
Ovarian cancer recurrence: is the definition of platinum resistance modified by PARPi and other intervening treatments? The evolving landscape in the management of platinum-resistant ovarian cancer


Abstract
Definitions of platinum resistance have been questioned and changed over the last five years, even though no predictive biomarker of resistance exists. These have sculpted how we approach platinum retreatment and, consequently, how we devise new treatment strategies for those patients with tumour progression on platinum therapy. Platinum-non-eligible ovarian cancer is treated with single-agent non-platinum drugs. When bevacizumab can be added to chemotherapy, progression-free survival improves significantly. For patients with a BRCA mutation, PARP inhibitor monotherapy is an option compared to chemotherapy. There is currently no clearly identified role for immune-checkpoint inhibition in this patient population. This review describes some of the challenges in treating patients with platinum resistance and suggests refinements in the selection of patients most likely to benefit from targeting a DNA damage response, angiogenesis or immune modulation. It also describes novel agents of interest and possible mechanisms of the synergy of therapeutic combinations.
Keywords
INTRODUCTION
Background of platinum retreatment
The idea of platinum rechallenge was introduced in the 1980s at a moment in history when few treatments were available for recurrent ovarian cancer. Across several Phase II studies, the treatment-free interval (TFI) was one of the most important variables predicting response to second-line chemotherapy[1]. Later, Markman and Hoskins proposed that trials of new agents be stratified into primary platinum-resistant, secondary platinum-resistant, potentially platinum-sensitive and those with indeterminate sensitivity[2]. These definitions underwent further refinement, with variation in the cut-offs of TFI between 4 and 12 months for intermediate platinum-sensitive disease and ultimately 6 months for being considered as platinum-sensitive, with this latter definition being used for the next 3 decades[3]. It was first rigorously questioned at the 2010 Gynecologic Cancer InterGroup (GCIG) Ovarian Cancer Consensus meeting, during which its use was criticized as the response to platinum gradually increases with TFIp (TFI after platinum) in a non-linear way[3]. During the fifth GCIG consensus meeting in 2015, the terminology: platinum-sensitive and platinum-resistant in clinical trials was replaced with TFIp considered as a continuous variable among others discussed below.
Current definitions of platinum resistance and clinicopathological predictors of platinum responsiveness
According to ESMO-ESGO consensus meeting guidelines for the management of recurrent ovarian cancer[4], platinum-non-eligible ovarian cancer (PNEOC) patients are those who progress on or immediately after their last platinum-based chemotherapy or have contraindications to platinum. Platinum-eligible ovarian cancer (PEOC) includes all other cases of relapse. This includes patients without evaluable or no residual disease after primary surgery or who have relapsed following stage I disease.
There is no biomarker of platinum resistance. However, research is ongoing to define predictive biomarkers of resistance as well as prognostic markers that may be used as tools to guide treatment selection in patients with PNEOC.
For example, Lee et al.[5] have developed a nomogram to refine prognostication in this group using six pre-treatment variables [TFIp, performance status, size of the largest tumour, cancer antigen-125 (CA-125), haemoglobin and the number of metastatic organ site]. This nomogram improved overall survival prediction in patients with PEOC compared to models with fewer prognostic factors or TFIp alone. This could have applications for stratification in clinical trials and counselling patients.
An important predictive variable of response to platinum is tumour biology and histology; for example, response rates are lower in patients with clear cell, low-grade serous and mucinous ovarian cancers[6]. Tumour molecular changes, including the presence of homologous recombination deficiency, increase the likelihood of a response to platinum[7].
Mechanisms of platinum resistance
DNA damage response detection and repair
The DNA damage response is utilised to detect DNA damage and initiate DNA repair in order to maintain genomic integrity[8]. It consists of a network of interrelated signalling pathways, which can be broadly divided into homologous recombination (HR) dependent and HR independent repair pathways [Figure 1].
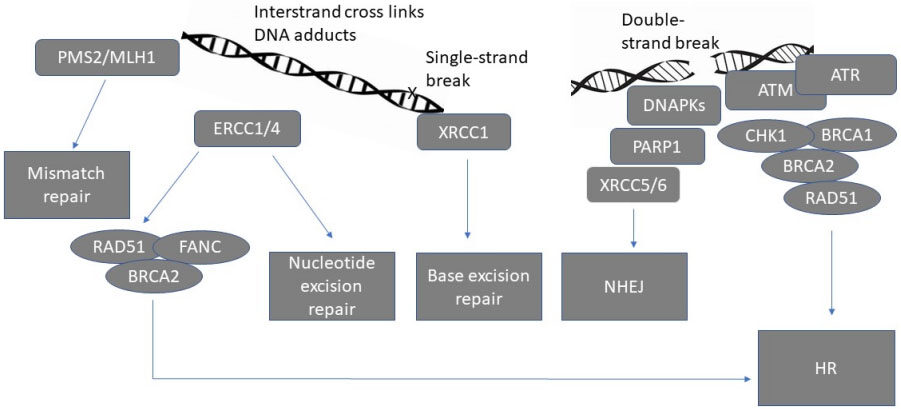
Figure 1. Schematic representation of major DNA repair pathways[8]. PMS2: postmeiotic segregation increased 2, MLH1: mutL homolog 1, ERCC1/4: Excision repair cross-complementing 1/4, RAD51: RAD51 recombinase, FANC: Fanconi anaemia complementation group, BRCA1/2 : Breast cancer gene 1/2, DNAPKs: DNA protein kinases, PARP1: Poly (ADP-ribose) polymerase, XRCC5/6: X-Ray Repair Cross Complementing 5/6, ATM: ATM Serine/Threonine Kinase, ATR: Ataxia telangiectasia and Rad3-related, CHK1: checkpoint kinase 1, HR: homologous recombination, NHEJ: non-homologous end joining.
HR dependent repair is designed to repair double-strand DNA breaks and interstrand crosslinks and, to a lesser extent, other kinds of DNA damage[9]. DNA repair, which is not dependent on HR, includes non-homologous end joining (NHEJ) for interstrand, double-strand breaks and intrastrand breaks. Other HR independent pathways that are less error-prone include mismatch repair, base excision repair and nucleotide excision repair (NER). These are typically recruited for the repair of single-strand breaks or damage induced by DNA adducts[10].
The ERCC1-XPF nuclease enzyme is translated from the mRNAs of the ERCC1 and ERCC4 genes[11]. This enzyme plays a key role in DNA damage repair chiefly via NER and also that caused by interstrand crosslinks and double-strand breaks by HR and NHEJ[11]. ERCC1-XPF targeting is a strategy being explored to increase the sensitivity of cancer cells to some DNA-damaging chemotherapeutic agents.
The master sensors (ATM, ATR, and DNA-PKs) are large kinases that sense DNA damage and initiate repair signalling cascades by phosphorylating key proteins, which are primarily involved in HR dependent repairs such as BRCA1, CHK1, CHK2, and RAD51[12]. The activation of signalling transduction pathways, including PI3K/AKT, promotes the activation of DNA damage response (DDR) cell checkpoints that halt cell cycle progression, allowing more time for DNA repair[8].
Correlation between platinum sensitivity and PARPi (PARP inhibitor) sensitivity
BRCA-deficient ovarian cancers have increased platinum sensitivity[13-14]. In vitro, reversion of BRCA mutations confers platinum and PARPi resistance[15-16]. In clinical studies, response to olaparib correlates with TFIp[17]. There is also a correlation between deficiency in other HR genes, ex vivo PARPi sensitivity, and platinum sensitivity in patients[18]. Multiple resistance mechanisms to platinum and PARPi have been described independently, although, following on from the above, significant mechanistic crossover exists.
Mechanisms of platinum and PARPi resistance
HR-dependent mechanisms of resistance include restoration of BRCA function by secondary or reversion mutations, or restoration of HR by loss of 53BP1, RIF1 or the shieldin complex amongst others [Table 1]. One major limitation of standard HR assays is that they are mostly insensitive to the detection of homologous recombination deficiency (HRD) reversion[16]. HR functional assays require viable cancer cells to be exposed to DNA damaging agents ex vivo, which therefore limits the access to samples and assay reproducibility.
Mechanisms of PARP/platinum resistance
| Mechanism | Proteins involved |
| PARP activity alteration loss of PARG increased stabilisation of replication forks | RAS PI3K/AKT PARP |
| Altered ion channel drug accumulation upregulation of drug efflux pumps Intracellular drug inactivation | VRAC MRP 2 |
| Restoration of BRCA function through secondary reversion mutation modification of other HR proteins | BRCA1/2 53BP1 RIF1 shieldin complex |
Mechanisms independent of HR include increased stabilisation of replication forks, upregulation of drug efflux pumps, PARP activity alteration, loss of PARG and RAS/PI3K/AKT pathway activation. Associated overexpression of STAT5B and RELA, two transcription factors associated with platinum resistance, is less well understood[19].
Platinum resistance may also emerge due to reduced intracellular drug accumulation, for example, through reduced intracellular drug uptake, intracellular drug inactivation, enhanced DNA repair or altered apoptotic signalling pathways[20].
Refining biomarkers of resistance to platinum and PARPi
HRD is a useful biomarker for predicting the initial response to both platinum chemotherapy and PARPi, though biomarkers of resistance require much refinement.
Standard tests for HRD, including the Myriad genomic instability score and Foundation Medicine loss of heterozygosity test, predict the presence of HRD based on genomic features[21]. These and other genomic tests vary in terms of the genomic features measured and the threshold definitions for identifying patients considered to have HRD. Clinically, HRD test results and PARPi responses can be discordant. This may be because tumours with reversion mutations that restore HR function still exhibit evidence of HRD on these assays or that alternative HR independent PARPi resistance mechanisms may be playing a predominant role. Functional assays of HR genes may overcome some of these challenges in predicting the presence of HRD[21]. The measurement of somatic mutations, such as a BRCA reversion mutation in ctDNA, is non-invasive and warrants further development[22].
Approximately 40% of high-grade serous HR proficient ovarian tumours, demonstrate increased Cyclin E expression by CCNE1 gene amplification, increased copy numbers or enhanced protein expression[23]. These CCNE1 high tumours are associated with platinum resistance and poor survival[24].
Other tumour factors contributing to treatment resistance
Tumour microenvironment
Together with genomic alterations in the DNA damage response, the tumour microenvironment is an increasingly recognised contributor to our understanding of resistance mechanisms in ovarian cancer[19].
The increased infiltration of immunosuppressive regulatory T cells has been correlated with enhanced tumour growth[25], whereas the presence of CD8+ tumour infiltrating lymphocytes is correlated with enhanced survival[26]. The most predominant immune cells associated with ovarian cancer are macrophages. Tumour-associated macrophages (TAMs) are easily polarised by tumour-cell-producing colony-stimulating factor-1 into an immunosuppressive M2-like phenotype[27]. The main pro-tumoural function of M2-like TAMs is the secretion of cytokines and exosomes that induce microRNAs, which directly promote the survival, invasion potential and chemoresistance of ovarian cancer cells[27]. PD-1, PD-L1 expression and Tumour Mutational Burden have not shown consistent validity as predictive biomarkers for immune checkpoint inhibition in ovarian cancer[19]. Retinoic acid-inducible gene-I overexpression is correlated with platinum-resistant ovarian and other refractory cancers[28]. Its overexpression is associated with local immunosuppressive changes and a distinct immune signature. Extensive stromal desmoplasia has also been associated with platinum resistance[29].
Altered metabolism in cancer tissues
Accumulating evidence suggests that tumour metabolism differs from that of matched normal tissues[30], and metabolic reprogramming may cause therapy resistance. Of relevance to platinum resistance, in one cisplatin-resistant PDX ovarian cancer model glycolysis, the tricarboxylic acid and urea cycle pathways were deregulated with higher mitochondrial respiration. This may suggest a role for therapies that modulate metabolism, such as metformin. Other drugs used in non-cancer indications and new small molecule inhibitors of mitochondrial complexes are being increasingly utilised to target cancer metabolism[31].
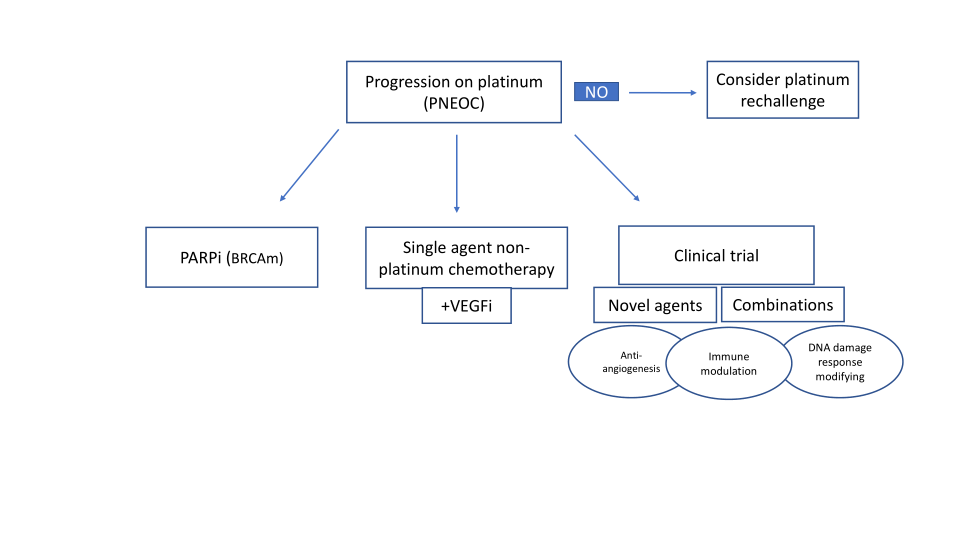
CURRENT APPROACHES TO THE MANAGEMENT OF PNEOC
Chemotherapy
Highlighting the clinical relevance of the arbitrariness of TFIp to decide on subsequent platinum retreatment, Lindemann et al.[32] compared second-line platinum vs. non-platinum regimens in a cohort of patients who would have traditionally been regarded as platinum-resistant, i.e., those with a TFIp < 6 mo. They found a greater CA-125 response rate of 51 vs. 21% (P < 0.001) in those treated with a platinum-based therapy compared to a non-platinum regimen; and in those patients with TFIp between 3 and 6 months, improved overall survival.
Using the new and modified definition of resistance, patients with PNEOC, i.e., those progressing on platinum, are typically offered non-platinum-based chemotherapy such as weekly paclitaxel, pegylated liposomal doxorubicin (PLD) or topotecan with or without bevacizumab[3]. There have been comparatively few randomised phase III trials in this setting. Table 2 summarises the key data. In the CORAIL trial, comparing lurbinectedin to PLD or topotecan in patients with a TFI < 6 mo, the PFS was similar across all groups[33]. In the AURELIA trial, patients who relapsed after 1-2 prior lines of platinum were randomised between topotecan, PLD, or weekly paclitaxel with or without bevacizumab[34]. The combination of bevacizumab with PLD, weekly paclitaxel or topotecan improved mPFS compared to chemotherapy alone. Alternative non-platinum options include oral etoposide, tamoxifen, gemcitabine and treosulfan[3].
Phase III trials in PNEOC
| Trial | Treatment Arms | mPFS | Reference |
| CORAIL | Lurbinectedin vs. control arm (PLD vs. topotecan) | 3.5 vs. 3.6 mo HR = 1.057 P = 0.6294 | Gaillard et al. (2018)[33] |
| ARIEL4 | Rucaparib vs. weekly paclitaxel (TFIp 1-6 months) | 6.4 vs. 5.7 mo HR = 0.78 | Oza (2021)[36] |
| AURELIA | Bevacizumab plus chemotherapy (PLD or topotecan or weekly paclitaxel) vs. chemotherapy alone | 6.7 vs. 3.4 mo HR = 0.48 P < 0.001 | Pujade-Lauraine |
| JAVELIN Ovarian 200 | Avelumab plus PLD vs. PLD vs. Avelumab | 3.7 vs. 3.5 vs. 1.9 mo HR one-sided P = 0.03 HR one sided P =0.99 | Pujade-Lauraine |
PARP-inhibitors
There is a role for single-agent PARP inhibitors, particularly in those with BRCA mutations that have become resistant to platinum. This is demonstrated in the single-arm Phase II QUADRA trial, in which patients were treated with niraparib after more than three lines of therapy that did not include a previous PARPi. The clinical benefit rates in patients with a BRCA1 or two mutations and TFIp < 6 months were 38% and 33%, and in the group that was platinum-refractory, 50% and 31%, at 16 and 24 weeks, respectively[35]. Although this is a single-arm Phase 2 study, the data does suggest an important role for niraparib in PNEOC patients with a BRCAm. ARIEL 4 (NCT02855944) is a Phase 3 study evaluating rucaparib vs. standard of care chemotherapy in patients with BRCA-mutated, relapsed ovarian cancer. Approximately half of the patients included in the trial had a TFIp of between 1 and 6 months, and the mPFS in this group was 6.4 months for rucaparib and 5.7 months for chemotherapy (HR 0.78, 95% CI 0.54-1.13)[36].
VEGF inhibitors
Angiogenesis is a hallmark of cancer[30], with neo-angiogenesis abundantly present in ovarian cancer. Antiangiogenic therapy plus chemotherapy has shown an improvement in responses and PFS in PEOC compared to chemotherapy alone[3], and improvements in PFS have also been demonstrated in patients with a TFIp < 6 months in the Phase III AURELIA trial using the VEGF-A monoclonal antibody, bevacizumab, plus chemotherapy[34], or in smaller randomised Phase II trials of VEGF-R small molecule inhibitors, pazopanib with weekly paclitaxel (MITO-11)[37] or sorafenib with topotecan (TRIAS)[38].
In AURELIA, the combination of bevacizumab with PLD, weekly paclitaxel or topotecan improved mPFS compared to chemotherapy alone in patients who relapsed after 1-2 prior lines of platinum[34] [Table 2]. However, it remains unclear based on these data when it might be appropriate to stop chemotherapy in those continuing to respond to the combination.
Immune checkpoint inhibitors
The results of trials of Immune checkpoint inhibitors (ICPI) monotherapy in ovarian cancer have been disappointing. In two Phase II trials of programmed cell death protein-1/ligand-1 (PD-1/PD-L1) inhibitors, pembrolizumab and avelumab showed little benefit in ovarian cancer cohorts[39-40]; however, it was hoped that in subgroups of patients including PNEOC patients, they may have a niche role.
Avelumab, either alone or in combination with PLD in platinum-resistant ovarian cancer (JAVELIN Ovarian 200), failed to show a significant OS benefit compared to PLD alone [Table 2]. However, exploratory analyses suggest there may have been a benefit of the combination in those with an initial response to earlier lines of chemotherapy[41]. As a role for bevacizumab has been demonstrated in AURELIA, the question of whether ICPI enhances this benefit is relevant. NRG-Gy009 study (NCT02839707), which has completed recruitment, compared the combinations of PLD and bevacizumab vs. PLD and atezolizumab vs. PLD and atezolizumab and bevacizumab; the results are awaited [Table 3].
Combination trials of interest
| clinicaltrials.gov identifier | Treatment arms | Proposed mechanism of synergy |
| NCT02502266 (NRG-Gy005) | Olaparib vs. cediranib vs. olaparib-cediranib vs. investigator’s choice of chemotherapy (paclitaxel/topotecan/PLD). | antiangiogenic therapy induces a hypoxic tumour microenvironment, thereby enhancing synthetic lethality by downregulation of HR genes |
| NCT02839707 (NRG-Gy009) | PLD and bevacizumab vs. PLD and atezolizumab vs. PLD and atezolizumab and bevacizumab | VEGF targeting reduces inhibition of tumour immune cell suppression which permits increased efficacy of PD-L1 inhibition and chemotherapy cytotoxicity |
| NCT04065269 (ATARI) | ceralasertib and olaparib vs. ceralasertib | ATR plus PARP inhibition overcomes PARPi resistance by inducing increases in replication fork stalling, double-strand breaks, and apoptosis |
Combining two ICPIs, such as anti-PD-1 and anti-cytotoxic T lymphocyte-associated-4 antibodies, may increase the activity of immunotherapy with evidence that nivolumab and ipilimumab showed a longer PFS than nivolumab alone (mPFS of 3.9 vs. 2 months), albeit with greater toxicity[42]. However, these figures are notably comparable to those seen for single-agent non-platinum-based chemotherapies.
Other trials are exploring the use of maintenance immunotherapy after chemotherapy to improve PFS. One such study is the PROMPT Phase II trial, in which patients receive pembrolizumab after 4-6 cycles of weekly paclitaxel (NCT03430700).
NEWER STRATEGIES FOR OVERCOMING PLATINUM RESISTANCE
The above data show that with current regimens, mPFS is short, and tools to select patients likely to benefit most are required.
Refinements in patient selection for bevacizumab
There are currently no predictive biomarkers for bevacizumab response available in the clinic. Angiogenic markers, including micro-vessel density, CD31 expression and tumour VEGF-A levels, may provide prognostic information in recurrent ovarian cancer. These were identified in the Gynecologic Oncology Group (GOG) 218 study as potential predictive biomarkers for the use of bevacizumab[43]. Another retrospective analytical study showed that a signature comprising alpha-1 acid glycoprotein, mesothelin, FLT4 and CA-125 identified those patients more likely to benefit from bevacizumab[44]. In a concordance exploratory study of ICON7 samples, plasma concentrations of several angiogenesis-associated factors were determined using multiplex ELISAs, with high Ang1 and low Tie2 levels correlating best with PFS.
Tie1 and 2 are receptor tyrosine kinases that function as key regulators of blood vessel development and pathological processes including angiogenesis[45]. One observational biomarker study (VALTIVE) is currently recruiting to determine the clinical value of measuring plasma Tie2 concentrations in ovarian cancer patients who are receiving bevacizumab (NCT04523116).
Novel treatments
Targeting Ataxia telangiectasia and Rad3-related
Targeting Ataxia telangiectasia and Rad3-related (ATR) is an important kinase regulating the DDR. It is responsible for sensing replication stress and signalling to cell cycle checkpoints to initiate repair[46]. ATR inhibitors have been shown to reduce the rate of DNA repair in cells, thereby increasing DNA damage and apoptosis[47]. Single-agent ATR inhibition appears to show some efficacy in PNEOC[46].
G-Quadruplex (G4) stabilisation
G4 structures can form at thousands of sequences in the human genome and increase the propensity for DNA damage by impeding DNA polymerase, and thereby DNA damage repair processes[48]. CX-5461 is a small molecule RNA polymerase transcription inhibitor that selectively kills HR deficient cancer cells by stabilising G4 structures[49]. Phase 1 studies of CX-5461 are being investigated in solid tumours, including in a platinum/PARPi resistant ovarian cancer cohort (NCT04890613).
Cell cycle checkpoint inhibition
The cell cycle checkpoint regulators CHK1 and CHK2 halt cell division to allow DNA damage to be repaired before DNA replication[50]. Cell cycle checkpoint inhibition may thereby prevent the progression of cancer cells through the cell cycle, halting replication and tumour progression. WEE-1 inhibitors block the activity of WEE-1 kinase, a G2 cell-cycle checkpoint, and enhance cancer cell apoptosis[51].
Prexasertib is one example of a CHK1 inhibitor, which demonstrated responses in a phase II trial that were most marked in patients with platinum-resistant ovarian cancer[52]. A phase II study of the combination of AZD1775, a WEE-1 inhibitor and carboplatin in platinum-resistant ovarian cancer, demonstrated an ORR of 43%[53].
Epigenetic re-sensitisation
Treatment resistance is often associated with the accumulation of epigenetic changes[54]. It has therefore been hypothesised that epigenetic modulation may re-sensitise tumours to platinum chemotherapy.
The DNA methyltransferase (DNMT) and Histone Deacetylase inhibitors have shown little activity as single agents in platinum-resistant ovarian cancer. However, in combination, they may enhance sensitivity to platinum by altering epigenetic regulation of gene expression. In a randomised phase II study assessing the DNMT inhibitor, guadecitabine in combination with carboplatin vs. investigator’s choice of chemotherapy, the PFS rate at 6 months was 37% vs. 11%[55]. The DNA damage initiated by DNMT inhibitors is repaired by the BER pathway, in which PARP1 plays a key role, and therefore there may also be a rationale to combine DNMT and PARP inhibition.
Combination approaches
Angiogenesis and PARPi
Antiangiogenic therapy has been shown to induce a hypoxic tumour microenvironment associated with the downregulation of HR genes[56], providing the rationale to enhance the synthetic lethality of PARPi with angiogenesis inhibitors which also separately work to interfere with angiogenesis.
The Phase II AVANOVA2 trial compared the combination of niraparib and bevacizumab vs. single-agent niraparib in patients with PEOC[57]. Niraparib plus bevacizumab significantly improved mPFS compared with niraparib alone (11.9 mo vs. 5.5 mo; HR 0.35, P < 0.0001) and has provided a rationale to test this strategy in PNEOC.
EVOLVE was a phase II trial of cediranib-olaparib in ovarian cancer progressing on PARPi, recruiting a cohort of patients who were also defined as platinum-resistant, with 2/10 patients in this cohort demonstrating a PR[58]. The anti-tumour activity of this combination continues to be assessed in the randomised Phase III NRG-Gy005 (NCT02502266) trial currently recruiting patients with platinum-resistant ovarian cancer to receive either olaparib, cediranib, olaparib-cediranib or investigator’s choice of chemotherapy (paclitaxel/topotecan/PLD) [Table 3].
DDR response
In a large panel of acquired and de novo PARPi- and platinum-resistant CCNE1 amplified in vitro and PDX models, ATR and PARPi synergy was demonstrated[59]. This, amongst other data, has provided the rationale for the combination of ceralasertib and olaparib for recurrent platinum-resistant ovarian cancer in CAPRI[60]. Although no objective responses were demonstrated, the combination was well tolerated, and in two patients with BRCA1 mutations, a 50% fall in CA-125 was seen.
It will be interesting to see the data from the combination arm of ATARI (NCT04065269) in platinum-resistant ovarian clear cell cancer and carcinosarcomas, which may provide insights into how the combination of these drugs may alter the DDR in those subgroups of patients that do not classically display responsiveness to chemotherapy and PARPi [Table 3].
There is a clear rationale to combine other drugs regulating the DDR described earlier in this review, for example, WEE1 inhibitors, with platinum-based chemotherapy and PARPi.
Immunotherapeutic combinations
An alternative strategy is the combination of an ICPI with a PARPi, chemotherapy or other DDR modifying drugs. One possible mechanism of synergy is the observation that PARPi can activate the STING (stimulator of interferon genes) pathway to increase T-cell tumour infiltration[61]. TOPACIO was a Phase 1/2 trial testing niraparib plus pembrolizumab in platinum-resistant ovarian and triple-negative breast cancer patients. A subgroup analysis of the ovarian cancer cohort showed that the combination was promising for patients without HR deficiency[62]. There was a small cohort of patients in this group, and therefore other larger studies will need to focus on immunogenomic profiling to select patients most likely to benefit from this strategy.
CONCLUSION
Although the definition of true resistance to platinum-based chemotherapy has changed over the last four decades, few treatments have significantly changed outcomes in the vast majority of patients in this cohort. Next-generation sequencing has become faster and more affordable due to automation, which is permitting standardisation of techniques for analysing liquid biopsies and immunogenomic profiling. These refinements may lead to an improvement in patient selection for some of the novel strategies and combinations discussed in this review. Biomarker-driven trial designs will accelerate the better selection of and sequencing of treatment lines, including those targeting immune modulation, modification of the DNA damage response and angiogenesis inhibition.
DECLARATIONS
Authors’ contributionsWriting and review of the manuscript: Flynn MJ, Ledermann JA.
Availability of data and materialsNot applicable.
Financial support and sponsorshipGrants: AstraZeneca; Merck/MSD.
Conflicts of interestMJF. Nothing to declare.
JAL: Advisory Boards and Lecture Fees: AstraZeneca; Clovis Oncology; Tesaro/GSK; Merck/MSD; Neopharm Advisory Boards: Pfizer; Artios Pharma; Eisai; VBL Therapeutics; Bristol Myers Squibb; Nuvation. IDMC: Regeneron.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Blackledge G, Lawton F, Redman C, Kelly K. Response of patients in phase II studies of chemotherapy in ovarian cancer: implications for patient treatment and the design of phase II trials. Br J Cancer 1989;59:650-3.
2. Markman M, Hoskins W. Responses to salvage chemotherapy in ovarian cancer: a critical need for precise definitions of the treated population. J Clin Oncol 1992;10:513-4.
3. Baert T, Ferrero A, Sehouli J, et al. The systemic treatment of recurrent ovarian cancer revisited. Ann Oncol 2021;32:710-25.
4. Colombo N, Sessa C, du Bois A, et al. ESMO-ESGO Ovarian Cancer Consensus Conference Working Group. ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease†. Ann Oncol 2019;30:672-705.
5. Lee CK, Simes RJ, Brown C, et al. A prognostic nomogram to predict overall survival in patients with platinum-sensitive recurrent ovarian cancer. Ann Oncol 2013;24:937-43.
6. Bamias A, Sotiropoulou M, Zagouri F, et al. Prognostic evaluation of tumour type and other histopathological characteristics in advanced epithelial ovarian cancer, treated with surgery and paclitaxel/carboplatin chemotherapy: cell type is the most useful prognostic factor. Eur J Cancer 2012;48:1476-83.
7. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 2014;20:764-75.
8. Yap TA, Plummer R, Azad NS, Helleday T. The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ Book 2019;39:185-95.
9. Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res-Fund Mol M 2001;477:131-53.
10. Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011;8:16-29.
11. Manandhar M, Boulware KS, Wood RD. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene 2015;569:153-61.
12. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol Cell 2017;66:801-17.
13. Taniguchi T, Tischkowitz M, Ameziane N, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 2003;9:568-74.
14. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006;66:8109-15.
15. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008;451:1116-20.
16. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008;68:2581-6.
17. Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol 2010;28:2512-9.
18. Mukhopadhyay A, Plummer ER, Elattar A, et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res 2012;72:5675-82.
19. McMullen M, Karakasis K, Madariaga A, Oza AM. Overcoming platinum and PARP-inhibitor resistance in ovarian cancer. Cancers (Basel) 2020;12:1607.
20. Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer 2021;21:37-50.
21. Stover EH, Fuh K, Konstantinopoulos PA, Matulonis UA, Liu JF. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol Oncol 2020;159:887-98.
22. Giannopoulou L, Zavridou M, Kasimir-Bauer S, Lianidou ES. Liquid biopsy in ovarian cancer: the potential of circulating miRNAs and exosomes. Transl Res 2019;205:77-91.
23. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 2015;5:1137-54.
24. Chan AM, Enwere E, McIntyre JB, et al. Combined CCNE1 high-level amplification and overexpression is associated with unfavourable outcome in tubo-ovarian high-grade serous carcinoma. J Pathol Clin Res 2020;6:252-62.
25. Ojalvo LS, Thompson ED, Wang TL, et al. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum Pathol 2018;74:135-47.
26. Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol 2012;124:192-8.
27. Nowak M, Klink M. The role of tumor-associated macrophages in the progression and chemoresistance of ovarian cancer. Cells 2020;9:1299.
28. Wolf D, Fiegl H, Zeimet AG, et al. High RIG-I expression in ovarian cancer associates with an immune-escape signature and poor clinical outcome. Int J Cancer 2020;146:2007-18.
29. Patch AM, Christie EL, Etemadmoghadam D, et al. Australian Ovarian Cancer Study Group. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489-94.
31. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov 2022;21:141-62.
32. Lindemann K, Gao B, Mapagu C, et al. Australian Ovarian Cancer Study Group. Response rates to second-line platinum-based therapy in ovarian cancer patients challenge the clinical definition of platinum resistance. Gynecol Oncol 2018;150:239-46.
33. Gaillard S, Oaknin A, Ray-coquard I, et al. Phase III trial of lurbinectedin versus PLD or topotecan in platinum-resistant ovarian cancer patients: results of CORAIL trial. Annals of Oncology 2018;29:viii332.
34. Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol 2014;32:1302-8.
35. Moore KN, Secord AA, Geller MA, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. The Lancet Oncology 2019;20:636-48.
36. Oza AM, Lisyanskaya AS, Fedenko AA, et al. Subgroup analysis of rucaparib versus chemotherapy as treatment for BRCA-mutated, advanced, relapsed ovarian carcinoma: effect of platinum sensitivity in the randomized, phase 3 study ARIEL4. JCO 2021;39:5517-5517.
37. Pignata S, Lorusso D, Scambia G, et al. Pazopanib plus weekly paclitaxel versus weekly paclitaxel alone for platinum-resistant or platinum-refractory advanced ovarian cancer (MITO 11): a randomised, open-label, phase 2 trial. The Lancet Oncology 2015;16:561-8.
38. Chekerov R, Hilpert F, Mahner S, et al. Sorafenib plus topotecan versus placebo plus topotecan for platinum-resistant ovarian cancer (TRIAS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 2018;19:1247-58.
39. Matulonis UA, Shapira-Frommer R, Santin AD, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol 2019;30:1080-7.
40. Disis ML, Taylor MH, Kelly K, et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: phase 1b results from the JAVELIN solid tumor trial. JAMA Oncol 2019;5:393-401.
41. Pujade-lauraine E, Fujiwara K, Ledermann JA, et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): an open-label, three-arm, randomised, phase 3 study. The Lancet Oncology 2021;22:1034-46.
42. Zamarin D, Burger RA, Sill MW, et al. Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: an NRG oncology study. J Clin Oncol 2020;38:1814-23.
43. Bais C, Mueller B, Brady MF, et al. NRG Oncology/Gynecologic Oncology Group. Tumor microvessel density as a potential predictive marker for bevacizumab benefit: GOG-0218 biomarker analyses. J Natl Cancer Inst 2017:109.
44. Collinson F, Hutchinson M, Craven RA, et al. Predicting response to bevacizumab in ovarian cancer: a panel of potential biomarkers informing treatment selection. Clin Cancer Res 2013;19:5227-39.
45. Leppänen VM, Saharinen P, Alitalo K. Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proc Natl Acad Sci U S A 2017;114:4376-81.
46. Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol Ther 2020;207:107450.
47. Foote KM, Nissink JWM, McGuire T, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem 2018;61:9889-907.
48. Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol 2015;33:877-81.
49. Khot A, Brajanovski N, Cameron DP, et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: results of a phase I dose-escalation study. Cancer Discov 2019;9:1036-49.
50. Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol Cell 2015;59:1011-24.
51. Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013;12:3159-64.
52. Lee J, Nair J, Zimmer A, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. The Lancet Oncology 2018;19:207-15.
53. Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol 2016;34:4354-61.
54. Li M, Balch C, Montgomery JS, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics 2009;2:34.
55. Oza AM, Matulonis UA, Alvarez Secord A, et al. A randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin Cancer Res 2020;26:1009-16.
56. Kaplan AR, Gueble SE, Liu Y, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med 2019;11:eaav4508.
57. Mirza MR, Åvall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. The Lancet Oncology 2019;20:1409-19.
58. Lheureux S, Oaknin A, Garg S, et al. EVOLVE: A Multicenter open-label single-arm clinical and translational phase II trial of cediranib plus olaparib for ovarian cancer after PARP inhibition progression. Clin Cancer Res 2020;26:4206-15.
59. Kim H, Xu H, George E, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020;11:3726.
60. Shah PD, Wethington SL, Pagan C, et al. Combination ATR and PARP inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol 2021;163:246-53.
61. Wang Z, Sun K, Xiao Y, et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep 2019;9:1853.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Flynn MJ, Ledermann JA. Ovarian cancer recurrence: is the definition of platinum resistance modified by PARPi and other intervening treatments? The evolving landscape in the management of platinum-resistant ovarian cancer. Cancer Drug Resist 2022;5:424-35. http://dx.doi.org/10.20517/cdr.2022.13
AMA Style
Flynn MJ, Ledermann JA. Ovarian cancer recurrence: is the definition of platinum resistance modified by PARPi and other intervening treatments? The evolving landscape in the management of platinum-resistant ovarian cancer. Cancer Drug Resistance. 2022; 5(2): 424-35. http://dx.doi.org/10.20517/cdr.2022.13
Chicago/Turabian Style
Flynn, Michael J., Jonathan A. Ledermann. 2022. "Ovarian cancer recurrence: is the definition of platinum resistance modified by PARPi and other intervening treatments? The evolving landscape in the management of platinum-resistant ovarian cancer" Cancer Drug Resistance. 5, no.2: 424-35. http://dx.doi.org/10.20517/cdr.2022.13
ACS Style
Flynn, MJ.; Ledermann JA. Ovarian cancer recurrence: is the definition of platinum resistance modified by PARPi and other intervening treatments? The evolving landscape in the management of platinum-resistant ovarian cancer. Cancer Drug Resist. 2022, 5, 424-35. http://dx.doi.org/10.20517/cdr.2022.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 6
likes
Like This Article 6
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.