
Overcoming drug resistance by targeting protein homeostasis in multiple myeloma

 , ...
, ... Abstract
Multiple myeloma (MM) is a plasma cell disorder typically characterized by abundant synthesis of clonal immunoglobulin or free light chains. Although incurable, a deeper understanding of MM pathobiology has fueled major therapeutical advances over the past two decades, significantly improving patient outcomes. Proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies are among the most effective anti-MM drugs, targeting not only the cancerous cells, but also the bone marrow microenvironment. However, de novo resistance has been reported, and acquired resistance is inevitable for most patients over time, leading to relapsed/refractory disease and poor outcomes. Sustained protein synthesis coupled with impaired/insufficient proteolytic mechanisms makes MM cells exquisitely sensitive to perturbations in protein homeostasis, offering us the opportunity to target this intrinsic vulnerability for therapeutic purposes. This review highlights the scientific rationale for the clinical use of FDA-approved and investigational agents targeting protein homeostasis in MM.
Keywords
INTRODUCTION
Multiple myeloma (MM) is a clonal proliferation of malignant plasma cells, the product of the terminal differentiation of B cells. Clinically, MM is suspected based on the presence of laboratory abnormalities such as hypercalcemia, anemia, renal failure, or radiological evidence of lytic bone disease[1].
The clinical use of molecularly targeted agents, including bortezomib, the first in class proteasome inhibitor (PI), immunomodulatory drugs (IMiDs) like thalidomide, lenalidomide and pomalidomide, and monoclonal antibodies targeting CD38 and SLAMF7 has dramatically impacted the life expectancy of MM patients. All these drugs have successfully passed regulatory approvals and are used in patients with newly diagnosed and/or relapsed, or refractory disease, in all stages of treatment, contributing to prolonging median overall survival to 7-8 years.
Despite an improved understanding of the pathobiology of myeloma and a significant drug-development effort in the past 2 decades, MM remains incurable and therapeutic resistance represents a major clinical concern. Even in patients whose disease initially responds to treatment, the acquisition of resistance to chemo-immunotherapy over time is a common phenomenon. The molecular mechanisms underlying such acquired resistance are only partially understood, thus limiting therapeutic success.
One pathognomonic feature of myeloma cells is the exuberant production of clonal, intact immunoglobulin and/or free light chains. MM cells contain a well-developed endoplasmic reticulum (ER) and Golgi apparatus, tailored for such sustained protein synthesis and secretion effort[2,3]. By virtue of their nature as factories of protein production, MM cells are especially prone to improper protein folding of nascent Ig and baseline proteotoxic stress[4,5]. Drugs that further dysregulate protein quality control and proteostasis have shown to be specifically toxic against MM by exacerbating proteotoxic stress and causing apoptosis.
In recent years, extensive research has focused on understanding the role that protein homeostasis (proteostasis) plays in supporting efficient protein synthesis, folding, secretion, and degradation and on the identification of putative molecular targets within this complex network[6-9].
In this review, we will outline the current understanding of protein metabolism and homeostasis in MM and the rationale for translating laboratory discovery in proteostasis into patient-focused therapies.
BIOLOGICAL RATIONALE FOR TARGETING THE PROTEOSTASIS NETWORK IN MM
Eukaryotic cells maintain a balanced proteome through the function of the proteostasis network, composed of a large number of proteins regulating four interlinked pathways: protein synthesis, folding, secretion, and degradation[5].
After synthesis, secreted proteins undergo an intricate process of folding involving sequential steps of post-translational modifications, such as glycosylation and disulphide bond formation, taking place in the ER[10]. Molecular chaperones assist client proteins during folding, exerting a stringent quality control that allows only proteins that reach a native conformation to progress along the secretory pathway [Figure 1].
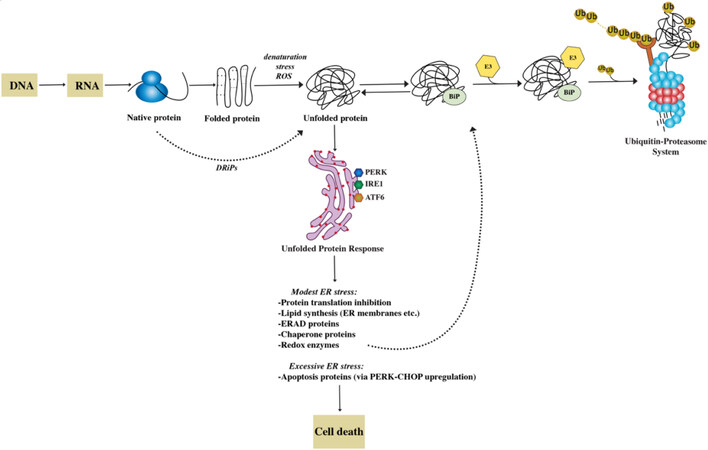
Figure 1. The proteostasis network. The folding of newly synthesized proteins is a complex mechanism that involves multiple steps. ~30% of nascent proteins, named DRiPs, have an inherent inefficiency of protein folding and undergo degradation within minutes from synthesis. Most proteins achieve a functional folded state. However, many are the causes that trigger spontaneous unfolding. These changes in conformation are recognized by the cell’s protein quality control machinery with activation of an unfolded protein response (UPR). The three branches of the UPR (PERK, IRE, ATF6) help restore protein homeostasis partially by increasing the synthesis of chaperone proteins. By association with exposed hydrophobic domains, chaperones like BiP (GRP78), favor refolding. Alternatively, they can facilitate the recognition of abnormal proteins, leading to their ubiquitylation by E3 and their degradation through the proteasome. If the ER stress cannot be mitigated and homeostasis cannot be reestablished, UPR induces cell death.
Nascent proteins that fail to do so and remain misfolded are diverted to proteasome-mediated degradation in a process called ER-associated degradation (ERAD). Thus, the proteostasis network also ensures that superfluous and misfolded proteins are removed by the ubiquitin-proteasome system (UPS). Alternative proteolytic mechanisms cooperate with the UPS in guaranteeing proteostasis, such as the macroautophagy (autophagy-lysosome) system and the aggresome pathway[5,11]. In fact, the accumulation of misfolded proteins is sensed as toxic by cells and highly conserved adaptive responses, such as the unfolded protein response (UPR) and the heat shock response (HSR), exist with the goal of restoring protein homeostasis. The UPR is a tripartite protein homeostasis mechanism triggered by the accumulation of misfolded proteins within the ER (ER stress). The ultimate goal of the UPR is to reduce protein toxicity by decreasing total protein synthesis while selectively upregulating chaperone transcription and translation[11]. However, if homeostasis cannot be restored, the UPR activates a terminal pathway, triggering C/EBP homologous protein (CHOP) and GADD34 transcription and ultimately apoptosis. The HSR similarly induces upregulation of chaperons via the master-regulator heat shock factor 1 (HSF1) to counteract protein misfolding[12,13].
The UPS plays a central role in maintaining protein homeostasis as it is the main proteolytic mechanism responsible for the degradation of misfolded proteins. Clearance of these specimens is critical to both avoid toxicity and recycle ubiquitin and amino acids.
Studies have shown that treatment of MM cells with PI perturbs proteostasis adaptive responses, impairs DNA repair, and eventually triggers apoptosis. Indeed, increased proteasomal workload (polyubiquitinated proteins), decreased proteasomal capacity, or a combination of both is a key determinant of PI sensitivity in MM[14]. Consistently, drugs that increase proteasome cargo such as ER stressors and heat shock protein inhibitors synergize with PIs[15]. Further works assessing combination therapies targeting one or more of these pathways in MM are currently ongoing.
THE UBIQUITIN-PROTEASOME SYSTEM
Protein translation and folding are imperfect mechanisms, and it is estimated that up to 30% of nascent proteins have an intrinsic inability to achieve stably folded conformations and never reach a functional state. These unstable proteins are termed defective ribosomal products (DRiPs)[16]. It is thought that such faulty protein species are even more abundant in highly secretory, malignant cells such as myeloma PC that synthesize extensive amounts of immunoglobulins. DRiPs contribute to proteotoxic stress by inducing the UPR and potentially overwhelming the UPS and ERAD, resulting in the accumulation of polyubiquitinated proteins[17,18].
The UPS is primarily responsible for the degradation of misfolded polypeptides, accounting for over 80% of total proteolysis[19,20]. Secreted and membrane proteins are used by cells to communicate with their environment and account for approximately 30% of the cellular proteome[10]. Before they are inserted into cellular membranes or released into the extracellular space, they must achieve their native conformation in the ER lumen. If they fail to do so, they are retro-translocated via the Sec61 translocon from the ER to the cytosol for ubiquitination and degradation by the proteasome in a cellular pathway termed ERAD[21].
The proteasome is an ATP-dependent, multi-catalytic protease mediating degradation of senescent and/or misfolded proteins that are generally tagged for degradation via K48-linked polyubiquitin chains[19]. Protein ubiquitination is controlled by a three-enzyme cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes. On the other hand, the large family of deubiquitinating enzymes (DUBs) remove ubiquitin, critically contributing to recycling ubiquitin and maintaining a steady-state pool of free ubiquitin[22,23]. Polyubiquitinated proteins are delivered to the 26S proteasome, a barrel-like complex composed of a 20S catalytic core, associated with 19S regulatory caps[24]. The regulatory subunits mediate recognition, binding, unfolding, and facilitate engagement of targeted proteins with the 20S catalytic core. Together with DUBs (e.g., UCH37, USP14), they also engage in the removal of polyubiquitin chains, which would otherwise impede transferring the cargo proteins through the tight pore created by the 19S cap on the 2 sides of the 20S core[25]. The 20S core contains β1, β2, β5 main catalytic subunits, which are responsible for caspase-like, trypsin-like, and chymotrypsin-like activities, respectively[24].
In addition to the constitutive proteasome, immune cells can be equipped with immunoproteasome, typically in the setting of infection or pro-inflammatory stimuli[26]. The immunoproteasome subunits β1i (LMP2), β2i (MECL-1), and β5i (LMP7) replace the constitutive catalytic subunits[26,27]. It has been previously shown that MM cells are equipped with large amounts of immunoproteasome, and selective inhibition of these catalytic activities is an appealing therapeutic strategy for MM and potentially other hematologic malignancies[28,29].
Proteasome inhibitors
PIs were initially developed in the mid-1990s as a research tool to investigate proteasome-mediated proteolysis mechanisms[30,31]. The clinical use of PI has radically changed the natural history of MM and now, along with immunomodulatory agents and monoclonal antibodies, form the backbone of MM treatment. These agents paved the way for therapies targeting protein homeostasis in cancer. The PIs bortezomib, carfilzomib, ixazomib, and oprozomib show inhibitory activity mainly against the β5 chymotrypsin-like subunit, while marizomib targets all three β-subunits [Figure 2A][32,33].
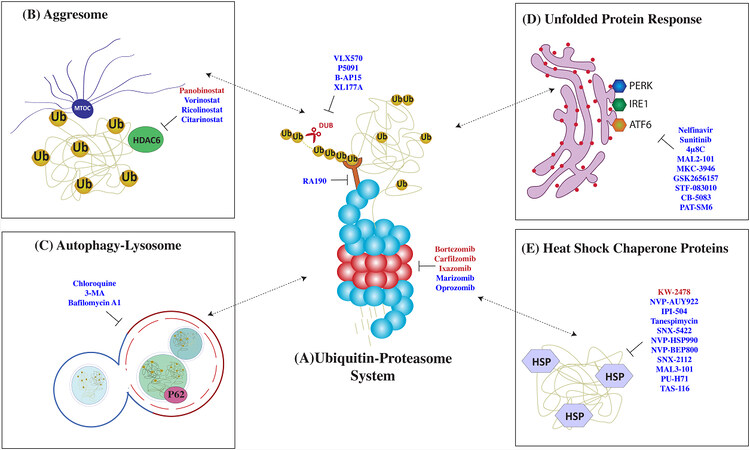
Figure 2. Therapies targeting protein control pathways in multiple myeloma (MM). MM cells are highly dependent on a strictly regulated network of protein quality control pathways such as (A) the ubiquitin-proteasome system (UPS), (B) aggresome formation, (C) autophagy, (D) unfolded protein response, and (E) the heat shock response. Drugs that target these pathways are listed here: FDA-approved drugs (in red) and experimental drugs (in blue).
Although the clinical use of PI was predicted to have significant toxic effects related to the ubiquitous expression of the proteasome, PIs are generally well tolerated, with distinct toxicities across different generations consisting of hematologic, gastrointestinal toxicities, and peripheral neuropathy, usually readily manageable[34].
The proposed molecular mechanisms of bortezomib anti-MM activity imply pleiotropic levels of actions, including direct actions on both the tumor cells and the BM microenvironment. In the context of the BM microenvironment, bortezomib modifies the cytokine milieu, has anti-angiogenic activity, impairs stroma-tumor crosstalk, and induces apoptosis of osteoclasts while supporting osteoblastogenesis[2,35,36].
The proposed mechanistic explanations for the direct anti-myeloma plasma cell effect include inhibition of pro-survival via NF-κB pathway regulation, impairment of DNA damage response, apoptosis via both caspase 8 and 9 cleavage and regulation of various members of the B-cell lymphoma protein family[37,38]. Induction of HSR, UPR, c-Jun NH2-Terminal Kinase, and TP53 are additional hypothesized mechanisms of PI action[39].
Importantly, despite initial fear of intolerable side effects, considering the ubiquitous and essential expression of the proteasome, MM are exquisitely sensitive to PI, with several lines of evidence suggesting that this can be attributed to baseline proteotoxicity in MM plasma cells[2,40,41]. In fact, both Ig synthesis and retention correlate with apoptotic sensitivity to PI, and manipulating Ig synthesis alters sensitivity[14]. The abundant production of Ig, paired with insufficient proteostasis mechanisms, leads to baseline proteotoxic stress that can be further exacerbated by PI, resulting in apoptosis. Increasing protein misfolding via ER stressors results in increased sensitivity to PI, while augmenting proteasome activity or inducing alternative proteolytic pathways mediates PI resistance[14]. These preclinical data suggest potential mechanisms of acquired resistance to PI.
Resistance to PI is ultimately inevitable in most MM patients, leading to refractory disease and negatively impacting outcomes. Research efforts have focused on identifying the molecular mechanisms of PI resistance to develop novel therapies able to overcome it. We and others have recently shown that targeting the compensatory proteasome stress response (PSR) is of therapeutic utility in MM and can overcome acquired or de novo PI resistance[42]. The transcription factor Nuclear Factor Erythroid 2 Like 1 (NFE2L1 or NRF1) is the master regulator of the PSR[42]. Under homeostatic conditions, NRF1 is continuously translated, inserted into the ER, glycosylated, retro-translocated to the cytosol, and targeted for proteasomal degradation via ERAD. Though, when proteasome activity is partially inhibited, NRF1 is deglycosylated by N-glycanase 1 (NGL1) and cleaved by the aspartic protease DNA-damage inducible 1 homolog 2 (DDI2). NRF1 in its active form translocates to the nucleus and dimerizes with small MAF proteins, leading to a complex transcriptional program that includes the biogenesis of new proteasome subunits[43-47]. Recent studies show that the genetic or pharmacologic blockade of NGL1 increases sensitivity to PI-mediated cytotoxicity, suggesting its therapeutic potential[42]. Recently, similar results were obtained via knock out of DDI2 or NRF1 directly[48].
A different strategy to increase PI activity has focused on developing next-generation PI characterized by increased potency, irreversible catalytic activity blockade, and/or broader catalytic subunits inhibition.
The first-in-class PI bortezomib is a peptide boronic acid that reversibly inhibits the β5 subunit. It is approved for administration via intravenous (i.v.) or subcutaneous (s.c.) injection[7]. By contrast, carfilzomib is an epoxyketone, a second-generation agent that irreversibly inhibits the β5 subunit. Preclinical studies showed that carfilzomib has a higher potency than bortezomib, and clinical trials showed durable responses to a single agent and combination therapy in patients relapsed/refractory to bortezomib and lenalidomide treatment[32,33,49]. These positive results led to its FDA approval in combination with lenalidomide and dexamethasone (Rd), as the second line of treatment in MM, by i.v. administration. Interestingly, the pattern of side effects of carfilzomib is quite distinct from bortezomib. While the latter can cause cardiovascular side effects, including tachyarrhythmia, hypertension, and systolic heart failure, the latter is often responsible for sensory peripheral neuropathy.
Ixazomib and oprozomib are orally bioavailable PIs chemically related to bortezomib and carfilzomib, respectively. The former is currently FDA approved for the treatment of RRMM, while the latter showed promising activity as a single agent and in combination with IMiDs in RRMM[50,51].
Finally, the irreversible agent marizomib has a unique β-lactone warhead, and, unlike all other clinically available PIs, it inhibits all three catalytic subunits within the 20S core[52]. Recent preclinical studies show potent activity of marizomib even in bortezomib-resistant MM cells, suggesting that broader blockade of proteasome subunits may increase effectiveness[53,54]. However, this potent and universal proteasomal subunits inhibition may result in a narrowing of the therapeutic index. Indeed, early phase clinical trials show that the potent inhibition of proteasome subunits was similarly accompanied by renal and central nervous system toxicity, hampering clinical development of this derivative in MM[54].
Deubiquitinating enzymes and ubiquitin receptors inhibitors
Potential molecular targets within the UPS include enzymes involved in ubiquitination (E1, E2, E3) and deubiquitination (DUBs) of proteins destined for proteasomal degradation[55,56]. The development of therapeutically effective inhibitors in this area can be challenging considering the numerous members of ubiquitinating and DUBs, their distinct structure, and the potential for systemic toxicities related to the nature of client proteins. Similar to PI, DUB inhibitors induce apoptosis in preclinical MM models preceded by accumulation of polyubiquitinated proteins and in a manner that is independent from the inhibition of the proteasome catalytic activity[57]. Therefore, there is a scientific rationale to predict that DUBs would overcome PI resistance by more generically targeting proteostasis. In fact, small molecule compounds P5091, B-AP15, and VLX1570 successfully induced apoptosis in multiple myeloma cell lines and primary cells, including those resistant to bortezomib [Figure 2A][58].
RPN13 is a polyubiquitin receptor (UbR) within the 19S subunit. Rpn13 binds with high-affinity ubiquitin carboxyl-terminal hydrolase L5 (UCHL37), the deubiquitinating enzyme that helps with ubiquitin (Ub) hydrolysis[59]. RPN13 and UCHL37 have been found to be relevant for cell cycle progression in vitro, and increased expression of the gene encoding RPN13 (ADRM1) has been reported in MM cells when compared to normal plasma cells[60]. Preclinical studies of RA190, a specific, small molecule inhibitor of RPN13, showed robust antitumor activity[61]. RA190 decreased the viability of MM cell line and patient-derived MM cells by inducing caspase-dependent apoptosis and UPR. Combination of RA190 with bortezomib, lenalidomide, or pomalidomide induces synergistic anti-MM activity, providing the framework for clinical evaluation of RPN13 inhibitors[60]. WL40 is a first in class, RPN13 degrader through proteolysis-targeting chimeric molecule[62]. WL40 was synthesized by fusing RA190 with cereblon (CRBN) binding ligand thalidomide, triggering degradation of cellular RPN13. Importantly, WL40 not only decreases the viability of patient MM cells, even those resistant to bortezomib, but prolongs the survival of xenografted human MM models. As predicted, WL40 induces cytotoxicity by activating ER stress response, p53/p21 signaling, and ultimately caspase apoptotic cascade[62].
The first in human UCHL5 inhibitor VLC1570 similarly showed promising and potent anti-MM activity in vitro; however, clinical development was halted due to fatal lung toxicity observed in a first in human, phase I study[63].
Based on the pattern of client proteins, USP7 has been proposed as a therapeutic target across numerous, distinct cancers[64,65]. P5091, a USP7 inhibitor, showed promising results in overcoming PI-resistance in MM via direct anti-MM activity alone or in combination with anti-MM agents. Similarly, a novel USP7 inhibitor, XL177A, was cytotoxic against MM in preclinical models and appeared to target supporting plasmacytoid dendritic cells to restore anti-MM immunity[58,66]. Recently, XL177A showed p53-dependent cytotoxicity against Ewing sarcoma and malignant rhabdoid tumor[67].
Ubiquitin enzymes
The process of ubiquitin conjugation to target proteins is highly dynamic and involves the regulated, sequential activity of three classes of enzymes: E1, E2, and E3 Ub enzymes[68]. Despite the diversity of E2 and E3 ubiquitin enzymes, there are only two E1 ubiquitin-activating enzymes. Thus, inhibiting the E1 ubiquitin-activating enzyme would theoretically block all ubiquitin-dependent pathways in cells. Recently, TAK-243, a potent and selective inhibitor of E1 Ub activating enzyme, suppressed myeloma cell line and primary cells viability through activation of protein kinase RNA-like ER kinase (PERK) arm of the ER stress response pathway, as well as induction of oxidative stress[69]. Comparable anti-MM activity was reported in murine myeloma models, supporting a potential clinical use of this strategy in RRMM, although the therapeutic index may not be favorable[70].
Since E3 proteins determine substrate specificity, it is not surprising that over 600 E3 enzymes are encoded by the human genome. These E3 ligases are generally classified into three large families with distinct catalytic domains: really interesting new gene (RING), homology to E6-Ap carboxyl terminus (HECT), and RING-in-between-RING (RBR)[71,72].
The RING class of E3 enzymes acts as a docking site to bring together the targeted substrate designated for degradation with the E2-Ub, thus working as an allosteric activator[73]. The HECT and RBR E3 classes catalyze substrate ubiquitination by undergoing a cysteine-dependent transthiolation reaction with E2-Ub, forming a covalent E3-Ub intermediate, and later the Ub moiety is transferred to a lysine on the target substrate[74]. The E3 ligase cereblon (CRBN) is the main target for anti-myeloma activity of IMiDs thalidomide, lenalidomide, and pomalidomide[75]. Lenalidomide has been shown to bind to the E3 ubiquitin-ligase complex composed of damage-specific DNA binding protein 1 and CRBN, enhancing its activity and facilitating ubiquitination and proteasome-mediated degradation of the Ikaros family of transcription factors[76,77].
Cullin-RING E3 are post-translationally activated by NEDD8 activating enzyme (NAE) in a process called neddylation[78]. Therefore, inhibiting neddylation would result in cullin-RING E3 ubiquitin ligase blockade. Inhibitors of NAE have been developed and have been under clinical investigation for a variety of cancer cell types. A Phase I clinical study on pevonedistat (MLN4924), an NAE inhibitor, showed modest activity in lymphoma, but no significant activity in MM[79]. Currently, a clinical trial is evaluating the efficacy of the Ixazomib-Pevonedistat combination in RRMM patients.
AGGRESOME PATHWAY
Targeting alternative proteolytic pathways, such as aggresomes and autophagy, in combination with PI has shown preclinical efficiency by increasing proteotoxic stress[80]. HDACs are a group of enzymes responsible for deacetylation of histone and non-histone proteins, resulting in inhibition of gene transcription with contributory effect on critical cellular events such as survival, proliferation, and crosstalk with the surrounding microenvironment[81]. HDAC6 coordinates the formation of perinuclear protein aggregates in structures called aggresomes, contributing to maintaining protein homeostasis[80,82]. Preclinical studies showed that aggresome formation is a possible mechanism of resistance to PI, and the combination of HDAC inhibitors (HDACi) with PI is synergistic in preclinical MM models[83].
Despite having limited activity as single agents, HDACi have proven to achieve durable responses when in combination with PI and IMiD. Panobinostat, in combination with bortezomib and dexamethasone, was recently approved as third-line therapy in MM patients with prior bortezomib and IMiDs exposure, based on 4-month prolongation of progression-free survival (PFS), near doubling of very good partial response (VGPR), and evidence of response in bortezomib-resistant patients when combined with bortezomib[84]. A distinct HDAC inhibitor, vorinostat, was investigated in the phase III, randomized, placebo-controlled, Vantage 008 trial in combination with bortezomib and dexamethasone [Figure 2B][85]. While the experimental group showed a prolonged overall response rate (ORR) and PFS, the clinical relevance of this outcome is not clear.
Frequent and often severe, gastrointestinal and hematologic side effects are the major limitation to the clinical development of HDACi[86,87]. Efforts to maintain efficacy and limit toxicities have led to the development of isoform-specific HDACi, focusing on the inhibition of HDAC6 and the aggresome pathway. Two selective HDAC6 inhibitors, ricolinostat (ACY-1215) and citarinostat (ACY-241), are currently being evaluated in clinical studies. A phase Ib trial on ricolinostat in combination with bortezomib/dexamethasone reported a 37% ORR in RRMM, and ricolinostat in combination with lenalidomide/dexamethasone had an ORR of 55% in RRMM[86]. Ricolinostat in combination with pomalidomide/dexamethasone is currently evaluated in clinical trials.
BG45, an HDAC3 inhibitor, has also shown promising preclinical results, showing direct and bone marrow microenvironment-mediated anti-MM activity alone and in combination with bortezomib and translation to early phase clinical trial is anticipated soon[88].
AUTOPHAGY
Autophagy is an evolutionarily conserved mechanism that plays a crucial function in maintaining cellular homeostasis as products of autophagic digestion can be re-utilized in anabolic processes, guaranteeing energy supply[89]. Autophagy critically participates in protein homeostasis by sequestering polyUb proteins in autophagic vacuoles that are later degraded upon fusion with lysosomes through an SQSTM1/p62-dependent mechanism[90]. While autophagy is typically seen as a pro-survival mechanism, autophagic cell death has been described, attesting to the complexity of this pathway. Studies have shown that a close interaction exists between autophagy, UPR, ERAD, HSR, and UPS[91-93].
Autophagy maintains quality control of newly synthesized proteins, potentially explaining the high levels of basal autophagy in MM[94]. The current consensus is that this process is essential for MM survival as an alternative proteolytic pathway, particularly when other proteostasis pathways are overwhelmed, thus providing a rationale for the combination of autophagy inhibitors with PI in MM[95].
Notably, many agents with proven anti-MM activity, like mTORC1 inhibitor rapamycin and bortezomib itself, were noticed to induce autophagy that thus may represent a potential escape mechanism[91,96,97].
Preclinical studies have shown that p62 contributes to protein homeostasis in MM cells by clearance of redundant misfolded proteins. Importantly, p62 is increased after PI treatment, suggesting a role as an escape mechanism and potential mechanism of resistance to PI[95]. Consistently, knocking out p62 increases sensitivity to PI, suggesting this may be a novel, attractive molecular target to overcome PI resistance in MM.
Other mechanisms of bortezomib resistance, in the context of autophagy, include the upregulation of a cytoskeleton protein, profilin-1, which promotes autophagy by binding Beclin-1 complex[98].
Clinical translation of these findings has been attempted by pharmacologic inhibition of this pathway with 3-methyladenine and chloroquine [Figure 2C][96]. Chloroquine inhibits autolysosome-mediated proteolysis by alkalinizing the lysosomal pH. Preclinical studies showed that chloroquine potentiated carfilzomib cytotoxicity and was able to overcome carfilzomib resistance in vitro[99]. In a phase II clinical trial, the combination of chloroquine and bortezomib/cyclophosphamide showed a modest 14% ORR in patients with refractory myeloma who progressed on a combination of bortezomib and cyclophosphamide[100].
Importantly, given the complexity of the autophagic pathway as well as non-selective targeting of autophagy, combined inhibition of autophagy and proteasome system in preclinical studies yielded conflicting and highly variable results, ranging from synergism to antagonism[94]. For instance, a specific study reported that the autophagy inhibitors 3-methyladenine and chloroquine had antagonistic effects when used in combination with bortezomib[91]. One potential explanation for these conflicting results is that autophagic cell death can contribute to the anti-MM effects of several agents, including bortezomib, thus representing a double-edge sword.
ER STRESS AND UNFOLDED PROTEIN RESPONSE
The UPR is a highly conserved cell response to stress, elicited by the accumulation of unfolded proteins in the ER[11]. The primary goal of the UPR is to maintain protein homeostasis by specifically halting de novo protein synthesis at the level of translation, while selectively inducing the transcription of chaperone molecules to aid in protein folding. The UPR integrates with the UPS via the ERAD in an attempt to resolve proteotoxicity via protein degradation. If homeostasis cannot be achieved, prolonged UPR activation ultimately leads to apoptosis, outlining the double-edged nature of this stress response pathway[101].
The UPR is a tripartite system relying on three distinct stress sensors: inositol-requiring enzyme 1 (IRE1), PERK, and activating transcription factor 6 (ATF6)[11]. The three branches of the UPR operate in parallel as feedback loops that mitigate ER stress. Proof-of-principle studies have shown that ER stressors, such as tunicamycin, thapsigargin, and brefeldin A, potentially synergize with PI in vitro [Figure 2D][14]. However, the clinical translation of these drugs is limited by the narrow therapeutic index and potential organ toxicity.
Activated IRE1 functions as an endonuclease, resulting in the mRNA splicing and activation of the transcription factor X-box binding protein (XBP1)[102]. Together with ATF6, spliced XBP1 (sXBP1) induces lipid biogenesis to sustain ER expansion, chaperone proteins to support nascent ER protein folding, or alternatively initiates ERAD to reduce ER stress[40]. High sXBP1 expression in primary MM cells was shown to correlate with poor overall survival, and expression of sXBP1 in B cells reproduces MM phenotype in mice, suggesting that sustained IRE1-XBP1 activation may contribute to MM pathogenesis[103]. Recently, decreased XBP1 splicing was shown to associate with de-differentiation from plasma cells to plasmablasts alongside decreased immunoglobulin production, decreased proteasome load, and reduced sensitivity to PI[14,104]. MKC-3946, a specific IRE1 endoribonuclease inhibitor, demonstrated substantial anti-MM activity alone or in combination with PI in preclinical studies[105,106]. By inhibiting the IRE1 branch of the UPR, MKC-3946 resulted in activation of PERK, downstream eIF2α phosphorylation, ATF4 cleavage and CHOP expression, thereby leading to a terminal UPR and apoptosis[107].
PERK phosphorylates eIF2α, resulting in repression of global protein synthesis, however, if the stress is persistent over time, PERK selectively cleaves and activates the transcription factor ATF4, resulting in the expression of the pro-apoptotic protein CHOP[2]. The combination of the selective PERK inhibitor GSK2606414 with bortezomib resulted in synergistic anti-MM activity in preclinical models[108].
Most recently, the HIV protease inhibitor nelfinavir was shown to block ER protein export, triggering a terminal UPR and consequent apoptosis in preclinical MM models, including bortezomib-resistant cell lines[109,110]. Importantly, the combination of nelfinavir with bortezomib and dexamethasone showed an impressive ORR of 65% in patients with lenalidomide resistant, bortezomib refractory MM in a phase II, single-arm study[111,112].
Targeting ERAD directly has recently emerged as a way to disrupt intracellular protein metabolism within MM cells. Of note, VCP/p97 is a cytosolic AAA-ATPase necessary for retro-translocation of misfolded proteins from the ER to undergo proteasome-mediated proteolysis as part of ERAD[113]. Preclinical studies on CB-5083 have shown robust activity against myeloma cell lines and a number of in vivo MM models. Anti-MM synergistic activity of CB-5083 and PI is likely explained by the p97-dependent retro-translocation of the transcription factor NRF1, which transcribes proteasome subunit genes following proteasome activity insufficiency[114]. Minor toxicity was observed in untransformed, non-secretory control cells[114]. However, a phase I clinical trial of p97 inhibitor CB-5083 was arrested due to off-target effects and retinal toxicities[115].
Finally, PAT-SM6 is a fully human immunoglobulin antibody targeting glucose-regulated protein 78 (GRP78)[116]. GRP78 is a major ER chaperone that facilitates protein assembly and regulates ER stress signaling[117,118]. PAT-SM6 treatment induces cytotoxicity of MM cells through induction of apoptosis as the main mechanism of action and activation of complement-dependent cytotoxicity as a second hypothesized mechanism. Further, GRP78 is an interesting target in MM due to its sensor function in the UPR activation. PAT-SM6 showed modest clinical activity as a single agent in RRMM, with 33% of patients enrolled in phase Ib trial reaching stable disease[119]. Further trials exploring the combination with existing myeloma drugs are planned.
HEAT SHOCK CHAPERONE PROTEINS
Heat shock proteins (HSP) are molecular chaperones that play a key role in de novo protein synthesis, protein folding, multiprotein complex assembly, and protein sorting[120]. HSP70 and HSP90 participate in chaperone-mediated autophagy, and they support the redirection of misfolded proteins for prompt degradation[121-123]. Compared to normal cells, MM cells are dependent on the HSP chaperone machinery because of the excessive load of misfolded proteins and high levels of DRiPs production[124]. HSPs, therefore, help alleviate proteotoxic stress, prevent terminal UPR, and support MM survival[124].
Among HSP, HSP70 and HSP90 are promising therapeutic targets and the two most widely studied HSPs in cancer, with a central role in supporting the folding of proto-oncogenes[125]. Specifically, both these proteins have been found to stabilize mutant p53 in an inactive form, thereby allowing cancer cells to evade anti-growth signals[126]. In preclinical MM models, HSP70 and HSP90 inhibition result in UPR activation and apoptosis [Figure 2E][127-129]. A combination of HSP90 inhibitors 17-AAG, KW-2478, and retaspimycin with bortezomib showed synergistic MM killing in vitro[130-132]. Several other HSP90 inhibitors such as NVP-HSP990, PU-H71, SNX5422, and NVP-AUY922 have been tested and showed promising preclinical results in MM[133-136]. Although many of these HSP90 inhibitors have completed phase I clinical trial, the narrow therapeutic index and modest clinical significance have hampered further clinical use[137,138]. When tested as monotherapy in phase I clinical trial, only Retaspimycin showed modest anti-MM activity, suggesting that a deeper understanding is necessary to overcome drug resistance in a clinical scenario[139]. Tanespimycin (17-AAG, KOS-953), an HSP90 small molecule inhibitor, proved effective against MM in vitro and has shown encouraging results in phase I/II clinical trials in combination with bortezomib[140-142].
HSP70 inhibitors, such as PER-16, Ver-155008, MAL3-101, have been developed as an alternative therapeutic strategy to HSP90 inhibitors with encouraging preclinical activity[128,129,136,143].
Finally, HSF1, the “master regulator” of the heat shock response, controlling expression of both HSP90 and HSP70, has been investigated as a potential therapeutic target[124]. In preclinical studies, several HSF1 inhibitors (e.g., CCT251236, KRIBB11) were found to induce MM cytotoxicity, with associated induction of the UPR[144].
CONCLUSION
Over the past twenty years, major progress has been made in understanding MM biology. A pathognomonic hallmark of MM is the intense synthesis of Ig, coupled with insufficient proteolytic mechanisms, resulting in pervasive, baseline proteotoxic stress. This intrinsic vulnerability makes MM cells the ideal target of novel therapies designed to disrupt protein synthesis, folding, and degradation. Although disrupting proteostasis via PI has been successful in MM, resistance is inevitable in most patients over time. In this review, we have described potential therapeutic avenues to overcome PI resistance by targeting the protein homeostasis network. Biology-based, clinical use of these agents holds promise to help overcome PI resistance in MM, with the goal of achieving prolonged remission and functional cure for most MM patients [Table 1].
Investigational agents targeting protein homeostasis in MM. This table outlines promising agents targeting protein homeostasis in advanced preclinical or early clinical development in MM, in different phases of a clinical trial
| Drug name | Target molecule/mechanism of action | Status | Study design | Clinical trial identifier/PMID |
| Proteasome inhibitors | ||||
| Marizomib (NPI-0052) | • Targets all three proteasomal subunits • PolyUb protein accumulation • Caspase 8- and 9-mediated apoptosis | Phase I completed | Marizomib + POM + DEX in RRMM | NCT02103335 |
| Phase II completed | Marizomib alone in RRMM | NCT00461045 | ||
| Phase I completed | Marizomib + Vorinostat in RRMM | NCT00667082 | ||
| Phase I completed | Marizomib + DEX | NCT02103335 | ||
| Oprozomib (ONX 0912, PR-047) | • Inhibits PSMB5 (proteasome), LMP7 (immunoproteasome) • PolyUb protein accumulation and terminal UPR induction • Caspase 8- and 9-mediated apoptosis • p53 and p21 upregulation • miR33b upregulation • PIM1 downregulation • Anti-angiogenesis | Phase Ib/II completed | Oprozomib alone | NCT01416428 |
| Phase I/II completed | Oprozomib + DEX + LEN/CPM in NDMM | NCT01881789 | ||
| Phase Ib/II terminated | Oprozomib + DEX in RRMM | NCT01832727 | ||
| Phase I/II completed | Oprozomib + POM + DEX in RRMM | NCT01999335 | ||
| Phase I active | Oprozomib IR or GR formulations + DEX + POM in RRMM | NCT02939183 | ||
| Phase Ib/II completed | Oprozomib + Melphalan + Prednisone in NDMM (transplant-ineligible) | NCT02072863 | ||
| DUBs and ubiquitin receptors inhibitors | ||||
| VLX1570 | • Inhibits proteasome USP14 activity | Phase I/II terminated | VLX1570 + DEX in RRMM | NCT02372240 |
| P5091 | • Inhibits proteasome USP7 activity | Preclinical | N/A | PMID: 22975377[58] |
| B-AP15 | • Blocks USP14 and UCHL5 • Growth arrest via downregulation of CDC25C, CDC2, and cyclin B1 • Induction of caspase-dependent apoptosis • Activation of UPR | Preclinical | N/A | PMID: 24319254[145] |
| RA190 | • Inhibits RPN13 and UCHL37 | Preclinical | N/A | PMID: 27118409[60] |
| XL177A | • Inhibits USP7 | Preclinical | N/A | PMID: 32210275[67] |
| HDAC inhibitors | ||||
| Ricolinostat (ACY-1215) | • Inhibits HDAC6 • Caspase 8 and 9 mediated apoptosis • PolyUb protein accumulation and terminal UPR induction • Aggresome disruption | Phase Ib/II active | Ricolinostat + POM + DEX in RRMM | NCT02400242 |
| Phase I/II active | Ricolinostat + LEN + DEX in RRMM | NCT01583283 | ||
| Phase I/II completed | Ricolinostat + BTZ + DEX in RRMM | NCT01323751 | ||
| Citarinostat (ACY-241) | • Inhibits HDAC6 • Downregulation of MYC and IRF4 • Aggresome disruption | Phase I | Citarinostat + POM + DEX in RRMM | NCT02400242 |
| Autophagy inhibitors | ||||
| Chloroquine | • Inhibits autophagy alkalinizing the lysosomal pH and inhibiting autophagosome and lysosome fusion | Phase I | Chloroquine + BTZ + CPM in RRMM | NCT01438177 |
| 3-MA | • Inhibits autophagy at the level of PI3K Class III | Preclinical | N/A | PMID: 19648108[146] |
| Bafilomycin A1 | • Inhibits autophagosome and lysosome fusion | Preclinical | N/A | PMID: 21174067[93] |
| Unfolded protein response modulators | ||||
| Nelfinavir | • Triggers pro-apoptotic PERK pathway • Inhibition of AKT phosphorylation | Phase I/II active | Nelfinavir + LEN + DEX in progressive MM | NCT01555281 |
| Phase I active | Nelfinavir + BTZ + Metformin in RRMM | NCT03829020 | ||
| Phase II completed | Nelfinavir + BTX + DEX in refractory MM | NCT02188537 | ||
| Sunitinib | • Inhibition of IRE1 activity | Phase II | Sunitinib malate in relapsed MM | NCT00514137 |
| Lovastatin, zolendronic acid, digeranyl bisphosphonate | • Inhibition of isoprenoid biosynthetic pathway and Rab geranyl • Ig light chain accumulation in the ER - activation of UPR | Preclinical | N/A | PMID: 20828814[147] |
| 4μ8C | • Inhibition of XBP1 mRNA splicing | Preclinical | N/A | PMID: 22315414[148] |
| MAL3-101 | • Induction of XBP1 mRNA splicing following inhibition of HSP70 | Preclinical | N/A | PMID: 22750096[149] |
| MKC-3946 | • Inhibition of XBP1 mRNA splicing | Preclinical | N/A | PMID: 14559994[40] |
| STF-083010 | • Inhibition of XBP1 mRNA splicing | Preclinical | N/A | PMID: 21081713[106] |
| GSK2656157 | • Inhibition of PERK and eIF2α phosphorylation, ATF4 translation, and CHOP mRNA expression | Preclinical | N/A | PMID: 23333938[150] |
| CB-5083 | • p97 inhibition - polyUb protein accumulation - UPR induction and apoptosis | Phase I terminated | CB-5083 + DEX | NCT02243917 |
| PAT-SM6 | • Inhibition of GRP78-UPR induction • Complement-dependent cytotoxicity | Phase I completed | PAT-SM6 single agent in RRMM | NCT01727778 |
| Heat Shock Protein (HSP) Inhibitors | ||||
| KW-2478 | • HSP90 inhibitor • Apoptosis | Phase II completed | KW-2478 + BTZ in RRMM | NCT01063907 |
| NVP-AUY922 | • HSP90 inhibitor • Apoptosis • Downregulation of survival pathways | Phase I/II completed | NVP-AUY922 +/- BTZ +/- DEX in RRMM | NCT00708292 |
| IPI-504 | • HSP90 inhibitor • Inhibition of UPR | Phase I completed | IPI-504 in RRMM | NCT00113204 |
| Tanespimycin (17-AAG, KOS-953) | • HSP90 inhibitor • Inhibition of downstream signaling pathways • Induction of UPR | Phase II/III completed | Tanespimycin + BTZ in RRMM | NCT00546780 |
| SNX-5422 | • HSP90 inhibitor • Apoptosis | Phase I completed | SNX-5422 in RRMM | NCT00595686 |
| NVP-HSP990 | • HSP90 inhibitor • Apoptosis • Cell cycle arrest | Preclinical | N/A | PMID: 22309072[133] |
| NVP-BEP800 | • HSP90 inhibitor • Apoptosis • Inhibition of STAT3, ERK, AKT pathways | Preclinical | N/A | PMID: 19686236[151] |
| SNX-2112 | • HSP90 inhibitor • Cell cycle arrest and cytotoxicity • Inhibition of ERK, AKT pathways • Inhibition of angiogenesis and osteoclastogenesis | Preclinical | N/A | PMID: 18948577[152] |
| MAL3-101 | • Inhibition of HSP70 • Induction of XBP1 mRNA splicing • Apoptosis and cell cycle arrest | Preclinical | N/A | PMID: 22750096[149] |
| PU-H71 | • Inhibition of HSP90 • Apoptosis and cell cycle arrest, UPR and apoptosis | Preclinical | N/A | PMID: 20977755[134] |
| TAS-116 | • HSP90 inhibitor • Induction of apoptosis • Disruption of ERK, AKT pathways | Preclinical | N/A | PMID: 25306900[153] |
| Selective degradors | ||||
| Phthalimide conjugated degraders | • Bind to CRBN E3 complex on one hand and to specific protein targets on the other to elicit proteasome-mediated degradation | Preclinical | N/A | PMID: 25999370[154] |
Only through a deep understanding of the fundamental mechanisms of protein homeostasis, novel targets can be identified to overcome PI resistance and improve patient outcome, resulting in long-term control, if not cure, for most MM patients.
DECLARATIONS
Authors’ contributionsWrote and edited the manuscript: Moscvin M, Ho M, Bianchi G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006;107:4907-16.
3. Cenci S, Mezghrani A, Cascio P, et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J 2006;25:1104-13.
4. Sontag EM, Vonk WIM, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol 2014;26:139-46.
5. Benbrook DM, Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp Oncol 2012;34:286-97.
6. Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell 2020;77:446-60.
7. Richardson PG, Sonneveld P, Schuster MW, et al. Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005;352:2487-98.
8. Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001;61:3071-6.
9. San Miguel JF, Schlag R, Khuageva NK, et al; VISTA Trial Investigators. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906-17.
10. Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 2004;15:3-16.
11. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081-6.
12. De Los Rios P, Ben-Zvi A, Slutsky O, Azem A, Goloubinoff P. Hsp70 chaperones accelerate protein translocation and the unfolding of stable protein aggregates by entropic pulling. Proc Natl Acad Sci U S A 2006;103:6166-71.
13. Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006;440:551-5.
14. Bianchi G, Oliva L, Cascio P, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009;113:3040-9.
15. Bianchi G, Richardson PG, Anderson KC. Promising therapies in multiple myeloma. Blood 2015;126:300-10.
16. Yewdell JW, Schubert U, Bennink JR. At the crossroads of cell biology and immunology: DRiPs and other sources of peptide ligands for MHC class I molecules. J Cell Sci 2001;114:845-51.
17. Cenci S, Sitia R. Managing and exploiting stress in the antibody factory. FEBS Lett 2007;581:3652-7.
18. Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000;404:770-4.
19. Goldberg AL. Functions of the proteasome: the lysis at the end of the tunnel. Science 1995;268:522-3.
21. Orlowski RZ. Why proteasome inhibitors cannot ERADicate multiple myeloma. Cancer Cell 2013;24:275-7.
22. Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009;137:133-45.
23. Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol 2011;12:605-20.
24. Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 1996;65:801-47.
25. Bhattacharyya S, Yu H, Mim C, Matouschek A. Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol 2014;15:122-33.
26. Ferrington DA, Gregerson DS. . Immunoproteasomes. The proteasomal system in aging and disease. Elsevier; 2012. p. 75-112.
27. Kimura H, Caturegli P, Takahashi M, Suzuki K. New insights into the function of the immunoproteasome in immune and nonimmune cells. J Immunol Res 2015;2015:541984.
28. Kuhn DJ, Orlowski RZ, Bjorklund CC. Second generation proteasome inhibitors: carfilzomib and immunoproteasome-specific inhibitors (IPSIs). Curr Cancer Drug Targets 2011;11:285-95.
29. Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M. The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol 2015;33:S74-9.
30. Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001;8:739-58.
31. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004;5:417-21.
32. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27-38.
33. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142-52.
34. Bianchi G, Ghobrial IM. Molecular mechanisms of effectiveness of novel therapies in multiple myeloma. Leuk Lymphoma 2013;54:229-41.
35. Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003;101:1530-4.
36. Hideshima T, Ikeda H, Chauhan D, et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009;114:1046-52.
37. Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood 2003;101:2377-80.
38. Fennell DA, Chacko A, Mutti L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2008;27:1189-97.
39. Hideshima T, Hayashi T, Chauhan D, Akiyama M, Richardson P, Anderson K. Biologic sequelae of c-Jun NH(2)-terminal kinase (JNK) activation in multiple myeloma cell lines. Oncogene 2003;22:8797-801.
40. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448-59.
41. Meister S, Schubert U, Neubert K, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res 2007;67:1783-92.
42. Sha Z, Goldberg AL. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 2014;24:1573-83.
43. Vangala JR, Sotzny F, Krüger E, Deshaies RJ, Radhakrishnan SK. Nrf1 can be processed and activated in a proteasome-independent manner. Curr Biol 2016;26:R834-5.
44. Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 2010;38:17-28.
45. Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014;3:e01856.
46. Koizumi S, Irie T, Hirayama S, et al. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016;5:e18357.
47. Tomlin FM, Gerling-Driessen UIM, Liu YC, et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent Sci 2017;3:1143-55.
48. Chen T, Ho M, Briere J, et al. Multiple myeloma cells depend on the DDI2/NRF1-mediated proteasome stress response for survival. Blood Adv 2021:bloodadvances.2020003820.
49. Kuhn DJ, Chen Q, Voorhees PM, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007;110:3281-90.
50. Ghobrial IM, Vij R, Siegel D, et al. A phase Ib/II study of oprozomib in patients with advanced multiple myeloma and waldenström macroglobulinemia. Clin Cancer Res 2019;25:4907-16.
51. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol 2014;15:1503-12.
52. Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005;8:407-19.
53. Weyburne ES, Wilkins OM, Sha Z, et al. Inhibition of the proteasome β2 site sensitizes triple-negative breast cancer cells to β5 inhibitors and suppresses Nrf1 activation. Cell Chem Biol 2017;24:218-30.
54. Richardson PG, Zimmerman TM, Hofmeister CC, et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 part 1. Blood 2016;127:2693-700.
55. Crawford LJ, Irvine AE. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev 2013;27:297-304.
56. Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Invest 2014;124:6-12.
57. D'Arcy P, Brnjic S, Olofsson MH, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011;17:1636-40.
58. Chauhan D, Tian Z, Nicholson B, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012;22:345-58.
59. Besche HC, Sha Z, Kukushkin NV, et al. Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J 2014;33:1159-76.
60. Song Y, Ray A, Li S, et al. Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 2016;30:1877-86.
61. Anchoori RK, Karanam B, Peng S, et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 2013;24:791-805.
62. Song Y, Park PMC, Wu L, et al. Development and preclinical validation of a novel covalent ubiquitin receptor Rpn13 degrader in multiple myeloma. Leukemia 2019;33:2685-94.
63. Rowinsky EK, Paner A, Berdeja JG, et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Invest New Drugs 2020;38:1448-53.
64. Kategaya L, Di Lello P, Rougé L, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017;550:534-8.
65. Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017;550:481-6.
66. Ray A, Ting DU, Song Y, et al. Blockade of deubiquitylating enzyme USP7 in plasmacytoid dendritic cells stimulates anti-myeloma immunity. Blood 202;136:43.
67. Schauer NJ, Liu X, Magin RS, et al. Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci Rep 2020;10:5324.
68. Ciechanover A, Finley D, Varshavsky A. The ubiquitin-mediated proteolytic pathway and mechanisms of energy-dependent intracellular protein degradation. J Cell Biochem 1984;24:27-53.
69. Hyer ML, Milhollen MA, Ciavarri J, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 2018;24:186-93.
70. Zhuang J, Shirazi F, Singh RK, et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019;133:1572-84.
71. Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol 2016;17:626-42.
72. Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol 2015;16:30-44.
73. Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol 2005;23:4776-89.
74. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995;373:81-3.
75. Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011;118:4771-9.
76. Krönke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014;343:301-5.
77. Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014;343:305-9.
78. McMillin DW, Jacobs HM, Delmore JE, et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol Cancer Ther 2012;11:942-51.
79. Shah JJ, Jakubowiak AJ, O'Connor OA, et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 2016;22:34-43.
80. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003;115:727-38.
81. Harada T, Hideshima T, Anderson KC. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int J Hematol 2016;104:300-9.
82. Catley L, Weisberg E, Kiziltepe T, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006;108:3441-9.
83. Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012;119:2579-89.
84. San-miguel JF, Hungria VTM, Yoon S, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014;15:1195-206.
85. Dimopoulos M, Siegel DS, Lonial S, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol 2013;14:1129-40.
86. Yee AJ, Bensinger WI, Supko JG, et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol 2016;17:1569-78.
87. Vogl DT, Raje N, Jagannath S, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res 2017;23:3307-15.
88. Ho M, Chen T, Liu J, et al. Targeting histone deacetylase 3 (HDAC3) in the bone marrow microenvironment inhibits multiple myeloma proliferation by modulating exosomes and IL-6 trans-signaling. Leukemia 2020;34:196-209.
90. Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 2005;280:40282-92.
91. Hoang B, Benavides A, Shi Y, Frost P, Lichtenstein A. Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 2009;8:1974-84.
92. Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett 2009;456:15-9.
93. Kawaguchi T, Miyazawa K, Moriya S, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol 2011;38:643-54.
94. Milan E, Fabbri M, Cenci S. Autophagy in plasma cell ontogeny and malignancy. J Clin Immunol 2016;36 Suppl 1:18-24.
95. Milan E, Perini T, Resnati M, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015;11:1161-78.
96. Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007;171:513-24.
97. Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007;14:230-9.
98. Lu Y, Wang Y, Xu H, Shi C, Jin F, Li W. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci 2018;109:2706-16.
99. Baranowska K, Misund K, Starheim KK, et al. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016;7:70845-56.
100. Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014;10:1380-90.
101. Woo CW, Cui D, Arellano J, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol 2009;11:1473-80.
102. Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997;90:1031-9.
103. Carrasco DR, Sukhdeo K, Protopopova M, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007;11:349-60.
104. Leung-Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013;24:289-304.
105. Ali MM, Bagratuni T, Davenport EL, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J 2011;30:894-905.
106. Papandreou I, Denko NC, Olson M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011;117:1311-4.
107. Mimura N, Fulciniti M, Gorgun G, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012;119:5772-81.
108. Bagratuni T, Patseas D, Mavrianou-Koutsoukou N, et al. Characterization of a PERK kinase inhibitor with anti-myeloma activity. Cancers (Basel) 2020;12:2864.
109. Kraus M, Bader J, Overkleeft H, Driessen C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J 2013;3:e103.
110. Besse A, Besse L, Stolze SC, et al. Nelfinavir blocks export of newly synthesized protein from the ER and interacts with ER-resident and mitochondrial proteins in an activity-dependent fashion. Blood 2017;130:3074.
111. Driessen C, Kraus M, Joerger M, et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: a phase I trial (SAKK 65/08). Haematologica 2016;101:346-55.
112. Driessen C, Müller R, Novak U, et al. Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma. Blood 2018;132:2097-100.
113. Hebert DN, Bernasconi R, Molinari M. ERAD substrates: which way out? Semin Cell Dev Biol 2010;21:526-32.
114. Auner HW, Moody AM, Ward TH, et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS One 2013;8:e74415.
115. Le Moigne R, Aftab BT, Djakovic S, et al. The p97 inhibitor CB-5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther 2017;16:2375-86.
116. Rasche L, Duell J, Morgner C, et al. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS One 2013;8:e63414.
117. Auner HW, Cenci S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol 2015;168:14-25.
118. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 2014;14:263-76.
119. Rasche L, Menoret E, Dubljevic V, et al. A GRP78-directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res 2016;22:4341-9.
121. Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 1997;137:825-34.
122. Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989;246:382-5.
123. Wang B, Chen Z, Yu F, et al. Hsp90 regulates autophagy and plays a role in cancer therapy. Tumour Biol 2016;37:1-6.
124. Zhang L, Fok JH, Davies FE. Heat shock proteins in multiple myeloma. Oncotarget 2014;5:1132-48.
125. Garcia-carbonero R, Carnero A, Paz-ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 2013;14:e358-69.
127. Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol 2002;22:8506-13.
128. Chatterjee M, Andrulis M, Stühmer T, et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013;98:1132-41.
129. Braunstein MJ, Scott SS, Scott CM, et al. Antimyeloma effects of the heat shock protein 70 molecular chaperone inhibitor MAL3-101. J Oncol 2011;2011:232037.
130. Ishii T, Seike T, Nakashima T, et al. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J 2012;2:e68.
131. Sydor JR, Normant E, Pien CS, et al. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A 2006;103:17408-13.
132. Mimnaugh EG, Xu W, Vos M, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther 2004;3:551-66.
133. Lamottke B, Kaiser M, Mieth M, et al. The novel, orally bioavailable HSP90 inhibitor NVP-HSP990 induces cell cycle arrest and apoptosis in multiple myeloma cells and acts synergistically with melphalan by increased cleavage of caspases. Eur J Haematol 2012;88:406-15.
134. Usmani SZ, Bona RD, Chiosis G, Li Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J Hematol Oncol 2010;3:40.
135. Stühmer T, Zöllinger A, Siegmund D, et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008;22:1604-12.
136. Bailey CK, Budina-Kolomets A, Murphy ME, Nefedova Y. Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol Ther 2015;16:1422-6.
137. Seggewiss-Bernhardt R, Bargou RC, Goh YT, et al. Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer 2015;121:2185-92.
138. Richardson PG, Chanan-Khan AA, Alsina M, et al. Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase 1 dose-escalation study. Br J Haematol 2010;150:438-45.
139. Siegel D, Jagannath S, Vesole DH, et al. A phase 1 study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 2011;52:2308-15.
140. Altun M, Galardy PJ, Shringarpure R, et al. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res 2005;65:7896-901.
141. Jain S, Diefenbach C, Zain J, O'Connor OA. Emerging role of carfilzomib in treatment of relapsed and refractory lymphoid neoplasms and multiple myeloma. Core Evid 2011;6:43-57.
142. Nawrocki ST, Carew JS, Maclean KH, et al. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood 2008;112:2917-26.
143. Zhang L, Fok JJ, Mirabella F, et al. Hsp70 inhibition induces myeloma cell death via the intracellular accumulation of immunoglobulin and the generation of proteotoxic stress. Cancer Lett 2013;339:49-59.
144. Fok JHL, Hedayat S, Zhang L, et al. HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin Cancer Res 2018;24:2395-407.
145. Tian Z, D'Arcy P, Wang X, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014;123:706-16.
146. Shanmugam M, McBrayer SK, Qian J, et al. Targeting glucose consumption and autophagy in myeloma with the novel nucleoside analogue 8-aminoadenosine. J Biol Chem 2009;284:26816-30.
147. Holstein SA, Hohl RJ. Isoprenoid biosynthetic pathway inhibition disrupts monoclonal protein secretion and induces the unfolded protein response pathway in multiple myeloma cells. Leuk Res 2011;35:551-9.
148. Cross BC, Bond PJ, Sadowski PG, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 2012;109:E869-78.
149. Goloudina AR, Demidov ON, Garrido C. Inhibition of HSP70: a challenging anti-cancer strategy. Cancer Lett 2012;325:117-24.
150. Atkins C, Liu Q, Minthorn E, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 2013;73:1993-2002.
151. Stühmer T, Chatterjee M, Grella E, et al. Anti-myeloma activity of the novel 2-aminothienopyrimidine Hsp90 inhibitor NVP-BEP800. Br J Haematol 2009;147:319-27.
152. Okawa Y, Hideshima T, Steed P, et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood 2009;113:846-55.
153. Suzuki R, Hideshima T, Mimura N, et al. Anti-tumor activities of selective HSP90α/β inhibitor, TAS-116, in combination with bortezomib in multiple myeloma. Leukemia 2015;29:510-4.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Moscvin M, Ho M, Bianchi G. Overcoming drug resistance by targeting protein homeostasis in multiple myeloma. Cancer Drug Resist 2021;4:1028-46. http://dx.doi.org/10.20517/cdr.2021.93
AMA Style
Moscvin M, Ho M, Bianchi G. Overcoming drug resistance by targeting protein homeostasis in multiple myeloma. Cancer Drug Resistance. 2021; 4(4): 1028-46. http://dx.doi.org/10.20517/cdr.2021.93
Chicago/Turabian Style
Moscvin, Maria, Matthew Ho, Giada Bianchi. 2021. "Overcoming drug resistance by targeting protein homeostasis in multiple myeloma" Cancer Drug Resistance. 4, no.4: 1028-46. http://dx.doi.org/10.20517/cdr.2021.93
ACS Style
Moscvin, M.; Ho M.; Bianchi G. Overcoming drug resistance by targeting protein homeostasis in multiple myeloma. Cancer Drug Resist. 2021, 4, 1028-46. http://dx.doi.org/10.20517/cdr.2021.93
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 20 clicks
Cite This Article 20 clicks

 Like This Article 2
likes
Like This Article 2
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.