
Auranofin and its analogs as prospective agents for the treatment of colorectal cancer

Abstract
Today colorectal cancer (CRC) is one of the leading causes of cancer death worldwide. This disease is poorly chemo-sensitive toward the existing medical treatments so that new and more effective therapeutic agents are urgently needed and intensely sought. Platinum drugs, oxaliplatin in particular, were reported to produce some significant benefit in CRC treatment, triggering the general interest of medicinal chemists and oncologists for metal-based compounds as candidate anti-CRC drugs. Within this frame, gold compounds and, specifically, the established antiarthritic drug auranofin with its analogs, form a novel group of promising anticancer agents. Owing to its innovative mechanism of action and its favorable pharmacological profile, auranofin together with its derivatives are proposed here as novel experimental agents for CRC treatment, capable of overcoming resistance to platinum drugs. Some encouraging results in this direction have already been obtained. A few recent studies demonstrate that the action of auranofin may be further potentiated through the preparation of suitable pharmaceutical formulations capable of protecting the gold pharmacophore from unselective reactivity or through the design of highly synergic drug combinations. The perspectives of the research in this field are outlined.
Keywords
INTRODUCTION
Colorectal cancer (CRC) is one of the most common cancers in Western countries and is a major worldwide health problem being one of the primary causes of cancer death[1]. Advances in the diagnosis and treatment of CRC brought a major effect in the management of this disease. Notably, in the last decades, screening programs and early diagnosis, together with new therapeutic options, allowed reducing CRC mortality considerably[2]. Despite this progress and the remarkable increase of knowledge about CRC biology and treatment, metastatic cases are still associated with a poor prognosis. In fact, the percentage of survival drops from ~65% to ~10% in the presence of metastatic CRC disease[3]. At least half of patients with colorectal cancer develop metastases[4], resulting in a poor outcome. The primary site for metastasis development is liver, representing the only site of metastasis in about one third of patients. Other common organs for distant metastases are lung, peritoneum, bone, brain, and spleen[4,5]. In CRC patients, the first therapeutic option is surgical resection, but the role of adjuvant chemotherapy (and chemoradiotherapy), in terms of disease-free survival and overall survival, is well recognized[6,7].
CRC is reputed as poorly chemo-sensitive, and for more than 30 years fluorouracil has been the reference drug. Fluorouracil can be administered both through continuous infusion line and through bolus administration. Recent reports evidenced however no significant differences in terms of prolonged survival rate[8,9]. Many efforts have been made to improve these results. Biochemical modulation of fluorouracil is one of the most interesting approaches to increase the therapeutic index of this compound. In metastatic disease confined to the liver, locoregional therapy through implantable pumps may be taken into account as well[10].
Until today, the standard therapy for CRC has relied on fluorouracil plus levamisole and/or calcium folinate (folinic acid)[6]. More recently, other treatment options have been introduced in the clinical practice. At present, there is increasing attention for the role of monoclonal antibodies (Mab) in CRC therapy. Among the various Mabs approved so far for CRC treatment, bevacizumab, panitumumab, and ramucirumab are the most commonly used[11]. Other clinical strategies rely on portal vein infusion of fluorouracil, alone or in combination with systemic therapy[12]. In rectal cancer, the best results are achieved by combining radiotherapy and chemotherapy. Recent reports support the use of induction or consolidation chemotherapy before surgery in locally advanced forms as well as total neoadjuvant treatment as the best therapeutic options[13]. In advanced colorectal cancer, a standard treatment has not been established yet, and different therapeutic options can be proposed according to the tumor staging and features. Typically, the first-line chemotherapy relies on the FOLFOX (leucovorin, 5-fluorouracil, and oxaliplatin) or CAPOX (capecitabine and oxaliplatin) protocol alone or in combination with Mab. At variance, the second-line approach also relies on compounds such as irinotecan and raltitrexed now entered in the clinical practice with encouraging results. For instance, FOLFIRI-based (leucovorin, 5-fluorouracil, and irinotecan) regimens are a common clinical practice, even in combination with antiangiogenic agents[14].
PLATINUM DRUGS IN CRC TREATMENT
Pt drugs play an important role in CRC treatment. Oxaliplatin is the only platinum anticancer drug in the clinical use for colorectal cancer, while cisplatin is not effective in the treatment of this tumor. The resistance to cisplatin in CRC is determined by several factors mostly related to the processing of cisplatin-induced DNA lesions by various biological actors. The tumor suppressor protein p53 is a key player in the cellular response to cisplatin. It is responsible for controlling the cascade of events leading to cell cycle arrest/repair or apoptosis through transcription and activation of numerous p53-dependent DNA damage response genes. Accordingly, deactivation of p53 in CRC is involved in cisplatin chemoresistance. In this view, the DNA mismatch repair (MMR) process has a relevant role as well. Indeed, cisplatin-induced lesions are recognized by MMR. Conversely, oxaliplatin-induced lesions to DNA are not recognized. As a consequence, oxaliplatin shows cytotoxic and anticancer effects that are basically independent of the MMR process[15,16]. However, beyond the formation of adducts with DNA, which are extensively recognized as a key step for the pharmacological activity of platinum-based anticancer drugs[17,18], the different therapeutic indication between cisplatin and oxaliplatin was discussed by Bruno et al.[19] in a seminal paper published in 2017. In this article, the authors reported on the mechanisms behind the anticancer effects of platinum-based drugs. Although it is not clear to what extent the effect of DNA-independent pathways contributes to the anticancer profile compared to the toxic effect of DNA adducts formation, the above authors pointed at the ribosome biogenesis stress induced by oxaliplatin as a likely mechanism also contributing to the overall pharmacological activity. This finding is relevant because might be involved in the distinct clinical implementation of oxaliplatin compared with cisplatin[19].
As outlined above, Pt compounds, and oxaliplatin in particular, play a major role in the current medical treatment of CRC. Although being featured by a higher tolerability compared with cisplatin, oxaliplatin manifests a few relevant limitations such as relevant systemic toxicity and the frequent insurgence of resistance that may lead to eventual treatment failure. These limitations imply that the discovery of new drugs for colorectal cancer is absolutely mandatory and urgent. Here, we analyze specifically the chances of discovering new effective anticancer agents within the field of metal-based drugs.
METAL BASED DRUGS AS A SOURCE OF NOVEL ANTICANCER AGENTS CAPABLE OF OVERCOMING DRUG RESISTANCE: THE CASE OF GOLD COMPOUNDS
Platinum-based compounds are very effective only against a relatively limited number of tumor types and manifest at the same time some severe side effects (e.g., gastrointestinal toxicity, nervous system toxicity, and bone marrow suppression) that heavily limit their use[20-22]. In addition, intrinsic and acquired drug resistance may greatly reduce the efficacy of platinum drugs with a consequent poor prognosis[23]. For this reason, intense efforts are warranted to explore and identify novel anti-tumor metallodrugs that may replace platinum compounds for specific therapeutic goals and for treatment improvement. Accordingly, many new metal compounds exploiting various transition metals have been prepared and evaluated, and some of them (e.g., gold, silver, copper, ruthenium, and other active metals) turned out to manifest very encouraging antitumor effects. In particular, coinage metals (especially Au and Ag) revealed a greater application potential as they are, on average, less toxic to humans than other transition metals. From a chemical point of view, gold compounds deserve particular attention owing to the unique position of gold in the periodic table, which ultimately leads to a larger electronegativity, a higher electron affinity, and a rich and peculiar redox profile. Several gold compounds, including both gold(III) and gold(I) compounds [i.e., gold(I) carbene, gold sodium thiomalate, gold thiolates, or gold compounds with bipyridyl-type ligands], were thus considered for cancer treatment, and many of them were found to possess remarkable antiproliferative properties in vitro against several human cancer cell lines[24].
Several studies have pointed out that gold compounds cause their outstanding cytotoxic effects by taking advantage of multiple molecular and cellular mechanisms. The most credited mechanisms involve inhibition of thiol-containing enzymes, especially thioredoxin reductase (TrxR)[25-27], direct mitochondrial damage[28-31], or alteration of DNA functions[32,33], all of which may contribute importantly to the observed anticancer actions. Although no non-platinum metal compound has been approved so far for clinical use, some gold drug candidates are being actively investigated. A few gold compounds have shown very promising results in preclinical research[34], and two of them even reached clinical trials.
AURANOFIN AND ITS ANALOGS AS ANTICANCER AGENTS: CHEMICO-BIOLOGICAL ASPECTS AND MECHANISTIC INFERENCES
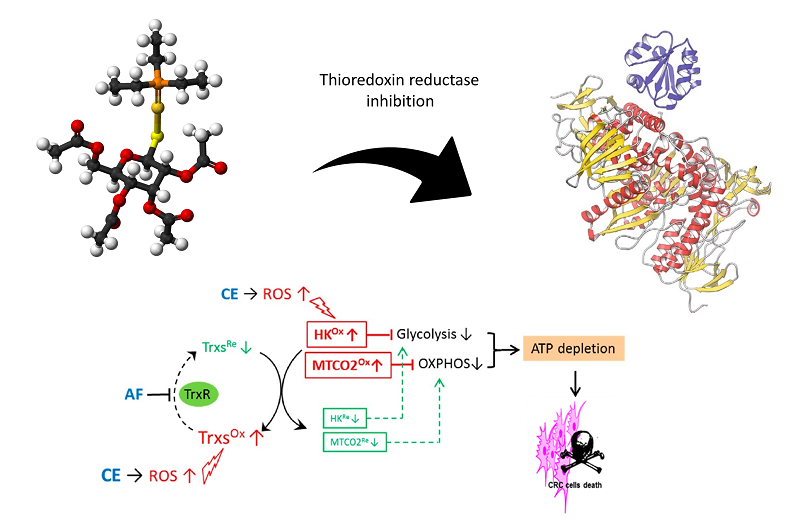
In the field of gold-based drugs, auranofin (AF) undoubtedly occupies a pivotal position. From the chemical point of view auranofin, i.e., [2,3,4,6-tetra-o-acetyl-l-thio-β-d-glyco-pyrano-sato-S-(triethyl- phosphine)-gold(I)], is a mixed ligand gold(I) complex with a linear geometry having a triethylphosphine molecule and a thioglucose derivative as gold(I) ligands [Figure 1].
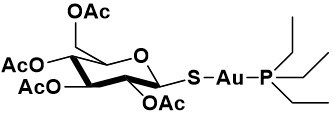
Figure 1. Chemical structure of auranofin.
Recently, various research groups have found that auranofin[35,36], beyond its known anti-inflammatory actions, also exhibits prominent anticancer, antibacterial, and antiparasitic properties[37-43]. Accordingly, during the past few years, auranofin has attracted a lot of attention in the medicinal chemistry scientific community as a prospective anticancer and anti-infective agent on the ground of drug repurposing strategies. As a matter of fact, AF has entered several different clinical trials as an anticancer, antiviral, or antiparasitic drug (ClinicalTrials.gov Identifier: NCT02089048, NCT01747798, NCT03456700, NCT01419691, NCT02063698, NCT02736968, NCT01737502, NCT02961829, NCT02770378, NCT03975790, and NCT01557348).
Auranofin was initially prepared in the late 1970s and found to manifest remarkable antiarthritic properties. Owing to its favorable pharmacological profile, auranofin was eventually approved for clinical use against rheumatoid arthritis in 1985, although in the absence of a precise understanding of its mode of action.
Moreover, it was found that auranofin was able to inhibit the growth of tumor cells in vitro and arrest the growth of an in vivo model tumor (leukemia P388) in mice. In fact, Mirabelli et al.[44], starting from auranofin and changing systematically the phosphine ligand, the sulfur ligand, or both ligands, obtained 62 distinct Au(I) complexes. These novel complexes were tested both in vitro, against B16 melanoma and P388 leukemia, and in vivo, against mouse P388 intraperitoneal leukemia, with encouraging results. Mechanistically, it could be ascertained that auranofin behaves as a prodrug capable of releasing its two originals ligands; in any case, it is well documented that the thiosugar ligand is a better leaving group than the phosphine and is the first ligand to be released. The resulting empty coordination position on the gold(I) center becomes available for coordination to biomolecules. Typically, gold compounds with the general formula Et3PAuX manifest relevant and roughly similar anticancer profiles. This finding implies that the thiosugar ligand is not fundamental for the cytotoxic activity and that the [Et3PAu]+ moiety is likely to be the true pharmacophore. The role of the X ligand is probably related to the cellular uptake: a more lipophilic nature of the ligand might determine a more favorable pharmacokinetic and biodistribution profile.
The chemical behavior of auranofin and its reactions with biomolecules have been intensely investigated. The reaction of auranofin with biomolecules has now been studied in detail: it could be established that auranofin binds proteins tightly by forming strong coordinative bonds to free cysteine or selenocysteine residues[45,46]. This type of reactivity may well account for its molecular mechanisms; for instance, there is now a general consensus that the tight binding of auranofin to the free selenocysteine group in the active site of TrxR is primarily responsible for its relevant actions at the cellular level, eventually leading to severe intracellular redox dysregulation and associated apoptotic cancer cell death [Figure 2].
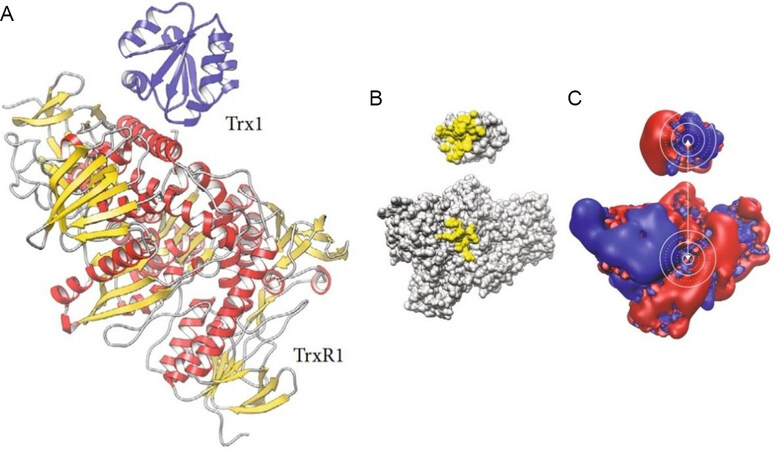
Figure 2. (A) Cartoon representation of the secondary structures of human thioredoxin-1-thioredoxin reductase 1 (Trx1-TrxR1) complex PDB: 3qfa. (B) The contact patches with direct molecular contacts are highlighted in yellow. (C) The isosurfaces of the electrostatic potential are depicted in blue (positive) and red (negative). The active site cysteinyl residues and interaction surfaces in the immediate contact area in both proteins are encircled in white lines. Reproduced and adapted from Hossain et al.[47] under the Creative Commons Attribution License 4.0 International (CC BY 4.0).
However, the interpretation of the mechanism of the anticancer actions of auranofin is still controversial and not conclusive. Although thioredoxin reductase inhibition is believed to be a central trait of its mode of action, other likely targets have been proposed and partially validated. As a matter of fact, some interesting proteomics data highlight, upon auranofin treatment, a few differentially expressed proteins belonging to different cellular processes, namely cell redox homeostasis, metabolism, and cell structure. Specifically, the main altered proteins were peroxiredoxins 1 and 6, linked to cell redox balance; triosephosphate isomerase 1, which plays a key role in the glycolysis and gluconeogenesis pathways; ezrin, essential for cell structure and cell migration; and the heterogeneous nuclear ribonucleoprotein H, whose increased cleavage leads to the caspase 3 activation triggering apoptosis[24-48].
In addition, a few recent studies suggested that AF produces important immunomodulatory effects[49]. In particular, it was found that AF induces ICD (immunogenic cell death) in cancer cells as a consequence of endoplasmic reticulum stress and reactive oxygen species (ROS) production. Notably,
Overall, these arguments demonstrate that auranofin, as already hypothesized for this compound as well as other gold(I) compounds, possesses a multi-target mode of action; indeed, auranofin interacts in cells and blood with several targets, mainly proteins with key functions, involving multiple cellular pathways and altering different biological networks.
AURANOFIN AND ITS ANALOGS SHOW PROMISE FOR COLORECTAL CANCER TREATMENT: IN VITRO EVIDENCE
The favorable chemical and pharmacological profile of auranofin as a prospective anticancer agent prompted researchers to synthesize and characterize some auranofin derivatives and assess their action in vitro against a few CRC cell lines. In particular, in recent studies, five analogs of auranofin were prepared where the thiosugar ligand was substituted by different anionic ligands (these compounds are depicted in Figure 3)[51,52]. The synthesis of these analogs is rather straightforward and starts from the commercially available Et3PAuCl. More precisely, the iodide analog can be prepared through a simple chloride-displacing reaction carried out with an excess of potassium iodide[51]; the cyanide derivative can be synthesized reacting the two ionic species K[Au(CN)2] and [Au(PEt3)2]Cl in a biphasic reaction[52]; and the thiocyanate and azido derivatives can be prepared by reacting the Et3PAuCl species, respectively, with potassium thiocyanate and sodium azide after its activation with silver nitrate[52].
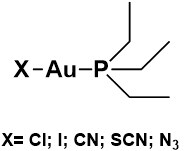
Figure 3. Chemical structures of auranofin analogs bearing different ligands in place of thiosugar moiety.
Then, the antiproliferative properties of two of these analogs, i.e., Et3PAuCl and Et3PAuI, as well as auranofin itself were comparatively assayed in vitro against four representative colorectal cancer lines, i.e., HCT8, HCT116, HT29, and Caco2, and two healthy cell lines, i.e., HDF (human fibroblast, adult) and HEK293 (human embryonic kidney), by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test[51]. As displayed in Table 1, all tested compounds produced very notable cytotoxic effects on all the selected CRC cell lines with half-maximal inhibitory concentration (IC50) values always falling in the 100-700 nM range. Et3PAuI was slightly less cytotoxic than the other two gold complexes. In line with expectations, the presence of the thiosugar ligand turned out not to be an essential feature for the cytotoxic action. Moreover, by considering the close similarity in the measured IC50 values, it can be inferred that the cellular uptake of the three compounds should not be very different. Remarkably, when measuring the cytotoxic effects on two healthy cell lines, HDF human fibroblast cells (adult) and HEK293 human embryonic kidney, no appreciable cytotoxic effects were detected for the three study complexes in the concentration range 0-5000 nM. The latter finding is a good index of selectivity for cancer cells.
IC50 values (nM) determined for Et3PAuI, AF, and Et3AuP-Cl (24 h incubation)
| Complex | HCT8 | HCT116 | HT29 | Caco2 | HDF | HEK293 |
| AF | 132 ± 16 | 180 ± 17 | 359 ± 35 | 465 ± 53 | > 5000 | > 5000 |
| Et3PAuCl | 105 ± 11 | 154 ± 22 | 122 ± 15 | 560 ± 93 | > 5000 | > 5000 |
| Et3PAuI | 260 ± 28 | 290 ± 36 | 318 ± 90 | 706 ± 232 | > 5000 | > 5000 |
Afterwards, since TrxR is reputed to be a primary target for auranofin, the inhibitory potencies of the three gold compounds against this enzyme were analyzed comparatively. The results are summarized in Table 2. Interestingly, the obtained IC50 values for TrxR inhibition are roughly consistent with those determined for the cytotoxic effects in CRC cell lines [Table 1]. This probably implies that the observed cytotoxic effects are somehow linked to the ability of these gold complexes to inhibit TrxR. Moreover, the present results confirm that Et3PAuCl is not only the most potent cytotoxic agent but also the most potent TrxR inhibitor of the series; although the IC50 values measured for auranofin and Et3PAuI are only slightly higher, they still fall in the nanomolar range.
Thioredoxin reductase activity assay. IC50 values (nM) were determined treating 2 U/L of TrxR with aliquots of AF, Et3AuPCl, and Et3PAuI (from 1 µM to 1 nM)
| Complex | IC50 (nM) |
| AF | 105 ± 17.3 |
| Et3AuPCl | 51.3 ± 8.5 |
| Et3PAuI | 193 ± 22.2 |
Conversely, the three auranofin analogs in which the thiosugar was replaced with stronger ligands (i.e., Et3PAuCN, Et3PAuSCN, and Et3PAuN3) turned out to be completely inactive against HCT116 cell line for concentrations ranging up to 1000 nM. This observation strongly supports the occurrence of a reaction mechanism in which the anionic groups have to be displaced from the nucleophilic active site of TrxR (the SEC-CYS motif) to observe the pharmacological activity of the [Au(PEt3)]+ moiety[52]. In conclusion, the results of this study demonstrated that auranofin and its chloride, and the iodide analogs manifest potent cytotoxic effects in vitro against four selected CRC lines with the measured IC50 values always falling in the nanomolar range and no apparent cytotoxic effect on human fibroblast cell line and human embryonic kidney cells up to a 5 µM concentration. The TrxR activity assay revealed that both Et3PAuCl and Et3PAuI retain the potent inhibitory action of auranofin (nanomolar range), being consistent with their observed cytotoxic effects. Overall, these findings are consistent with the concept that TrxR remains the most probable and most relevant biomolecular target for these gold compounds. Moreover, these results, although obtained in vitro on cell cultures, support the idea that auranofin and its analogs are optimal drug candidates for further testing against more advanced and sophisticated CRC models. It should also be noted that the DNA-independent mode of action of AF and some of its analogs is a key aspect determining the high cytotoxicity toward in vitro CRC models. As an example, AF, Et3PAuCl, and Et3PAuI [Table 1] have been reported to exert anticancer effects significantly greater than cisplatin and even oxaliplatin on representative colorectal cancer cell lines [Table 3][51,52].
The IC50 values (µM) determined for cisplatin and oxaliplatin (24 h incubation) against HT29 and HCT116 lines are also included as reference
| Complex | HCT116 | HT29 |
| Cisplatin | 21.96 ± 1.11 | 16.39 ± 1.10 |
| Oxaliplatin | 49.2 ± 0.9 | 19.7 ± 1.2 |
Based on the above considerations, it could be inferred that the ability of auranofin and some of its analogs to exert a greater anticancer activity in CRC lines than cisplatin and oxaliplatin might depend on the different mechanisms underlying the pharmacological effects. In fact, in contrast to oxaliplatin[55], auranofin does not efficiently bind DNA, thus being its activity substantially unaffected by MMR, p53, and nucleotide excision repair functions. In fact, gold-based drugs produce the desired anticancer effects mainly through a DNA-independent mode, i.e., targeting specific enzymes such as the Trx system[51,52].
NEW PERSPECTIVES IN THE USE OF AURANOFIN AND ITS ANALOGS: ENCAPSULATION OF GOLD COMPOUNDS IN BIOCOMPATIBLE NANOPARTICLES
As stated above, Et3PAuCl is an auranofin derivative exhibiting very attractive biological and pharmacological properties. Similar to auranofin, Et3PAuCl possesses potent cytotoxic properties in vitro toward numerous cancer cell lines, thus being a promising anticancer drug candidate. In this frame, some investigators wondered whether Et3PAuCl encapsulation might lead to a better pharmacological profile, considering the expected reduction of unwanted side-reactions that are mainly responsible for the adverse effects and for drug inactivation. A reasonable option to achieve this goal consists in using biocompatible nanoparticles as nanocarriers to protect the gold complex from the biological environment. To achieve this goal, Menconi et al.[43] exploited organic polyethylene glycol-poly lactic acid-co-glycolic acid (PLGA-PEG)-based nanoparticles of intermediate size, which could host a certain number of metallodrug’s copies into their hydrophobic core [Figure 4]. Et3PAuCl was encapsulated in these biocompatible PLGA-PEG nanoparticles, and the new formulation was evaluated in colorectal HCT116 cancer cells in comparison to free Et3PAuCl.
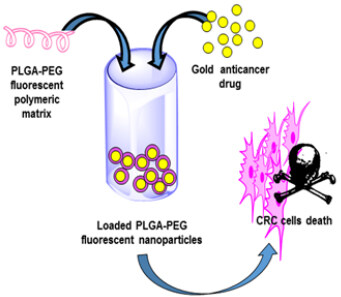
Figure 4. Schematic representation of the Et3PAuCl-loaded NPs tested against colorectal cancer models. Reproduced and adapted from Menconi et al.[43] under the Creative Commons Attribution License 4.0 International (CC BY 4.0).
Notably, the encapsulated Et3PAuCl mostly retains the cellular actions of the free complex and causes even larger cytotoxic effects in CRC cells, through apoptosis and autophagy. Moreover, a large inhibition of two crucial signaling pathways, namely extracellular signal-regulated kinase (ERK) and protein kinase B (AKT), by the encapsulated form of Et3PAuCl, was clearly evidenced by the fact that this inhibition was not found in cells treated with the free drug. Overall, these results point out that encapsulation of Et3PAuCl in PLGA-PEG nanoparticles does not significantly affect the antiproliferative properties of this gold complex. However, some changes in the biological effects of the studied gold complex could be detected, which were specifically evidenced by the differential effects produced on the ERK and AKT signaling pathways. It would be of interest to extend such experimentation to appropriate in vivo models of CRC.
NEW PERSPECTIVES IN THE USE OF AURANOFIN AND ITS ANALOGS: THE ROLE OF COMBINATION THERAPIES
Another valuable strategy in the use of gold compounds as anti-colorectal cancer agents is offered by the exploitation of so-called combination therapies. It is a common practice in cancer pharmacology to use anticancer drugs in association. Cocktails of drugs instead of single drugs are indeed very popular in current anticancer medical treatments for various reasons: (1) the application of lower concentrations of intrinsically toxic drugs with a narrow therapeutic index; (2) the opportunity to achieve a considerable synergism; and (3) the effective chance of reducing resistance insurgency. Combination therapy may also consist of the combination of an anticancer drug with a common non-cytotoxic drug that has nonetheless the important potential to modulate/enhance the cytotoxic effect of the first drug. This type of strategy was applied very recently by Han et al.[56] to auranofin for the treatment of CRC. In detail, these authors performed a high-throughput screening of a library of 1280 FDA-approved clinical drugs in the search for compounds that might enhance the anticancer activity of auranofin in vitro. Surprisingly, they found that the anti-inflammatory drug celecoxib (CE), a cyclooxygenase 2 inhibitor, strongly potentiated the anticancer activity of AF[56].
Notably, the promising in vitro results obtained for the AF + CE association were later supported by very encouraging in vivo results, as displayed in Figure 5. Since AF and CE are FDA-approved drugs that are used in the clinic, it is quite straightforward to translate the results of this study into an immediate clinical cancer treatment.
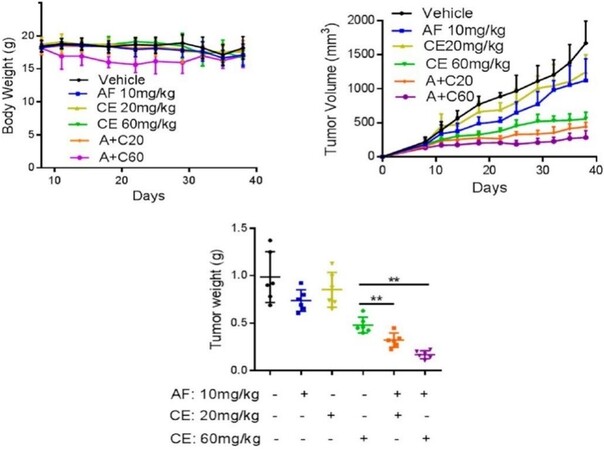
Figure 5. The effect of the combination of auranofin with celecoxib in mice. Athymic nude mice bearing DLD-1 xenografts were treated with the following drugs via oral injection (P.O.): olive oil (vehicle), AF 10 mg/kg, CE 20 mg/kg, CE 60 mg/kg, AF 10 mg/kg + CE
Mechanistically, the AF/CE combination induced severe oxidative stress, resulting in ROS-mediated hexokinase inhibition and disruption of mitochondrial redox homeostasis. Overall, these effects eventually caused a significant decrease of ATP generation. The CE-induced ROS increase together with AF-mediated inhibition of thioredoxin reductase determined a large shift of Trx2 to its oxidized form, producing a degradation of MT-CO2 (mitochondrially-encoded cytochrome C oxidase II) and a dysfunction of the electron transport chain (see Figure 6).
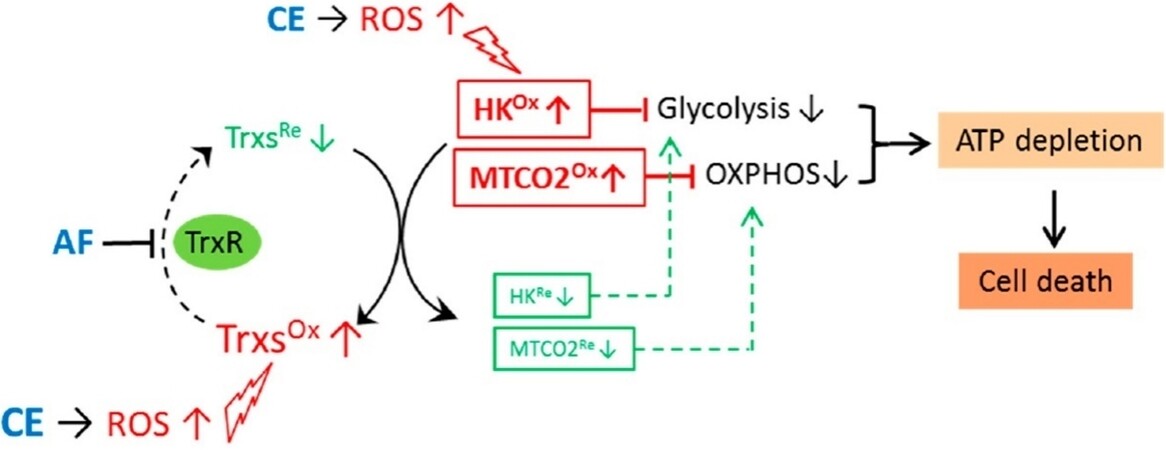
Figure 6. Synergy between auranofin and celecoxib against colon cancer in vitro and in vivo through a novel redox-mediated mechanism. CE induces ROS increase, which in turn causes oxidation of proteins (Trxs, HK, and MTCO2). AF inhibits TrxR, and thus keeps Trxs in oxidized form, which cannot reduce/repair the oxidized proteins (HK and MTCO2), leading to inhibition of both glycolysis and mitorespiration, ATP depletion, and cell death. Reproduced and adapted from Han et al.[56] under the Creative Commons Attribution License 4.0 International (CC BY 4.0). CE: Celecoxib; ROS: reactive oxygen species; HK: hexokinase.
CONCLUSIONS
CRC is the second most deadly cancer worldwide. Medical treatments for CRC are still largely insufficient and rarely curative; it follows that the inoperable metastatic disease results in most cases in patient’s death[57]. Metal-based drugs may play a significant and growing role in the therapeutics of CRC; oxaliplatin is already employed in the treatment of this disease, but its use is often limited by the insurgence of platinum resistance. Gold compounds are promising experimental anticancer drugs and might offer a valuable alternative to platinum drugs by overcoming resistance to Pt drugs[58]. Among the existing medicinal gold compounds, auranofin and its analogs - given the ease of their repurposing - may be the most appropriate and obvious drug candidates for CRC[59]. Auranofin has indeed manifested relevant anticancer actions and already entered clinical trials for other types of cancer, in particular ovarian cancer and various hematological malignancies. Surprisingly, auranofin has been scarcely tested so far for CRC treatment. However, just a few years ago, it was demonstrated that AF is very effective in vitro against four representative CRC lines while being far less toxic for healthy cells, thus showing some degree of selectivity. Moreover, a few studies in the recent literature suggest new valuable strategies to improve the pharmacological profiles of AF and its analogs. We refer specifically to a couple of studies that delineate feasible strategies for therapeutic intervention[43,56]. In a first study, encapsulation of Et3PAuCl in PLGA nanoparticles proved to bring about some favorable pharmacological effects such as retention of the cytotoxic activity, attenuation of the general reactivity of the gold center, and expected reduction of the drug’s adverse effects. Alternatively, a second study showed that the anticancer action of AF in CRC may be greatly potentiated through appropriate drug combinations. In fact, a systematic screening procedure applied to a large library of 1280 FDA-approved drugs revealed a strong synergism between auranofin and the anti-inflammatory drug celecoxib. This synergism was well documented both in vitro and in vivo. In view of these initial yet very encouraging results, we propose that the testing of auranofin and its analogs toward suitable CRC models is further expanded and encouraged taking advantage of new pharmaceutical formulations and appropriate drug combinations. Additionally, extensive in vivo testing of these gold compounds against suitable animal models of CRC would be highly desirable at this stage to reinforce and validate the concepts presented here.
DECLARATIONS
Authors’ contributionsConception and writing of the manuscript: Massai L, Cirri D, Marzo T, Messori L
All the authors approved the final version of the paper.
Availability of data and materialsNot applicable.
Financial support and sponsorshipMassai L, Cirri D, Marzo T and Messori L thank Ente Cassa Risparmio Firenze (ECR), and AIRC for funding the project “Advanced mass spectrometry tools for cancer research: novel applications in proteomics, metabolomics and nanomedicine” (Multi-user Equipment Program 2016, Ref. code 19650). Marzo T gratefully acknowledges the Beneficentia Stiftung, Vaduz (BEN2019/48 and University of Pisa (Rating Ateneo 2019-2020) for the financial support. This work is supported by the University of Pisa under the “PRA - Progetti di Ricerca di Ateneo” Institutional Research Grants - Project no. PRA_2020_58 “Agenti innovativi e nanosistemi per target molecolari nell’ambito dell’oncologia di precisione” to Marzo T. Cirri D gratefully acknowledges AIRC (Associazione Italiana per la Ricerca sul Cancro) for the financial support (two-year fellowship for Italy “Marcello e Rosina Soru” - Project Code: 23852).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Ferlizza E, Solmi R, Sgarzi M, Ricciardiello L, Lauriola M. The roadmap of colorectal cancer screening. Cancers (Basel) 2021;13:1101.
2. Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther 2020;5:22.
4. Vatandoust S, Price TJ, Karapetis CS. Colorectal cancer: metastases to a single organ. World J Gastroenterol 2015;21:11767-76.
5. Valderrama-Treviño AI, Barrera-Mera B, Ceballos-Villalva JC, Montalvo-Javé EE. Hepatic metastasis from colorectal cancer. Euroasian J Hepatogastroenterol 2017;7:166-75.
6. Petrelli F, Coinu A, Lonati V, Barni S. A systematic review and meta-analysis of adjuvant chemotherapy after neoadjuvant treatment and surgery for rectal cancer. Int J Colorectal Dis 2015;30:447-57.
7. Mukkamalla SKR, Huynh DV, Somasundar PS, Rathore R. Adjuvant chemotherapy and tumor sidedness in stage ii colon cancer: analysis of the national cancer data base. Front Oncol 2020;10:568417.
8. Tamura T, Kuwahara A, Kadoyama K, et al. Effects of bolus injection of 5-fluorouracil on steady-state plasma concentrations of 5-fluorouracil in Japanese patients with advanced colorectal cancer. Int J Med Sci 2011;8:406-12.
9. Larsson PA, Carlsson G, Gustavsson B, Graf W, Glimelius B. Different intravenous administration techniques for 5-fluorouracil. Pharmacokinetics and pharmacodynamic effects. Acta Oncol 1996;35:207-12.
10. Tomlinson SK, Melin SA, Higgs V, et al. Schedule-selective biochemical modulation of 5-fluorouracil in advanced colorectal cancer - a phase II study. BMC Cancer 2002;2:1-7.
11. Françoso A, Simioni PU. Immunotherapy for the treatment of colorectal tumors: focus on approved and in-clinical-trial monoclonal antibodies. Drug Des Devel Ther 2017;11:177-84.
12. Laffer U, Metzger U, Aeberhard P, et al. Adjuvant perioperative portal vein or peripheral intravenous chemotherapy for potentially curative colorectal cancer: long-term results of a randomized controlled trial. Int J Colorectal Dis 2008;23:1233-41.
13. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA 2021;325:669-85.
14. Shinozaki E, Makiyama A, Kagawa Y, et al. Treatment sequences of patients with advanced colorectal cancer and use of second-line FOLFIRI with antiangiogenic drugs in Japan: a retrospective observational study using an administrative database. PLoS One 2021;16:e0246160.
15. Köberle B, Schoch S. Platinum complexes in colorectal cancer and other solid tumors. Cancers (Basel) 2021;13:2073.
16. Tomicic MT, Steigerwald C, Rasenberger B, Brozovic A, Christmann M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch Toxicol 2019;93:2265-77.
17. Mirakhorli M, Shayanfar N, Rahman SA, Rosli R, Abdullah S, Khoshzaban A. Lack of association between expression of MRP2 and early relapse of colorectal cancer in patients receiving FOLFOX-4 chemotherapy. Oncol Lett 2012;4:893-7.
18. Huang MY, Tsai HL, Lin CH, et al. Predictive value of ERCC1, ERCC2, and XRCC1 overexpression for stage III colorectal cancer patients receiving FOLFOX-4 adjuvant chemotherapy. J Surg Oncol 2013;108:457-64.
19. Bruno PM, Liu Y, Park GY, et al. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med 2017;23:461-71.
21. McQuade RM, Stojanovska V, Abalo R, Bornstein JC, Nurgali K. Chemotherapy-induced constipation and diarrhea: pathophysiology, current and emerging treatments. Front Pharmacol 2016;7:414.
22. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans 2018;47:6645-53.
23. Martinez-Balibrea E, Martínez-Cardús A, Ginés A, et al. Tumor-related molecular mechanisms of oxaliplatin resistance. Mol Cancer Ther 2015;14:1767-76.
24. Gamberi T, Pratesi A, Messori L, Massai L. Proteomics as a tool to disclose the cellular and molecular mechanisms of selected anticancer gold compounds. Coord Chem Rev 2021;438:213905.
25. Fernández-moreira V, Herrera RP, Gimeno MC. Anticancer properties of gold complexes with biologically relevant ligands. Pure Appl Chem 2019;91:247-69.
26. Liu W, Gust R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem Soc Rev 2013;42:755-73.
27. Kim JH, Ofori S, Parkin S, Vekaria H, Sullivan PG, Awuah SG. Anticancer gold(iii)-bisphosphine complex alters the mitochondrial electron transport chain to induce in vivo tumor inhibition. Chem Sci 2021;12:7467-79.
28. Rigobello MP, Scutari G, Boscolo R, Bindoli A. Induction of mitochondrial permeability transition by auranofin, a gold(I)-phosphine derivative. Br J Pharmacol 2002;136:1162-8.
29. Citta A, Schuh E, Mohr F, et al. Fluorescent silver(I) and gold(I)-N-heterocyclic carbene complexes with cytotoxic properties: mechanistic insights. Metallomics 2013;5:1006-15.
30. Gordon AN, Tonda M, Sun S, Rackoff W. Doxil Study 30-49 Investigators. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer. Gynecol Oncol 2004;95:1-8.
31. Rackham O, Nichols SJ, Leedman PJ, Berners-Price SJ, Filipovska A. A gold(I) phosphine complex selectively induces apoptosis in breast cancer cells: implications for anticancer therapeutics targeted to mitochondria. Biochem Pharmacol 2007;74:992-1002.
32. Messori L, Marcon G, Innocenti A, Gallori E, Franchi M, Orioli P. Molecular recognition of metal complexes by DNA: a comparative study of the interactions of the parent complexes [PtCl(TERPY)]Cl and [AuCl(TERPY)]Cl2 with double stranded DNA. Bioinorg Chem Appl 2005:239-53.
33. Alsaeedi MS, Babgi BA, Hussien MA, Abdellattif MH, Humphrey MG. DNA-binding and anticancer activity of binuclear gold(I) alkynyl complexes with a phenanthrenyl bridging ligand. Molecules 2020;25:1033.
34. Ott I, Gust R. Non platinum metal complexes as anti-cancer drugs. Arch Pharm (Weinheim) 2007;340:117-26.
36. Shaw CF. . Chrysotherapy:gold-drug metabolism and immunochemistry. In: Clarke MJ, Sadler PJ, editors. Metallopharmaceuticals II. Berlin: Springer Berlin Heidelberg; 1999. p. 187-216.
37. Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med 2007;42:872-81.
38. Harbut MB, Vilchèze C, Luo X, et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci U S A 2015;112:4453-8.
39. Thangamani S, Maland M, Mohammad H, et al. Repurposing approach identifies auranofin with broad spectrum antifungal activity that targets Mia40-Erv1 pathway. Front Cell Infect Microbiol 2017;7:4.
40. Díez-Martínez R, García-Fernández E, Manzano M, et al. Auranofin-loaded nanoparticles as a new therapeutic tool to fight streptococcal infections. Sci Rep 2016;6:19525.
41. Onodera T, Momose I, Kawada M. Potential anticancer activity of auranofin. Chem Pharm Bull (Tokyo) 2019;67:186-91.
42. Raninga PV, Lee AC, Sinha D, et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int J Cancer 2020;146:123-36.
43. Menconi A, Marzo T, Massai L, et al. Anticancer effects against colorectal cancer models of chloro(triethylphosphine)gold(I) encapsulated in PLGA-PEG nanoparticles. Biometals 2021;34:867-79.
44. Mirabelli CK, Johnson RK, Sung CM, Faucette L, Muirhead K, Crooke ST. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res 1985;45:32-9.
45. Zoppi C, Messori L, Pratesi A. ESI MS studies highlight the selective interaction of Auranofin with protein free thiols. Dalton Trans 2020;49:5906-13.
46. Tolbatov I, Cirri D, Marchetti L, et al. Mechanistic insights into the anticancer properties of the auranofin analog Au(PEt3)I: a theoretical and experimental study. Front Chem 2020;8:812.
47. Hossain MF, Bodnar Y, Klein C, et al. Molecular basis for the interactions of human thioredoxins with their respective reductases. Oxid Med Cell Longev 2021;2021:6621292.
48. Magherini F, Modesti A, Bini L, et al. Exploring the biochemical mechanisms of cytotoxic gold compounds: a proteomic study. J Biol Inorg Chem 2010;15:573-82.
49. Yue S, Luo M, Liu H, Wei S. Recent advances of gold compounds in anticancer immunity. Front Chem 2020;8:543.
50. Freire Boullosa L, Van Loenhout J, Flieswasser T, et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol 2021;42:101949.
51. Marzo T, Cirri D, Gabbiani C, et al. Auranofin, Et3PAuCl, and Et3PAuI are highly cytotoxic on colorectal cancer cells: a chemical and biological study. ACS Med Chem Lett 2017;8:997-1001.
52. Cirri D, Fabbrini MG, Massai L, et al. Structural and solution chemistry, antiproliferative effects, and serum albumin binding of three pseudohalide derivatives of auranofin. Biometals 2019;32:939-48.
53. Marzo T, Pillozzi S, Hrabina O, et al. cis-Pt I2(NH3)2: a reappraisal. Dalton Trans 2015;44:14896-905.
54. Cirri D, Pillozzi S, Gabbiani C, et al. PtI2(DACH), the iodido analogue of oxaliplatin as a candidate for colorectal cancer treatment: chemical and biological features. Dalt Trans 2017;46:3311-7.
55. Noordhuis P, Laan AC, van de Born K, Honeywell RJ, Peters GJ. Coexisting molecular determinants of acquired oxaliplatin resistance in human colorectal and ovarian cancer cell lines. Int J Mol Sci 2019;20:3619.
56. Han Y, Chen P, Zhang Y, et al. Synergy between Auranofin and Celecoxib against colon cancer in vitro and in vivo through a novel redox-mediated mechanism. Cancers (Basel) 2019;11:931.
57. Bradley C, Yabroff KR, Warren JL, Chawla N, Zeruto C, Lamont EB. Trends in chemotherapy-related treatment of advanced-stage colorectal (CRC) cancer. J Clin Oncol 2013;31:258-258.
58. Arena S, Corti G, Durinikova E, et al. A subset of colorectal cancers with cross-sensitivity to olaparib and oxaliplatin. Clin Cancer Res 2020;26:1372-84.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Massai L, Cirri D, Marzo T, Messori L. Auranofin and its analogs as prospective agents for the treatment of colorectal cancer. Cancer Drug Resist 2022;5:1-14. http://dx.doi.org/10.20517/cdr.2021.71
AMA Style
Massai L, Cirri D, Marzo T, Messori L. Auranofin and its analogs as prospective agents for the treatment of colorectal cancer. Cancer Drug Resistance. 2022; 5(1): 1-14. http://dx.doi.org/10.20517/cdr.2021.71
Chicago/Turabian Style
Massai, Lara, Damiano Cirri, Tiziano Marzo, Luigi Messori. 2022. "Auranofin and its analogs as prospective agents for the treatment of colorectal cancer" Cancer Drug Resistance. 5, no.1: 1-14. http://dx.doi.org/10.20517/cdr.2021.71
ACS Style
Massai, L.; Cirri D.; Marzo T.; Messori L. Auranofin and its analogs as prospective agents for the treatment of colorectal cancer. Cancer Drug Resist. 2022, 5, 1-14. http://dx.doi.org/10.20517/cdr.2021.71
About This Article
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 2
likes
Like This Article 2
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.