) and is more common in pretreated CRPC patients than in treatment-naïve patients. A study using liquid biopsies and ctDNA showed that 50% of CRPC patients, pretreated with either ENZ or orteronel (a CYP17A1 inhibitor), evidenced AR amplification[38,39]. One in vivo study utilizing CRPC xenografts treated with ABI showed a three-fold increase in AR expression[40]. In a separate in vitro study, a bicalutamide-resistant LNCaP cell line was shown to display an overexpressed AR and hyper-sensitivity to minimal levels of androgen[41]. In vitro studies using ENZ-resistant LNCaP cells showed an increase in AR levels compared to naïve LNCaP cells[42].
AR point mutations
AR point mutations are also more frequent in CRPC than in primary androgen-sensitive PCa, especially among patients pretreated with ADT. Several studies evidenced somatic AR point mutations in ~10% of CRPC tissues, whereas none were found in any of the primary PCa tissues examined[36,37,43]. This is consistent with results obtained in a previous study that performed a targeted AR sequencing in a cohort of 181 primary cancers and 37 CRPCs. The study revealed that somatic AR point mutations were found to occur only in CRPC and more frequently in patients subjected to prior antiandrogen treatments[44]. Supposedly, when AR signaling is more effectively suppressed, clonal selection of tumor cells will enhance AR somatic mutations, subsequently yielding aberrant transcriptomes[43]. There are > 100 clinically-relevant somatic point mutations detected in AR[44]; the majority are single-base substitutions and are clustered in the ligand-binding domain (LBD). The modifications in LBD alter the steroid-binding pocket and result in broadening ligand specificity and AR activation by alternative non-androgen ligands including progesterone, estrogen, and some AR antagonists[45].
Four main recurrent LBD point mutations are described in several studies: (1) L702H, a leucine to histidine substitution at amino acid 702; (2) W742C, a tryptophan to cysteine substitution at amino acid 742; (3) H875Y, a histidine to tyrosine substitution at amino acid 875; and (4) T878A, a threonine to alanine substitution at amino acid 878 (T878A). These mutations are present in approximately 15%-20% of CRPC cases[46,47]. The gain of function mutation “T878A” described in the LNCaP cell line was shown to confer an expansion of binding specificity to AR by both steroid hormones (e.g., progesterone) and first-generation antiandrogens (e.g., bicalutamide or flutamide)[48,49]. L702H, a mutation that causes glucocorticoid-mediated AR activation, is associated with primary resistance to ABI[50]. In another study, patients harboring T878A and L702H mutations showed poor prostate-specific antigen (PSA) response after ABI or ENZ treatment[38]. In addition, several AR mutations were shown to be capable of converting the AR antagonists into agonists, as seen with F877L and F876L mutations associated with resistance to second-generation antiandrogens ENZ and ARN-509[50,51]. In line with other studies, the data obtained validate the significance of AR mutations in deriving drug resistance.
AR post-translational modifications
AR post-translational modifications (PTMs), including serine/threonine and tyrosine phosphorylation, acetylation, methylation, ubiquitination, and sumoylation, have essential roles in enhancing AR activity through maintaining protein stability, nuclear localization, and transcriptional activity[52,53]. Thereby, PTMs may contribute to AR reactivation and acquisition of drug resistance in mCRPC[54]. A study indicated that the usage of hypomethylating drugs has the effect of reversing and delaying progression to mCRPC. One potential underlying mechanism is through downregulating DNA methyltransferase 1-dependent STAT3 activity[55]. Phosphorylation of AR residues plays a significant role in activating AR and promoting continued PCa cell growth. The treatment of the castrate-recurrent CWR-R1 cell line with EGF under low androgen conditions was shown to promote AR transcriptional activity through the phosphorylation of AR at serine 515 and serine 578, MAPK, and PKC consensus sites, respectively[56]. A prostate tissue microarray analysis indicated the intracellular non-receptor tyrosine kinase (NRTK) Ack1 to induce AR phosphorylation at Tyrosine 284 and correlate positively with disease progression and negatively with the survival of PCa patients. In activated Ack1-expressing prostate cells, treatment with antiandrogens failed to affect the expression and activation of AR[57]. In addition, the NTRK SRC proved clinically to be at high levels in mCRPC and to mediate AR tyrosine phosphorylation and subsequent pathway activation[58-61].
Ubiquitination is another PTM that was shown to have an important role in regulating AR activity. RNF6, a ubiquitin ligase with an AR enhancing role, acts by inducing AR ubiquitination and promoting AR transcriptional activity. RNF6 was detected t at elevated levels in hormone-refractory human PCa tissues[62]. Another study identified ubiquitin-specific protease 12 (Usp12) as a positive regulator of AR. Usp12, in complex with the cofactors Uaf-1 and WDR20, interacts with and deubiquitinates AR, resulting in increased protein stability and transcriptional activity of AR[63].
AR co-regulators: co-activators, co-repressors, and chromatin remodelers
A series of co-regulator protein factors such as co-activators, co-repressors, and chromatin remodelers are known to regulate AR transcriptional activity by being co-recruited to chromatin and binding directly or in a protein complex to AR to regulate gene activity. The balance of co-activator and co-repressors is required for the proper regulation of AR-mediated transcription. Misbalance in these factors leads to higher AR activity and less active antagonism by antiandrogens contributing to drug resistance and mCRPC progression[64,65]. FKBP51, a co-activator and a target gene of AR, was detected at upregulated levels in relapsed LAPC-4 tumors grown in castrated mice[66]. FKBP51 improved the formation of a super chaperone complex via recruiting p23 co-chaperone to ATP-bound Hsp90, which in line keeps AR with a high-affinity conformation for ligand-binding, consequently promoting androgen-mediated AR transcriptional activity and growth[66,67].
Elevation of AR signaling can also occur through the loss of AR regulatory repressing signals. For example, retinoblastoma (Rb), a well-known tumor suppressor, was shown to be highly reduced in mCRPC and to be associated with tumor recurrence. The loss or inactivation of Rb induced an E2F1-mediated increase in the levels of AR mRNA/protein and two relevant target genes, PSA and TMPRSS2, consequently promoting castrate-resistant growth and resistance to bicalutamide in PCa cells[68].
A recent study identified and verified myosin heavy chain 9 (MYH9) as a novel AR cofactor. The data show that inhibiting MYH9 in an androgen-independent cell line (LNCaP-AI) promoted AR nuclear translocation and enhanced the expression of PSA, indicative of the MYH9 role as a co-repressor to nuclear AR signaling and a novel factor mediating the progression of CRPC[69].
Other pioneering factors, i.e., FOXA1 and GATA2, act as chromatin remodelers at enhancer sites to regulate the expression and activity of AR in mCRPC. FOXA1 induces open chromatin conformation to allow the binding of other transcription factors, aiding in an enhanced AR transactivation and mCRPC progression[70]. CTCs isolated from patients with ABI- or ENZ-resistant metastasis showed amplification in the FOXA1 gene in > 30% of CRPC patients, pinpointing the crucial role of FOXA1 in AR regulation and tumor progression[71]. In addition, GATA2 was shown to boost AR activity and CRPC progression. Overexpression of GATA2 was correlated with more aggressiveness in PCa[72]. High expression of GATA2 in CRPC cell line “ARCaPM” brings about an increased IGF-2 expression and consequently confers chemotherapy resistance[73]. An additional important AR co-regulator and chromatin remodeling factor is homeobox B13 (HOXB13), which acts by interacting with AR and binding to its target loci[74]. Analysis of transcriptome expression from several databases revealed that the expression of HOXB13 is elevated during the progression of the primary PCa to CRPC[75]. In a separate study, HOXB13 was shown to be highly expressed in hormone-refractory tumors compared to tumors without PSA after initial treatment[76]. Interestingly, HOXB13 also mediates the oncogenic function of AR splice variants, mainly ARV7, and acts as a pivotal upstream regulator of AR-V7–driven transcriptomes[77]. These findings could explain HOXB13’s role in resistance towards antiandrogen treatment.
AR splice variants
Transcriptionally activated AR splice variants (ARVs) are truncated forms of AR that lack the C-terminal LBD region, the binding site for first- and second-generation drugs, hence remain constitutively activated independently of androgen or antagonist action. This ligand-independence feature makes ARVs potentially contributors to disease progression and treatment resistance in mCRPC. ARV1, ARV7, and ARV567 are the most common isoforms, while ARV7 is the most extensively studied one[78,79]. ARV7 is associated with resistance to both ABI and ENZ. Studying ARV7 expression in CTCs revealed its association with clinical resistance to both drugs in men receiving ENZ. Patients with detectable ARV7 levels had poorer PSA response, shorter progression-free survival (PFS), and reduced overall survival (OS) compared to ARV7-negative patients. Similar results were observed in patients treated with ABI. Markedly, the ARV7 level was higher in males pretreated with ENZ and ABI than in treatment-naïve males[80]. The selective siRNA-mediated knockdown of ARV7 in CWR22Rv1 cells inhibited androgen-independent growth and re-established the responsiveness to antiandrogen drugs[81]. In one study, ARV567es was identified and characterized as a contributor to PCa progression and resistance to castration in human PCa xenograft models and a biomarker for patients with early recurrence. In addition, AR-V567es transcripts detected in 23% of CRPC bone metastases were shown to be associated with poor prognosis and short survival[82]. These data strongly support the implication of ARVs as resistance-driving factors in CRPC.
Intra-tumoral and alternative androgen biosynthesis/altered steroidogenesis
Although CRPC develops in the presence of sub-physiological levels of circulating androgens, its progression is associated with the accumulation or synthesis of intra-tumoral androgen and accordingly maintained AR activity[83]. The low plasma levels of androgens following ADT, can be bypassed by the local conversion of adrenal androgens to testosterone and de novo local synthesis of androgens through increased expression of enzymes involved in steroidogenesis, such as CYP17A, in the prostatic tissue. The elevated intra-tumor level of androgens stimulates AR paracrine and autocrine activation, regardless of serum androgen levels[84,85].
The intra-tumor synthesis of steroid hormones was shown to be significantly increased in CRPC patients compared to naïve primary PCa. To compare the levels of androgen, a study utilized autopsies and tissue biopsies from hormone-sensitive and hormone-resistant patient tumors, and the latter was shown to possess high levels of continuous androgen production compared to primary tumors[83]. A primary source of these de novo intra-prostatic androgens are the adrenal androgens dehydroepiandrosterone (DHEA) and androstenedione (AD), generated by the action CYP17A1 enzyme. In the prostate, DHEA and AD can act on the AR axis once converted to testosterone and DHT. DHEA also exists predominantly in a sulfated form (DHEA-S). In CRPC, although ABI (CYP17 inhibitor) results in a significant decline of both DHEA and AD, the persistent DHEA-S can serve as a precursor for testosterone and DHT synthesis in prostate tissue, thus conferring resistance to ABI[86-88]. Studies in CRPC xenografts have shown that several genes involved in the androgen synthesis pathway are upregulated during hormonal therapy. The overexpression of AKR1C3, an enzyme implicated in the steroidogenesis pathway, drives resistance to ABI acetate via increasing androgen synthesis and signaling[89]. In line, downregulating AKR1C3 re-sensitizes resistant cells to ABI treatment. Consistently, AKR1C3 activation has been shown to be associated with ENZ resistance[90]. Other enzymes implicated in steroidogenesis, i.e., SRD5A1 and HSD3B2, were found to be overexpressed in samples from CRPC bone marrow metastases[91].
Glucocorticoid receptor overexpression
The role of glucocorticoids is complex in PCa because of their dual role. Despite that, it is shown that whenever adenosine diphosphate (ADP) is used to antagonize AR signaling, the GR is activated and overexpressed to confer resistance to the treatment used[92]. Both GR and AR have similar structures and mechanisms of action; thus, in mCRPC, whenever a treatment is used to block AR signaling[93], GR is usually activated to attach to nuclear AREs and activate genes that stimulate tumor progression and cell endurance[94]. A preclinical study showed that GR was overexpressed and conferred resistance in in vitro cell lines (VCaP and LNCaP/AR) that were treated with apalutamide and ENZ. However, it was shown that VCaP was desensitized to ENZ when GR was knocked out[93]. Another study also showed that CRPC was resistant to DOC when GR was overexpressed. In this study, GR antagonism was used to successfully re-sensitize the cells to DOC[95]. Puhr et al.[96] observed that GR is expressed minimally in primary PC tissue and the expression notably increases with long-term treatment with ENZ.
Neuroendocrine differentiation
While only about 1% of all primary PCa are diagnosed as neuroendocrine prostate cancer (NEPC), up to 30% of mCRPCs are NEPC[97]. This profound phenotypic shift, as a result of selection pressure from ADT or potent antiandrogens, from tumors with histological features of adenocarcinoma that express AR to AR-negative neuroendocrine prostate tumors has been termed lineage plasticity[98]. The loss or mutation of both tumor suppressors TP53 and RB1 has been found to be crucial in NEPC differentiation[99]. Tan et al.[100] observed Rb protein loss in 90% of NEPC with RB1 allelic loss in 85% of cases. A recent in vivo preclinical mouse study found that Rb1 loss promotes plasticity and metastasis in PCa with TP53, and tumor suppressor phosphatase and tensin homolog (Pten) loss causes secondary resistance to therapies that target the AR signal axis[101]. A different preclinical study of in vitro and in vivo models of human PCa showed evidence of lineage plasticity and a shift from androgen-dependent PCa to androgen-independent NEPC after treatment with ENZ[102]. This phenotypic shift was induced by the loss of RB1 and TP53 and was facilitated through overexpression of the transcription factor SOX2. It was also demonstrated that the inhibition of SOX2 restored the function of TP53 and RB1[102]. At the moment, there are no therapeutics in drug development studies that target the loss or mutations of TP53 or RB1. However, a Phase II study is currently ongoing to evaluate the potential use of alisertib (aurora kinase A inhibitor) in mCRPC and NEPC patients[103].
Tumor microenvironment
The ability of the tumor microenvironment to promote drug resistance is likely linked to the role of the cancer-associated stromal cells in tumor initiation and progression. Tumor microenvironment-derived neuregulin 1 (NRG1), identified in cancer-associated fibroblast (CAF) supernatant, was shown to induce resistance in tumor cells through activation of human epidermal growth factor receptor-3 (HER3) and the subsequent downstream signaling molecules such as MAPK to promote cell proliferation and survival[104]. Blocking the NRG1/HER3 axis was shown to re-sensitize tumors to hormone deprivation in vitro and in vivo. In addition, patients with mCRPC having increased tumor NRG1 activity showed an inferior response to second-generation antiandrogen therapy[105,106]. Zhang et al.[106] showed that ENZ drives in overproduction of NRG1. It is still unclear how AR inhibitors affect the production of NRG1, but many studies have shown that NRG1 overexpression is correlated with poor outcomes and reaction to ADT.
SPP1, an important extracellular matrix component secreted by multiple kinds of cell types including immune cells, fibroblasts, osteoclasts, smooth muscle, and epithelial cells, is overexpressed in many types of tumors[107,108]. In the GSE32269 dataset, SPP1 expression was shown to be significantly higher in the mCRPC group than in the primary PCa group. The Human Cancer Metastasis Database analysis showed that SPP1 expression levels were significantly higher in bone metastases than in lymph node and posterior peritoneum metastases, thus correlating the expression of SPP1 with the progression of mCRPC[109]. To investigate the relationship between SPP1 expressions and ENZ resistance in vivo, Pang et al.[108] studied the effect of ENZ treatment on SPP1 knock downed cells (22Rv1 cell line). The results show that SPP1 knockdown significantly inhibited 22Rv1 cell proliferation after ENZ treatment.
Other signaling alterations/implication of growth factors, kinases, cytokines, enzymes...
The aberrant activation of multiple signaling pathways plays a key role in deriving drug resistance in mCRPC. High levels of growth factors including IGF-1, EGF, and TGF-α/β have been reported in mCRPC[110]. A study showed that the overexpression and activation of EGFR mediate DOC resistance in CRPC by inducing AKT-dependent ABCB1 (MDR1) expression[111,112]. Besides, the stimulation of EGFR was shown to derive the activation of Ack1/Tnk2, which is known to correlate positively with the progression to the mCRPC stage[113]. In addition, PCa patients whose tumors showed moderate to strong staining of activated Ack1 displayed a poor prognosis[113].
Several studies assured the association of mCRPC with increased activation of the PI3K-AKT-mTOR pathway[114,115]. Phosphatase and Tensin Homolog (PTEN) has been attributed to the radical amelioration of the PI3K/AKT pathway in nearly 50% of PCa patients. This atypical augmentation of the latter pathway distorts the protein-serine/threonine kinases promoting castration-resistant growth. PI3K-AKT-mTOR pathway interacts and cooperates with several key oncogenic signaling cascades such as MAPK and WNT signaling to facilitate PCa growth and drug resistance[116]. Furthermore, augmented levels of ERKs activation have been reported in recurrent mCRPC patients, and correlative studies have further linked several mutations down the RAS isoforms with the tumorigeneses of PCa[117]. Although collateral activation of the PI3K/AKT and RAS/MAPK pathways have been implicated in several studies, cell proliferation and invasiveness of p63-expressing prostate tumors were solely attributed to the MAPK signaling pathway, which suggests its critical involvement in mCRPC[118].
Cytokines overexpression is also known to trigger tumor progression and drug resistance in CRPC[119,120]. A study reported that inhibiting STAT3, a downstream target of IL-6, results in decreased growth of ENZ-resistant cells. Consistently, blocking STAT3 signaling by inhibiting its phosphorylation was shown to re-sensitize ENZ-resistant cells, hence supporting the implication of the IL-6-STAT3 pathway in ENZ-acquired resistance[121]. Moreover, radically increased IL-6 expression is a direct consequence of the constitutive activation of the NF-κB pathway[122,123]. NF-κB levels influence the expression of an essential PCa biological marker for progression, PSA. Additionally, NF-κB is a key factor in the upregulation of IL-8 cytokine; the latter is involved in the regulation of prostate vasculature and apoptosis[124]. High levels of circulating IL-8 were detected in advanced PCa at a stage when the tumors no longer respond to antiandrogens[125,126]. PCa cells overexpressing IL-8 show reduced effectiveness of bicalutamide[124]. The pro-inflammatory cytokine TNF-α has also been reported to be elevated in CRPC, and such chronic stimulus is a prototypical inducer of NF-κB expression, thereby increasing metastasis, proliferation, and drug resistance[127].
Analysis of whole-exome and transcriptome sequencing of mCRPC biopsies revealed that alterations in DNA-damage repair (DDR) genes including BRCA2, BRCA1, and ATM occur at higher frequencies in mCRPC (19.3%) than in primary PCa, of which 12.7% of the samples were identified with loss of BRCA2[43]. Cells with deleterious mutations in BRCA1 or BRCA2 compensate for this loss by increasing their dependency on poly(ADP-ribose) polymerase (PARP) activity for DNA repair[128,129].
Prostate-specific membrane antigen (PSMA) is a type II transmembrane glycoprotein that is normally expressed in the prostate epithelium. Although it is expressed in other tissues (such as the salivary glands, proximal tubules of the kidney, and small intestine), its levels there are minimal. Importantly, in PCa tissues, PSMA is significantly overexpressed, having the highest expression in advanced PCa and mCRPC. Moreover, following androgen deprivation and hormonal therapy, PSMA expression seems to be increased[130-132].
EMERGING TREATMENTS TARGETING mCRPC
Based on the above, there is a crucial need for novel alternative approaches and drugs that could overcome resistance in advanced PCa stages. In fact, several treatments, which are under development and trials, could emerge as promising therapies for patients with mCRPC and become the next-generation standards of care. Table 2 summarizes the evolving targeted treatments in mCRPC along with their relevant clinical trials[15,133-156].
Summary of the completed and ongoing clinical trials of emerging therapies in mCRPC
| Therapy | Agent(s) | Trial name/Clinicaltrials.gov identifier | Trial phase | Main findings/Comments |
| PARPi | Olaparib | TOPARP-A/NCT01682772[133] | II | Olaparib improved the RR specifically in patients with DDR gene defects |
| Olaparib | TOPARP-B/NCT01682772[134] | II | Olaparib improved the RR specifically in patients with BRCA1/2 aberrations | |
| Olaparib or ENZ/ABI | PROfound/NCT02987543[15,135] | III | Olaparib increased PFS and OS | |
| Rucaparib | TRITON2/NCT02952534[136,137] | II | Rucaparib improved RR and PSA RR in patients with BRCA alterations | |
| Rucaparib or physician’s choice of ABI/ENZ/DOC | TRITON3/NCT02975934 | III | Ongoing trial | |
| Niraparib | GALAHAD/NCT02854436[138,139] | II | Ongoing trial; interim results show that niraparib improved RR in patients with BRCA1/2 biallelic alterations | |
| Talazoparib | TALAPRO-1/NCT03148795[140] | II | Ongoing trial; interim results show that talazoparib improved RR in patients with BRCA1/2 alterations | |
| Talazoparib + ENZ or ENZ | TALAPRO-2/NCT03395197 | III | Ongoing trial | |
| PSMA radioligand therapy | LuPSMA | [141,142] | II | LuPSMA showed high RR, low toxicity, and reduction of pain in patients LuPSMA showed a better response than other therapies when rechallenged upon progression |
| LuPSMA or cabazitaxel | TheraP/NCT03392428[143] | II | The percentage of patients who achieved PSA50 is higher in the LuPSMA group The percentage of patients with AEs is lower in the LuPSMA group | |
| LuPSMA + best supportive/best standard of care or best supportive/best standard of care | VISION/NCT03511664[144] | III | LuPSMA improved PFS and OS | |
| PSMA-targeted immunotherapy | CAR+ T cells | NCT01140373[145] | I | 3 107 CAR+ T cells/kg was safe with persisting CAR-T cells in peripheral blood for up to two weeks One patient exhibited a long-term response with stable disease status for more than 16 months |
| PSMA-targeted/TGFβ-resistant CAR-T cells | NCT03089203[146] | I | Ongoing trial | |
| Fully humanized anti-PSMA monoclonal IgG1 antibody conjugated to MMAE PSMA-ADC | NCT01695044[147] | II | Discontinued trial as 40% and 31% of the participants showed progressive disease and AEs, respectively | |
| MEDI3726 (PSMA-ADC linked to pyrrolobenzodiazepine) | NCT02991911[148] | I | AEs occurred in 91% of the patients and 33% discontinued MEDI3726 exhibited responses at higher doses, but treatment was discontinued | |
| MLN2704 (PSMA-ADC with a humanized monoclonal antibody linked to the maytansinoid DM) | [149] | I/II | MLN2704 had a low PSA50 response Neurotoxicity was dose-limiting | |
| Pasotuxizumab (AMG 212 or BAY 2010112) | NCT01723475[150] | I | AMG 212 had dose-dependent clinical efficacy and manageable safety Relatively long-term response was seen in two patients | |
| Acapatamab (AMG 160) | NCT03792841[151,152] | I | Acapatamab had an acceptable safety profile, promising PSA50 response, stable disease in 8/15 patients, and 14% of the patients continued the treatment for more than 6 months | |
| Androgen receptor degraders | ARV-110 | NCT03888612[153] | I | Ongoing trial |
| PI3K pathway inhibitors | Ipat (GDC-0068) + ABI or placebo + ABI | NCT01485861[154,155] | Ib/II | Ipat improved PFS and OS Patients with PTEN loss had prolonged PFS |
| Ipat + ABI or placebo + ABI | NCT03072238[156] | III | Ongoing trial; interim results show that Ipat prolonged PFS Two treatment-related deaths occurred in both groups |
PARP inhibitors
The PARP superfamily, with at least 18 members, comprises nuclear enzymes involved in the DNA repair machinery of single-stranded breaks (SSBs), cell proliferation, and death[157]. Inhibition of PARP proteins impedes the ligation of SSBs that consequently convert into double-stranded breaks (DSBs). When DDR genes are defective, PARP inhibition leads to the accumulation of DSBs, promoting an enhanced lethal effect known as the “synthetic lethality” that ultimately induces cell death. According to this rationale, PARP inhibitors (PARPi) emerged as a therapeutic approach targeting malignancies with defective DDR genes, particularly BRCA1/2[158-160]. In fact, PARPi application showed success in BRCA-deficient breast and ovarian cancer and promising efficacy in clinical trials involving mCRPC patients[129,161]. Several PARPi have been tested and approved in mCRPC including olaparib, rucaparib, niraparib, talazoparib, and veliparib.
Olaparib (Lynparza) is a small, oral, and bioavailable molecule that selectively binds and inhibits PARP[162]. Mateo et al.[134] published two Phase II clinical trials, “TOPARP-A” (NCT01682772) and “TOPARP-B” (NCT01682772), involving patients with mCRPC who had previously received standard treatments and who were treated with olaparib. Both studies demonstrated that patients with defective DDR genes and who were no longer responding to the current treatments exhibited a high response rate to olaparib. Importantly, response to treatment seemed dependent on specific DDR gene defects, as the highest response was achieved in the BRCA1/2 aberrant subgroup[133,134,163].
In May 2020, based on the results of the “PROfound” (NCT02987543) clinical study, olaparib was granted a “breakthrough” FDA approval for the treatment of men with mCRPC harboring germline and/or somatic mutations in DDR genes and who formerly received second-generation antiandrogens[164]. PROfound is a prospective, randomized, Phase III trial. Patients with mCRPC and DDR genes alterations whose disease progressed while receiving ENZ or ABI were randomly assigned to receive either olaparib or ENZ/ABI (control). The results show that patients receiving olaparib had significantly longer median imaging-based PFS than the control group[135]. Moreover, this study was the first to show that patients on opalarib had a significantly longer duration of survival compared to the control[15].
Recently, Marshall et al.[165] performed a retrospective, observational study involving patients with mCRPC having somatic or germline mutations in BRCA1, BRCA2, or ATM who were treated with olaparib. This study aimed to assess whether responses to olaparib are different between men with mutations in BRCA1/2 vs. ATM. Indeed, patients with BRCA1/2 alterations exclusively achieved > 50% decline in prostate-specific antigen (PSA50 response). Moreover, these patients had a longer median PFS. Thus, this study demonstrated that men with BRCA1/2 respond better to opalarib than those harboring ATM mutations, and better alternatives should be considered for patients with ATM alterations[165].
These clinical trials helped to establish the efficacy and safety of opalarib as a monotherapy. Other trials investigating opalarib as a monotherapy outside the scope of DDR genes are underway[166]. Moreover, several preclinical and clinical studies testing the synergistic effects of opalarib with other cytotoxic drugs (chemotherapy, AR-directed therapy, immune checkpoint inhibitors, radioligand therapy, radiotherapy, etc.) on mCRPC are still ongoing or completed.
Rucaparib is a small, oral, bioavailable PARPi that was evaluated in clinical trials due to its chemosensitization, radiosensitization, and antineoplastic potency[167]. Rucaparib elicits a cytotoxic effect comparable to opalarib[168]. Results of “TRITON2” (NCT02952534) Phase II trial prompted an “accelerated” FDA approval of rucaparib for men with mCRPC and BRCA mutations who were previously treated with AR-directed therapy and taxane-based chemotherapy. In this study, rucaparib treatment on patients with BRCA alterations showed promising response rates, particularly in comparison to patients with non-BRCA DDR gene alteration[136,137,169].
Based on the ClinicalTrials.gov website, a Phase III clinical trial, “TRITON3” (NCT02975934), is ongoing. Patients with mCRPC harboring BRCA1, BRCA2, or ATM mutations who progressed on AR-directed therapy but did not receive chemotherapy are actively being recruited. This study aims to assess the patients’ response to rucaparib monotherapy vs. a treatment of the physician’s choice of ABI, ENZ, or DOC to verify the clinical benefit of rucaparib. In addition to TRITON3, other studies are currently active to evaluate the efficacy of rucaparib in combination with nivolumab or chemotherapeutic treatments[160].
Niraparib (Zejula) is a small, oral, once-daily, bioavailable, potent, and highly selective Poly (ADP-ribose) Polymerase Inhibitors(PARPi) with antineoplastic activity[170]. “GALAHAD” (NCT02854436) is an active, ongoing, Phase II clinical trial that aims to assess the efficacy, safety, and pharmacokinetics of niraparib in men with treatment-refractory mCRPC and DNA repair alterations (biallelic alterations). Interim analysis shows that niraparib may have promising efficacy as a monotherapy for men with mCRPC, specifically those harboring BRCA1/2 biallelic alteration[138,139]. Niraparib is also being tested in several trials as a combination therapy with other cytotoxic drugs (ABI and radium-223).
Talazoparib (Talzenna) is a small, oral, bioavailable molecule that was demonstrated to be the most potent among PARPi in terms of in vitro activity and trapping of PARP on DNA SSBs[171,172]. Between October 2017 and March 2020, participants were recruited and enrolled in “TALAPRO-1” (NCT03148795), an ongoing Phase II clinical trial whereby patients with mCRPC and DDR gene alterations who progressed on standard treatments were eligible and provided with talazoparib. In addition, different clinical trials assessing the efficacy of combining talazoparib with other therapeutics are ongoing including the Phase III TALAPRO-2 (NCT03395197) trial, which compares the combination of talazoparib and ENZ vs. ENZ alone[140].
PSMA-based therapies
The overexpression of PSMA, particularly in mCRPC, makes it a potential therapeutic target for PCa. Upon binding to their receptors, PSMA ligands are internalized, leading to persistent retention of the ligand intracellularly. This signaling process seems specific to cancer cells, as in normal cells, a relatively rapid washout takes place. Abusing this feature in cancer cells that have increased PSMA ligand uptake, targeting PSMA by radioligand therapy or by immunotherapy has become an emerging therapeutic approach to treat mCRPC[173,174].
PSMA radioligand therapy
The radioligand targeting PSMA, which has come the farthest in development, is lutetium-177, [177Lu]-PSMA-617 (LuPSMA). It is a small, radiolabeled beta-emitter with a high binding affinity to PSMA. LuPSMA is advantageous by the short-range path length of the emitted beta particle, allowing efficient delivery of the radiation to the target cells while minimizing off-target effects on surrounding tissues[141]. Since 2014, many retrospective and prospective studies have been performed to assess the efficacy and safety of LuPSMA in treating mCRPC.
The German Society of Nuclear Medicine performed the largest multicenter retrospective data analysis that studied the toxicity and efficacy of LuPSMA on 145 mCRPC patients. This study showed promising results for LuPSMA in terms of high efficacy and desirable safety, whereby 40% of the patients responded after a single treatment cycle[175].
Hofman et al.[141] performed a Phase II prospective trial involving men with mCRPC who progressed after receiving conventional PCa treatments and had high expression of PSMA. This trial’s sample size was expanded, and long-term outcomes were assessed by Violet et al.[142]. Analysis of the results of those studies showed a high response rate, low toxicity, reduction of pain in patients with mCRPC who received LuPSMA, and better response over other therapies when rechallenged upon progression.
The results of two recent trials, the “TheraP” (NCT03392428) Phase II prospective study and the “VISION” (NCT03511664) Phase III trial, involving mCRPC patients treated with LuPSMA proved that the latter is a potential novel efficient therapeutic for mCRPC and possible alternative to the approved chemotherapeutic drug CBZ and taxane-based regimens[143,144].
Finally, although LuPSMA is the most studied PSMA radioligand therapeutic agent in recent years, other molecules are also being investigated, such as the alpha-emitting actinium-225 (225Ac-PSMA-617). This is an alpha-emitter with higher potency and a shorter range than beta-emitters[176]. A meta-analysis was recently published in September 2021 that summarizes the effects of 225Ac-PSMA-617 in mCRPC from nine studies with 263 patients; the authors concluded that 225Ac-PSMA-617 might be a potentially effective therapeutic option for mCRPC patients[177].
PSMA-targeted immunotherapy
In addition to its significantly high expression in mCRPC, PSMA has a large extracellular domain which makes it an ideal target for immune agents[174]. Several approaches for PSMA-targeted immunotherapy exist including chimeric antigen receptor T cells, antibody-drug conjugates, bispecific T-cell engagers, and PSMA-directed vaccines.
a. Chimeric antigen receptor T cells
Chimeric antigen receptor (CAR) is a genetically engineered transgenic T-cell receptor. It consists of an antigen recognition moiety allowing T cells to identify intact specific tumor-associated antigens and T-cell signaling domains that activate T cells. Thus, CAR-T cells combine the properties of antibodies that recognize particular antigens with the cytolytic killing of T cells[178-180].
A Phase I study (NCT01140373) was performed by Slovin et al.[145] to assess the safety, tolerability, and efficiency of escalating doses of PSMA-targeted CAR-T cells. Seven patients included in this study received from 107 to 3 107 CAR-T cells/kg. The highest given dose was shown to be safe with persisting CAR-T cells in peripheral blood for up to two weeks. Importantly, one patient exhibited a long-term response having a stable disease status for more than 16 months[145].
Moreover, a first-time Phase I clinical trial (NCT03089203) to test the safety, feasibility, and efficacy of PSMA-targeted/TGFβ-resistant CAR-T cells is ongoing. The experimental approach strives to overcome the immunosuppressive tumor microenvironment (TME) in mCRPC, to which high levels of TGFβ are a contributing factor. This rationale is based on in vivo disseminated PCa models results, where PSMA-redirected CAR-T cells expressing a dominant-negative TGFβ receptor had an increased T-cell proliferation and cytokine release, long-term persistence, and greater tumor elimination[146]. The results of this clinical trial are pending.
b. Antibody-drug conjugate
Antibody-drug conjugate (ADC) technology is an emerging therapeutic approach that is still in its early phases. ADCs are highly specific monoclonal antibodies targeted against specific tumor antigens and chemically conjugated to cytotoxic agents[181]. Target selection is one of the key considerations in ADCs design. The target needs to be highly expressed on the surface of tumor cells vs. low to no expression in normal cells. Moreover, the target preferably needs to have internalization characteristics that allow the transport of the antibody to carry the cytotoxic payloads into cancerous cells. These requirements are met in PSMA, making it an attractive target; therefore, several ADCs targeting PSMA in PCa are in clinical development[182].
A Phase II trial (NCT01695044) was performed by Petrylak et al.[147], a Phase I trial (NCT02991911) by de Bono et al.[148], and a Phase I/II multiple escalating dose trial by Milowsky et al.[149] tested different PSMA-ADCs. The different trials showed that, although these drug conjugates had some activity in mCRPC, their effect was accompanied by significant adverse events (such as neutropenia and neuropathy), which led to the discontinuation of some of these treatments. Hence, it was noticed that toxicity related to the long circulation in the system of PSMA-targeted ADCs limited the success of the clinical trials[183].
c. Bispecific T-cell engagers
Bispecific T-cell engagers (BiTEs) are bispecific antibodies that simultaneously target a T-cell-specific molecule (almost always CD3 chain due to its conserved property) and a TAA, which could be PSMA in the case of PCa[184]. BiTEs showed success in several types of cancer, such as acute lymphoblastic leukemia, where blinatumomab was the first FDA-approved BiTE[185,186].
Hummel et al.[150] conducted a first-in-human, dose-escalation, Phase I clinical trial (NCT01723475) to primarily test PSMA CD3 first-generation BiTE pasotuxizumab (known as AMG 212 or BAY 2010112) in patients with mCRPC refractory to standard therapy. In this study, data analysis demonstrated the dose-dependent clinical efficacy and manageable safety of pasotuxizumab in mCRPC, with 3 out of 16 patients exhibiting ≥ 50% PSA response and two patients having a relatively long-term response[150].
Another first-in-human, Phase I study (NCT03792841) to assess acapatamab (AMG 160), a novel next-generation PSMA CD3 BiTE with an extended half-life, in mCRPC was conducted[151]. AMG 160 gave a promising PSA50 response and acceptable safety profile in treated patients. This study prompted testing AMG 160 as a combination therapy with other agents (pembrolizumab, ENZ, ABI, AMG 404, and etanercept prophylaxis) in ongoing trials[152,187,188].
d. PSMA-directed vaccines
Cancer vaccines, intended to enhance the immune response against tumor cells by increasing the pool of TAA-specific host T cells, have failed to demonstrate a considerable benefit in PCa thus far (such as the Phase III clinical trials “PROSTVAC” that targets PSA and “GVAX”, where both failed to meet their primary endpoint of enhancing the overall survival)[189,190]. However, this approach continues to be investigated, given its potential efficacy, minimal side effects, limited cost, and easy synthesis. Specifically, PSMA is being considered as an appropriate target for the development of PCa vaccines[191]. Using new computational tools to select suitable B-cell epitopes with high antigenicity, the “673RHVIYAPSSHNKYAGE25” peptide was predicted as having the best binding affinity. Future steps involve synthesis of the peptide and testing its in vivo efficacy as a potential PSMA-directed vaccine[192].
Androgen receptor degraders
Androgen receptor degraders emerged as an alternative therapeutic strategy to manage mCRPC. One of the tools that allow targeting of AR to degradation is using proteolysis-targeting chimera (PROTAC) novel technology. PROTACs, also known as bivalent chemical protein degraders, are heterobifunctional compounds consisting of two recruiting ligands joined by a linker. One ligand moiety binds to the protein of interest, while the other is specific to E3 ubiquitin ligase (E3). Thus, PROTACs form a ternary complex with the target protein and E3 and consequently promote target ubiquitination and degradation[193].
Salami et al.[194] published a study in 2018 that aimed to assess whether ARCC-4, a low-nanomolar PROTAC ARD, was better at targeting AR signaling in CRPC cells than the currently approved competitive antagonist ENZ. Interestingly, ARCC-4 promoted the degradation of about 95% of cellular ARs, especially the clinically relevant AR with point mutations resistant to ENZ. Moreover, ARCC-4 induced apoptosis and maintained its antiproliferative effect in a hyperandrogenic environment that impedes ENZ activity. This study showed promising results where PROTAC-mediated AR degradation might overcome the mCRPC AR-dependent mechanism of drug resistance[194].
To date, several PROTACs reached clinical trials in different types of cancer, including ARV-110 in mCRPC. The oral and bioavailable ARV-110 was tested earlier in vitro and in vivo, where it led to the degradation of more than 95% of ARs in the tested PCa cell lines and demonstrated antitumor effects in both ENZ-naïve and -resistant PCa xenograft models. ARV-110, similar to ARCC-4, promoted the degradation of clinically relevant AR mutants and maintained its activity even in a milieu with high androgen levels. The first-in-human, Phase I clinical trial (NCT03888612) testing ARV-110 in mCRPC patients who received more than two prior treatments was performed. This trial is still ongoing; however, early data show that ARV-110 has a reasonable safety profile and a promising antitumor activity in mCRPC[153].
The results from these preclinical and clinical trials, among others, show that ARD might be a novel alternative therapeutic option to treat mCRPC.
PI3K pathway inhibitors
As mentioned above, loss of PTEN and the atypical activation of the PI3K signaling network are one of the possible mechanisms driving mCRPC growth. This fact supported the development of several PI3K pathway inhibitors including ipatasertib (Ipat) (GDC-0068), which is an oral, bioavailable, AKT non-ATP-competitive inhibitor that impedes the PI3K/AKT pathway and subsequently tumorigenesis[195].
Two clinical trials, a Phase Ib/II (NCT01485861) involving patients who formerly received DOC and an ongoing Phase III trial (NCT03072238) with treatment-naïve patients, tested the combination of Ipat and ABI in mCRPC. In general, both studies demonstrated enhanced antitumor effects when combining Ipat with ABI compared to ABI monotherapy, with increased benefit for patients with PTEN loss[154-156]. Thus, the results of those trials suggest that a combination of AKT and AR signaling inhibitors might provide a promising therapeutic approach to treating men with mCRPC with a poor prognosis due to PTEN loss.
CONCLUSION
mCRPC remains a challenge for PCa disease management. AR signaling comprises a fundamental role in PCa pathogenesis even in the advanced androgen-insensitive stages. AR signaling, along with other various molecular pathways, is subjected to diverse modifications and aberrations that can be used as biomarkers to personalize treatment for mCRPC patients and overcome drug resistance to the standard therapy. Thus, further understanding of the mechanisms mediating drug resistance in mCRPC is crucial for identifying future targeted therapeutic modalities.
DECLARATIONS
Authors’ contributionsConceptualized, outlined, and drafted the review: Abou-Kheir W, Mukherji D
Made substantial contributions to study design, drafting the paper, generating tables and figures: Yehya A, Ghamlouche F, Zahwe A
Contributed to drafting parts of the paper: Zeid Y, Wakimian K
Reviewed the paper and approved the final version: Yehya A, Ghamlouche F, Zahwe A, Zeid Y, Wakimian K, Mukherji D, Abou-Kheir W
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestDM reports institutional funding from Astellas, Personal travel support/honoraria from Astellas, Janssen, Bayer, Ipsen, BMS, and MSD. All authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Barsouk A, Padala SA, Vakiti A, et al. Epidemiology, staging and management of prostate cancer. Med Sci (Basel) 2020;8:28.
4. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res 2006;66:7783-92.
5. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA 2017;317:2532-42.
6. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013;32:5501-11.
7. Bono JS, Logothetis CJ, Molina A, et al; COU-AA-301 Investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995-2005.
8. Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2012;13:983-92.
9. Ryan CJ, Smith MR, de Bono JS, et al. COU-AA-302 Investigators. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138-48.
10. Scher HI, Fizazi K, Saad F, et al. AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187-97.
11. Massard C, James N, Culine S, et al. Arades trial: a first-in-man, open-label, phase I/II safety, pharmacokinetic, and proof-of-concept study of ODM-201 in patients (PTS) with progressive metastatic castration-resistant prostate cancer (MCRPC). Ann Oncol 2012;23:ixe16.
12. Tannock IF, de Wit R, Berry WR, et al. TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-12.
13. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. The Lancet 2010;376:1147-54.
14. Parker C, Nilsson S, Heinrich D, et al. ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med 2013;369:213-23.
15. Hussain M, Mateo J, Fizazi K, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med 2020;383:2345-57.
16. Kantoff PW, Higano CS, Shore ND, et al. IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411-22.
17. Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. In: Li JJ, Li SA, Mohla S, Rochefort H, Maudelonde T, editors. Hormonal Carcinogenesis V. New York: Springer; 2008. p. 223-34.
18. Tucci M, Scagliotti GV, Vignani F. Metastatic castration-resistant prostate cancer: time for innovation. Future Oncol 2015;11:91-106.
19. Sonpavde G, Attard G, Bellmunt J, et al. The role of abiraterone acetate in the management of prostate cancer: a critical analysis of the literature. Eur Urol 2011;60:270-8.
20. Pia A, Vignani F, Attard G, et al. Strategies for managing ACTH dependent mineralocorticoid excess induced by abiraterone. Cancer Treat Rev 2013;39:966-73.
21. Sydes M, Mason M, Spears M, et al. Adding abiraterone acetate plus prednisolone (AAP) or docetaxel for patients (pts) with high-risk prostate cancer (PCa) starting long-term androgen deprivation therapy (ADT): directly randomised data from STAMPEDE (NCT00268476). Ann Oncol 2017;28:v619.
22. Sternberg CN, Petrylak DP, Madan RA, Parker C. Progress in the treatment of advanced prostate cancer. Am Soc Clin Oncol Educ Book 2014:117-31.
23. Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol 2017;71:151-4.
24. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787-90.
25. Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res 1997;57:229-33.
26. Giannakakou P, Nakano M, Nicolaou KC, et al. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc Natl Acad Sci USA 2002;99:10855-60.
27. Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res 2012;72:4611-5.
28. Santis M, Saad F. Practical guidance on the role of corticosteroids in the treatment of metastatic castration-resistant prostate cancer. Urology 2016;96:156-64.
29. Teply BA, Luber B, Denmeade SR, Antonarakis ES. The influence of prednisone on the efficacy of docetaxel in men with metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis 2016;19:72-8.
30. Hirayama Y, Sadar MD. Does increased expression of glucocorticoid receptor support application of antagonists to this receptor for the treatment of castration resistant prostate cancer? AME Med J 2018;3:66-66.
31. Rice MA, Malhotra SV, Stoyanova T. Second-generation antiandrogens: from discovery to standard of care in castration resistant prostate cancer. Front Oncol 2019;9:801.
32. Aggarwal RR, Thomas G, Youngren J, et al. Androgen receptor (AR) amplification in patients (pts) with metastatic castration resistant prostate cancer (mCRPC) resistant to abiraterone (Abi) and enzalutamide (Enz): preliminary results from the SU2C/PCF/AACR West Coast Prostate Cancer Dream Team (WCDT). J Clin Oncol 2015;15:5068.
33. Kim EH, Cao D, Mahajan NP, Andriole GL, Mahajan K. ACK1-AR and AR-HOXB13 signaling axes: epigenetic regulation of lethal prostate cancers. NAR Cancer 2020;2:zcaa018.
34. Lakshmana G, Baniahmad A. Interference with the androgen receptor protein stability in therapy-resistant prostate cancer. Int. J. Cancer 2019;144:1775-9.
35. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33-9.
36. Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239-43.
37. Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011-25.
38. Conteduca V, Wetterskog D, Sharabiani MTA, et al. PREMIERE Collaborators. , Spanish Oncology Genitourinary Group. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol 2017;28:1508-16.
39. Romanel A, Gasi Tandefelt D, Conteduca V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med 2015;7:312re10.
40. Mostaghel EA, Marck BT, Plymate SR, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 2011;17:5913-25.
41. Kawata H, Ishikura N, Watanabe M, Nishimoto A, Tsunenari T, Aoki Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate 2010;70:745-54.
42. Yamamoto Y, Loriot Y, Beraldi E, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res 2015;21:1675-87.
43. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215-28.
44. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11-22.
45. Lorente D, Mateo J, Zafeiriou Z, et al. Switching and withdrawing hormonal agents for castration-resistant prostate cancer. Nat Rev Urol 2015;12:37-47.
46. Beltran H, Yelensky R, Frampton GM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 2013;63:920-6.
47. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat 2012;33:887-94.
48. Culig Z, Hobisch A, Cronauer MV, et al. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol 1993;7:1541-50.
49. Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer 2002;100:309-17.
50. Balbas MD, Evans MJ, Hosfield DJ, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013;2:e00499.
51. Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 2013;3:1020-9.
52. Coffey K, Robson CN. Regulation of the androgen receptor by post-translational modifications. J Endocrinol 2012;215:221-37.
53. Wen S, Niu Y, Huang H. Posttranslational regulation of androgen dependent and independent androgen receptor activities in prostate cancer. Asian J Urol 2020;7:203-18.
54. Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol 2012;352:70-8.
55. Chen MF, Chen WC, Chang YJ, Wu CF, Wu CT. Role of DNA methyltransferase 1 in hormone-resistant prostate cancer. J Mol Med (Berl) 2010;88:953-62.
56. Ponguta LA, Gregory CW, French FS, Wilson EM. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem 2008;283:20989-1001.
57. Mahajan K, Challa S, Coppola D, et al. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate 2010;70:1274-85.
58. Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res 2009;15:3540-9.
59. Migliaccio A, Varricchio L, De Falco A, et al. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene 2007;26:6619-29.
60. Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res 2011;71:862-72.
61. Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006;10:309-19.
62. Xu K, Shimelis H, Linn DE, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 2009;15:270-82.
63. Burska UL, Harle VJ, Coffey K, et al. Deubiquitinating enzyme Usp12 is a novel co-activator of the androgen receptor. J Biol Chem 2013;288:32641-50.
64. Senapati D, Kumari S, Heemers HV. Androgen receptor co-regulation in prostate cancer. Asian J Urol 2020;7:219-32.
65. Culig Z. Androgen receptor coactivators in regulation of growth and differentiation in prostate cancer. J Cell Physiol 2016;231:270-4.
66. Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol Cell Biol 2010;30:1243-53.
67. Chen S, Sullivan WP, Toft DO, Smith DF. Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaper 1998;3:118.
68. Sharma A, Yeow WS, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest 2010;120:4478-92.
69. Liu C, Liao Z, Duan X, et al. The MYH9 cytoskeletal protein is a novel corepressor of androgen receptors. Front Oncol 2021;11:641496.
70. Teng M, Zhou S, Cai C, Lupien M, He HH. Pioneer of prostate cancer: past, present and the future of FOXA1. Protein Cell 2021;12:29-38.
71. Gupta S, Li J, Kemeny G, et al. Whole genomic copy number alterations in circulating tumor cells from men with abiraterone or enzalutamide-resistant metastatic castration-resistant prostate cancer. Clin Cancer Res 2017;23:1346-57.
72. Büscheck F, Zub M, Heumann A, et al. The independent prognostic impact of the GATA2 pioneering factor is restricted to ERG-negative prostate cancer. Tumour Biol 2019;41:1010428318824815.
73. Vidal SJ, Rodriguez-Bravo V, Quinn SA, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer cell 2015;27:223-39.
74. Yao J, Chen Y, Nguyen DT, et al. The homeobox gene, HOXB13, regulates a mitotic protein-kinase interaction network in metastatic prostate cancers. Sci Rep 2019;9:9715.
75. Faisal* F, Alshalalfa M, Davicioni E, et al. MP68-10 hoxb13 expression and its role in prostate cancer progression and neuroendocrine differentiation. J Urol 2019:201.
76. Kim YR, Oh KJ, Park RY, et al. HOXB13 promotes androgen independent growth of LNCaP prostate cancer cells by the activation of E2F signaling. Mol Cancer 2010;9:124.
77. Navarro HI, Goldstein AS. HoxB13 mediates AR-V7 activity in prostate cancer. Proc Natl Acad Sci USA 2018;115:6528-9.
78. Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009;69:2305-13.
79. Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009;69:16-22.
80. Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028-38.
81. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013;73:483-9.
82. Hörnberg E, Ylitalo EB, Crnalic S, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 2011;6:e19059.
83. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447-54.
84. Geller J, Albert J, Loza D. Steroid levels in cancer of the prostate-markers of tumour differentiation and adequacy of anti-androgen therapy. J Steroid Biochem 1979;11:631-6.
85. Kumagai J, Hofland J, Erkens-Schulze S et al. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. The Prostate 2013;73:1636-50.
86. Yin L, Hu Q. CYP17 inhibitors-abiraterone, C17, 20-lyase inhibitors and multi-targeting agents. Nat Rev Urol 2014;11:32-42.
87. Knuuttila M, Yatkin E, Kallio J, et al. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am J Pathol 2014;184:2163-73.
88. Tamae D, Mostaghel E, Montgomery B, et al. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact 2015;234:332-8.
89. Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer 2007;120:719-33.
90. Liu C, Lou W, Zhu Y, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res 2015;75:1413-22.
91. Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006;66:2815-25.
92. Crona DJ, Whang YE. Androgen receptor-dependent and -independent mechanisms involved in prostate cancer therapy resistance. Cancers (Basel) 2017;9:67.
93. Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013;155:1309-22.
94. Isikbay M, Otto K, Kregel S, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer 2014;5:72-89.
95. Kroon J, Puhr M, Buijs JT, et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr Relat Cancer 2016;23:35-45.
96. Puhr M, Hoefer J, Eigentler A, et al. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved antiandrogen therapy. Clin Cancer Res 2018;24:927-38.
97. Carver BS. Defining and targeting the oncogenic drivers of neuroendocrine prostate cancer. Cancer Cell 2016;29:431-2.
98. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
99. Aparicio AM, Shen L, Tapia EL, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res 2016;22:1520-30.
100. Tan HL, Sood A, Rahimi HA, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2014;20:890-903.
101. Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78-83.
102. Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84-8.
103. Lee JK, Phillips JW, Smith BA, et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016;29:536-47.
104. Orme JJ, Huang H. Microenvironment-mediated resistance to anti-androgen therapy. Cancer Cell 2020;38:155-7.
105. Gil V, Miranda S, Riisnaes R, et al. PCF/SU2C international prostate cancer dream team. HER3 is an actionable target in advanced prostate cancer. Cancer Res 2021;81:6207-18.
106. Zhang Z, Karthaus WR, Lee YS, et al. Tumor microenvironment-derived NRG1 promotes antiandrogen resistance in prostate cancer. Cancer Cell 2020;38:279-296.e9.
107. Pang X, Gong K, Zhang X, Wu S, Cui Y, Qian BZ. Osteopontin as a multifaceted driver of bone metastasis and drug resistance. Pharmacol Res 2019;144:235-44.
108. Pang X, Zhang J, He X, et al. SPP1 promotes enzalutamide resistance and epithelial-mesenchymal-transition activation in castration-resistant prostate cancer via PI3K/AKT and ERK1/2 pathways. Oxid Med Cell Longev 2021;2021:5806602.
109. Zheng G, Ma Y, Zou Y, Yin A, Li W, Dong D. HCMDB: the human cancer metastasis database. Nucleic Acids Res 2018;46:D950-5.
110. Harman SM, Metter EJ, Blackman MR, Landis PK, Carter HB. Baltimore Longitudinal Study on Aging. Serum levels of insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-3, and prostate-specific antigen as predictors of clinical prostate cancer. J Clin Endocrinol Metab 2000;85:4258-65.
111. Hour TC, Chung SD, Kang WY, et al. EGFR mediates docetaxel resistance in human castration-resistant prostate cancer through the Akt-dependent expression of ABCB1 (MDR1). Arch Toxicol 2015;89:591-605.
112. Liao Y, Guo Z, Xia X, et al. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res 2019;38:157.
113. Mahajan NP, Liu Y, Majumder S, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci U S A 2007;104:8438-43.
114. Pearson HB, Li J, Meniel VS, et al. Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with pten loss to accelerate progression and castration-resistant growth. Cancer Discov 2018;8:764-79.
115. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer 2013;20:R83-99.
116. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci 2020;21:4507.
117. Rybak AP, Bristow RG, Kapoor A. Prostate cancer stem cells: deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015;6:1900-19.
118. Jeong JH, Wang Z, Guimaraes AS, et al. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS One 2008;3:e3949.
119. Culig Z. Interleukin-6 function and targeting in prostate cancer. In: Birbrair A, editor. Tumor microenvironment. Cham: Springer International Publishing; 2021. p.1-8.
120. Bishop JL, Thaper D, Zoubeidi A. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers (Basel) 2014;6:829-59.
121. Liu C, Lou W, Zhu Y, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res 2014;20:3198-210.
122. Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. Co-operative functions between nuclear factors NFkappaB and CCAT/enhancer-binding protein-beta (C/EBP-beta) regulate the IL-6 promoter in autocrine human prostate cancer cells. Prostate 2004;61:354-70.
123. Miguel MP, Royuela M, Bethencourt FR, Santamaría L, Fraile B, Paniagua R. Immunoexpression of tumour necrosis factor-alpha and its receptors 1 and 2 correlates with proliferation/apoptosis equilibrium in normal, hyperplasic and carcinomatous human prostate. Cytokine 2000;12:535-8.
124. Araki S, Omori Y, Lyn D, et al. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res 2007;67:6854-62.
125. Aalinkeel R, Nair MP, Sufrin G, et al. Gene expression of angiogenic factors correlates with metastatic potential of prostate cancer cells. Cancer Res 2004;64:5311-21.
126. Lehrer S, Diamond EJ, Mamkine B, Stone NN, Stock RG. Serum interleukin-8 is elevated in men with prostate cancer and bone metastases. Technol Cancer Res Treat 2004;3:411.
127. Bouraoui Y, Ricote M, García-Tuñón I, et al. Pro-inflammatory cytokines and prostate-specific antigen in hyperplasia and human prostate cancer. Cancer Detect Prev 2008;32:23-32.
128. Aly A, Ganesan S. BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol 2011;3:66-74.
129. Adashek JJ, Jain RK, Zhang J. Clinical development of PARP inhibitors in treating metastatic castration-resistant prostate cancer. Cells 2019;8:860.
130. Bakht MK, Oh SW, Youn H, Cheon GJ, Kwak C, Kang KW. Influence of androgen deprivation therapy on the uptake of PSMA-targeted agents: emerging opportunities and challenges. Nucl Med Mol Imaging 2017;51:202-11.
131. O’Keefe DS, Bacich DJ, Huang SS, Heston WDW. A perspective on the evolving story of PSMA biology, PSMA-based imaging, and endoradiotherapeutic strategies. J Nucl Med 2018;59:1007-13.
132. Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 1997;3:81-5.
133. Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697-708.
134. Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 2020;21:162-74.
135. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020;382:2091-102.
136. Abida W, Patnaik A, Campbell D, et al. TRITON2 investigators. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a. BRCA1 2020;38:3763-72.
137. Abida W, Campbell D, Patnaik A, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the Phase II TRITON2 study. Clin Cancer Res 2020;26:2487-96.
138. Smith M, Sandhu S, Kelly W, et al. Pre-specified interim analysis of GALAHAD: a phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann Oncol 2019;30:v884-5.
139. Smith MR, Sandhu SK, Kelly WK, et al. GALAHAD Investigators. Phase II study of niraparib in patients with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): preliminary results of GALAHAD. JCO 2019;37:202-202.
140. de Bono JS, Mehra N, Scagliotti GV, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol 2021;22:1250-64.
141. Hofman MS, Violet J, Hicks RJ, et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): a single-centre, single-arm, phase 2 study. Lancet Oncol 2018;19:825-33.
142. Violet J, Sandhu S, Iravani A, et al. Long-term follow-up and outcomes of retreatment in an expanded 50-patient single-center phase II prospective trial of 177Lu-PSMA-617 theranostics in metastatic castration-resistant prostate cancer. J Nucl Med 2020;61:857-65.
143. Hofman MS, Emmett L, Sandhu S, et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. The Lancet 2021;397:797-804.
144. Sartor O, de Bono J, Chi KN, et al. VISION Investigators. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med 2021;385:1091-103.
145. Slovin SF, Wang X, Hullings M, et al. Chimeric antigen receptor (CAR + ) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). JCO 2013;31:72-72.
146. Narayan V, Gladney W, Plesa G, et al. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. JCO 2019;37:TPS347-TPS347.
147. Petrylak DP, Vogelzang NJ, Chatta K, et al. PSMA ADC monotherapy in patients with progressive metastatic castration-resistant prostate cancer following abiraterone and/or enzalutamide: efficacy and safety in open-label single-arm phase 2 study. Prostate 2020;80:99-108.
148. de Bono JS, Fleming MT, Wang JS, et al. Phase I study of MEDI3726: a prostate-specific membrane antigen-targeted antibody-drug conjugate, in patients with mCRPC after failure of abiraterone or enzalutamide. Clin Cancer Res 2021;27:3602-9.
149. Milowsky MI, Galsky MD, Morris MJ, et al. Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol Oncol 2016;34:530.e15-21.
150. Hummel H, Kufer P, Grüllich C, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting bispecific T cell engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). JCO 2019;37:5034-5034.
151. Tran B, Horvath L, Dorff TB, et al. Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE) immune therapy targeting prostate-specific membrane antigen (PSMA), in patients with metastatic castration-resistant prostate cancer (mCRPC). JCO 2020;38:TPS261-TPS261.
152. Tran B, Horvath L, Rettig M, et al. Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE immune therapy) targeting prostate-specific membrane antigen, in patients with metastatic castration-resistant prostate cancer (mCRPC). JCO 2020;38:TPS5590-TPS5590.
153. Petrylak DP, Gao X, Vogelzang NJ, et al. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). JCO 2020;38:3500.
154. de Bono JS, De Giorgi U, Rodrigues DN, et al. Randomized phase II study evaluating akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res 2019;25:928-36.
155. De Bono JS, De Giorgi U, Massard C, et al. Randomized phase II study of AKT blockade with ipatasertib (GDC-0068) and abiraterone (Abi) vs. Abi alone in patients with metastatic castration-resistant prostate cancer (mCRPC) after docetaxel chemotherapy (A. MARTIN Study). JCO 2016;34:5017.
156. Sweeney C, Bracarda S, Sternberg CN, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. The Lancet 2021;398:131-42.
158. Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood 2011;117:6074-82.
159. Rescigno P, Chandler R, de Bono J. Relevance of poly (ADP-ribose) polymerase inhibitors in prostate cancer. Curr Opin Support Palliat Care 2018;12:339-43.
160. Xu C, Mao S, Jiang H. Recent advances in DNA repair pathway and its application in personalized care of metastatic castration-resistant prostate cancer (mCRPC). In: Huang T, editor. Precision Medicine. New York: Springer US; 2020. p. 75-89.
161. Kamel D, Gray C, Walia JS, Kumar V. PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: an update of clinical trials. Curr Drug Targets 2018;19:21-37.
162. PubChem. PubChem Compound Summary for CID 23725625, Olaparib. 2021. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Olaparib [Last accessed on 5 May 2022].
163. Nientiedt C, Duensing A, Zschäbitz S, et al. PARP inhibition in prostate cancer. Genes Chromosomes Cancer 2021;60:344-51.
164. Food U, Administration D. FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer. silver spring, MD, US FDA; 2020
165. Marshall CH, Sokolova AO, McNatty AL, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol 2019;76:452-8.
166. Jang A, Sartor O, Barata PC, Paller CJ. Therapeutic potential of PARP inhibitors in the treatment of metastatic castration-resistant prostate cancer. Cancers (Basel) 2020;12:3467.
167. PubChem. PubChem compound summary for CID 9931954, rucaparib. 2021. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Rucaparib [Last accessed on 5 May 2022].
168. Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 2014;13:433-43.
169. Anscher MS, Chang E, Gao X, et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA-mutated metastatic castrate-resistant prostate cancer. Oncologist 2021;26:139-46.
170. PubChem. PubChem compound summary for CID 24958200, Niraparib. 2021. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Niraparib [Last accessed on 5 May 2022].
171. PubChem. PubChem compound summary for CID 135565082, Talazoparib. 2021. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Talazoparib [Last accessed on 5 May 2022].
172. Takebe N, Quinn M, Gupta G, Chen AP. PARP inhibition to enhance response to chemotherapy. Targeting cell survival pathways to enhance response to chemotherapy. Elsevier 2019:231-57.
173. Eiber M, Fendler WP, Rowe SP, et al. Prostate-specific membrane antigen ligands for imaging and therapy. J Nucl Med 2017;58:67S-76S.
174. Giraudet AL, Kryza D, Hofman M, et al. PSMA targeting in metastatic castration-resistant prostate cancer: where are we and where are we going? Ther Adv Med Oncol 2021;13:17588359211053898.
175. Rahbar K, Ahmadzadehfar H, Kratochwil C, et al. German multicenter study investigating 177Lu-PSMA-617 radioligand therapy in advanced prostate cancer patients. J Nucl Med 2017;58:85-90.
176. Poty S, Francesconi LC, McDevitt MR, Morris MJ, Lewis JS. α-Emitters for radiotherapy: from basic radiochemistry to clinical studies-part 1. J Nucl Med 2018;59:878-84.
177. Lee DY, Kim YI. Effects of 225Ac-labeled prostate-specific membrane antigen radioligand therapy in metastatic castration-resistant prostate cancer: a meta-analysis. J Nucl Med 2022;63:840-6.
178. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev 2019;34:45-55.
179. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020;38:473-88.
180. Schepisi G, Cursano MC, Casadei C, et al. CAR-T cell therapy: a potential new strategy against prostate cancer. J Immunother Cancer 2019;7:258.
181. Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani AH. Antibody-drug conjugates: possibilities and challenges. Avicenna J Med Biotechnol 2019;11:3-23.
182. Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody-drug conjugates: a comprehensive review. Mol Cancer Res 2020;18:3-19.
183. Wang X, Shirke A, Walker E, et al. Small molecule-based prodrug targeting prostate specific membrane antigen for the treatment of prostate cancer. Cancers (Basel) 2021;13:417.
184. Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. Biomark Res 2021;9:38.
185. Franquiz MJ, Short NJ. Blinatumomab for the treatment of adult B-cell acute lymphoblastic leukemia: toward a new era of targeted immunotherapy. Biologics 2020;14:23-34.
186. Nie S, Wang Z, Moscoso-Castro M, et al. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib Ther 2020;3:18-62.
187. Subudhi SK, Siddiqui BA, Maly JJ, et al. Safety and efficacy of AMG 160, a half-life extended BiTE immune therapy targeting prostate-specific membrane antigen (PSMA), and other therapies for metastatic castration-resistant prostate cancer (mCRPC). JCO 2021;39:TPS5088-TPS5088.
188. Dorff T, Rettig M, Machiels J-P, et al. 340 Phase 1 study of AMG 160, a half-life extended BiTE® (bispecific T-cell engager) therapy targeting prostate-specific membrane antigen, in patients with metastatic castration-resistant prostate cancer. J Immunother Cancer 2020;8:A207-A8.
189. Gulley JL, Borre M, Vogelzang NJ, et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol 2019;37:1051-61.
190. Antonarakis ES, Eisenberger MA. Phase III trials with docetaxel-based combinations for metastatic castration-resistant prostate cancer: time to learn from past experiences. J Clin Oncol 2013;31:1709-12.
191. Olson WC, Heston WD, Rajasekaran AK. Clinical trials of cancer therapies targeting prostate-specific membrane antigen. Rev Recent Clin Trials 2007;2:182-90.
192. Hashemzadeh P, Ghorbanzadeh V, Valizadeh Otaghsara SM, Dariushnejad H. Novel predicted B-Cell epitopes of PSMA for development of prostate cancer vaccine. Int J Pept Res Ther 2020;26:1523-5.
193. Toure M, Crews CM. Small-molecule PROTACS: new approaches to protein degradation. Angew Chem Int Ed Engl 2016;55:1966-73.
194. Salami J, Alabi S, Willard RR, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol 2018;1:100.
195. PubChem. PubChem compound summary for CID 24788740, Ipatasertib. 2021. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Ipatasertib [Last accessed on 5 May 2022].
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yehya A, Ghamlouche F, Zahwe A, Zeid Y, Wakimian K, Mukherji D, Abou-Kheir W. Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo. Cancer Drug Resist 2022;5:667-90. http://dx.doi.org/10.20517/cdr.2022.15
AMA Style
Yehya A, Ghamlouche F, Zahwe A, Zeid Y, Wakimian K, Mukherji D, Abou-Kheir W. Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo. Cancer Drug Resistance. 2022; 5(3): 667-90. http://dx.doi.org/10.20517/cdr.2022.15
Chicago/Turabian Style
Yehya, Amani, Fatima Ghamlouche, Amin Zahwe, Yousef Zeid, Kevork Wakimian, Deborah Mukherji, Wassim Abou-Kheir. 2022. "Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo" Cancer Drug Resistance. 5, no.3: 667-90. http://dx.doi.org/10.20517/cdr.2022.15
ACS Style
Yehya, A.; Ghamlouche F.; Zahwe A.; Zeid Y.; Wakimian K.; Mukherji D.; Abou-Kheir W. Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo. Cancer Drug Resist. 2022, 5, 667-90. http://dx.doi.org/10.20517/cdr.2022.15
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 2
likes
Like This Article 2
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.