
Potential mechanisms of resistance to current anti-thrombotic strategies in Multiple Myeloma
 0
0Abstract
Multiple Myeloma (MM) is a common haematological malignancy that is associated with a high rate of venous thromboembolism (VTE) with almost 10% of patients suffering thrombosis during their disease course. Recent studies have shown that, despite current thromboprophylaxis strategies, VTE rates in MM remain disappointingly high. The pathophysiology behind this consistently high rate of VTE is likely multifactorial. A number of factors such as anti-thrombin deficiency or raised coagulation Factor VIII levels may confer resistance to heparin in these patients, however, the optimal method of clinically evaluating this is unclear at present, though some groups have attempted its characterisation with thrombin generation testing (TGT). In addition to testing for heparin resistance, TGT in patients with MM has shown markedly varied abnormalities in both endogenous thrombin potential and serum thrombomodulin levels. Apart from these thrombin-mediated processes, other mechanisms potentially contributing to thromboprophylaxis failure include activated protein C resistance, endothelial toxicity secondary to chemotherapy agents, tissue factor abnormalities and the effect of immunoglobulins/“M-proteins” on both the endothelium and on fibrin fibre polymerisation. It thus appears clear that there are a multitude of factors contributing to the prothrombotic milieu seen in MM and further work is necessitated to elucidate which factors may directly affect and inhibit response to anticoagulation and which factors are contributing in a broader fashion to the hypercoagulability phenotype observed in these patients so that effective thromboprophylaxis strategies can be employed.
Keywords
INTRODUCTION
Background
Multiple Myeloma (MM) is a common haematological malignancy that remains a significant therapeutic challenge despite major advances in both treatments and biological understanding of the disease. Almost all patients will suffer relapse and in conjunction with this, MM is associated with a high rate of venous thromboembolism (VTE). It has long been acknowledged that there is a strong association between cancer and thrombosis but the risk of these events varies widely across the spectrum of cancer subgroups. VTE rates remain high in patients with MM with almost 10% of patients suffering from thrombosis during their disease course[1,2].
The occurrence of a thrombotic event in patients with MM can be associated with significant morbidity and can lead to interruption of treatment, requirement for anticoagulation use and indeed may preclude the use of certain agents in future treatment regimens. Certain studies have also suggested that patients with MM who suffer a thrombotic event are also more likely to have poorer overall survival though further confirmatory data is awaited in this area[3,4]. In light of these issues, optimisation of thromboprophylaxis and further understanding of the mechanisms conferring resistance to and failure of current anti-thrombotic strategies is an ongoing critical area of research [Figure 1].
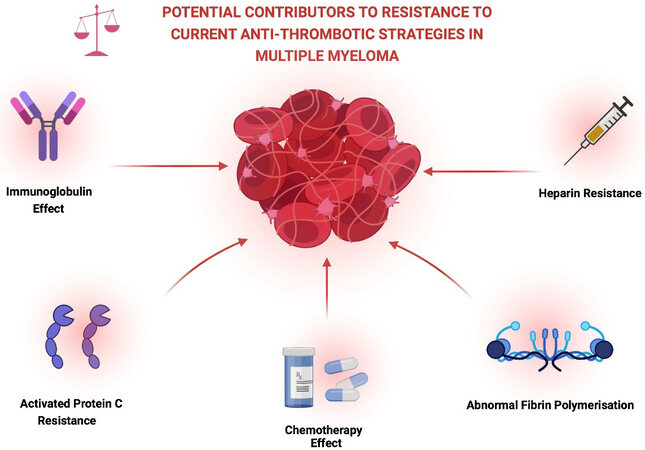
Figure 1. Description of some of the possible contributory factors to the persistently elevated VTE rates seen in multiple myeloma. Created with BioRender.com.
Treatment of MM
The treatment of MM has rapidly evolved over the past number of years with many therapeutic options now available. Triplet/three drug combinations are the initial standard of care for induction therapy though choice of treatment will depend on factors such as patient fitness, local treatment guidelines and availability/funding of therapy[5,6]. Generally, in younger, fitter patients who may be eligible for autologous transplantation (ASCT), initial therapy will comprise of a proteasome inhibitor such as bortezomib and either an immunomodulatory (IMiD) agent such as lenalidomide or an alkylating agent such as cyclophosphamide. These drugs are usually combined with dexamethasone, a corticosteroid, for maximum efficacy[5]. There are also very efficacious treatments available for older patients or those who may not be fit for ASCT including a combination of lenalidomide and dexamethasone or VMP therapy (bortezomib, melphalan and prednisolone)[5,7]. Upon relapse of disease, treatment options include carfilzomib (a second generation PI), alternative IMiDs such as pomalidomide and anti-CD38 monoclonal antibodies including daratumumab and isatuximab[5,7]. It is well recognised that IMiD therapy confers a high thrombotic risk to the patient, particularly when combined with high doses of steroids, but the exact thrombotic risks of many other agents have been less well characterised[8,9].
Risk assessment of venous thromboembolism
Patients with MM have a multitude of risk factors for development of thrombosis. These can generally be categorised into patient related factors such as prior VTE and increased body mass index (BMI), MM related factors (e.g., renal impairment, hyperviscosity, disease activity, reduced mobility) and therapy-related risk factors. There are a number of models and predictive scores that have attempted to stratify individual risk for VTE. The Khorana score is widely used to identify risk for cancer-associated VTE in patients with malignancy who are initiating systemic therapy. It is comprised of five clinical and laboratory indices, including type of cancer, platelet count, haemoglobin level and white cell count (WCC) as well as BMI[10]. However, given that it is largely based on solid organ tumour data, it has been recognised that this score likely has limitations in the MM population with several studies observing that it does not accurately predict VTE risk within this cohort[2,11,12]. In fact, in a study of over 300 patients with MM, Barrett et al.[2] noted that WCC was the only single variable from the Khorana score that retained predictive significance for VTE in their cohort.
The current International Myeloma Working Group (IMWG) has provided guidance, based on stratification by Palumbo et al.[13], on thromboprophylaxis in MM by compiling a VTE risk assessment incorporating individual, therapy- and myeloma-related factors. This guidance has also been incorporated into the National Comprehensive Cancer Network (NCCN) guidelines but several groups have observed that these guidelines do not fully capture VTE risk and thus have sought to develop novel and more rigorous VTE risk assessment tools [Table 1]. Sanfilippo et al.[12] explored individual risk factors for VTE and combined the relevant factors to develop a risk prediction model called the “IMPEDE VTE” score which attributes scores to each contributory risk factor. Similarly, Li et al.[14] evaluated the performance of the current NCCN guidelines in a large population based cohort while simultaneously developing their own VTE risk-assessment model specifically for patients undergoing therapy with IMiDs. They concluded that their own model, the SAVED score, which incorporated recent surgery, Asian ethnicity, prior history of VTE, age > 80 and dexamethasone dose had a greater discriminative power than the current consensus recommended by the NCCN guidelines[14]. While these scoring algorithms have advanced VTE-risk discrimination in MM, some concerns have been raised that these scores were designed using retrospective data which was based upon therapies that are now less commonly used[15]. Incorporation of additional biomarkers may further improve the accurate risk prediction of VTE for this vulnerable cohort.
Recently developed risk stratification tools for VTE in multiple myeloma
| IMPEDE VTE score Variable | Score | SAVED score Variable | Score |
| IMiD (immunomodulatory agent) | 4 | Recent surgery (within last 90 days) | 2 |
| Body mass index (>/ 25 kg/m2) | 1 | Ethnicity: Asian race | -3 |
| Fracture (pelvic, hip or femur) | 4 | Prior history of VTE | 3 |
| Use of erythropoietin-stimulating agent | 1 | Age “eight” > 80 | 1 |
| Use of doxorubicin chemotherapy | 3 | Dexamethasone use (standard or high) | 1 or 2 |
| Use of dexamethasone (high-dose/low-dose) | 4/2 | Low risk: </ 1 High risk: >/ 2 | |
| Ethnicity = Pacific Islander/Asian | -3 | ||
| Prior VTE (preceding MM) | 5 | ||
| Presence of tunneled line | 2 | ||
| Current/existing use of therapeutic LMWH/warfarin | -4 | ||
| Current/existing use of prophylactic LMWH/aspirin | -3 | ||
| Low risk: </ 3 High risk: 4-7 Very high risk: >/ 8 | |||
Current anti-thrombotic regimens in MM
It is well recognised that due to the unacceptably high thrombosis rates in MM, pharmacological thromboprophylaxis is often necessitated to try mitigate this risk. The IMWG recommend the use of aspirin in lower risk patients and either prophylactic-dose low molecular weight heparin or treatment-dose warfarin in patients at higher risk[13]. In more recent times, focus has switched to evaluating the effectiveness of the direct oral anticoagulant medications (DOACs) including the factor Xa inhibitors (apixaban, rivaroxaban) and factor II inhibitors (dabigatran) in prevention of VTE in MM. While DOACs have been shown in several large trials to be effective and safe in both treating and preventing VTE in patients with malignancy, very few studies have sought to confirm this effect specifically in cohorts of MM patients[16-18]. Despite this, a number of small scale studies have cautiously reported their experiences of the safe and effective use of DOACs in MM.
Storrar et al.[19] have described their experience of apixaban thromboprophylaxis in 70 patients with MM, all receiving IMiDs as part of induction therapy. These patients were treated with apixaban 2.5 mg twice daily and there were no reports of VTE during the first six months of therapy with low rates of major bleeding also observed. They concluded that apixaban appeared to be a safe and attractive option for this cohort of patients. Similarly, Cornell et al.[20] reported their experiences of apixaban thromboprophylaxis in a more heterogenous group of 50 patients with MM undergoing IMiD-based therapy at various timepoints of the disease course. Again, no instances of VTE were observed in the first six months of observation and, reassuringly there were also no instances of major haemorrhage during this time period. They too concluded that low-dose apixaban was safe and well-tolerated as primary prevention of VTE in patients receiving IMiD therapy. These are, however, small-scale studies and larger, prospective studies will be required to provide conclusive evidence for the long term use of DOACs in MM.
VTE rates despite thromboprophylaxis: immunomodulatory agents
The rate of VTE occurrence remains unacceptably high in MM patients with approximately 10% of patients developing a thrombosis during the course of their disease[21,22]. It has been demonstrated that thrombosis rates in patients with malignancy are highest in the first year following diagnosis and in particular, in MM, thrombosis rates appear to be highest in the first 3 months following diagnosis[1,13,22,23]. It is hypothesised that these initial high rates are due to both accelerated neoplastic activity, increased tumour burden and, with treatment initiation, there may be overwhelming release of procoagulant factors and cytokines once apoptosis ensues. Given the particularly high risk of VTE associated with IMiDs, adjuvant pharmacological thromboprophylaxis is now widely in use. However, despite this, many groups have reported that VTE rates remain persistently elevated. Leclerc et al.[22] reported their observations from a cohort of 213 patients who had received IMiD therapy where thrombotic events were reported in 18.3% of patients with more than half of these events occurring during the first three months of therapy, during which time the vast majority of patients were receiving thromboprophylaxis. VTE occurred more often with anti-platelet use rather than with anticoagulation, an observation that was also seen in the MELISSE study[22,24].
Bradbury et al.[25] also recently described the thrombosis rates during the Myeloma IX and Myeloma XI trials. Both trials are phase 3 trials incorporating the upfront use of an IMiD and corticosteroid-containing regimens as induction therapy with the Myeloma IX study containing data from 1936 patients and the Myeloma XI study containing data from 4358 patients. Due to the interim development of the aforementioned IMWG risk assessment guidelines, there were higher rates of thromboprophylaxis (80.5% vs. 22.3%) in the later Myeloma XI study but despite this, there was only a modest reduction in thrombosis rates in the latter trial. In the Myeloma IX trial, the 6 months cumulative thrombosis incidence rate was 20.7% in the CVAD (cyclophosphamide, vincristine, adriamycin, dexamethasone) group (non IMiD based) and 15.0% in the CTD (cyclophosphamide, thalidomide, dexamethasone) group (IMiD containing) while in the Myeloma XI trial, 6 months cumulative incidence of VTE was 10.7% in the CRD (cyclophosphamide, lenalidomide, dexamethasone) group and 11.7% in the CTD group, thus showing that despite adherence to current thromboprophylaxis guidelines, VTE rates remain disappointingly high[25].
High VTE rates with other anti-myeloma therapies
Despite IMiDs often being regarded as the culpable prothrombotic agent, consideration has to be given to the thrombotic risk posed by other classes of drugs used to treat MM and worryingly, some of the more novel agents appear to be increasingly linked with thrombosis. Carfilzomib is a second generation proteasome inhibitor with reported VTE rates of 5%-14% in clinical trials and although thromboprophylaxis is recommended for MM patients receiving carfilzomib-containing regimens, the most effective thromboprophylaxis strategy remains to be determined, nor has the mechanism underlying carfilzomib-associated thrombosis been adequately studied[26-29]. The findings of Piedra et al.[29] suggest that these patients might benefit from a more enhanced thromboprophylaxis regimen. They recently reported their observations from a retrospective study of VTE incidence across different induction regimens used in newly diagnosed MM (NDMM). They compared rates between KRD (carfilzomib, lenalidomide, dexamethasone) + aspirin, RVD (lenalidomide, bortezomib, dexamethasone) + aspirin and KRD + rivaroxaban and found significantly differing rates of VTE depending on thromboprophylaxis agent used with rates of 16.1%, 4.8% and 4.8% observed in the groups respectively. This suggests that carfilzomib may be more thrombogenic than bortezomib (when combined with lenalidomide), a first generation proteasome inhibitor, though this risk appeared to be somewhat mitigated by using a DOAC rather than an anti-platelet agent[29]. In addition to this, recent advances in cellular therapy have led to the availability of anti-BCMA chimeric antigen receptor T (CAR-T) cell therapy in MM. CAR T-cell therapy is associated with high rates of VTE with
POSSIBLE MECHANISMS OF RESISTANCE: THROMBIN-MEDIATED
Heparin resistance
Potential mechanisms contributing to heparin resistance
As discussed above, many patients with MM who are deemed at high risk of VTE are administered thromboprophylaxis with LMWH and it is also commonly utilised at therapeutic doses for treatment of thrombotic episodes in MM. Heparin binds to antithrombin and accelerates the interaction between antithrombin and thrombin or antithrombin and factor Xa, both of which should result in inhibition of prothrombotic activity. Heparin resistance is often defined as the need for higher heparin doses to achieve a targeted level of anticoagulation but it may be multifactorial and its identification is complicated by the lack of consensus on the appropriate target levels in conjunction with the optimal method for measuring heparin effect[31].
One of the important mechanisms responsible for heparin resistance is deficiency of antithrombin (formerly known as antithrombin III). Many disease states such as liver failure, sepsis and treatments, such as asparaginase use in acute leukaemia, are associated with reductions in antithrombin levels but the prevalence of antithrombin deficiency in MM is far less well known and has not been evaluated on a large scale basis[31,32]. It is however, well described that significant proteinuria can contribute to thrombotic potential and that reduced serum albumin levels are associated with increased urinary protein loss, including that of antithrombin[33]. This may be of particular significance both in patients with MM and amyloidosis given the high rates of kidney impairment seen in these conditions. Thus, certain groups have highlighted the need for consideration for testing for antithrombin deficiency in patients with MM in the context of thrombosis and significant proteinuria[21].
Aside from antithrombin deficiency, an additional factor that may contribute to heparin resistance is presence of high levels of coagulation Factor VII (FVIII). Increased doses of heparin have been necessitated for effective thromboprophylaxis in patients who exhibit high levels of FVIII such as patients suffering with acute infective/inflammatory conditions such as Covid-19[31]. Increased FVIII levels in MM have been reported by several groups with particular elevations seen in those undergoing IMiD therapy[34-36]. This raises the question that perhaps higher doses of heparin are necessitated in such patients to achieve adequate protective anticoagulation levels and thus reduce the high rates of VTE seen with current standard dosage.
Evaluating heparin resistance with thrombin generation testing
Thrombin generation assays (TGAs) are global haemostasis assays that seek to assess the entire coagulation process and are thought to be more sensitive than a standard coagulation profile given that the endpoint is conversion of fibrinogen to fibrin. They are in vitro assays that utilise tissue factor (TF) and phospholipids to activate the coagulation cascade. The concentration of thrombin is thus measured over a defined time period and a curve is generated to illustrate this production. Various measurements can then be extrapolated from the curve to describe thrombin activity such as lag time (time until thrombin initiation), peak thrombin generation and endogenous thrombin potential (ETP) which reflects the thrombin formation capacity of the plasma[37]. Several groups have postulated that patients with MM may be “resistant” to treatment with heparin and have sought to evaluate this resistance using TGAs.
Chalayer et al.[38] describe their observations where they sought to characterise the effect of heparin thromboprophylaxis on thrombin generation in a cohort of patient with MM with a view to exploring potential mechanisms of heparin resistance. They compared a group of patients with MM with intermediate/high VTE risk factors with a group of patients hospitalised with respiratory illness who were deemed to have a low thrombotic risk. They observed similar ETP levels in both of the groups and concluded that there was no in vitro evidence of specific resistance to LMWH in this particular cohort of MM patients[38]. Gracheva et al.[39] also assessed a variety of global haemostasis assays (including TGAs, thromboelastography and thrombodynamics) in a cohort of 59 patients, 25 with NDMM and 34 patients undergoing stem cell mobilisation. They compared their results with a cohort of healthy individuals and found that that both thrombodynamics and thromboelastography showed hypercoagulability in both sets of MM patients when compared with the healthy cohort. They then assessed TGAs in the patients undergoing stem cell mobilisation who were receiving heparin therapy and found widely varied results for each individual with no demonstration of hypocoagulability in some patients and more worryingly, a shift towards hypercoagulability in over 20% of patients. The authors concluded that the use of heparin therapy, at its current dose, was ineffective in these patients and that it may either indicate inadequate dosage or heparin resistance in this cohort[39]. Given the stark discrepancy between these studies, further research in larger, more heterogenous groups of MM patients would be necessitated to evaluate if heparin resistance is prevalent and clinically relevant in MM.
Predicting VTE risk with thrombin generation assay testing
Thrombin generation has been studied extensively in malignancy and a large prospective observational study, the Vienna Cancer and Thrombosis Study, assessed the predictive value of thrombin generation for VTE in over 1000 patients with a variety of malignancies. They concluded that patients with an elevated peak thrombin (>/ 611 nM) had an increased risk for development of VTE with a hazard ratio of 2.1[37]. However, the numbers of MM patients included in this study were small thus several groups have sought to establish the predictive value of TGAs specifically in MM.
Leiba et al.[40] tested thrombin generation in a cohort of 36 patients with MM at various timepoints over a 2.5-year period with the main objective of determining if thrombin generation has a predictive value for VTE. They observed significantly higher ETPs and peak height values in those who developed thrombotic events compare with those who did not. Interestingly, they also noted a gradual increase in thrombin generation parameters in the time period preceding the thrombotic event. Unlike the findings seen by Gracheva et al.[39] they noted that anticoagulation therapy was associated with a significant decrease in ETP and peak height values. As previously discussed, there is an excess incidence of VTE in MM in the first year following diagnosis and the authors observed a moderate in increase in TG parameters concomitant with commencement of MM treatment suggesting that this may partially explain the timing of this excess incidence[40]. Overall, they concluded that thrombin generation testing, both at baseline and during therapy, could serve as a predictive tool for thromboembolic events and perhaps may also have a role in monitoring individual response to heparin[40].
Fotiou et al.[41] subsequently carried out a larger prospective study of patients with NDMM and sought to explore the hypercoagulability characteristics in the period of treatment naivety pre-induction therapy. They recruited 144 patients with NDMM and compared their results to a group of healthy individuals. One of their outcomes was measurement of thrombin generation and, in direct contrast to the above findings, they found that, overall, thrombin generation was attenuated in the MM patients compared to the healthy individuals with longer lag-times and lower peak values observed. They also found significantly reduced ETPs in the MM cohort indicating an overall reduced enzymatic activity of thrombin. Interestingly, certain lower ETPs appeared to correlate with a higher risk of VTE and the authors concluded that they had identified clinically relevant biomarkers for VTE risk stratification with their hypothesis being that attenuation of thrombin generation should be interpreted as a reflection of endothelial cell activation i.e., “exhausted thrombin generation”[41]. Legendre et al.[42] also showed a potentially hypocoagulable state, with reduced peak thrombin and ETP values in a small group of 14 patients who were both chemotherapy and anticoagulant naïve.
Thus, there appears to be a marked discrepancy in the evaluation of thrombin generation in MM with both hyper- and hypo-coagulable states being observed by different groups, depending on timing of the testing and exposure of the patient to chemotherapy agents. Thrombin generation also increases with age and thus matching against younger, healthy control subjects may confound results[42]. Further evaluation in larger studies with stringent age-matching is necessary to determine both the possible predictive value of thrombin generation for VTE and the clinical utility of such testing in monitoring response and resistance to anticoagulant use (see Table 2 for result of thrombin generation testing).
Results of thrombin generation evaluation in patients with MM
| Author(s) | Multiple Myeloma patient cohort evaluated | Evaluated heparin resistance | Evaluated predictive potential for VTE | Findings |
| Chalayer et al.[38] 2019 | Patients on first line therapy with high VTE risk (n = 6) | Yes | No | No evidence for an in vitro LMWH resistance in those with MM compared to patients without MM |
| Gracheva et al.[39] 2015 | Patients with primary MM (n = 25) and patients with MM in remission (n = 34) undergoing blood stem cell mobilization | Yes | No | Possible “heparin resistance” with no heparin effect seen in 22% of patients and hypercoagulability seen in certain patients |
| Leiba et al.[40] 2017 | Patients with newly diagnosed disease (n = 13) and patients receiving therapy upon relapse (n = 23) | No | Yes | Patients who had a thrombotic event exhibited significantly higher ETP and peak height values than those who did not have a thrombotic event |
| Fotiou et al.[41] 2018 | Patients with newly diagnosed disease (n = 144) | No | Yes | Lower ETP values were associated with VTE occurrence |
| Legendre et al.[42] 2017 | Patients with newly diagnosed disease (before treatment, including anti-coagulation) (n = 14) | No | Yes | Hypocoagulable profiles including decreased ETP values |
The potential role of thrombomodulin in pathogenesis of thrombosis
Thrombomodulin is a transmembrane glycoprotein which is located on the luminal surface of endothelial cells. It has roles in both regulation of coagulation and inflammation with its main function being to bind to thrombin. Binding of thrombin to thrombomodulin results in loss of thrombin’s procoagulant and profibrogenic properties and acquisition of the ability to activate protein C. It has thus been hypothesised that a reduction in thrombomodulin levels in MM may confer a hypercoagulable profile in patients.
Corso et al.[43] sought to serially evaluate the changes in coagulation profiles in patients with RRMM both before and during/after treatment with thalidomide. While many markers were not found to significantly vary, they did observe significant variations in thrombomodulin levels. Thrombomodulin levels appeared to be slightly reduced in most patients at baseline but underwent further reductions during initial treatment with thalidomide therapy with the authors thus concluding that reductions in thrombomodulin levels may have a pathogenic role in thalidomide-related thrombosis. Fotiou et al.[41] also noted a reduction in thrombomodulin levels in their cohort of 144 patients with NDMM. Zappasodi et al.[44] subsequently sought to confirm the effect of thalidomide on thrombomodulin levels and evaluated the serial behaviour of thrombomodulin in 26 patients with RRMM who commenced thalidomide and dexamethasone therapy. Unlike their initial study where serum thrombomodulin levels fell during the first month of thalidomide therapy, they did not observe any significant modifications of thrombomodulin levels from baseline during the early stages of therapy. They did however note that the cohort of patients had low median basal values of thrombomodulin and thus concluded that this could perhaps suggest a thrombophilic state intrinsic to the MM disease process rather than then anti-myeloma therapy[44]. Again, these observations are limited not only by low patient numbers but also by lack of evaluation in patients being treated with the newer therapeutic agents that are currently in widespread use.
OTHER POSSIBLE MECHANISMS OF RESISTANCE
APC resistance
Many groups have sought to unravel the prothrombotic phenotype in patients with MM with common observations including significant elevations in von Willebrand Factor (VWF) antigen, Factor VIII, D-dimer and fibrinogen levels in patients with active disease. However, the contributory effect of these abnormalities towards VTE occurrence has not been fully disentangled thus far[15,36,45-49]. Interestingly, abnormalities of activated protein C (APC) appear to be a relatively common phenomenon exhibited in patients with MM and, unlike the above, do appear to significantly contribute to VTE occurrence. The normal biological role of APC is to inactivate Factor Va and Factor VIIIa with APC resistance (APCR) leading to non-cleavage of these factors and resultant increased thrombin generation which can potentially lead to a hypercoagulable state. In the Caucasian population, resistance to APC is one of the most common coagulation abnormalities associated with VTE and generally correlates with the presence of the Factor V Leiden R506Q mutation[50]. However, acquired APCR has been described in association with certain malignancies including MM[51,52].
Given this association, Elice et al.[53] sought to fully characterise the incidence of acquired APCR and its correlation with VTE in a large cohort of patients with MM (n = 1178). They recorded APCR in 109 patients (9%) but interestingly, this did not necessarily translate into the presence of factor V Leiden mutations with over half of these patients testing negatively for this mutation. Those with an abnormal APCR ratio but negative DNA testing for the factor V Leiden mutation were considered to have acquired APCR. Within this cohort of patients with acquired APCR, a higher incidence of VTE was observed when compared with controls (31% vs. 12%) and furthermore, they also exhibited a lower thrombosis-free survival. The authors thus concluded that the presence of acquired APCR was statistically associated with an increased VTE risk[51,53].
Following on from this, other groups have sought to further unravel the mechanisms behind APCR and isolate the exact causative single nucleotide polymorphism (SNPs) in the endothelial protein C receptor (EPCR). EPCR has a key regulatory role in protein C activity via binding and facilitating the interaction with the thrombin-thrombomodulin complex. Several polymorphisms have been reported in EPCR with the 4678G/C SNP thought to be associated with high levels of circulating APC and reduced risk of thrombosis. Dri et al.[54] sought to evaluate the presence of this polymorphism in a cohort of patients with MM who had developed thrombosis and found a significantly lower frequency of the 4678C allele of the EPCR gene in MM patients compared with the known frequency in a healthy adult population. The authors concluded that the reduced frequency of this protective allele could be contributing to overall disease hypercoagulability and that it may be useful to test for same to risk stratify MM patients. However, this was a small study and thus further evaluation of larger series of patients would be necessitated to determine that there is in fact an increased prevalence of this SNP in the MM population and if it indeed has a contributory effect to overall rates of VTE[54].
Endothelial dysfunction and toxicity related to the effect of chemotherapy
With the widespread introduction of several novel therapies for MM, there has been a focus on emergent cardiovascular complications in conjunction with the persistence of high rates of VTE in the MM population[26,55]. Many of the hypotheses surrounding this VTE risk centre on the toxicity that may be caused to the endothelium by certain anti-myeloma agents. Dexamethasone is a corticosteroid that is used in many of the treatment regimens for MM. Studies have shown that combining dexamethasone with other agents increases a patients risk of VTE[8,9]. The reasons behind this are not well understood but there is some in vitro evidence that dexamethasone can stimulate the endothelium to increase expression of both VWF and TF[56]. Similarly, IMiD agents and carfilzomib are both thought to activate and cause direct toxicity to the endothelium with a downstream pro-thrombotic effect occurring following release of coagulation factors and cytokines[57,58].
A small number of studies have been carried out specifically evaluating the toxic effects of anti-myeloma chemotherapy agents on endothelial cells (ECs). Early in vitro studies of IMiD agents noted that in a previously compromised/injured endothelium, as which would be seen with prior chemotherapy use, thalidomide could alter the expression of thrombin receptor PAR-1 and thus induce endothelial dysfunction and potentially a hypercoagulable state[59]. In a more recent study, Sanchez et al.[60] have reported their observations on exposure of ECs to some of the newer MM induction chemotherapy agents and they have shown that these agents can in fact lead to increased VWF and ICAM-1 expression and also an increase in cell permeability, exhibited by reduced VE-cadherin expression and cell monolayer integrity. Interestingly, this effect appeared to be somewhat counteracted by addition of the endothelial protectant agent defibrotide[60], thus raising the hypothesis that perhaps endothelial protection agents could have some anti-thrombotic utility in MM.
The direct effects of M-protein/immunoglobulin on the prothrombotic environment and fibrin clot formation
MM is characterised by high levels of circulating clonal immunoglobulins or “M-proteins” and these immunoglobulins are hypothesised to have the ability to contribute directly to thrombotic risk, not only by direct vascular wall injury but also by interference with fibrinolysis[61]. Fibrinolysis is the breakdown of fibrin within blood clots and is a highly regulated enzymatic process that, if functioning correctly, prevents unnecessary accumulation of intravascular fibrin. Clot stability is dependent upon many factors including local factors such as calcium concentration, thrombin concentration, pH and platelets numbers[62]. It is also influenced by the geometric compilation of the fibrin network and the composition and diameter of the fibrin fibres from which it is constructed[62]. Abnormal fibrin clot structure can lead to prolonged resolution/retraction of a clot[63].
Carr et al.[64] originally sought to evaluate the influence of high levels of immunoglobulin on fibrin polymerisation and clot structure in vitro, noting that in congenital dysfibrinogenemia, recurrent thrombosis were associated with the presence of abnormally thin fibres of fibrin. They hypothesised that, given that fibrin structures are sensitive to the environment in which they are formed, that high levels of immunoglobulin would interfere with fibrin monomer polymerisation. They characterised the clot formation in MM and , as seen in congenital dysfibrinogenemia, they noted that the clots were associated with the presence of abnormally thin fibrin fibres[64]. They subsequently sought to evaluate the resistance of such clots to fibrinolysis and observed that these clots dissolved at a slower rate and they thus concluded that they were associated with inhibition of fibrinolysis[65].
Undas et al.[66] subsequently sought to evaluate fibrin clot properties and their determinants ex vivo in a cohort of 106 MM patients. Following collection of peripheral blood from these patients, they performed fibrin clot analysis, measuring variables such as clot permeation, clot compaction, turbidity measurements and also the efficiency of fibrinolysis using plasma clot lysis assays. They observed that plasmin clot variables differed significantly between the MM patients and a cohort of healthy control subjects with changes including higher fibrin fibre densities and reduced tPA-mediated lysability. They hypothesised that higher thrombin generation potentials were likely a major mechanism in these alterations in clot properties and that this abnormal fibrin clot structure likely exhibited a prothrombotic phenotype[66].
The concept that local thrombin concentrations can impact upon the structure of clots with higher thrombin concentrations generating more stable clots which are more resistant to fibrinolysis and may promote thrombosis has been well described[62,63,67,68]. It thus appears possible that in MM, given the duel pathology of both high circulating immunoglobulin levels and potentially high thrombin generation, clots produced could be less susceptible to fibrinolysis and thus perhaps more resistant to the current anticoagulant strategies that are employed.
Tissue factor
TF is an essential glycoprotein that serves as principal initiator of coagulation. TF-mediated conversion of Factor IX to its activated form is pivotal for effective haemostasis while disruption of the endothelium causes exposure of TF-expressing endothelial cells and thus facilitates binding of factor VII[69]. Apart from its roles in coagulation, TF has also been implicated in both tumour spread and angiogenesis and in conjunction with this, high levels of TF have also been described in several solid organ malignancies with conflicting reports on its potential utility as a biomarker for VTE occurrence[70]. TF can also be demonstrated on circulating microparticles (MPs) which are small, procoagulant membrane vesicles that are released from cells during activation or during apoptosis. These MPs are found in low steady state concentrations in healthy individuals and increase during times of inflammation. MPs carrying TF express platelet selectin glycoprotein ligand-1 which binds p-selectin on the surface of activated endothelial cells[70].
In light of the above, several groups have sought to establish the role of TF in MM disease.
Other groups sought to evaluate the TF activity in patients with MM with Auwerda et al.[72] evaluating this in a cohort of 122 patients with MM. They found that MP-TF activity levels were increased in the MM cohort in comparison to healthy volunteers. They performed serial evaluation of these activity levels and found that MP-TF activity levels showed a reduction post-induction therapy in many of the patients, but interestingly, levels remained elevated in those who had suffered a thrombotic episode[72]. Nielsen et al.[73] also sought to explore the role of TF in MM by isolating extracellular vesicles (EVs) from the peripheral blood of 20 patients with MM and subsequently demonstrating substantially higher thrombin generation and TF activity when compared with healthy control subjects. Similar to the above, they also performed serial activity levels and noted that the procoagulant activity of the EVs diminished after treatment, though of note, neither of the treatment groups that they evaluated contained the more thrombogenic IMiD agents (treatment groups = bortezomib, cyclophosphamide and dexamethasone and melphalan, prednisolone and bortezomib)[73].
All groups concluded that their findings may, at least in part, explain why there is an increased risk of VTE in MM but the prothrombotic contribution of TF in MM would need to be confirmed in larger cohorts of patients. In addition to this, TF activity is not routinely measured in clinical practice and a standardised assay would have to be developed and validated before it could be considered for widespread use as a biomarker.
CONCLUSION
It is clear that rates of thrombosis remain high in MM and, unfortunately, current thromboprophylaxis regimens do not entirely abrogate this risk of VTE. Thus the gap in knowledge between the persistently high levels of VTE and the inadequacies in current preventative measures needs to be bridged.
It is also increasingly evident that there are a multitude of factors contributing to the prothrombotic milieu seen in MM, many of which cannot be evaluated in a clinical setting. Further work is necessitated to elucidate specific pathways and factors contributing to the hypercoagulable phenotype observed in these patients. Additional clinical studies should focus on identifying novel biomarkers of VTE risk in MM and developing effective screening methods for anticoagulant resistance. Taken together, these approaches may help inform therapeutic strategies to overcome these mechanisms of resistance and provide effective thromboprophylaxis for patients with MM.
DECLARATIONS
Authors’ contributionsWriting of the manuscript: Comerford C, Glavey S, O’Sullivan JM, Quinn J
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Kristinsson SY, Fears TR, Gridley G, et al. Deep vein thrombosis after monoclonal gammopathy of undetermined significance and multiple myeloma. Blood 2008;112:3582-6.
2. Barrett A, Quinn J, Lavin M, et al. Validation of risk-adapted venous thromboembolism prediction in multiple myeloma patients. J Clin Med 2021;10:3536.
3. Schoen MW, Luo S, Gage B, Carson KR, Sanfilippo KM. Association of venous thromboembolism with increased mortality in patients with multiple myeloma. J Clin Oncol 2018;36:8051.
4. Kristinsson SY, Pfeiffer RM, Björkholm M, Schulman S, Landgren O. Thrombosis is associated with inferior survival in multiple myeloma. Haematologica 2012;97:1603-7.
6. Gandolfi S, Prada CP, Richardson PG. How I treat the young patient with multiple myeloma. Blood 2018;132:1114-24.
7. Moreau P, San Miguel J, Sonneveld P, et al. ESMO Guidelines Committee. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28:iv52-61.
8. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma: a systemic review. N Engl J Med 2007;357:2123-32.
9. Zonder JA, Crowley J, Hussein MA, et al. Lenalidomide and high-dose dexamethasone compared with dexamethasone as initial therapy for multiple myeloma: a randomized Southwest Oncology Group trial (S0232). Blood 2010;116:5838-41.
10. Khorana AA, DeSancho MT, Liebman H, Rosovsky R, Connors JM, Zwicker J. Prediction and prevention of cancer-associated thromboembolism. Oncologist 2021;26:e2-7.
11. Snyder PB, Groebner RJ, Leonard AW, Osborne TH, Wilson HR. Development and validation of a predictive model for the pedestal height. Phys Plasmas 2009;16:056118.
12. Sanfilippo KM, Luo S, Wang TF, et al. Predicting venous thromboembolism in multiple myeloma: development and validation of the IMPEDE VTE score. Am J Hematol 2019;94:1176-84.
13. Palumbo A, Rajkumar SV, Dimopoulos MA, et al. International Myeloma Working Group. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 2008;22:414-23.
14. Li A, Wu Q, Luo S, et al. Derivation and validation of a risk assessment model for immunomodulatory drug-associated thrombosis among patients with multiple myeloma. J Natl Compr Canc Netw 2019;17:840-7.
15. Santoro M, Romano A, Mancuso S, et al. Prevention of venous thromboembolic events occurring in myeloma patients treated with second-generation novel agents. Panminerva Med 2021;63:1-6.
16. McBane RD 2nd, Wysokinski WE, Le-Rademacher JG, et al. Apixaban and dalteparin in active malignancy-associated venous thromboembolism: the ADAM VTE trial. J Thromb Haemost 2020;18:411-21.
18. Raskob GE, van Es N, Verhamme P, et al. Hokusai VTE Cancer Investigators. Edoxaban for the treatment of cancer-associated venous thromboembolism. N Engl J Med 2018;378:615-24.
19. Storrar NPF, Mathur A, Johnson PRE, Roddie PH. Safety and efficacy of apixaban for routine thromboprophylaxis in myeloma patients treated with thalidomide- and lenalidomide-containing regimens. Br J Haematol 2019;185:142-4.
20. Cornell RF, Goldhaber SZ, Engelhardt BG, et al. Primary prevention of venous thromboembolism with apixaban for multiple myeloma patients receiving immunomodulatory agents. Br J Haematol 2020;190:555-61.
21. Swan D, Rocci A, Bradbury C, Thachil J. Venous thromboembolism in multiple myeloma - choice of prophylaxis, role of direct oral anticoagulants and special considerations. Br J Haematol 2018;183:538-56.
22. Leclerc V, Karlin L, Herledan C, et al. Thromboembolic events and thromboprophylaxis associated with immunomodulators in multiple myeloma patients: a real-life study. J Cancer Res Clin Oncol 2021; doi: 10.1007/s00432-021-03693-5.
23. Lapietra G, Serrao A, Fazio F, Petrucci MT, Chistolini A. Venous thromboembolism prophylaxis in patients with multiple myeloma: where are we and where are we going? J Thromb Thrombolysis 2021;52:584-9.
24. Leleu X, Rodon P, Hulin C, et al. MELISSE, a large multicentric observational study to determine risk factors of venous thromboembolism in patients with multiple myeloma treated with immunomodulatory drugs. Thromb Haemost 2013;110:844-51.
25. Bradbury CA, Craig Z, Cook G, et al. Thrombosis in patients with myeloma treated in the Myeloma IX and Myeloma XI phase 3 randomized controlled trials. Blood 2020;136:1091-104.
26. Patel VG, Cornell RF. Cardiovascular complications associated with multiple myeloma therapies: incidence, pathophysiology, and management. Curr Oncol Rep 2019;21:29.
27. Atrash S, Tullos A, Panozzo S, et al. Cardiac complications in relapsed and refractory multiple myeloma patients treated with carfilzomib. Blood Cancer J 2015;5:e272.
28. Landgren O, Sonneveld P, Jakubowiak A, et al. Carfilzomib with immunomodulatory drugs for the treatment of newly diagnosed multiple myeloma. Leukemia 2019;33:2127-43.
29. Piedra K, Peterson T, Tan C, et al. Comparison of venous thromboembolism incidence in newly diagnosed multiple myeloma patients receiving bortezomib, lenalidomide, dexamethasone (RVD) or carfilzomib, lenalidomide, dexamethasone (KRD) with aspirin or rivaroxaban thromboprophylaxis. Br J Haematol 2022;196:105-9.
30. Parks AL, Kambhampati S, Fakhri B, et al. Incidence, management and outcomes of arterial and venous thromboembolism after chimeric antigen receptor modified T cells for B-cell lymphoma and multiple myeloma. Blood 2020;136:7-7.
31. Levy JH, Connors JM. Heparin resistance - clinical perspectives and management strategies. N Engl J Med 2021;385:826-32.
32. Wang TF, Makar RS, Antic D, et al. Management of hemostatic complications in acute leukemia: guidance from the SSC of the ISTH. J Thromb Haemost 2020;18:3174-83.
33. Kumar S, Chapagain A, Nitsch D, Yaqoob MM. Proteinuria and hypoalbuminemia are risk factors for thromboembolic events in patients with idiopathic membranous nephropathy: an observational study. BMC Nephrol 2012;13:107.
34. Auwerda JJA, Sonneveld P, De Maat MPM, Leebeek FWG. Prothrombotic coagulation abnormalities in patients with newly diagnosed multiple myeloma. Haematologica 2007;92:279-80.
35. van Marion AM, Auwerda JJ, Lisman T, et al. Prospective evaluation of coagulopathy in multiple myeloma patients before, during and after various chemotherapeutic regimens. Leuk Res 2008;32:1078-84.
36. Minnema MC, Fijnheer R, De Groot PG, Lokhorst HM. Extremely high levels of von Willebrand factor antigen and of procoagulant factor VIII found in multiple myeloma patients are associated with activity status but not with thalidomide treatment. J Thromb Haemost 2003;1:445-9.
37. Ay C, Dunkler D, Simanek R, et al. Prediction of venous thromboembolism in patients with cancer by measuring thrombin generation: results from the Vienna Cancer and Thrombosis Study. J Clin Oncol 2011;29:2099-103.
38. Chalayer E, Tardy-Poncet B, Montmartin A, Boussoualim K, Genthon A, Tardy B. Effect of heparin thromboprophylaxis on thrombin generation in multiple myeloma patients. Br J Haematol 2019;186:337-9.
39. Gracheva MA, Urnova ES, Sinauridze EI, et al. Thromboelastography, thrombin generation test and thrombodynamics reveal hypercoagulability in patients with multiple myeloma. Leuk Lymphoma 2015;56:3418-25.
40. Leiba M, Malkiel S, Budnik I, et al. Thrombin generation as a predictor of thromboembolic events in multiple myeloma patients. Blood Cells Mol Dis 2017;65:1-7.
41. Fotiou D, Sergentanis TN, Papageorgiou L, et al. Longer procoagulant phospholipid-dependent clotting time, lower endogenous thrombin potential and higher tissue factor pathway inhibitor concentrations are associated with increased VTE occurrence in patients with newly diagnosed multiple myeloma: results of the prospective ROADMAP-MM-CAT study. Blood Cancer J 2018;8:102.
42. Legendre P, Verstraete E, Martin M, et al. Hypocoagulability as assessed by thrombin generation test in newly-diagnosed patients with multiple myeloma. Blood Cells Mol Dis 2017;66:47-9.
43. Corso A, Lorenzi A, Terulla V, et al. Modification of thrombomodulin plasma levels in refractory myeloma patients during treatment with thalidomide and dexamethasone. Ann Hematol 2004;83:588-91.
44. Zappasodi P, Mangiacavalli S, Terulla V, et al. Thrombomodulin levels are not modified during thalidomide treatment. Eur J Haematol 2006;77:453-4.
45. Robak M, Treliński J, Chojnowski K. Hemostatic changes after 1 month of thalidomide and dexamethasone therapy in patients with multiple myeloma. Med Oncol 2012;29:3574-80.
46. Gomperts ED, Shulman G, Lynch SR. Factor VIII and factor-VIII-related antigen in multiple myelomatosis and related conditions. Br J Haematol 1976;32:249-55.
47. Crowley MP, Kevane B, O’Shea SI, et al. Plasma thrombin generation and sensitivity to activated protein C among patients with myeloma and monoclonal gammopathy of undetermined significance. Clin Appl Thromb Hemost 2016;22:554-62.
48. Auwerda JJ, Sonneveld P, de Maat MP, Leebeek FW. Prothrombotic coagulation abnormalities in patients with newly diagnosed multiple myeloma. Haematologica 2007;92:279-80.
49. Patmore S, Dhami SPS, O'Sullivan JM. Von Willebrand factor and cancer; metastasis and coagulopathies. J Thromb Haemost 2020;18:2444-56.
51. Jiménez-Zepeda VH, Domínguez-Martínez VJ. Acquired activated protein C resistance and thrombosis in multiple myeloma patients. Thromb J 2006;4:11.
52. De Lucia D, Devita F, Orditura M, et al. Hypercoagulable state in patients with advanced gastrointestinal cancer: evidence for an acquired resistance to activated protein C. Tumori 1997;83:948-52.
53. Elice F, Fink L, Tricot G, Barlogie B, Zangari M. Acquired resistance to activated protein C (aAPCR) in multiple myeloma is a transitory abnormality associated with an increased risk of venous thromboembolism. Br J Haematol 2006;134:399-405.
54. Dri AP, Politou M, Gialeraki A, Bagratuni T, Kanellias N, Terpos E. Decreased incidence of EPCR 4678G/C SNP in multiple myeloma patients with thrombosis. Thromb Res 2013;132:400-1.
55. Kistler KD, Kalman J, Sahni G, et al. Incidence and risk of cardiac events in patients with previously treated multiple myeloma versus matched patients without multiple myeloma: an observational, retrospective, cohort study. Clin Lymphoma Myeloma Leuk 2017;17:89-96.e3.
56. Kerachian MA, Cournoyer D, Harvey EJ, et al. Effect of high-dose dexamethasone on endothelial haemostatic gene expression and neutrophil adhesion. J Steroid Biochem Mol Biol 2009;116:127-33.
57. Bird JM, Owen RG, D’Sa S, et al. Haemato-oncology Task Force of British Committee for Standards in Haematology (BCSH) and UK Myeloma Forum. Guidelines for the diagnosis and management of multiple myeloma 2011. Br J Haematol 2011;154:32-75.
58. Li W, Garcia D, Cornell RF, et al. Cardiovascular and thrombotic complications of novel multiple myeloma therapies: a review. JAMA Oncol 2017;3:980-8.
59. Kaushal V, Kaushal GP, Melkaveri SN, Mehta P. Thalidomide protects endothelial cells from doxorubicin-induced apoptosis but alters cell morphology. J Thromb Haemost 2004;2:327-34.
60. Martinez-Sanchez J, Palomo M, Torramade-Moix S, et al. The induction strategies administered in the treatment of multiple myeloma exhibit a deleterious effect on the endothelium. Bone Marrow Transplant 2020;55:2270-8.
61. Eby C. Pathogenesis and management of bleeding and thrombosis in plasma cell dyscrasias. Br J Haematol 2009;145:151-63.
62. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation NIH Public Access. Blood Rev 2015;29:17-24.
64. Carr ME Jr, Zekert SL. Abnormal clot retraction, altered fibrin structure, and normal platelet function in multiple myeloma. Am J Physiol 1994;266:H1195-201.
65. Carr ME, Dent RM, Carr SL. Abnormal fibrin structure and inhibition of fibrinolysis in patients with multiple myeloma. J Lab Clin Med 1996;128:83-8.
66. Undas A, Zubkiewicz-Usnarska L, Helbig G, et al. Altered plasma fibrin clot properties and fibrinolysis in patients with multiple myeloma. Eur J Clin Invest 2014;44:557-66.
67. Wolberg AS, Monroe DM, Roberts HR, Hoffman M. Elevated prothrombin results in clots with an altered fiber structure: a possible mechanism of the increased thrombotic risk. Blood 2003;101:3008-13.
68. Undas A, Ariëns RA. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol 2011;31:e88-99.
69. López-Pedrera C, Barbarroja N, Dorado G, Siendones E, Velasco F. Tissue factor as an effector of angiogenesis and tumor progression in hematological malignancies. Leukemia 2006;20:1331-40.
70. Hanna DL, White RH, Wun T. Biomolecular markers of cancer-associated thromboembolism. Crit Rev Oncol Hematol 2013;88:19-29.
71. Papageorgiou L, Alhaj Hussen K, Thouroude S, et al. Modelization of blood-borne hypercoagulability in myeloma: a tissue-factor-bearing microparticle-driven process. TH Open 2019;3:e340-7.
72. Auwerda JJ, Yuana Y, Osanto S, et al. Microparticle-associated tissue factor activity and venous thrombosis in multiple myeloma. Thromb Haemost 2011;105:14-20.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Comerford C, Glavey S, O’Sullivan JM, Quinn J. Potential mechanisms of resistance to current anti-thrombotic strategies in Multiple Myeloma. Cancer Drug Resist 2022;5:214-28. http://dx.doi.org/10.20517/cdr.2021.115
AMA Style
Comerford C, Glavey S, O’Sullivan JM, Quinn J. Potential mechanisms of resistance to current anti-thrombotic strategies in Multiple Myeloma. Cancer Drug Resistance. 2022; 5(1): 214-28. http://dx.doi.org/10.20517/cdr.2021.115
Chicago/Turabian Style
Comerford, Claire, Siobhan Glavey, Jamie M. O’Sullivan, John Quinn. 2022. "Potential mechanisms of resistance to current anti-thrombotic strategies in Multiple Myeloma" Cancer Drug Resistance. 5, no.1: 214-28. http://dx.doi.org/10.20517/cdr.2021.115
ACS Style
Comerford, C.; Glavey S.; O’Sullivan JM.; Quinn J. Potential mechanisms of resistance to current anti-thrombotic strategies in Multiple Myeloma. Cancer Drug Resist. 2022, 5, 214-28. http://dx.doi.org/10.20517/cdr.2021.115
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 4
likes
Like This Article 4
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.