
Emerging actionable targets to treat therapy-resistant colorectal cancers


Abstract
In the last two decades major improvements have been reached in the early diagnosis of colorectal cancer (CRC) and, besides chemotherapy, an ampler choice of therapeutic approaches is now available, including targeted and immunotherapy. Despite that, CRC remains a “big killer” mainly due to the development of resistance to therapies, especially when the disease is diagnosed after it is already metastatic. At the same time, our knowledge of the mechanisms underlying resistance has been rapidly expanding which allows the development of novel therapeutic options in order to overcome it. As far as resistance to chemotherapy is concerned, several contributors have been identified such as: intake/efflux systems upregulation; alterations in the DNA damage response, due to defect in the DNA checkpoint and repair systems; dysregulation of the expression of apoptotic/anti-apoptotic members of the BCL2 family; overexpression of oncogenic kinases; the presence of cancer stem cells; and the composition of the tumoral microenvironment and that of the gut microbiota. Interestingly, several mechanisms are also involved in the resistance to targeted and/or immunotherapy. For example, overexpression and/or hyperactivation and/or amplification of oncogenic kinases can sustain resistance to targeted therapy whereas the composition of the gut microbiota, as well as that of the tumoral niche, and defects in DNA repair systems are crucial for determining the response to immunotherapy. In this review we will make an overview of the main resistance mechanisms identified so far and of the new therapeutic approaches to overcome it.
Keywords
INTRODUCTION
Notwithstanding widespread, effective measures of preventive screening, early detection and major advances in treatment options, colorectal cancer (CRC) is still the fourth cause of cancer-related death worldwide[1,2]. In fact, due to the relatively asymptomatic progression of the disease in the early stages, patients are frequently diagnosed with metastatic CRC (mCRC), with a 5-year survival rate of around 14%[3]. Neo-adjuvant or adjuvant chemotherapy, together with surgery, represent the backbone of the therapeutic approach for both early-stage and metastatic cancer patients. Depending on tumor site and progression of the disease, chemotherapy can be associated with radiotherapy[4-6] and, in selected cases, it can be possibly integrated with targeted therapy and/or immunotherapy. However, despite a variety of therapeutic approaches, resistance - both intrinsic and acquired - to drug treatment(s) remains one of the greatest challenges in the long-term management of incurable mCRC and eventually contributes to death[7,8]. In the last two decades, remarkable progresses have been made in the understanding of the mechanisms underlying drug resistance and in the unraveling of the feedback mechanisms fueling acquired resistance to targeted and/or immunotherapy. In addition, several phenotypes and synthetic lethality screens uncovered important liabilities that can be pharmacologically targeted in resistant tumors and in specific mutational settings[9]. Accordingly, hundreds of clinical trials are currently undergoing to test new drugs and new combinations for the treatment of mCRC. This review will pinpoint the most promising targets emerged in the last decade.
RESISTANCE TO CHEMOTHERAPY
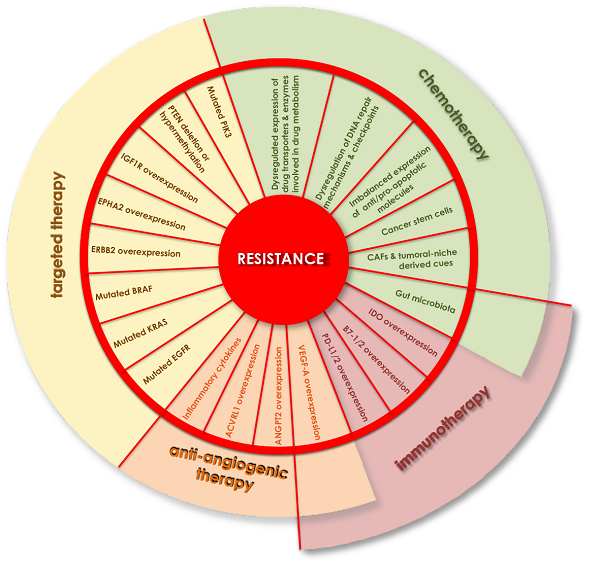
According to the American Society of Clinical Oncology (ASCO) guidelines (https://www.asco.org/), the main drugs used for chemotherapy are 5-fluorouracil (5-FU), irinotecan, and oxaliplatin, either alone or in combinations such as in the FOLFOX (5-FU + leucovorin + oxaliplatin) or FOLFIRI (5-FU + leucovorin + irinotecan) regimens. 5-FU-based chemotherapy is the main approach for advanced CRC and, when used in combination, initial responses are up to 55%-65%. When administered sequentially, median overall survival (OS) can range from 18 to 20 months[10]. Although 5-FU initially de-bulks the tumor mass, recurrence usually occurs, thus pinpointing how acquired drug resistance, and consequent tumor re-growth, represents the main obstacle to effective clinical outcomes for CRC patients. Several different mechanisms have been identified accounting for resistance to 5-FU-based chemotherapy[11] and increasing number of strategies are being explored to increase the responsiveness of resistant tumors to chemotherapy [Figure 1].
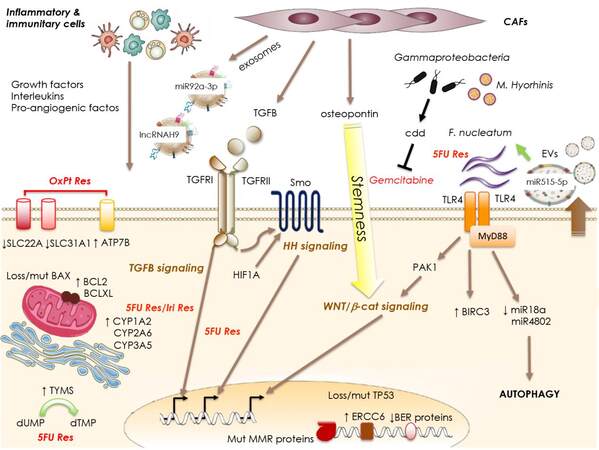
Figure 1. Mechanisms of resistance to chemotherapy. Tumor cell-intrinsic mechanisms may involve: dysregulated expression of drug transporters (SLC family members or ATP7B) and enzymes involved in drug metabolism (CYP family members), imbalanced expression of anti-/pro-apoptotic molecules (BAX mutation/loss or increased BCL2 expression), or dysregulation of DNA repair mechanisms and checkpoints (TP53 mutation/loss, MMR proteins mutation, diminished expression of BER proteins, or increased ERCC6 expression). External signals acting on the tumor cells to trigger resistance may derive from the cells populating the tumoral niche such as the CAFs releasing TGFB, osteopontin and exosomes containing specific lncRNAs and miRs. In addition, inflammatory and immunitary cells of the niche release a number of interleukins, growth factors, pro-angiogenic factors. Also, specific components of the microbiota can contribute to the resistance to chemotherapy by directly inactivating the drug (Gammaproteobacterial or M. hyorhinis) or by engaging the TLR4/MyD88 system to transduce pro-survival and autophagic signals. SLC: Solute carrier; MMR: mismatch-repair system; CAFs: cancer-associated fibroblast.
Dysregulated expression of drug transporters and enzymes involved in drug metabolism
Plasma membrane transporters are pivotal for the uptake into and the efflux out of the cells of endogenous and exogenous molecules. They are divided in two big families: the ATP-binding cassette (ABC) family and the solute carrier (SLC) family. Members of both families serve a range of physiological roles but some of them also are determinants of drug disposition via affecting absorption, distribution, and excretion of drugs [Table 1].
Dysregulated expression of drug transporters and enzymes involved in drug metabolism
| Gene | Protein function | Correlation with drug resistance | Ref. |
| SLC22A3 | Uptake of platinum compounds | Lower SLC22A3 expression correlates with worst PSF in patients receiving FOLFOX6 regimen | [20] |
| Post-treatment intracellular concentration of OxPt is higher in SLC22A3-overexpressing cells; upregulation of SLC22A3 in mouse xenografts rendered tumors more responsive to OxPt treatment | [21] | ||
| SLC31A1 | Copper influx transporter, involved also in OxPt intake | SLC31A1 level predicts prolonged survival and enhanced response to platinum-based regimens in cancer patients with several epithelial cancers | [24] |
| ATP7B | Copper efflux transporter, involved also in OxPt efflux | Increased levels of ATP7B are associated with poor outcome in CRC patients receiving oxaliplatin-based chemotherapy | [23] |
| CYP1A2/CYP2A6 CYP3A5 | Cytochrome P450 enzymes involved in drug metabolism | Increased expression in 5-FU-resistant HCT116 CRC cell line; addition of the CYP450 inhibitor phenylpyrrole enhanced 5-FU-induced cytotoxicity in 5-FU-resistant cells | [25] |
| Higher intratumoral expression of CYP3A5 in patients with CRC who do not respond to irinotecan-based chemotherapy | [29] | ||
| TYMS | Enzyme that maintains the dTMP pool critical for DNA replication and repair and is inhibited by 5-FU | Increased expression of TYMS in pretreatment tumor biopsies identified tumors non-responsive to 5-FU-based therapy | [26] |
Since the mid-1980s several members of the ABC family have been deemed to be play an important role in the resistance to chemotherapy, given that their dysregulated expression may lead to a decreased uptake or an increased efflux of anticancer agents[12-14]. Even though roughly half of the members have been shown to efflux anticancer agents in some context, the use of cell lines with high ABC transporter expression levels might have led to an overestimation of their role in cancer[15]. In addition, transporters are usually parts of more complex networks usually under the same transcriptional regulation; therefore, their upregulation may be more of an epi-phenomenon than of a cause-related effect[16]. A paradigmatic example of this are the data reported by Gao et al.[17]. They first showed that, in colon cancer cell lines made resistant to 5-FU, drug treatment induced the expression of different ABC transporters as well as the activation of IRE1, an enzyme involved in the unfolded protein response ensuing from endoplasmic reticulum stress[18]. Disabling the enzyme, by both a specific inhibitor and by RNA silencing, they observed a decrease in the expression of the transporters as well as the sensitization to 5-FU. Given that the unfolded protein response is a complex response where several genes are regulated downstream of IRE1[19], their experiments do not rule out that the sensitization might be due to the suppression (or activation) of other components of the response. Therefore, the decrease in the expression of the transporters simply correlates with the sensitization effect without a clear cause-effect relationship having been established.
For decades now plenty of experimental data have been produced - both in in vitro and in vivo systems - showing that the increased expression of several members correlate with increased resistance to chemotherapeutic agents as well as their inhibition by specific inhibitors could restore drug sensitivity[12,13]. Although the substrates and key roles for most of these transporters have been identified, the extent to which these transporters play an effective role in clinical multidrug resistance has not yet been clarified[15,16], not in general nor in the specific case of CRC. In fact, ex-vivo data are still pretty inconclusive: for example ABCB1 expression is generally low in tumors, but for few exceptions, and specific inhibitors have clinically failed[16].
On the other hand, some members of the SLC family 22 shown to be involved in the uptake of platinum compounds have been more directly linked to drug efficacy. For example, in a retrospective study
A change in the expression or activation of enzymes involved in drug metabolism by cancer cells can enhance the catabolism of the drugs or result in diminished activation of prodrugs. In both cases, the result is an impairment of the pharmacological action of the chemotherapeutic agent [Table 1]. For example, Untereiner et al.[25] produced a 5-FU-resistant HCT116 CRC cell line characterized by a significant increase in the expression of the drug-metabolizing cytochrome P450 enzymes CYP1A2 and CYP2A6. Accordingly, they reported that the addition of the CYP450 inhibitor phenylpyrrole enhanced 5-FU-induced cytotoxicity in 5-FU-resistant cells[25]. Also, increased expression of thymidylate synthase (the enzyme inhibited by 5-FU) in pretreatment tumor biopsies could identify tumors that would be non-responsive to 5-FU-based therapy[26]. At variance, downregulation of thymidine phosphorylase (which plays an essential role in activating the oral prodrug capecitabine to 5-FU) causes resistance via insufficient drug activation[27].
Irinotecan undergoes extensive metabolism in both the liver and the intestine; it is converted to inactive metabolites by CYP3A4 and CYP3A5[28]. Studying a CRC patient cohort, Buck et al.[29] recently reported an association between the response to irinotecan and the expression CYP3A5; in fact, they found a significantly higher intratumoral expression of CYP3A5 in patients with CRC who do not respond to irinotecan-based chemotherapy and thus suggested a causal role of CYP3A5 in tumor resistance.
Dysregulation of DNA repair mechanisms and checkpoints
5-FU and all the most used drugs for treating CRC act by inducing, directly or indirectly, DNA damage and consequently activate cell’s DNA damage response. Depending on the entity of the damage and on the functionality of the DNA damage response, apoptosis can be the outcome. In fact, several DNA repair mechanisms can intervene to remove or repair drug-induced damage thus avoiding cytotoxicity, and their dysregulation can contribute to the drug-resistant phenotype [Table 2]. For example, ERCC6 - a member of the excision repair cross-complementation (ERCC) family of enzymes, involved in the nucleotide excision repair pathway (NER) - is upregulated in CRC tissues compared to matched non-tumoral adjacent tissues and is also upregulated in patients resistant to 5-FU treatment. Conversely, low ERCC6 expression is associated with better response to chemotherapy and survival in CRC[30].
Dysregulation of DNA repair mechanisms and checkpoints
| Gene/system | Protein function | Correlation with drug resistance | Ref. |
| ERCC6 | Member of ERCC family of enzymes involved in the NER | Upregulated in CRC tissues compared to matched non-tumoral adjacent tissues Higher levels in patients resistant to 5-FU treatment low expression associated with better response to chemotherapy and survival | [30] |
| BER | Members of the BER system repairs bulky helix-distorting lesions | CRC patients with defects in components of the mismatch-repair system and expressing high levels of BER proteins have more aggressive tumors and poor outcomes after chemotherapy | [31] |
| MMR | Members of MMR are involved in recognizing and repairing erroneous insertion, deletion, and mis-incorporation of bases | dMMR patients with stages II and III CRC are less responsive, if not at all, to 5-FU-based chemotherapy whereas a better response is achievable in pMMR patients | [33,34] |
| TP53 | DNA damage checkpoint | Loss or impairment of TP53 affects the response to chemotherapy in several in vitro and in vivo systems | [37] |
| Loss or impairment of TP53 predict the poor response of patients with mCRC treated with chemotherapy | [38] | ||
| Loss or impairment of TP53 associated with poor survival after FOLFOX therapyLoss or impairment of TP53 associated with poor survival after FOLFOX therapy | [39,40] |
Activity of the base-excision repair (BER) system is pivotal in determining 5-FU-induced cytotoxicity in cells concomitantly defective for components of the mismatch-repair system (dMMR), as shown by the fact that CRC patients expressing high levels of BER proteins have more aggressive tumors and poor outcomes after chemotherapy[31]. Of note, zelpolib, a specific inhibitor of DNA polymerase δ an essential component of both NER and BER pathways - has been recently synthesized and shown to render cells sensitive to PARP inhibitors[32]. Therefore, it would be of interest to assess whether PARP inhibitors are cytotoxic in ERCC6-overexpressing and MMR/BER defective 5-FU-resistant models.
MMR status is particularly important in CRC given that around 15% of patients have one or more components (MLH1, MLH3, MSH2, or MSH3) mutated or silenced, often with a microsatellite instability (MSI) phenotype. Notably, patients with stages II and III CRC are less responsive, if not at all, to 5-FU-based chemotherapy whereas a better response is achievable in patients with MMR-proficient (pMMR) tumors[33,34]. In addition, MMR phenotype is also predictive of resistance to oxaliplatin-based chemotherapy[35]. Notably, collections of evidence are accumulating that dMMR and MSI-high (MSI-H) heavily pre-treated patients (at least two prior lines of therapy for metastatic disease) show a durable clinical benefit when treated with programmed cell death protein 1 (PD-1) inhibitors, particularly in the metastatic setting[36].
The response to DNA damage is tightly controlled by DNA damage checkpoints and their misfunctioning may allow the survival of damaged cells and the selection of new mutations. In this case, the resistance to chemotherapy as well as the tumor progression are favored characteristics. The best-known DNA damage checkpoint, mutated or lost in 50% to 70% of CRC cases, is TP53 which makes it the gene with the highest mutation rate in CRC[37,38]. Loss or impairment of TP53 have been shown to affect the response to chemotherapy in several in vitro and in vivo systems[37], to predict the poor response of patients with mCRC treated with chemotherapy[38], and be associated with poor survival after FOLFOX therapy[39,40].
Many promising synthetic lethal vulnerabilities, whose targeting kills p53-null drug resistant-resistant CRC cells, have been identified by an shRNA screen performed in our lab[41]. Several of them have successively been validated by other labs such as PIM-1[42], TRIB3[43], EPHA2[44,45], CHK1[46], and VEGFR2[47]. We focused our studies particularly on two targets: GSK3B and p65BTK. We showed that GSK3B is significantly more activated in drug-resistant vs. responsive CRC patients and is associated with cancer progression, poor response to therapy, and worse OS. Experimentally, we demonstrated that in the absence of p53, GSK3B activity allows cells to survive, despite treatment with DNA-damaging drugs, by sustaining DNA repair and that its downregulation restored sensitivity to 5-FU of p53-null colon cancer cells - both in in vitro and in in vivo models - via induction of regulated necrosis[48]. Moreover, we also demonstrated that GSK3A is redundant with GSK3B modulating drug resistance and chemotherapy-induced regulated necrosis[49]. A general role for GSK3B in sustaining resistance to chemotherapy has been validated also in other types of cancers and 9-ING-41, a novel GSK3B inhibitor developed by actuate therapeutics, is currently undergoing phase 1 and phase 2 trials, as a single agent and in combination with cytotoxic agents, in patients with refractory cancers, including CRCs[9]. p65BTK is a novel isoform of the Bruton’s tyrosine kinase we isolated from the screening and subsequently characterized. Compared to the known Bruton’s tyrosine kinase (BTK) expressed in bone marrow-derived cell lineages, p65BTK lacks a stretch of 86 amino acids at the N-term and is translated from an mRNA containing a different first exon. Protein abundance is regulated at the post-transcriptional level and under the control of RAS/ERK pathway. Moreover, p65BTK is endowed with strong transforming activity that depends on ERK1/2 and its inhibition abolishes RAS transforming activity. Accordingly, p65BTK overexpression in CRC tissues correlates with ERK1/2 activation[50], histotype, and cancer progression[51]. p65BTK silencing or chemical inhibition affects the growth of CRC cells and overcomes the 5-FU resistance of p53-null CRC cells by abolishing a 5-FU-elicited TGFB1 protective response and triggering E2F-dependent apoptosis. In addition, the combination of BTK inhibitors with
Interestingly, its targeting is effective in cells with p53 loss and defects along the RAS/MAPK pathway, making it an actionable target for a broad range of solid tumors resistant to SOC chemotherapy. Notably several drugs targeting BTK are already in clinic for certain types of B-cell malignancies (e.g., ibrutinib, acalabrutinib, and zanubrutinib), whereas several others are in clinical trial for B-cell malignancies (e.g., orelabrutinib, spebrutinib, pirtobrutinib, and BGB-3111) and for different type of diseases characterized by excessive B-cell activation (e.g., remibrutinib, renebrutinib, tolebrutinib, and rilzabrunib). Thus, assessing their efficacy in drug-resistant CRCs and other solid tumors would be feasible and advantageous.
Imbalanced expression of anti-/pro-apoptotic molecules
Drug resistance has been demonstrated to be associated with low levels of the pro-apoptotic BAX protein or its loss[55,56], which can occur in CRC patients because of inactivating mutation or somatic frameshift mutation in the dMMR setting[57]. In a low-expressing experimental CRC model, its re-induction by using andrographolide, a natural diterpenoid, restored the apoptotic response to 5-FU of drug-resistant CRC cells[58]. Drug resistance has also been associated with increased levels of the anti-apoptotic BCL-XL protein, which largely occurs in CRC either by amplification or by overexpression[59]. Several molecules antagonizing BCL-XL, the so-called BH3-mimetics, are currently being tested in experimental models in combination with chemo- or targeted therapy. For example, ABT-737 has been shown to improve the response to oxaliplatin in a TP53wt/KRASmut background[60]. Both ABT-737 and WEHI-539 have been shown to lower the apoptotic threshold by increasing the mitochondrial priming, thus sensitizing resistant CRC cells to chemotherapy; also, Ch282-5 potentiated the response to oxaliplatin both in vitro and in vivo[61]. On the other hand, ABT-263 has been proven to overcome hypoxia-driven radioresistance and improve radiotherapy[62]. Several BH-3 mimetics are currently being tested in clinical trials, even though not for CRC, but mainly for hematological malignancies and other solid tumors[59]. Hence, it may be hypothesized that in a near future these molecules will also be tested in CRC patients.
Cancer stem cells and the tumoral niche
It has been repeatedly demonstrated that in the bulk of the tumor population, a very small fraction is represented by cancer stem cells (CSCs) that are intrinsically resistant to chemotherapy. In addition, upon exposure to anticancer agents and radiotherapy they may enter a quiescent state, resistant to anti-proliferative drugs; once the therapy is suspended, they can self-renew and differentiate into heterogeneous lineages of cancer cells, thus driving tumor recurrence[63]. Initially, several data indicated that CSCs upregulate drug-efflux pumps, have a superior DNA-repair capacity, and have enhanced antioxidant defenses[64]. More recently, cell plasticity and, in particular, the ability of CSCs to adopt a quiescent state have also emerged as important drivers of drug resistance. In fact, lineage-tracing approaches have revealed that the potential of committed cells to move up and down the hierarchy of differentiation (“plasticity”) is more widespread than previously thought. Interestingly, several studies have provided evidence that both CSCs and non-CSCs are plastic and capable of undergoing phenotypic transitions in response to appropriate stimuli[64]. Therefore, under stimuli coming from the tumoral niche - composed of mesenchymal cells, tumor-infiltrating immune cells (TIIC), endothelial cells, extracellular matrix, and inflammatory mediators - the stemness phenotype can be acquired also by non-CSCs. In particular, cancer cells that can enter a reversible drug-tolerant “persister” state in response to treatment have been described[65]; this population is made of cycling and non-cycling persisters which arise from different cell lineages with distinct transcriptional and metabolic programs. Upregulation of antioxidant gene programs and a metabolic shift to fatty acid oxidation has been associated with persister proliferative capacity across multiple cancer types, including CRC. Notably, using different inhibitors to impede oxidative stress or metabolic reprogramming led to a significant reduction in the fraction of cycling persisters[66].
In CRC it has been shown that chemotherapy enriches for cells with a CSC phenotype. However, a pivotal role for a full definition of functional stem cell is concomitantly played by tumor microenvironment (TME) properties and in particular by osteopontin - a multifunctional protein that can also act as a cytokine - produced by key cancer-associated fibroblasts (CAFs)[67]. Recently, an escape mechanism leading to tumor re-growth after 5-FU treatment has been identified in a p53-mediated activation of the WNT/beta-catenin signaling, a pivotal pathway to sustain CRC CSCs. Accordingly, the addition of a WNT inhibitor to 5-FU effectively suppressed the CSCs and reduced tumor re-growth after discontinuing the treatment[68]. Also, CRC CSCs can escape immune surveillance by avoiding recognition by the innate immune system and shape the TME through the release of exosomes, cytokines, and chemokines to generate an immunosuppressive niche that facilitates cancer progression[69].
As mentioned above, TME cues from the tumoral niche can support drug resistance. Important players in this scenario are CAFs, which represent an essential component of tumoral stroma and produce several cytokines acting on tumor cells. Beyond osteopontin, another multifunctional cytokine released by CAFs is transforming growth factor beta (TGFB) that can act synergistically with hypoxia-induced tumor cell-expressed HIF1A to sustain 5-FU/oxaliplatin resistance via activation of the hedgehog pathway, as demonstrated by in vitro and in vivo experiments using patient-derived cells[70]. Interestingly, 5-FU-induced TGFB production occurs also in CRC cells as a mechanism of resistance and targeting TGFBRI restores the sensitivity of drug-resistant cells to 5-FU toxicity[71]. In addition, the 5-FU-elicited TGFB1 protective action can also be abolished by the use of BTK inhibitors[51]. Other than through cytokine release, CAFs can communicate with the cancer cell directly, by transferring exosomes. For example, it has been shown that exosomal transfer of miR-92a-3p to CRC cells activates the WNT/beta-catenin pathway and inhibits mitochondrial apoptosis, and contributes to cell stemness, epithelial-to-mesenchymal transition, metastasis, and resistance to 5-FU/oxaliplatin treatment[72]. Also, exosomal transfer of lncRNA H19 has been found to promote the stemness and chemo-resistance of CRC via activation of the WNT/beta-catenin pathway due to its action as a competing endogenous RNA sponge for miR-141[73]. Notably, these data further indicate WNT/beta-catenin pathway as a potentially actionable target to overcome chemotherapy resistance.
Exosomes can also be exchanged by cancer cells themselves either in normal conditions or upon chemotherapy. For example, it has been shown that miR-21 is significantly upregulated in exosomes purified from CRC cell lines and that adding them to the cells induces resistance against 5-FU through the downregulation of PDCD4[74]. Exosomes derived from 5-FU-resistant cells instead are enriched in growth/differentiation factor 15 (GDF15) and dipeptidyl peptidase IV (DPP4), both of which proved to be potent inducers of angiogenesis[75,76]. Accordingly, 5-FU-resistant CRC cells showed high microvascular density in vivo[75]. Taken together these data indicate that GDF15 and DPP4 may be novel targets for inhibiting angiogenesis in 5-FU-resistant colon cancers.
Finally, in the tumoral niche, different types of TIICs are present that interact with cancer cells and the other components of the niche through cytokine production, eventually altering tumor growth and its response to drug therapy[69,77]. TIICs in the TME have dual functions in cancer progression: TIIC-related inflammation facilitates tumorigenesis, but TIICs also harbor antitumor properties when appropriately activated. Cancer cell-secreted factors hijack TIIC functions to promote tumor development and metastasis and to suppress immune recognition[69]. Accordingly, one of the most important progress in cancer treatment has been the recent addition to chemotherapy of the so-called immune checkpoint (ICIs) inhibitors that act by interrupting the immunosuppressive signals within the TME and reactivating antitumor immunity[78].
The gut microbiota
Gut microbiota seems to be implicated in chemotherapy efficacy through numerous mechanisms [Table 3], including xenometabolism, immune interactions, and altered community structure. Evidence suggests a potential relationship between the presence of Fusobacterium nucleatum and resistance to 5-FU and oxaliplatin chemotherapy and no response to immunotherapy. For example, Yu et al.[79] reported that F. nucleatum was abundant in CRC tissues in patients with recurrence post-chemotherapy and associated with clinicopathological characteristics. Mechanistically, F. nucleatum targeted TLR4 and MYD88 innate immune signaling and specific microRNAs to activate the autophagy pathway and support drug resistance[79]. Other mechanisms by which F. nucleatum confers resistance to chemotherapy have been reported, such as the upregulation of BIRC3 expression (a member of the inhibitor of apoptosis family, IAPs)[80] and the activation of the WNT/beta-catenin pathway via a TLR4/P-PAK1 cascade[81]. In an animal model of CRC, the intratumor presence of bacteria belonging to the Gammaproteobacteria class and producing the long form of the bacterial enzyme cytidine deaminase, which mediates gemcitabine deamination, conferred resistance to gemcitabine; accordingly, the elimination of bacteria by ciprofloxacin treatment restored the response to chemotherapy. In addition, the presence of Mycoplasma hyorhinis determined resistance to gemcitabine[82]. At variance, responses to oxaliplatin were reduced in efficacy in tumor-bearing mice that lacked microbiota[83] indicating that the microbial population resident in the gut can affect both sensitivity and resistance to chemotherapy, being these effects modulated by different bacteria.
The gut microbiota and the response to chemotherapy/immunotherapy
| Bacterium | Correlation with drug resistance | Mechanism | Ref. |
| Fusobacterium nucleatum | Abundant in CRC tissues of patients with recurrence post-chemotherapy and associated with clinicopathological characteristics | Targeting of TLR4 and MYD88 innate immune signaling and targeting specific microRNAs to activate the autophagy pathway | [79] |
| Upregulation of BIRC3 expression | [80] | ||
| Activation of the WNT/beta-catenin pathway via a TLR4/P-PAK1 cascade | [81] | ||
| Gammaproteobacteria Mycoplasma hyorhinis | Resistance to gemcitabine reverted by eliminating bacteria by ciprofloxacin treatment | Production of the long form of the bacterial enzyme cytidine deaminase which mediates gemcitabine deamination | [82] |
| Whole microbiota | Reduction of the response to oxaliplatin in mice lacking microbiota | [83] | |
| Bacteroides thetaiotaomicron Bacteroides fragilis | Their introduction in SPF mice models of CRC restored the response to anti-CTLA-4 treatment, absent in germ-free or antibiotic treated mice | [147] |
Finally, it has been recently shown that commensal bacteria can produce extracellular vesicles (EVs) able to deliver to the human cells bacterial RNA molecules with gene expression regulatory ability, thus suggesting that they might potentially regulate the expression of genes involved in drug resistance. The communication between commensal bacteria and host cells have been shown to be bi-directional suggesting a reciprocal regulation of gene expression and accordingly, biological cell function. An example of this communication having an impact in the development of drug resistance is offered by the previously mentioned study by
RESISTANCE TO TARGETED THERAPY
According to ASCO guidelines, chemotherapy can be possibly integrated with anti-angiogenic agents such as bevacizumab or aflibercept (first-line and second-line treatment, for advanced CRC) and regorafenib (in patients with mCRC who have already received chemotherapy and other targeted therapies). In selected cases, chemotherapy may also be integrated with the addition of targeted therapy. For example, anti-EGFR (anti-epidermal growth factor receptor) monoclonal antibodies (e.g., cetuximab and panitumumab) are indicated only for RAS- and BRAF-wild type (wt) tumors since the mutation of these two genes confer resistance to the EGFR blockade. In addition, a combination using the BRAF inhibitor encorafenib and cetuximab may be used to treat patients with BRAF-mutant (mut) mCRC who have received at least one previous treatment. In the last decade, several tumor-expressed “immune checkpoints”, immunosuppressive molecules blocking antitumor immunity, have been identified and “checkpoint inhibitors” have been developed and entered the clinic. Among them are antibodies against programmed cell PD-1 or its ligand PD-L1 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4). So far, immunotherapy has a limited use in CRC since PD-1 targeting monoclonal antibodies can be used only in selected cases of mCRCs bearing defects along the DNA damage response pathways. For example, pembrolizumab is used for the therapy of mCRCs that are MSI-H or dMMR. In contrast, nivolumab can be administered to patients with dMMR or MSI-H mCRC that has grown or spread after treatment with chemotherapy, either alone or in combination with ipilimumab (anti-CTLA-4 monoclonal antibody). Notably, the FDA has issued its approval for pembrolizumab as the first-line treatment of patients with dMMR/MSI-H mCRC in June 2020, even though neither the Japanese Pharmaceuticals and Medical Devices Agency nor the European Medicines Agency has yet approved pembrolizumab as the frontline regimen[84].
To expand the range of possible combinations of precision drugs to add to chemotherapy several efforts have been made to better stratify patients and to understand the basis of resistance/insensitivity to targeted therapy.
Resistance to anti-EGFR blockade
Perhaps the best studied so far are the mechanisms and the determinants underlying the resistance to anti-EGFR drugs [Figure 2], given that they have been the first targeted drugs employed for the treatment of CRC, and are so far the most used [Tables 4 and 5]. The main genetic defects hampering the response to EGFR blockade are activating mutations leading to constitutive activation of signaling pathways downstream of EGFR such as mutations in KRAS (30%-40%), NRAS (4%), BRAF (7%-15%), or PIK3CA (17%-30%) genes and PTEN deletion or truncation (7%) which altogether account for unresponsiveness in around 70% of cases[85,86]. It has been reported that in approximately 50% of RAS-wt patients, sensitivity to EGFR blockade is lost because of secondary occurring mutations[87]. In a small fraction of the remaining resistant tumors two important biomarkers are emerging. Bardelli et al.[88] initially reported that certain patients harboring RAS-wt gene and resistant to anti-EGFR therapy presented an amplification of the MET proto-oncogene that proved to be responsible for de novo and acquired resistance to anti-EGFR therapy. In fact, functional studies showed that MET activation conferred resistance to anti-EGFR therapy both in vitro and in vivo and could be overcome using MET inhibitors[88]. Remarkably, examining circulating tumor DNA, it was possible to find MET amplification even before the recurrence became clinically evident. Much experimental evidence further supported the benefit of targeting MET in anti-EGFR-resistant CRCs and explored the mechanisms driven by MET. Among them, acquisition of cetuximab resistance was shown to be mediated by TGFA overexpression which in turn induced EGFR-MET interaction[89]. Also, SRC activation promoted cetuximab resistance by directly interacting with MET; accordingly, pretreatment with SRC inhibitors abolished cetuximab-mediated MET activation and rendered CRC cells sensitive to cetuximab[90]. Interestingly, concurrent inhibition of MET and SRC decreased viability and enhanced apoptosis in both RAS-wt and RAS-mut CRC cells. Therefore, combined inhibition of MET and SRC may be a promising strategy for treating of CRC, independent of anti-EGFR resistance[91]. In addition, MET inhibition, by enhancing the formation of DNA double-strand breaks and possibly alleviating tumor hypoxia, has been found to sensitize CRC cells also to irradiation[92], further expanding the actionability of MET in CRCs.
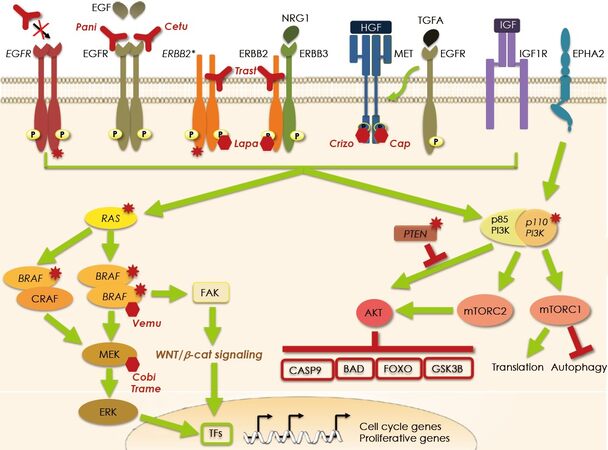
Figure 2. Mechanisms of resistance to targeted therapy against EGFR and mutated BRAF. Intrinsic resistance to EGFR blockade by panitumumab (Pani) and cetuximab (Cetu) occurs in case of activating mutations in the EGFR and its downstream effectors such as RAS, BRAF, and PIK3CA and when there is loss of PTEN either due to truncation or deletion. Acquired resistance stems from stimulation of collateral pathways impinging on the same downstream signaling activated by EGFR (i.e., the RAS/MAPK and the PIK3/AKT pathways). Hyperactivation of these pathways may be due to overexpression or mutation of ERBB2, heterodimerization of ERBB2 and ERBB3, engagement of the EGFR by TGFA which in turn activates MET, MET overexpression, EPHA2 overexpression, or IGF1R engagement. Strategies to overcome the insensitivity to EGFR blockade are directed at the inhibition of the collateral signaling such as using ERBB2 blocking agents trastuzumab (Trast) and lapatinib (Lapa) and the MET inhibitors crizotinib (Crizo) and capmatinib (Cap). Resistance to the mutated BRAF inhibitor vemurafenib (Vemu) originates from the shifting in the choice of the dimerization companion. In the resistant cells instead of mutated BRAF homodimers, mutated BRAF/CRAF heterodimers are formed which are insensitive to the inhibitor and thus MAPK activation is refueled. To prevent hetodimerization a novel inhibitor, that specifically bind mutated BRAF homodimers has been developed. Another route of escape to mutant BRAF blockade has been described where activation of the WNT/β-catenin signaling is triggered via FAK. Both MAPK and β-catenin signaling eventually activate a series of transcription factors (TFs) responsible for the transcription of proliferative genes. Currently, to prevent resistance to BRAF inhibition, a vertical blockade of the pathway is being tested in clinical trials using vemurafenib in combination with a MEK inhibitor such as cobimetinib (Cobi) or trametinib (Trame). Mutated proteins are indicated by the red stars and the italicized name.
Resistance to anti-EGFR blockade
| Treatment | Mutational background | Mechanism | Ref. |
| Cetuximab | Activating mutations in KRAS, NRAS, BRAF or PIK3CA genes and PTEN deletion, truncation, or epigenetic silencing | Constitutive activation of signaling pathways downstream of EGFR | [85,86] |
| Cetuximab Panitumumab | MET amplification in wtKRAS setting | Activation of signaling pathways converging onto PIK3CA and MAPK; resistance bypassed using MET inhibitors | [88] |
| Cetuximab | TGFA overexpression which in turn induced EGFR-MET interaction | [89] | |
| Cetuximab | Cetuximab-mediated MET activation via interaction with SRC and abolished by SRC inhibitors | [90] | |
| Cetuximab | ERBB2 amplification /mutations in quadruple (KRAS, NRAS, BRAF, and PIK3CA) wt setting | Constitutive activation of signaling pathways downstream of the receptor; resistance overcome by addition of anti-ERBB2 agents | [97,98] |
| Cetuximab | ERBB2 amplification and heregulin overexpression | ERBB2/ERBB3 dimerization; resistance bypassed by silencing ERBB2 and depleting heregulin | [101] |
| Intrinsic resistance to anti-EGFR agents | ERBB2 activating mutations in KRAS-wt mCRCs | Constitutive activation of signaling pathways downstream of the receptor; resistance bypassed by dual targeting of ERBB2 via trastuzumab and lapatinib | [103] |
| Cetuximab+Refametinib | KRAS-mut | PI3K-AKT pathway activation ensuing from autocrine loops and heterodimerization of multiple receptors (ERBB2, ERBB3, and IGF1R) | [112] |
| Cetuximab+FOLFIRI | RAS-wt mCRC | High levels of EPHA2 significantly correlated with worse PSF and increased progression rate in a cohort of patients; primary and acquired resistance to cetuximab reverted in in in vitro and in vivo models by adding a specific EPHA2 inhibitor | [114] |
Strategies to overcome resistance to EGFR inhibition
| Drug(s) | Mutational background | Mechanism | Ref. |
| Cetuximab + Crizotinib | MET amplification | Double targeting of EGFR and MET | [88] |
| Cetuximab + PHA665752 | Overexpression of TGF-α | Double targeting of EGFR and MET | [89] |
| Cetuximab + PHA665752 Cetuximab + Dasatinib | RAS-wt; MET activation | Double targeting of EGFR and MET Double targeting of EGFR and SRC | [90] |
| Cetuximab + Lapatinib Cetuximab + Pertuzumab | ERBB2 amplification | Double targeting of EGFR and ERBB2 | [97] |
| Trastuzumab + Neratinib Trastuzumab + Afatinib | ERBB2 amplification or mutation | Double targeting of ERBB2 | [98] |
| Trastuzumab + Lapatinib (clinical trial) | ERBB2 overexpression; RAS-wt | Double targeting of ERBB2 | [103] |
| Cetuximab + ALW-II-41-27 | EPHA2 overexpression | Double targeting of EGFR and EPHA2 | [114] |
Despite experimental efficacy, the clinical trials performed so far did not confirm the expectations. In a phase 2 study the addition of the specific MET inhibitor tivantinib to cetuximab allowed only 10% of MET-high, KRAS-wt mCRC patients to achieve objective response, even though in a difficult-to-treat setting (tumor progression on cetuximab or panitumumab after a first-line chemotherapy)[93].
In another phase 1/2 study addition of tivantinib to cetuximab + irinotecan was tested in KRAS-wt patients previously treated but who progressed or presented with mCRC. In these cases, the combination did not significantly improve progression-free survival (PFS), even though subgroup analyses trended in favor of tivantinib in patients with MET-high tumors, PTEN-low tumors, or those pretreated with oxaliplatin[94]. One possible explanation for these disappointing clinical results might be that clinical efficacy has been obscured by a lack of standardization in MET assessment for patient stratification. Accordingly, a very recent paper indicated that subtyping of MET expression may be required to identify MET-addicted malignancies in CRC patients who will truly benefit from MET inhibition[95].
In 2011 ERBB2 gene amplification was reported in 7% of patients with CRC[96]. Subsequently, ERBB2 amplification or somatic mutations have been described by several studies, with highly different reported rates of positivity (up to 50%) likely due to several factors such as cohort heterogeneity, a small study population, antibody clone selection, staining platform, and different scoring system. Using a molecularly annotated platform of 85 mCRC patient-derived xenografts, Bertotti et al.[97] identified ERBB2 amplification as an actionable liability in cetuximab-resistant CRCs harboring wt KRAS/NRAS/BRAF/PIK3CA. Moreover, they showed that the addition of anti-ERBB2 agents to anti-EGFR monoclonal antibodies overcame resistance and inhibited tumor growth[97]. The same group also showed that tumor growth was reduced by single targeting of ERBB2 in cetuximab-resistant, quadruple (i.e., KRAS, NRAS, BRAF, and PIK3CA) wt CRC patient derived xenografts with ERBB2 mutations. However, only double targeting of EGFR and ERBB2 led to durable tumor regression[98].
Later, the role of ERBB2 amplification/mutation in predicting response to anti-EGFR treatment was confirmed by different groups. Cremolini et al.[99], conducted a prospective, case-control study to demonstrate the negative predictive impact of a panel of rare genomic alteration including ERRB2 amplification/activating mutations, MET amplification, ROS1/NTRK1-3/RET rearrangements, and PIK3CA exon 20, PTEN, and ALK mutations. Of the resistant cases, 51.1% were associated with one of these candidate molecular alterations, ERBB2 amplification being the most frequent (14.9%), followed by MET amplification (8.5%)[99]. A retrospective study performed in RAS/BRAF-wt mCRC patients reported that anti-EFGR therapy allowed a PFS significantly longer in patients without ERBB2 amplification than in those bearing the alteration[100]. Shorter PFS and OS were demonstrated also in cetuximab-treated CRC patients with ERBB2-amplified tumors and higher serum heregulin levels. In addition, in vitro experiments proved that silencing ERBB2 overexpression, as well as depleting heregulin - thus disrupting of ERBB2/ERBB3 heterodimerization - restored the response to cetuximab[101]. Notably, ERBB2 amplification could be identified non only in tissues but also in circulating tumor DNA of CRC patients non-responsive to anti-EGFR therapy[102]. Sartore-Bianchi et al.[103], demonstrated that ERBB2 activating mutations in KRAS-wt mCRCs predicted intrinsic resistance to anti-EGFR agents that could be bypassed by combining dual targeting ERBB2 via trastuzumab (anti-ERBB2 monoclonal antibody) and lapatinib (dual EGFR and ERBB2 tyrosine kinase inhibitor). A phase 2 basket trial recently obtained an overall response rate of 32% when using the dual targeting approach (pertuzumab and trastuzumab) in pretreated ERBB2-amplified mCRC patients[104]. In addition, two other phase 2 clinical trials also identified ERBB2 as a druggable target in refractory KRAS-wt mCRCs with ERBB2 amplification[105,106], and ERBB2-targeted combinations have been recently included in international guidelines for mCRC[107].
Besides the above-mentioned clinical studies, several trials are currently undergoing where ERRB2-targeted antibody-drug conjugates (i.e., TDM-1, DS-8201, A166, ZW25, and ZW49) and novel tyrosine kinase inhibitors (i.e., tucatinib, sapitinib, neratinib, pyrotinib, poziotinib, and ceralasertib) are being tested alone or in different combinations[108]. A recent study pinpointed that an optimal combination may require triple targeting. Mangiapane et al.[109], using 60 CRC specimens obtained a collection of CSC-derived spheres which was then used to perform preclinical experiments to validate different therapeutic options. They found that high expression levels of ERBB2 occurred in spheres showing hyperactive PI3K/AKT pathway and proved that triple targeting of ERBB2, MEK and PI3K induces CSC death and regression of anti-EGFR-resistant tumor xenografts, including those carrying KRAS and PIK3CA mutation[109].
Even though MAPK deregulation plays a pivotal role in both intrinsic and secondary resistance to anti-EGFR agents, other mechanisms (sometimes co-occurring) contribute or can account for secondary resistance, due to CRC molecular heterogeneity[110]. For example, stimulation of EGFR results not only in the RAS/RAF/MAPK pathway but also in the PTEN-PI3K-AKT cascade activation[111].
Recently, Vitiello et al.[112] used four different KRAS-mut CRC cell lines rendered resistant to a combination of cetuximab and refametinib (a selective MEK-inhibitor) and demonstrated that PI3K-AKT pathway activation acts as an escape mechanism in this setting. They showed that autocrine loops and heterodimerization of multiple receptors lead to the activation of several receptor tyrosine kinase, such as ERBB2, ERBB3 and IGF1R, whose signaling onto the PI3K/AKT pathway allows to bypass the block imposed by the double targeting of EGFR and MEK[112]. The effect on the response to cetuximab in the setting of the three most common AKT1 activating mutations found in CRC patients (i.e., E17K, E49K, and L52R) was recently investigated by overexpressing them in a cetuximab-sensitive KRAS-wt CRC cell line. Interestingly, all of them impaired the cytotoxic response not only toward cetuximab but also to chemotherapy. In addition, also the most common mutations of CTNNB1 (i.e., T41A, S45F, and S33P) were found to sustain resistance to the same agents[113]. These data indicate that possible therapeutic strategies to counteract the resistance to EGFR-targeted therapy would therefore be the use of inhibitors of the PI3K/AKT pathway. Notably, trials to test AKT and PI3K inhibitors for CRC are currently undergoing.
A novel predictive biomarker of resistance and a potential therapeutic target for improving the response to anti-EGFR agents has recently been identified in EPHA2. Studying a cohort of 82 RAS-wt mCRC patients treated with FOLFIRI + cetuximab, Martini et al.[114] found that 62.6% of patients expressed the tyrosine kinase receptor and that high levels of EPHA2 significantly correlated with worse PFS and increased progression rate. In in vitro models a specific EPHA2 inhibitor reverted primary and acquired resistance to cetuximab causing cell growth inhibition, inducing apoptosis and cell-cycle G1-G2 arrest. In xenografts experiments the treatment with the inhibitor upon progression with cetuximab significantly inhibited tumor growth[114].
Resistance to BRAF inhibitors
The BRAF kinase, operating downstream of RAS along the pathway leading to ERK activation, is considered an oncogenic driver in CRC and its mutation arises in 7%-15% of patients with mCRC. Over 95% of BRAF mutations in mCRC occur in codon 600 (V600E) whereas non-V600E BRAF mutations occur only in about 2% of patients and define a clinically distinct subtype with a better prognosis. In general, patients with
Resistance to BRAF inhibitors
| BRAFi | Mutational background | Mechanism | Ref. |
| Vemurafenib | BRAFV600E | Only BRAF monomers inhibited by vemurafenib; EGFR-mediated reactivation of MAPK signaling, due to the formation of active RAF and CRAF protein dimers | [118,119] |
| Vemurafenib | BRAFV600E | Upregulation of Wnt/β-catenin pathway through activation of FAK | [122] |
| Vemurafenib | BRAFV600E | Upregulation of NPM1 | [123] |
Strategies to overcome resistance to BRAF inhibitors
| Drug(s) | Mutational background | Mechanism | Ref. |
| PLX8394 | BRAFV600E | Specific inhibition of BRAF dimers; prevent EGFR-mediated reactivation of MAPK signaling | [120] |
| Cobimetinib + A-1210477 | BRAFV600E | Double targeting of MEK and MCL-1 | [121] |
| Vemurafenib + PF-562271 Vemurafenib + ICG-001 Vemurafenib + PF-562271 + trametinib Vemurafenib + ICG-001 + trametinib | BRAFV600E | Double targeting of BRAFV600E and FAK Double targeting of BRAFV600E and β-catenin Triple targeting of BRAFV600E, FAK, and MEK Triple targeting of BRAFV600E, β-cat, and MEK | [122] |
| Vemurafenib + NSC348884 | BRAFV600E | Double targeting of BRAFV600E and NPM1 | [123] |
| Encorafenib + Cetuximab (clinical trial) | BRAFV600E | Double targeting of BRAFV600E and EGFR | [136] |
Given the EGFR reactivation observed in BRAF inhibitor-treated CRC models, dual targeting has been tested. Adding cetuximab to vemurafenib in xenografts experiments resulted in increased antitumor activity and improved survival[124] and combined administration of BRAF and EGFR inhibitors induces tumor regression in most patients[116,125-127]. However, within 6 months, resistance inevitably occurs. Analyzing matched tumor tissues obtained from eight patients before treatment and at the time of disease progression after therapy, Yaeger et al.[128] found that in resistant tumors new alterations occurred in genes that encode components of the RAS/RAF pathway (activating mutations of KRAS or NRAS or amplification of
Preclinical and clinical data obtained in BRAFV600E melanoma models and patients indicated that simultaneous and vertical targeting of more than one node along the MAPK/ERK pathway, such as BRAF and MEK, could bypass the resistance to BRAF inhibitors[129,130] and delay the onset of relapse. However, also this combination did not live up to expectations in BRAFV600E CRCs. In fact, combination treatment with BRAF plus MEK inhibitors, dabrafenib and trametinib, led to a median PFS of 3.5 months, with 56% of patients achieving stable disease, 9.3% partial response, and only 2.3% complete response[131]. It has been recently shown that the combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib triggered a massive apoptotic response only in BRAFV600E CRC cells harboring also TERT promoter mutations but not in BRAFV600E cells lacking TERT mutations. Accordingly, the combination of inhibitors nearly completely abolished the growth of xenografted tumors BRAFV600E/TERT-mut but had little effect on tumors harboring only BRAFV600E. Notably, after the treatment was discontinued, doubly mutated tumors remained barely measurable whereas BRAFV600E tumors regrew rapidly[132]. These findings suggest that double testing for BRAFV600E and TERT mutations may stratify patients for combinatorial therapy with BRAF plus MEK inhibitors.
Recent clinical trials investigated the benefit of targeting different nodes along the MAPK pathway in BRAF-mut mCRC[133-135]. BEACON III was a three-arm phase 3 study that enrolled patients with BRAFV600E mCRC tumors who progressed on one or two prior regimens. In this setting the safety and efficacy of encorafenib (BRAF inhibitor) and cetuximab with or without binimetinib (MEK inhibitor) were evaluated and compared with the control arm with SOC therapy (irinotecan/cetuximab or FOLFIRI/cetuximab). Both triplet and doublet therapy induced an overall response rate that was 10 times more than in the control arm. In addition, PFS tripled and mean OS doubled for the doublet and triplet arms vs. control arm. Little difference was observed between doublet and triplet arms[136]. Based on these results, on April 2020, the FDA approved encorafenib in combination with cetuximab for mCRCs with a BRAFV600E mutation[137].
A final note, not directly related to resistance but pointing to alternative actionable targets in BRAFV600E CRCs, should be made about immunotherapy. As previously mentioned, therapy with ICIs is limited to mCRCs bearing defects along DNA damage response pathways. Notably, around 60% of BRAF-mut CRCs also are MSI-H, a condition associated with a higher proportion of tumor infiltrating lymphocytes present in the tumor[138]. Therefore, it is reasonable to suggest that ICIs could represent the new SOC for this subgroup[139].
Resistance to anti-angiogenic therapy
The first anti-angiogenic agent entering the clinic for the treatment of mCRC has been bevacizumab, a humanized monoclonal antibody anti-VEGF-A which binds circulating VEGF-A and blocks the interaction with its cell surface receptors (VEGFR1 and 2), thus reducing microvascular growth and inhibiting the blood supply to the tumor tissues[140]. In recent years the development of anti-angiogenic agents has increased, leading to the addition to of aflibercept (second-line treatment for advanced CRC) and regorafenib (in patients with mCRC who have already received chemotherapy and other targeted therapies). Aflibercept is a recombinant fusion protein consisting of VEGF-binding portions from the extracellular domains of human VEGFR1 and 2, that are fused to the Fc portion of the human IgG1 immunoglobulin and acts like a “VEGF trap”. Regorafenib is multi-kinase inhibitor targeting angiogenic, stromal, and oncogenic receptor tyrosine kinase that shows strong anti-angiogenic activity due to its dual targeted VEGFR2-TIE2 tyrosine kinase inhibition. In addition, other anti-angiogenic drugs are in preclinical and clinical phase I-III studies[141].
Despite initial promising preclinical results, anti-angiogenic monotherapies did not lead to many clinical benefits, due to primary or acquired resistance, through activation of alternative mechanisms that support vascularization and tumor growth [Figure 3]. Clinical data show that anti-angiogenesis therapies can prolong PFS but have a limited impact on OS and do not set a permanent cure in CRC[142]. This limited clinical significance is likely due to the fact that treatment with antiangiogenic agents causes a discontinuity in the blood vessel network, which leads to a new hypoxic condition with activation of vascular mimicry, alternative pro-angiogenic pathways and recruitment of endothelial cells derived from bone marrow[143].
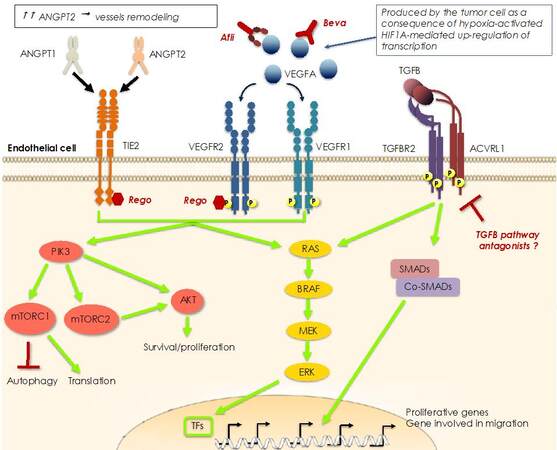
Figure 3. Mechanisms of resistance to anti-angiogenic therapy. Hyperproduction of VEGFA is a consequence of the hypoxic status in the center of the tumor which activates HIF1A, a transcription factor that regulates VEGFA transcription. Overproduction of VEGFA can be blocked by monoclonal antibodies (bevacizumab, Beva) or a VEGF-trap (aflibercept, Afli). Alternatively, the signal can be blocked downstream of the dedicated receptor by small kinase inhibitors like regorafenib (Rego), which can also inhibit the TIE2 receptor, thus imposing a double anti-angiogenic blockade. In fact, both receptors are critical in stimulating the proliferation and migration of the endothelial cells, necessary for the formation of new vessels into and around the tumor mass. In particular, TIE2 receptor is regulated by ANGPT1 and 2, the former inducing signals involved in maturation and stabilization of blood vessels, whereas the latter blocks this signaling and triggers remodeling of vascular sprouts. Overproduction of ANGPT2 can drive a parallel neo-angiogenic signaling when VEGR-driven signaling is impeded by Beva-based therapy. In addition, another parallel neo-angiogenic pathway that can act as an escape route to VEGFA blockade is initiated by TGFB binding to a multimeric receptor formed by a TGFB2 dimer in complex with an activin receptor-like kinase 1 (ACVRL1) dimer.
So far, the mechanisms of resistance to anti-VEGF therapy have not been fully elucidated. The main factor used by cancer cells to adapt to oxygen deprivation is hypoxia-inducible factor (HIF) of which three members are known (i.e., HIF1A, HIF2A, and HIF3A). During hypoxia, HIF1A translocates into the cell nucleus to form the activated HIF1 complex along with HIF1B. Hypoxic contexts induce upregulation of VEGF expression through the upstream transcription factor HIF1A. These factors induce tumors to acquire more angiogenic and invasive potentials, promoting metastasis[144]. Preclinical studies and clinical trials suggest that inhibition of a specific growth factor can induce the expression of others[145]. For instance, in a phase II study in which mCRC patients were treated with a combination of 5-FU, leucovorin and irinotecan (FOLFIRI) and bevacizumab, several angiogenic factors, including the placental growth factor and hepatocyte growth factor, were found to increase before disease progression[146]. Activin receptor-like kinase 1 (ACVRL1) is an emerging target for antiangiogenic therapy; it is a TGFB transmembrane receptor expressed preferentially from proliferating endothelial cells and is crucial to TGFB-mediated angiogenesis. Significantly, VEGF-A and ACVRL1 expression have been shown to regulate angiogenesis. Data support the hypothesis that ACVRL1 expression and function following anti-VEGF/VEGFR inhibitor treatment may contribute to vascular adaptation from the VEGF-driven neovascularization[147]. In particular, the TGFB axis may provide an escape pathway for tumor angiogenesis[148]. Thus, agents that inhibit TGFB signaling combined with VEGF/VEGFR- therapies may potentiate treatment response.
Moreover, alternative angiogenic pathway can contribute to the escape from anti-VEGF treatment such as the angiopoietin-TIE signaling system. TIE-1 and -2 are specific vascular receptor tyrosine kinase that control vascular permeability and the development and remodeling of blood vessels through angiopoietin-1 (ANGPT1) and angiopoietin-2 (ANGPT2). While ANGPT1 permits maturation or stabilization of blood vessels through TIE2, ANGPT2 blocks this pathway causing remodeling of vascular sprouts subsequent to exposure to VEGF[149]. High serum levels of ANGPT2 have been related to poor response to bevacizumab treatment[150], suggesting that ANGPT2 has a critical role in the resistance mechanism against anti-VEGF therapy[151].
In preclinical models, the double block of VEGF and ANGPT2 inhibited revascularization and tumor progression of tumors resistant to anti-VEGF therapy[152,153]. Finally, cytokines have also been involved in mediating resistance to anti-angiogenic therapy. IL-17 and its downstream factor, granulocyte colony-stimulating factor (G-CSF), are the leading players of inflammation; it has been reported that a high level of IL-17 induces cells to produce a paracrine network that confers resistance to anti-VEGF therapy. Serum G-CSF levels in CRC patients are higher than those in healthy volunteers and are associated with the stage of tumor progression[154,155].
Interestingly, a novel anti-angiogenic approach that might help in circumventing resistance to anti-angiogenic therapies could be represented by metronomic chemotherapy, a schedule where low doses of chemotherapy are administered chronically. In fact, several studies demonstrated that the administration of 5-FU under metronomic chemotherapy schedule induces an antiangiogenic effect behind the cytotoxic effect[156]. In addition, it has been reported a reasonable disease control with minimal side effects and good quality of life in elderly or heavily pre-treated patients under metronomic chemotherapy schedule. Moreover, a phase III randomized trial has shown that maintenance therapy with metronomic capecitabine plus bevacizumab following cycles of conventional treatment with capecitabine + oxaliplatin + bevacizumab significantly improved PFS compared to the observation group with no worsening of quality of life[157].
Resistance to immune checkpoint inhibitors
Compared to pMMR/microsatellite stable CRCs, dMMR/MSI-H CRCs - because of the defects in repairing DNA damage/insertion of microsatellites - present a high mutation burden and produce a plethora of immunogenic neoantigens on the MHC, thus recruiting more tumor infiltrating lymphocytes. However, dMMR/MSI-H CRCs also highly upregulate the expression of immune checkpoints - (i.e., PD-1 ligands, CTLA-4 ligand, LAG-3, and IDO1) that account for the suppression of an active immune response despite the high tumor infiltrating lymphocyte count [Figure 4][158]. Because of this scenario dMMR/MSI-H CRCs have been shown to be sensitive to treatment with ICIs in several phase 2 trials[84,158-160]. Notably, the KEYNOTE-177 phase 3 trial compared the effect of anti-PD-1 pembrolizumab vs. SOC chemotherapy given as a first line therapy. A significant clinical benefit of pembrolizumab resulting in the doubling of PSF was evident and 83% of pembrolizumab responders were still responding after 2 years or longer, compared to the 35% of the chemotherapy responder. However, 30% of pembrolizumab-treated patients had primary resistance[161] indicating that additional therapeutic strategies are needed in the field of immunotherapy. Interestingly, Checkmate-142 phase 2 clinical trial reported that a double targeting strategy given as a first-line therapy led to significant and clinically meaningful improvements in patients’ outcomes. Specifically, anti-PD-1 nivolumab was administered together with anti-CTLA4 ipilumab, and the PSF rate at 12-month was 77% compared to 55% reported with single PD-1 blockade in the KEYNOTE-177 study[84]. At variance with patients with dMMR/MSI-H CRC those with microsatellite stable mCRC had very limited clinical benefit when receiving the combination with CTLA-4 and PD-L1 inhibitors[160]. Given that about 95% of mCRC cases are pMMR/microsatellite stable tumors and resistant to ICIs, several efforts are focused on the understanding of the mechanisms of resistance to immunotherapy for the development of new therapeutic approaches. Notably, another promising inhibitor to be added to the combination of ICIs is relatlimab, a monoclonal antibody that binds to LAG-3, a checkpoint molecule that functions to negatively regulate homeostasis of T- and B-cells and NK cells[162]. So far, it has been tested in clinical trials for treating melanomas and, interestingly, in a phase 3 trial in patients with newly diagnosed inoperable or metastatic melanomas, its combination with nivolumab doubled PSF[162]. In addition, a newly characterized monoclonal anti-LAG-3 antibody resulted in a significant delay in tumor growth when combined with
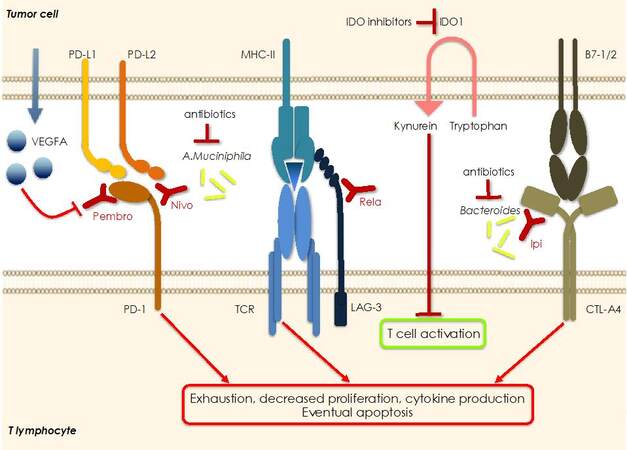
Figure 4. Mechanisms of resistance to immuno-therapy. Cancer cells can express several immunosuppressive molecules that can be blocked using specific inhibitors in order to re-activate the anti-tumor immune response. Often, they are redundant so that the re-activation of one pathway may be compensated by hyperactivation of a different immunosuppressive route so that resistance to therapy is the result. Resistance to anti-PD1 blockade may be due to overproduction and release of VEGFA, as well as by overexpression of IDO1 with consequent increased levels of kynurein in the microenvironment. Specific components of the microbiota are also necessary for the successful blockade of PD-1 and CTL-A4 indicating that antibiotics may represent a resistance factor to ICIs. Finally, concurrent blockade of two or more immune checkpoints has been shown to improve the response and overcome the resistance to ICIs. Pembro: Pembrolizumab; Nivo: nivolumab; Rela: relatlimab; Ipi: ipilumab; IDO1: indoleamine 2, 3-dioxygenase 1.
An immunosuppressive role has been reported for VEGF, which can act via different mechanisms such as by decreasing the number of TILs and activating immune checkpoint molecules[167]. Notably, in a CRC mouse model it has been reported that addition of anti-angiogenic therapy reduced intrinsic resistance to an anti-PD-1 antibody via normalizing tumor vascularization and reducing hypoxia[168]. Several trials are ongoing to evaluate the efficacy of combining ICIs with an anti-angiogenic strategy plus chemotherapy, being the molecules tested bevacizumab and regorafenib.
Recently, several studies have shown that the gut microbiota plays a key role in regulating the efficacy of ICIs for cancer treatment of different solid tumors [Table 3, Figure 4][169]. For example, the treatment with an anti-CTLA-4 monoclonal antibody controlled the growth of different tumor models (including a CRC model) in mice maintained in specific pathogen-free conditions and in the presence of specific Bacteroides, but not in germ-free mice and mice previously treated with antibiotics. Therefore, it appears that the gut microbiota might regulate the efficacy of immunotherapy with anti-CTLA-4. Accordingly, the introduction of two specific Bacteroides species (i.e., B. thetaiotaomicron and B. fragilis) in germ-free mice and antibiotics-treated mice restored the response to anti-CTLA-4 treatment[170]. Notably, a series of studies in melanoma mouse models and patients uncovered other microbiota components as instrumental in determining the response to ICIs. For example, in a melanoma mouse model the addition of the commensal gut Bifidobacterium to PD-L1 checkpoint blockade nearly abolished the growth of scarcely sensitive tumors[171]. Comparative analysis of the oral and gut microbiome of melanoma patients undergoing anti-PD-1 immunotherapy revealed significant differences in the diversity and composition of the microbiome of responders vs. non-responders. Similarly, the analysis of patient fecal microbiome samples showed significant diversity and relative abundance of bacteria of the Ruminococcaceae family in responding patients[172]. Analysis of baseline stool samples from metastatic melanoma patients before immunotherapy uncovered significant association between commensal microbial composition and clinical response. Bacterial species more abundant in responders included Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium. Accordingly, in a mouse model fecal microbiota transplantation from responding patients in germ-free mice improved tumor control, augmented T cell responses, and increased efficacy of anti-PD-L1 therapy[173]. Routy et al.[174] showed that fecal microbiota transplantation from cancer patients who responded to ICIs into germ-free or antibiotic-treated mice improved the efficacy of anti-PD-1 therapy, whereas fecal microbiota transplantation from non-responding patients was ineffective. Profiling patients’ stool samples revealed low levels of the bacterium Akkermansia muciniphila in non-responding patients with lung and kidney cancer. Accordingly, oral supplementation of the bacteria after fecal microbiota transplantation with non-responders feces in antibiotic-treated mice restored the response to PD-1 blockade[174]. Regulation of gut microbiota composition or addition of the “right” bacteria to therapy seems therefore a very promising approach to overcome resistance, or at least significantly improve the response to ICIs.
CONCLUSION
Intrinsic and acquired resistance to chemo-, targeted, and immune-therapy remain the core problem(s) to tackle for ameliorating the outcome of CRC patients, especially in the metastatic setting. Significant improvements and longer expectancy of life have been reached using a combination/integration of the different types of therapy, but only the identification of additional targetable liabilities to be exploited in specific settings will allow to concomitantly attack the tumor from different angles, hopefully leading to its eradication. In this sense, promising approaches are: the development of novel nanoparticles able to vehicle chemotherapy directly inside the cells avoiding the intake/efflux systems, often deregulated in resistant cells; the induction of synthetic lethality via the use of inhibitors of specific DNA damage checkpoints or DNA repair enzymes in tumors presenting defects in the DNA repair mechanisms; the use of BH-3 mimetics in tumors highly expressing anti-apoptotic members of the BCL2 family; the targeting of several kinases shown to sustain resistance to classic chemotherapy and also resistance to target therapy via activation of feed-back loops; the targeting of pathways crucial for cancer stem cells maintenance and viability; the targeting of several components of the TME, recognized to be responsible for an immune-privileged environment, among which the so-called immune checkpoints; and the modulation of the microbiota. For the most of these approaches an unavoidable task will be patient stratification according to specific biomarkers and/or genetic defects, indicating that precision medicine is the ultimate path to cover in order to identify effective therapies for each patient. In addition, given the high tumoral heterogenicity and the Darwinian pressure exerted by anti-cancer treatments, to prevent and/or overcome resistance it will be also essential to combine different types of therapies and using vertical approaches to target multiple nodes along the same pathway.
DECLARATIONS
Authors’ contributionsDrafting the article, revising it, and giving final approval prior to submission: Grassilli E, Cerrito MG
Availability of data and materialNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177-93.
2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017;66:683-91.
3. Colorectal cancer: statistics. Available from: https://www.cancer.net/cancer-types/colorectal-cancer/statistics [Last accessed on 23 Dec 2021].
4. Yoshino T, Arnold D, Taniguchi H, et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol 2018;29:44-70.
5. Glynne-Jones R, Wyrwicz L, Tiret E, et al. ESMO Guidelines Committee. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29:iv263.
6. Benson AB, Venook AP, Al-Hawary MM, et al. NCCN Guidelines insights: colon cancer, version 2.2018. J Natl Compr Canc Netw 2018;16:359-69.
7. Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol 2016;8:57-84.
8. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6:1769-92.
9. Cerrito MG, Grassilli E. Identifying novel actionable targets in colon cancer. Biomedicines 2021;9:579.
10. Cremolini C, Schirripa M, Antoniotti C, et al. First-line chemotherapy for mCRC-a review and evidence-based algorithm. Nat Rev Clin Oncol 2015;12:607-19.
11. Marin JJG, Macias RIR, Monte MJ, et al. Cellular mechanisms accounting for the refractoriness of colorectal carcinoma to pharmacological treatment. Cancers (Basel) 2020;12:2605.
12. Grant CE, Valdimarsson G, Hipfner DR, et al. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res 1994;54:357-61.
13. Cao D, Qin S, Mu Y, Zhong M. The role of MRP1 in the multidrug resistance of colorectal cancer. Oncol Lett 2017;13:2471-6.
14. Zhao J, Li W, Zhu D, et al. Association of single nucleotide polymorphisms in MTHFR and ABCG2 with the different efficacy of first-line chemotherapy in metastatic colorectal cancer. Med Oncol 2014;31:802.
15. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer 2018;18:452-64.
16. Borst P. Looking back at multidrug resistance (MDR) research and ten mistakes to be avoided when writing about ABC transporters in MDR. FEBS Lett 2020;594:4001-11.
17. Gao Q, Li XX, Xu YM, et al. IRE1α-targeting downregulates ABC transporters and overcomes drug resistance of colon cancer cells. Cancer Lett 2020;476:67-74.
19. Park SM, Kang TI, So JS. Roles of XBP1s in transcriptional regulation of target genes. Biomedicines 2021;9:791.
20. Gu J, Dong D, Long E, et al. Upregulated OCT3 has the potential to improve the survival of colorectal cancer patients treated with (m)FOLFOX6 adjuvant chemotherapy. Int J Colorectal Dis 2019;34:2151-9.
21. Gu J, Wang L, Li T, et al. Role and mechanism of organic cation transporter 3 in oxaliplatin treatment of colon cancer in vitro and in vivo. Oncol Rep 2019;42:1355-64.
22. Safaei R, Howell SB. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hematol 2005;53:13-23.
23. Martinez-Balibrea E, Martínez-Cardús A, Musulén E, et al. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int J Cancer 2009;124:2905-10.
24. Sun S, Cai J, Yang Q, Zhao S, Wang Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: a meta-analysis of literatures and datasets. Oncotarget 2017;8:16036-51.
25. Untereiner AA, Pavlidou A, Druzhyna N, Papapetropoulos A, Hellmich MR, Szabo C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem Pharmacol 2018;149:174-85.
26. Leichman CG, Lenz HJ, Leichman L, et al. Quantitation of intratumoral thymidylate synthase expression predicts for disseminated colorectal cancer response and resistance to protracted-infusion fluorouracil and weekly leucovorin. J Clin Oncol 1997;15:3223-9.
27. Salonga D, Danenberg KD, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 2000;6:1322-7.
28. Santos A, Zanetta S, Cresteil T, et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res 2000;6:2012-20.
29. Buck E, Sprick M, Gaida MM, et al. Tumor response to irinotecan is associated with CYP3A5 expression in colorectal cancer. Oncol Lett 2019;17:3890-8.
30. Zhao Z, Zhang G, Li W. Elevated expression of ERCC6 confers resistance to 5-fluorouracil and is associated with poor patient survival in colorectal cancer. DNA Cell Biol 2017;36:781-6.
31. Leguisamo NM, Gloria HC, Kalil AN, et al. Base excision repair imbalance in colorectal cancer has prognostic value and modulates response to chemotherapy. Oncotarget 2017;8:54199-214.
32. Mishra B, Zhang S, Zhao H, et al. Discovery of a novel DNA polymerase inhibitor and characterization of its antiproliferative properties. Cancer Biol Ther 2019;20:474-86.
33. Jover R, Zapater P, Castells A, et al. Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur J Cancer 2009;45:365-73.
34. Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010;28:3219-26.
35. Alex AK, Siqueira S, Coudry R, et al. Response to chemotherapy and prognosis in metastatic colorectal cancer with DNA deficient mismatch repair. Clin Colorectal Cancer 2017;16:228-39.
36. Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 inhibitors in metastatic dMMR/MSI-H colorectal cancer. Front Oncol 2019;9:396.
37. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol 2019;20:199-210.
38. Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015;149:1177-90.e3.
39. Kandioler D, Mittlböck M, Kappel S, et al. p53 Research Group and the Austrian Breast and Colorectal Study Group (ABCSG). TP53 mutational status and prediction of benefit from adjuvant 5-fluorouracil in stage III colon cancer patients. EBioMedicine 2015;2:825-30.
40. Pilat N, Grünberger T, Längle F, et al. Assessing the TP53 marker type in patients treated with or without neoadjuvant chemotherapy for resectable colorectal liver metastases: a p53 Research Group study. Eur J Surg Oncol 2015;41:683-9.
41. Grassilli E, Narloch R, Federzoni E, et al. A kinase-directed RNAi screen identifies GSK-3b/a as a target to overcome drug resistance of p53-null colon cancer cells. Clin Cancer Res 2010;16 supplement:A5.
42. Panchal NK, Sabina EP. A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy. Life Sci 2020;255:117866.
43. Ferreira BI, Santos B, Link W, De Sousa-Coelho AL. Tribbles pseudokinases in colorectal cancer. Cancers (Basel) 2021;13:2825.
44. Xiao T, Xiao Y, Wang W, Tang YY, Xiao Z, Su M. Targeting EphA2 in cancer. J Hematol Oncol 2020;13:114.
45. Buckens OJ, El Hassouni B, Giovannetti E, Peters GJ. The role of Eph receptors in cancer and how to target them: novel approaches in cancer treatment. Expert Opin Investig Drugs 2020;29:567-82.
46. Reilly NM, Novara L, Di Nicolantonio F, Bardelli A. Exploiting DNA repair defects in colorectal cancer. Mol Oncol 2019;13:681-700.
47. Fathi Maroufi N, Rashidi MR, Vahedian V, Akbarzadeh M, Fattahi A, Nouri M. Therapeutic potentials of Apatinib in cancer treatment: possible mechanisms and clinical relevance. Life Sci 2020;241:117106.
48. Grassilli E, Narloch R, Federzoni E, et al. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res 2013;19:3820-31.
49. Grassilli E, Ianzano L, Bonomo S, et al. GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS One 2014;9:e100947.
50. Grassilli E, Pisano F, Cialdella A, et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene 2016;35:4368-78.
51. Lavitrano M, Ianzano L, Bonomo S, et al. BTK inhibitors synergise with 5-FU to treat drug-resistant TP53-null colon cancers. J Pathol 2020;250:134-47.
52. Sala L, Cirillo G, Riva G, et al. Specific expression of a new bruton tyrosine kinase isoform (p65BTK) in the glioblastoma gemistocytic histotype. Front Mol Neurosci 2019;12:2.
53. Grassilli E, Cerrito MG, Bonomo S, Giovannoni R, Conconi D, Lavitrano M. p65BTK is a novel biomarker and therapeutic target in solid tumors. Front Cell Dev Biol 2021;9:690365.
54. Giordano F, Vaira V, Cortinovis D, et al. p65BTK is a novel potential actionable target in KRAS-mutated/EGFR-wild type lung adenocarcinoma. J Exp Clin Cancer Res 2019;38:260.
55. Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science 2000;290:989-92.
56. Manoochehri M, Karbasi A, Bandehpour M, Kazemi B. Down-regulation of BAX gene during carcinogenesis and acquisition of resistance to 5-FU in colorectal cancer. Pathol Oncol Res 2014;20:301-7.
57. Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997;275:967-9.
58. Wang W, Guo W, Li L, et al. Andrographolide reversed 5-FU resistance in human colorectal cancer by elevating BAX expression. Biochem Pharmacol 2016;121:8-17.
59. Ramesh P, Medema JP. BCL-2 family deregulation in colorectal cancer: potential for BH3 mimetics in therapy. Apoptosis 2020;25:305-20.
60. Raats DA, de Bruijn MT, Steller EJ, Emmink BL, Borel-Rinkes IH, Kranenburg O. Synergistic killing of colorectal cancer cells by oxaliplatin and ABT-737. Cell Oncol (Dordr) 2011;34:307-13.
61. Wang X, Zhang C, Yan X, et al. A novel bioavailable BH3 mimetic efficiently inhibits colon cancer via cascade effects of mitochondria. Clin Cancer Res 2016;22:1445-58.
62. Ritter V, Krautter F, Klein D, Jendrossek V, Rudner J. Bcl-2/Bcl-xL inhibitor ABT-263 overcomes hypoxia-driven radioresistence and improves radiotherapy. Cell Death Dis 2021;12:694.
63. Kim YS, Lee HJ, Park JM, et al. Targeted molecular ablation of cancer stem cells for curing gastrointestinal cancers. Expert Rev Gastroenterol Hepatol 2017;11:1059-70.
65. Vallette FM, Olivier C, Lézot F, et al. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem Pharmacol 2019;162:169-76.
66. Oren Y, Tsabar M, Cuoco MS, et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021;596:576-82.
67. Lenos KJ, Miedema DM, Lodestijn SC, et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat Cell Biol 2018;20:1193-202.
68. Cho YH, Ro EJ, Yoon JS, et al. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun 2020;11:5321.
69. Lin CC, Liao TT, Yang MH. Immune adaptation of colorectal cancer stem cells and their interaction with the tumor microenvironment. Front Oncol 2020;10:588542.
70. Tang YA, Chen YF, Bao Y, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A 2018;115:E5990-9.
71. Romano G, Santi L, Bianco MR, et al. The TGF-β pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget 2016;7:22077-91.
72. Hu JL, Wang W, Lan XL, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer 2019;18:91.
73. Ren J, Ding L, Zhang D, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018;8:3932-48.
74. Sun LH, Tian D, Yang ZC, Li JL. Exosomal miR-21 promotes proliferation, invasion and therapy resistance of colon adenocarcinoma cells through its target PDCD4. Sci Rep 2020;10:8271.
75. Zheng X, Ma N, Wang X, et al. Exosomes derived from 5-fluorouracil-resistant colon cancer cells are enriched in GDF15 and can promote angiogenesis. J Cancer 2020;11:7116-26.
76. Zheng X, Liu J, Li X, et al. Angiogenesis is promoted by exosomal DPP4 derived from 5-fluorouracil-resistant colon cancer cells. Cancer Lett 2021;497:190-201.
77. Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013;218:1402-10.
78. Tolba MF. Revolutionizing the landscape of colorectal cancer treatment: the potential role of immune checkpoint inhibitors. Int J Cancer 2020;147:2996-3006.
79. Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017;170:548-63.e16.
80. Zhang S, Yang Y, Weng W, et al. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J Exp Clin Cancer Res 2019;38:14.
81. Chen Y, Peng Y, Yu J, et al. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017;8:31802-14.
82. Geller LT, Barzily-Rokni M, Danino T, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357:1156-60.
83. Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013;342:967-70.
84. Ooki A, Shinozaki E, Yamaguchi K. Immunotherapy in colorectal cancer: current and future strategies. J Anus Rectum Colon 2021;5:11-24.
85. Rankin A, Klempner SJ, Erlich R, et al. Broad detection of alterations predicted to confer lack of benefit from EGFR antibodies or sensitivity to targeted therapy in advanced colorectal cancer. Oncologist 2016;21:1306-14.
86. Raskov H, Søby JH, Troelsen J, Bojesen RD, Gögenur I. Driver gene mutations and epigenetics in colorectal cancer. Ann Surg 2020;271:75-85.
87. Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov 2014;4:1269-80.
88. Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013;3:658-73.
89. Troiani T, Martinelli E, Napolitano S, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 2013;19:6751-65.
90. Song N, Liu S, Zhang J, et al. Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int J Mol Sci 2014;15:5838-51.
91. Song N, Qu X, Liu S, et al. Dual inhibition of MET and SRC kinase activity as a combined targeting strategy for colon cancer. Exp Ther Med 2017;14:1357-66.
92. Jia Y, Dai G, Wang J, et al. c-MET inhibition enhances the response of the colorectal cancer cells to irradiation in vitro and in vivo. Oncol Lett 2016;11:2879-85.
93. Rimassa L, Bozzarelli S, Pietrantonio F, et al. Phase II study of tivantinib and cetuximab in patients with KRAS wild-type metastatic colorectal cancer with acquired resistance to EGFR inhibitors and emergence of MET overexpression: lesson learned for future trials with EGFR/MET dual inhibition. Clin Colorectal Cancer 2019;18:125-32.e2.
94. Eng C, Bessudo A, Hart LL, et al. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int J Cancer 2016;139:177-86.
95. Craig SG, Mende S, Humphries MP, et al. Orthogonal MET analysis in a population-representative stage II-III colon cancer cohort: prognostic and potential therapeutic implications. Mol Oncol 2021;15:3317-28.
96. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;48:330-7.
97. Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 2011;1:508-23.
98. Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov 2015;5:832-41.
99. Cremolini C, Morano F, Moretto R, et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: the PRESSING case-control study. Ann Oncol 2017;28:3009-14.
100. Bregni G, Sciallero S, Sobrero A. HER2 amplification and Anti-EGFR sensitivity in advanced colorectal cancer. JAMA Oncol 2019;5:605-6.
101. Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011;3:99ra86.
102. Mohan S, Heitzer E, Ulz P, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet 2014;10:e1004271.
103. Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:738-46.
104. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol 2018;36:536-42.
105. Nakamura Y, Okamoto W, Kato T, et al. TRIUMPH: primary efficacy of a phase II trial of trastuzumab (t) and pertuzumab (p) in patients (pts) with metastatic colorectal cancer (mCRC) with HER2 (ERBB2) amplification (amp) in tumor tissue circulating tumor DNA (ctDNA): a GOZILA sub-study. Ann Oncol 2019;30:v199-200.
106. Strickler J, Zemla T, Ou F, et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): initial results from the MOUNTAINEER trial. Ann Oncol 2019;30:v200.
107. Mauri G, Arena S, Siena S, Bardelli A, Sartore-Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol 2020;31:1135-47.
108. Guarini C, Grassi T, Pezzicoli G, Porta C. Beyond RAS and BRAF: HER2, a new actionable oncotarget in advanced colorectal cancer. Int J Mol Sci 2021;22:6813.
109. Mangiapane LR, Nicotra A, Turdo A, et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut 2022;71:119-28.
110. Pietrantonio F, Vernieri C, Siravegna G, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res 2017;23:2414-22.
111. Baselga J. The EGFR as a target for anticancer therapy - focus on cetuximab. Eur J Cancer 2001;37:16-22.
112. Vitiello PP, Cardone C, Martini G, et al. Receptor tyrosine kinase-dependent PI3K activation is an escape mechanism to vertical suppression of the EGFR/RAS/MAPK pathway in KRAS-mutated human colorectal cancer cell lines. J Exp Clin Cancer Res 2019;38:41.
113. Hasbal-Celikok G, Aksoy-Sagirli P, Altiparmak-Ulbegi G, Can A. Identification of AKT1/β-catenin mutations conferring cetuximab and chemotherapeutic drug resistance in colorectal cancer treatment. Oncol Lett 2021;21:209.
114. Martini G, Cardone C, Vitiello PP, et al. EPHA2 is a predictive biomarker of resistance and a potential therapeutic target for improving antiepidermal growth factor receptor therapy in colorectal cancer. Mol Cancer Ther 2019;18:845-55.
115. Kopetz S, Desai J, Chan E, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol 2010;28:3534-3534.
116. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726-36.
117. Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015;33:4032-8.
118. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-3.
119. Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227-35.
120. Yao Z, Gao Y, Su W, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 2019;25:284-91.
121. Kawakami H, Huang S, Pal K, Dutta SK, Mukhopadhyay D, Sinicrope FA. Mutant BRAF upregulates MCL-1 to confer apoptosis resistance that is reversed by MCL-1 antagonism and cobimetinib in colorectal cancer. Mol Cancer Ther 2016;15:3015-27.
122. Chen G, Gao C, Gao X, et al. Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther 2018;17:806-13.
123. Grbčić P, Fučkar Čupić D, Gamberi T, Kraljević Pavelić S, Sedić M. Proteomic profiling of BRAFV600E mutant colon cancer cells reveals the involvement of nucleophosmin/c-Myc axis in modulating the response and resistance to BRAF inhibition by vemurafenib. Int J Mol Sci 2021;22:6174.
124. Yang H, Higgins B, Kolinsky K, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 2012;72:779-89.
125. Atreya CE, Van Cutsem E, Bendell JC, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J Clin Oncol 2015;33:103.
126. Tabernero J, Geel RV, Guren TK, et al. Phase 2 results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol ;34:3544.
127. Yaeger R, Cercek A, O'Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF mutant metastatic colorectal cancer patients. Clin Cancer Res 2015;21:1313-20.
128. Yaeger R, Yao Z, Hyman DM, et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res 2017;77:6513-23.
129. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877-88.
130. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30-9.
131. Halle BR, Johnson DB. Defining and targeting BRAF mutations in solid tumors. Curr Treat Options Oncol 2021;22:30.
132. Tan J, Liu R, Zhu G, Umbricht CB, Xing M. TERT promoter mutation determines apoptotic and therapeutic responses of BRAF-mutant cancers to BRAF and MEK inhibitors: achilles heel. Proc Natl Acad Sci U S A 2020;117:15846-51.
133. Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 2019;381:1632-43.
134. Kopetz S, Guthrie KA, Morris VK, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol 2021;39:285-94.
135. Van Cutsem E, Huijberts S, Grothey A, et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal cancer: safety lead-in results from the phase III BEACON colorectal cancer study. J Clin Oncol 2019;37:1460-9.
136. Tabernero J, Grothey A, Van Cutsem E, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol 2021;39:273-84.
138. Lapeyre-prost A, Terme M, Pernot S, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol 2017;330:295-342.
139. Grassi E, Corbelli J, Papiani G, Barbera MA, Gazzaneo F, Tamberi S. Current therapeutic strategies in BRAF-mutant metastatic colorectal cancer. Front Oncol 2021;11:601722.
140. Rosen LS, Jacobs IA, Burkes RL. Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target Oncol 2017;12:599-610.
141. Torben Frøstrup H, Qvortrup C, Pfeiffer P. Angiogenesis inhibitors for colorectal cancer. A review of the clinical data. Cancers 2021:13,1031.
142. de Gramont A, Van Cutsem E, Schmoll H, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225-33.
143. Belotti D, Pinessi D, Taraboletti G. Alternative vascularization mechanisms in tumor resistance to therapy. Cancers 2021;13:1912.
144. Knopik-skrocka A, Kręplewska P, Jarmołowska-jurczyszyn D. Tumor blood vessels and vasculogenic mimicry - current knowledge and searching for new cellular/molecular targets of anti-angiogenic therapy. Adv Cell Biol 2017;5:50-71.
145. Lai E, Cascinu S, Scartozzi M. Are all anti-angiogenic drugs the same in the treatment of second-line metastatic colorectal cancer? Front Oncol 2021;11:637823.
146. Hecht JR, Cohn A, Dakhil S, et al. SPIRITT: a randomized, multicenter, phase II Study of panitumumab with FOLFIRI and bevacizumab with FOLFIRI as second-line treatment in patients with unresectable wild type KRAS metastatic colorectal cancer. Clin Colorectal Cancer 2015;14:72-80.
147. Hu-Lowe DD, Chen E, Zhang L, et al. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res 2011;71:1362.
148. Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol 2020;31:1336-49.
150. Goede V, Coutelle O, Neuneier J, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer 2010;103:1407-14.
151. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep 2014;8:696-706.
152. Kienast Y, Klein C, Scheuer W, et al. Ang-2-VEGF-A crossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res 2013;19:6730-40.
153. Park JS, Kim IK, Han S, et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 2016;30:953-67.
154. Krzystek-Korpacka M, Diakowska D, Kapturkiewicz B, Bębenek M, Gamian A. Profiles of circulating inflammatory cytokines in colorectal cancer (CRC), high cancer risk conditions, and health are distinct. Possible implications for CRC screening and surveillance. Cancer Lett 2013;337:107-14.
155. Łukaszewicz-Zając M, Mroczko B. Circulating biomarkers of colorectal cancer (CRC)-their utility in diagnosis and prognosis. J Clin Med 2021;10:2391.
156. Cazzaniga ME, Cordani N, Capici S, Cogliati V, Riva F, Cerrito MG. Metronomic chemotherapy. Cancers (Basel) 2021;13:2236.
157. Simkens LHJ, van Tinteren H, May A, et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): a phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015;385:1843-52.
158. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43-51.
159. Giannakis M, Mu XJ, Shukla SA, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep 2016;15:857-65.
160. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet oncol 2017;18:1182-91.
161. André T, Shiu KK, Kim TW, et al. KEYNOTE-177 Investigators. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med 2020;383:2207-18.
163. Yu X, Huang X, Chen X, et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs 2019;11:1139-48.
164. Tang K, Wu YH, Song Y, Yu B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol 2021;14:68.
165. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020;9:1777625.
166. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol 2018;336:175-203.
167. Voron T, Marcheteau E, Pernot S, et al. Control of the immune response by pro-angiogenic factors. Front Oncol 2014;4:70.
168. Wang Q, Gao J, Di W, et al. Anti-angiogenesis therapy overcomes the innate resistance to PD-1/PD-L1 blockade in VEGFA-overexpressed mouse tumor models. Cancer Immunol Immunother 2020;69:1781-99.
169. Anfossi S, Calin GA. Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy. Cancer Drug Resist 2020;3:356-70.
170. Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079-84.
171. Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084-9.
172. Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359:97-103.
173. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Grassilli E, Cerrito MG. Emerging actionable targets to treat therapy-resistant colorectal cancers. Cancer Drug Resist 2022;5:36-63. http://dx.doi.org/10.20517/cdr.2021.96
AMA Style
Grassilli E, Cerrito MG. Emerging actionable targets to treat therapy-resistant colorectal cancers. Cancer Drug Resistance. 2022; 5(1): 36-63. http://dx.doi.org/10.20517/cdr.2021.96
Chicago/Turabian Style
Grassilli, Emanuela, Maria Grazia Cerrito. 2022. "Emerging actionable targets to treat therapy-resistant colorectal cancers" Cancer Drug Resistance. 5, no.1: 36-63. http://dx.doi.org/10.20517/cdr.2021.96
ACS Style
Grassilli, E.; Cerrito MG. Emerging actionable targets to treat therapy-resistant colorectal cancers. Cancer Drug Resist. 2022, 5, 36-63. http://dx.doi.org/10.20517/cdr.2021.96
About This Article
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 26
likes
Like This Article 26
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.