
Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer

Abstract
The cancer stem cell (CSC) state and epithelial-mesenchymal transition (EMT) activation are tightly interconnected. Cancer cells that acquire the EMT/CSC phenotype are equipped with adaptive metabolic changes to maintain low reactive oxygen species levels and stemness, enhanced drug transporters, anti-apoptotic machinery and DNA repair system. Factors present in the tumor microenvironment such as hypoxia and the communication with non-cancer stromal cells also promote cancer cells to enter the EMT/CSC state and display related resistance. ATP, particularly the high levels of intratumoral extracellular ATP functioning through both signaling pathways and ATP internalization, induces and regulates EMT and CSC. The three of them work together to enhance drug resistance. New findings in each of these factors will help us explore deeper into mechanisms of drug resistance and suggest new resistance-associated markers and therapeutic targets.
Keywords
INTRODUCTION
Drug resistance in cancer is a very complicated process with numerous participating components. These include cancer cells in the tumor as well as stromal cells in the tumor microenvironment (TME) and their communications and interactions. Both cancer cells and TME are evolving and ever changing, making drug resistance a dynamic process. Within TME and cancer cells, proteins and small molecules are also involved in drug resistance, working as functional factors to regulate drug resistance. Our recent studies have shown that extracellular ATP (eATP) in TME is found in concentrations 103 to 104 times higher than eATP found in normal tissues[1-5]. The greatly elevated concentration of eATP plays much larger and diverse roles than previously realized in augmenting drug resistance in cancer cells through internalization[6-8], promoting cell survival[6-9], accelerating ATP-binding cassette (ABC) transporter activities[10,11], inducing epithelial-mesenchymal transition (EMT)[12] and possibly the formation of cancer stem cells (CSC). With these new advancements in mind, in this review, we summarize recent findings in these fields in an attempt to generate a clearer and more cohesive picture for CSC, EMT, ATP, and their distinctive and interactive relationships for creating increased drug resistance as a phenotype for cancer cells. Better anticancer therapies are likely to be developed by improved understanding of these factors and their roles in drug resistance.
CANCER STEM CELLS
CSCs refer to a subset of tumor cells that have an unlimited self-renewal capacity and an intense tumorigenic potential (stemness) to differentiate into their non-tumorigenic progenies which comprise the rest of the tumor bulk[13,14]. Among tumor cells, CSCs are a subpopulation that constitutes varied percentages of the total tumor cells, and this fraction can dramatically increase upon anticancer therapy correlating with therapy resistance and tumor rebound[13,15]. CSCs are believed to reside stochastically in a tumor but be predominantly induced in hypoxic, low pH, and fewer nutrient regions such as the tumor niche[16]. One study discovered that cancer cells can reprogram into CSCs when receiving DNA damage signals, suggesting the potential risk of genotoxic therapies in inducing CSC and tumor relapse[17]. Substantial studies have shown that CSCs have enhanced resistance to conventional radio-/chemo-therapy and targeted therapies and play essential roles in cancer relapse and metastasis[14,18]. Tumors with a high CSC signature tend to have a poorer prognosis compared with those with decreased CSC populations[19]. CSCs have the nature to be slow-cycling or quiescent, spending the majority of time in G0 and thus can circumvent the chemotherapy or radiotherapy targeting the rapidly dividing tumor cells[20]. Other cellular mechanisms of therapy resistance in CSCs will be discussed in the following sections.
The intratumoral heterogeneity and its implication in therapy resistance (tumor recurrence) was explained by two models: clonal evolution (CE or stochastic) and CSC (hierarchy). In the CE model, tumor cells all have equivalent potential to accumulate mutations over time to gain resistance-related traits. The selection of the traits leads to increased tumor heterogeneity and disease progression[21]. On the contrary, in the CSC model, tumors are initiated with a heterogeneous mixture of genetically distinct subclones of CSCs that each gives rise to their fast-dividing progeny cancer cells and ultimately leads to the functional and phenotypical heterogeneity of the tumor[22]. According to the CSC model, CSCs contribute to drug resistance and tumor progression by the intrinsic resistance possessed by CSCs combined with genetic changes that occur during therapy[22]. The main target of conventional cytotoxic therapies are the rapidly-dividing and apoptosis-sensitive cancer cells, but resistant CSCs can survive, enrich in number, accumulate oncogenic genetic changes during therapy and finally expand even under drug treatment, functioning as the precursors of new resistant tumor masses and ultimately leading to clinical relapse[22-24]. More recently, several studies questioned the unidirectional hierarchic CSC model by showing cancer cells are plastic[25]. A famous example is that cell population isolated from breast cancer cell lines displayed stem-, basal-, or luminal-like phenotypes[26]. Each phenotype was capable to convert into the other two phenotypes and over time produced all three phenotypes with the same proportions of cell types in the original cell line, but only stem-like cells could form tumors upon xenotransplantation[26]. One recent study showed that glioblastoma cells expressing CSC markers represented a plastic state that can be adopted by most of the cells in response to various microenvironmental cues, which may contribute to enhanced tumorigenic potential[27]. With the CSC plasticity, a more fluid hierarchic CSC model is now accepted, where cancer cells can undergo a dynamic transition from a non-CSC to CSC phenotype and vice versa to adapt to different stimuli in TME[25,28]. The activation of EMT serves as one of the mechanisms for switching phenotypes.
ABC transporters are functional markers of CSCs
Forty-nine ABC transporters have been identified in the human genome and are classified in seven subfamilies (ABC A-G) based on amino acid sequence similarities and protein structural organization[29]. Most ABC transporters are membrane-bound and function as active transporters that catalyze the transport of diverse substrates, including anticancer agents across the plasma and intracellular membrane, using ATP-provided energy. Three major ABC transporters are frequently implicated in multidrug resistance (MDR), namely, P-glycoprotein (P-gp/ABCB1), breast cancer resistance protein (BCRP/ABCG2), and multidrug resistance-associated proteins (MRPs/ABCCs). These ABC transporters can mediate resistance to a wide range of chemotherapeutics, EGF receptor-tyrosine kinase inhibitors (EGFR-TKIs) and other targeted drugs[30], thereby predicting recurrence and poor survival[31,32]. Moreover, co-expression of multiple ABC transporters correlates with decreased overall survival[33]. The robust cell-surface expression of ABC transporters, especially P-gp and BCRP, is recognized as a key feature of CSCs[13]. Patrawala et al.[34] first characterized “side population” cells from human cancer cells purified by flow cytometry-based technique isolating for the side population of cells which exclude dye via ABC transporters. These side population cells possessed stem cell properties mediated by BCRP[34]. As of now, several studies have demonstrated P-gp and BCRP play a key role in maintaining cell viability as well as stemness features[35-38]. On the other hand, the expression of ABC transporters is under the regulation of multiple signaling pathways associated with CSC phenotypes and EMT activation, contributing to CSC and EMT-related resistance. This will be described in later sections. Although potentially life-threatening adverse events remain a major obstacle for the first-, second- and third-generation of small molecule ABC transporter inhibitors, therapies combating ABC transporter-mediated drug efflux continue to be developed[39]. One advanced strategy is to use TKIs as ABC transporter inhibitors, because most TKIs (e.g., imatinib, erlotinib, nilotinib and lapatinib) can target more than one ABC transporter and such a multi-target approach might be promising[39]. Another strategy is to deplete intracellular ATP molecules, which cuts down the energy supply of ABC transporters and reduces MDR. Several approaches of this strategy will be discussed in section V.
ALDH-associated drug resistance
The ALDH family of proteins are cytosolic enzymes that scavenge intracellular highly toxic aldehydes to carboxylic acids. ALDH activity measured by Aldefluor assay is a hallmark of CSCs. Though these proteins have minimal metabolic activity in normal cancer cells, quiescent CSCs exhibit high ALDH activity to catalyze drug metabolism and turn the drugs into less toxic agents[13]. Among 19 members of ALDHs, aldehyde dehydrogenase 1 (ALDH1) is considered a CSC biomarker, which indicates stemness and resistance to chemotherapy[40,41] and predicts poor clinical outcomes in breast and prostate cancer[42]. High expression of ALDH in CSCs have been characterized to play key roles in resistance to chemotherapeutics (cisplatin, doxorubicin, etoposide, and fluorouracil), TKIs (Gefitinib and Erlotinib) and radiation in various cancer cell lines including breast, lung, gastric, and head/neck cancers[42]. High ALDH activity also correlates with EMT, tumor invasion and metastasis. Ovarian cancer stem-like cells sorted by high expression of ALDH are prone to activate stemness, invasiveness, EMT and anti-apoptotic properties compared to those with low ALDH[43]. Mechanistically, ALDH1 and aldehyde dehydrogenase 3 family member A1 (ALDH3A1) can act as a detoxifying enzyme and protect cells from increased reactive oxygen species (ROS) by direct scavenging of radiation-induced free radicals or by producing the antioxidant reduced nicotinamide adenine dinucleotide phosphate (NADPH), suggesting a pivotal role in resistance to radiation and chemotherapy[44]. High ALDH1 family member A1 (ALDH1A1) led to nuclear factor-erythroid 2-related factor 2 (Nrf2) activation via p62-associated pathway in ALDH1-high CSC-like ovarian cancer cells. Nrf2 activation in these cells contributed to CSC-like properties including high levels of P-gp and BCRP and chemo resistance[45]. Additionally, enhanced ALDH activity is associated with the activation of the pro-survival signaling pathways such as transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), Notch, and the mechanistic target of rapamycin (mTOR)[42]. For example, ALDH activity was regulated by another putative CSC marker Nanog, through the Notch1/Akt signaling pathway in breast cancer stem cells[46]. Additionally, the enhanced ALDH activity led to cellular radio-resistance by simulating double-strand break repair[46]. Inhibitors directly targeting isoforms of ALDH, such as ALDH1A1 and ALDH3A1 have been developed, which hold promise for eliminating CSCs particularly in gynecologic cancers[47]. Challenges of using ALDH inhibitors include limited in vivo activity and the need of using ALDH inhibitors in combination with other anticancer therapies[47].
Oxidative stress-associated resistance
ROS can induce therapy resistance at both high and low levels[13,48]. Compared to more differentiated cells, CSCs have a robust ROS scavenging system and maintain ROS at low levels, which may be attributable to an increase in ROS scavenging molecules or/and enhanced mitochondrial respiratory capacity[49-51]. On the one hand, the low ROS levels serve to prevent cell death from ROS overloading and maintain stemness, tumorigenic capacity and tumor radio- and chemo-resistance[48,50-53]. On the other hand, elevated ROS levels emerge as a potential factor to induce EMT particularly under hypoxia, and such induction of EMT can contribute to the acquisition of stemness features[48,54]. Whereas excessive oxidative stress can cause damage to DNA, proteins and lipids and lead to apoptosis[55].
Glutathione (GSH) is vitally important for the maintenance of cellular redox homeostasis and is important for various signaling processes in cell apoptosis and proliferation[56]. Survival through radio- and chemo-therapy may trigger the synthesis of GSH at high amounts, which can enhance cell survival[19] as well as function as a signaling molecule to induce CSC phenotype[57], thus serving as one way to integrate ROS with CSC signaling[57]. Interestingly, one study reported that exogenous administration of GSH can increase intracellular GSH levels and induce resistance to cisplatin by suppressing apoptosis[58]. GSH conjugation is the first step of the cell detoxifying mercapturic acid pathway, in which Glutathione S-transferases (GSTs) binds to GSH and cytotoxic compounds (include certain chemotherapeutics), forming conjugates readily excreted via membrane transporters[56]. Several members of the MRP transporter family are responsible for this excretion, including MRP1, MRP2, MRP3, MRP4, MRP5, MRP7[56]. Moreover, ABC transporters such as MRP1 are often upregulated in drug-resistant cancer cells along with GSH or GSTs[59,60], whereas some ABC transporters like ABCB6 can regulate ROS via porphyrin biosynthesis and diminish the ROS-inductive effect of chemotherapeutics[61]. Notably, the overexpression of GSTs and MRP1 has been recognized as one underlying mechanism for therapeutic resistance in various cancers (breast, colorectal, lung, ovarian, pancreatic), with overexpressed GST pi 1, a member of the GST family, showing intense correlation with CSCs-related resistance and being considered as a biomarker for cancer[56,62-65].
Metabolic reprograming, CSC plasticity, and hypoxia
Cancer cells display a rapid metabolic adaptation in response to low-nutrient and hypoxia in the surrounding tumor microenvironment. This adaptation, namely “metabolic reprogramming”, refers to the shift of cellular bioenergetics, a new hallmark of cancer[66] and is utilized by CSCs[67]. CSCs can fulfill their energy demands by shifting their phenotypes[13]. The CSC phenotype is believed to be a plastic state that can be adopted by most cancer cells[27] and is capable of transitioning between a stem, mesenchymal-like phenotype and a non-stem, epithelial-like one[68]. Different CSC phenotypes can correlate with metabolic differences and add to the heterogeneity of cancer cells, and thereby ease their adaptation to TME and limit the efficacy of conventional therapies[27,69]. Activation of EMT in non-CSCs is a predominant driver to convert them into the CSC state[70]. EMT induces metabolic reprogramming to accommodate cellular changes during EMT, and imparts cancer cells with the phenotypic and metabolic plasticity of CSC, which is required for drug resistance.
Hypoxia has been recognized as a key factor in TME that contributes to the plasticity and heterogeneity of CSCs[71]. Cancer cells localized in hypoxic regions tend to display CSC-like phenotypes and are more invasive than cells in oxygenated regions within tumors[72]. The non-CSCs grow and proliferate rapidly, which leads to hypoxia in the nearby milieu[13]. The poor oxygen availability may reasonably shift the metabolism from disadvantaged oxidative pathways to glycolysis[73].
Chemoresistant pancreatic cancer cells rely on glycolysis to maintain low ROS levels and consequent CSC and EMT phenotypes[74]. This correlation of hypoxia and CSC features have been demonstrated in various tumor types[73]. One central mediator of this adaptive metabolic shift is hypoxia-inducible factor 1
Furthermore, the metabolic reprogramming of CSCs is a flexible mechanism which allows CSCs to either rely on glycolysis or mitochondrial OXPHOS, depending on the tumor type and the adaptation to a particular condition (such as hypoxia) within the TME[13]. The metabolic switch to glycolysis was demonstrated in CD44+CD24lowEPCAM+ CSCs in breast cancer, radioresistant CSCs in nasopharyngeal carcinoma, and CD133+CD49f+ CSCs in hepatocellular carcinoma. The underlying mechanisms include Hif-1α-mediated enhanced glycolysis, STAT3-mediated induction of aerobic glycolysis[81], and c-Myc-driven glycolytic programme[82]. In contrast, CSCs in other cancer types adopt OXPHOS as the preferred process for energy production. This notion is exemplified in CD133+ CSCs in glioblastoma and pancreatic adenocarcinoma, low reactive oxygen species quiescent stem cells in leukemia, and side population of lung and breast cancer[83]. One study demonstrated diverse oncogene-addicted cancer cells and CSCs (lung, melanoma, leukemia, breast) showed selective reliance on OXPHOS and resistance to EGFR-TKIs[84]. Invasive ovarian cancer and ovarian CSCs were also recently found to prefer OXPHOS, and chemoresistant ovarian cancer cells exhibited adaptability to switch between OXPHOS and glycolysis under different stress[85]. In addition to glycolytic and OXPHOS phenotypes, CSCs can also rely on mitochondrial fatty acid oxidation for ATP and NADPH generation[83].
Given the crucial role of hypoxia in CSC metabolism and plasticity, targeting Hif-1α and downstream Hif signaling pathways to overcome hypoxia is a reasonable approach to reduce CSCs. Strategies inhibiting Hif-1α has been well summarized by Onnis et al.[86]. Inhibition of glycolysis also serves as a promising approach in combination with chemotherapy, which was shown to effectively eradicate chemoresistant glioblastoma CSCs which reside in their hypoxic niches and rely on glycolysis for ATP generation[87]. Additionally, inhibition of glycolysis by dichloroacetate may reverse the metabolic shift to OXPHOS, leading to increased ROS and promoted apoptosis in CSCs of several cancer types[72].
Acidic TME induces CSC phenotypes and chemoresistance
Acidosis, meaning extracellular pH ranging from 6.2 to 6.8, is a hallmark of TME in solid tumors[88]. Tumor acidosis is a consequence of the exacerbated aerobic glycolysis and hydration of CO2 derived from mitochondrial respiration[89]. Acidosis can activate EMT by modulating the expression of EMT-related proteins[90]. The adaptive CSC-like phenotypes are likely triggered and maintained by the EMT activation[91,92]. When exposed to acidic conditions, cancer cells have been reported to show reduced proliferation and even quiescence, and increased resistance to apoptosis[93]. Acidosis may regulate CSC-associated gene expression via hypoxia-inducible factor 2α (Hif-2α)[91], which is considered as a master regulator of gene expression and a marker of CSC in glioma[94]. Both the abundance and transcription activity of Hif-2α was documented to be increased by acute acidosis[91]. Acidosis is related to the metabolic shift towards a dependence on mitochondrial OXPHOS, which is an adaptative mechanism for efficient ATP generation[95], particularly in therapy-resistant CSCs of certain types of cancers as described above. Altered lipid metabolism is another result of acidosis and is correlated with chemoresistance. For instance, acidosis activated partial EMT and increased ATP generation through enhanced fatty acid uptake in various cancer types[96]. Additionally, under acidosis, increased lipid desaturation was found to be essential to the maintenance of stemness in ovarian cancer cells[97]. Acidosis can also contribute to resistance by enhancing drug efflux activities of P-gp[98] and BCRP[99]. Finally, cancer cells undergo autophagy as a survival mechanism when stimulated by acute acidosis[91], and this increased autophagic flux is associated with stemness, quiescence, and chemotherapy resistance in a variety of cancers[100].
The adaptive metabolism under tumor acidosis sustains resistance-related CSC phenotypes, which can be targeted by inhibiting key enzymes/transporters in the metabolic pathway. Moreover, many pH sensitive nanoparticles have been developed with the goal to selectively deliver anticancer drugs to TME[101]. A combination of such a delivery system and siRNA of key enzymes/transporters (e.g., ASCT2) might significantly inhibit tumor growth and reduce drug resistance[102].
Autophagy in CSCs and resistance
Autophagy is an arising player in the maintenance of CSC stemness[100]. Autophagy can be stimulated by the same range of stressors in CSC and EMT induction, including oxidative stress, hypoxia, nutrient-deprivation, as well as genotoxic therapies[48,100]. Many EMT inducers like TGF-β and CSC related transcription factors such as sex determining region Y-box 2 (SOX2), and Nanog are also capable of inducing autophagy[100]. On the other hand, CSCs show dependence on autophagy. Autophagy promotes the expression of stem cell markers CD44 and mesenchymal markers vimentin, as well as in vivo tumorigenesis and CSC related drug resistance[100]. Autophagy acts as a double-edged sword for resistance to therapies: it protects cancer cells from stressors by recycling metabolites degraded from damaged organelles and mediates MDR, thus protecting cancer cells from anticancer drugs. On the contrary, it may also induce autophagic cell death and thus kill MDR cancer cells with deficient apoptosis pathways[30,100].
Forkhead box (FOX) O3a, a transcription factor suppressing protective autophagy particularly in CSCs, was identified to form a negative feedback loop in which its turnover was controlled by autophagy. Upon autophagy inhibition, FOXO3a protein increased and accumulated in the nucleus, and transcriptionally activated the pro-apoptotic gene, p53 upregulated modulator of apoptosis, leading to p53-independent apoptosis and consequently sensitizing colorectal cancer (CRC) cells to chemotherapy drugs such as doxorubicin and etoposide[103]. This study suggested that a homeostasis of autophagy is maintained by FOXO3a and is crucial to cancer cell survival. In contradiction to what is described above, FOXO3a was reported to play a promoting role in both hypoxia-induced and sorafenib-induced autophagy and sorafenib resistance in hepatocellular carcinoma, while the knockout of FOXO3a significantly inhibited autophagy and restored sorafenib sensitivity[104].
Nrf2 is an important activator of protective autophagy. In response to chemotherapy-induced oxidative stress, the accumulation of Nrf2 transcriptionally upregulates genes involved in the activation of protective autophagy such as an autophagy cargo receptor p62. Such autophagy eliminates cellular cytotoxic products and thus promotes tumor survival and chemoresistance[105]. High CD44 expression in breast cancer stem-like cells led to p62-associated Nrf2 elevation, possibly due to the activation of autophagy. Nrf2 activation consequently contributed to the promotion of tumor initiating capability, migration and invasion, and anticancer drug resistance[106]. In leukemia stem cells, inhibition of histone methyltransferase G9a induced ROS and apoptosis, which was bypassed by the activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK)/Nrf2 pathway[107]. Autophagy was also found to be induced upon G9a inhibition and autophagy inhibitors significantly increased apoptosis, suggesting PERK/Nrf2 might suppress ROS-induced apoptosis by induction of pro-survival autophagy, although the autophagy was not abrogated by PERK/Nrf2 inhibition[107].
Mitophagy is one of the mitochondria quality control systems which selectively targets damaged mitochondria for autophagy-mediated degradation. Mitophagy can lead to reduced ROS and promote cell survival and resistance in cancer cells[108,109]. In CSCs, mitophagy is involved in the maintenance of stemness by suppressing mitochondria-mediated p53 phosphorylation and allows the expression of downstream CSC transcription factor Nanog[110]. Mitophagy also prompts a quiescent state by enhancing mitochondrial turnover and forces the energy metabolism from OXPHOS to glycolysis[100]. A recent study found that mitophagy can be induced by doxorubicin in CD133+/CD44+ CRC CSCs, and inhibition of mitophagy significantly enhanced doxorubicin sensitivity, suggesting a role of mitophagy in CSC-related chemoresistance[111]. Autophagy and mitophagy are believed to be maintained in equilibrium in CSCs to promote resistance, therefore either autophagy/mitophagy inhibition or overactivation in order to disrupt the equilibrium would be a therapeutic strategy[13], Table 1 summarizes drug resistance factors discussed in this section.
Major factors involved in cancer stem cell-related drug resistance
| Associated factor | Major normal biological function | Functions in cancer drug resistance | Major Ref. | Additional remarks |
| ABC transporters | Efflux toxic metabolites | Efflux anticancer drugs, reducing ROS | [30,31,33,35,38,56,62,64] [61] | Functional marker of CSC |
| Low oxidative stress | Anti-apoptosis, detoxification | Maintaining stemness, preventing therapy-induced apoptosis | [48,50,51] [19,49,50,53,56,58] | |
| ALDH | Oxidation of aldehydes, detoxification | Detoxification, reducing ROS, inducing CSC | [13,41,42] [42,44] [45] | Hallmark of CSC |
| Metabolic reprogramming | Not applicable | Reducing ROS, adaptation to TME | [73,75] [13,27,69,84,85] | Hallmark for cancer metabolism |
| Autophagy | Removal and recycling cellular components | Inducing CSC, promoting survival | [48,100] [30,100,103,105-109] | Mitophagy reduces ROS and promotes survival |
EPITHELIAL-MESENCHYMAL TRANSITION
The mechanisms taken by CSCs to confer drug resistance are believed to be largely attributable to the activation of EMT[70]. EMT is a development process during embryogenesis and epithelial wound healing. As hijacked by cancer cells, EMT endows epithelial cells with malignant traits such as loss of cell-cell adhesion and apical-basal polarity, transition to a mesenchymal phenotype, and enhanced migratory and invasive potentials. EMT can be triggered by hypoxia[112], mechanical stress[113], and inflammatory cytokines (e.g., TGF‐β, TNF‐α, IL‐1β, IL‐6)[114], among which TGF-β/SMAD signaling is established in multiple EMT models to play a central role[70]. Cells undergoing EMT display decreased expression levels of epithelial markers such as E-cadherin, zonula occludens-1 (ZO-1), occluding, laminin-1, claudin, and increased mesenchymal markers such as N-cadherin, vimentin and fibronectin[115]. Such a switch in gene expression is under the control of multiple key signaling pathways, including TGF-β, Notch, Wnt, Hedgehog (Hh), PI3K/Akt/mTOR, MAPK/ERK, NF-κB[115]. These signaling pathways ultimately converge on a relatively small group of transcription factors that orchestrate the changes in gene-expression associated with EMT[70]. These transcription factors are often referred to as “EMT-inducing transcription factors”, such as Snail, Slug, zinc finger E-box binding homeobox 1/2 (Zeb1/2), Twist1/2, FOXC2, and FOXM1. Besides transcriptional control, EMT is also regulated by post-transcriptional mechanisms, including microRNAs, long non-coding RNAs, alternative splicing, epigenetics, and post-translational protein stability[116].
An important notion brought by Shibue and Weinberg[70] is that the EMT program is usually partially activated and concurrently expresses epithelial and mesenchymal markers, and the partial EMT phenotypes are highly diverse even within individual tumors. Remarkably, cancer cells that undergo partial EMT are recognized to have a higher metastatic risk than complete EMT, probably due to cell migration by clusters and enhanced attachment to the extracellular matrix[70,116]. The induction of partial EMT phenotype is also associated with CSC expansion[117]. Jolly et al.[116] summarized evidence for the existence of the partial EMT phenotype and its implication in resistance to chemo- and targeted therapies in multiple carcinomas.
EMT induces CSC phenotypes and resistance
EMT is a critical regulator of CSC phenotypes, with emerging evidence indicating therapy-resistant cells enter the CSC state via the activation of EMT[70] and thus display resistance to radio- and chemo-therapy by the gained CSC properties. Though the elucidation of the precise mechanisms of how EMT induces stemness needs further efforts, current studies suggest several EMT-TFs can promote stemness via the activation of CSC regulatory signaling pathways (Notch, Hh, Wnt, Mitofusin) and epigenetic regulators such as microRNA (miR)-200 family members and polycomb complex protein BMI1, a key player in CSC maintenance[118]. For instance, Slug enhances CSC self-renewal capacity; Zeb1 can directly inhibit miR-200 family members and induce stemness by Zeb1/miR-200/BMI1 or Zeb1/miR-200/Notch axis; Twist transcriptionally activates BMI1[118]. One newly recognized connective player is the mitochondrial fusion pathway: upregulation of mitofusin proteins can be triggered by EMT and lead to segregation of the fused mitochondria into CSC upon asymmetric cell division, which provided CSCs with enhanced glutathione synthesis and ROS scavenging to maintain stemness[52]. In turn, CSCs can provoke EMT in non-CSCs via exosome-mediated mechanism and shape the latter to CSCs with an apoptosis-resistant feature[119].
The cellular mechanisms of resistance tightly linked to EMT are attributable to the roles of TGF-β and some EMT-TFs in activating pro-survival signaling, enhancing DNA damage repair, and promoting MDR by ABC transporters. TGF-β and EMT-TFs are also implicated in CSC induction, maintenance of stemness and modulation of metabolism, demonstrating the links among EMT, CSC, and drug resistance.
TGF-β associated drug resistance
TGF-β is a well-established EMT-inducing cytokine that functions as a double-edged sword in relation to drug resistance. At early stages of tumor progression, TGF-β functions as an important tumor suppressor in epithelial cells by inducing apoptosis; however, during later stages, the pro-metastatic role of TGF-β often contributes to resistance[120,121]. A recent study revealed one mechanism of opposing responses of cell survival and apoptosis mediated by TGF-β: by alternative splicing of TGF-β-activated kinase 1 (Tak1). The short isoform of Tak1 was constitutively active and supported TGF-β-induced EMT and NF-κB signaling, whereas the full-length isoform of Tak1 promoted TGF-β-induced apoptosis[122]. Chronic exposure to chemotherapy induced an increase in TGF-β, EMT markers, stem cell markers CD44+/CD24-, sphere formation, and anti-apoptosis protein Bcl-2 in triple negative breast cancer cells, indicating a role of TGF-β in chemo-resistance by regulating EMT, stemness, and apoptosis[123]. TGF-β prompts EMT in epithelial non-CSCs, and the activated EMT-regulatory genes can be crucial in driving cellular plasticity towards mesenchymal CSC states. This is exemplified by Zeb1: in non-CSCs the transcription of Zeb1 was maintained in a poised chromatin configuration which readily enabled the conversion to a stem-like state in response to TGF-β[124]. Moreover, TGF-β can modulate CSC metabolism and promote tumor heterogeneity, leading to drug resistance[125]. For instance, a high concentration of TGF-β near the tumor-vasculature bestowed slower-cycling properties to neighboring squamous cell carcinoma stem cells, which transcriptionally activates p21 and stabilized Nrf2 thereby markedly enhances glutathione metabolism to diminish the effectiveness of cisplatin[125]. In addition, TGF-β can induce EMT-mediated resistance to EGFR-TKI by activating the MAPK, which can be blocked by the MEK1/2 inhibitor[126].
Drug resistance induced by overexpression of EMT-TFs
EMT-TFs such as Snail, Slug, Twist, Zeb, FOXM1 and FOXC2 act as bridges connecting EMT and drug resistance. One major mechanism is the overexpression of EMT-TFs enhances the efflux activity of ABC transporters and thereby promotes MDR. EMT-TFs are known to have binding sites on the promoter regions of multiple ABC transporter genes[127]. In sorafenib-resistant HCC with increased metastasis and enhanced MDR by upregulated P-gp, MDR was suggested as a downstream event from EMT and directly triggered by EMT, as the siRNA knockdown of Snail blocked EMT and partially reversed MDR[128]. PI3K/Akt signaling pathway can induce EMT-associated resistance by the direct regulation on Snail, which then activates P-gp-mediated MDR[128,129]. Overexpression of Slug in HCC cells induced chemo-resistance via upregulation of BCRP but downregulation of P-gp, as well as by the increased expression of stem cell marker CD133 in both complete and partial EMT phenotypes[130]. Chemotherapeutics like doxorubicin can induce FOXM1, which directly activates BCRP to increase the drug efflux and resistance in bladder cancer cells[131].
Moreover, EMT-TFs can enhance antiapoptotic or suppress apoptotic signaling, either independent of or dependent on their role in the induction of CSC phenotype. For example, Twist1 induces resistance to EGFR-TKI by transcriptional suppression of pro-apoptotic gene BCL2L11 (BIM), and the addition of a pan-Bcl-2 inhibitor overcame the resistance[132]. Prostate cancer cells resistant to androgen deprivation (AD) therapy were characterized by elevated FOXC2, the associated EMT/CSC phenotype and increased drug resistance, which were related to the activation of p38 MAPK signaling, a key pathway in promoting cell survival and proliferation[133].The study of the mechanism suggested that FOXC2 augmented p38 phosphorylation[133].
Furthermore, EMT-TFs increase DNA damage repair and promote tumor cell survival under anticancer treatment. Overexpression of transcription factor 4 promoted doxorubicin resistance and stemness, likely by the upregulation of Zeb1 and Zeb2 in CRC cells[134]. Zeb1 promoted DNA repair via ATM- checkpoint kinase 1 (CHK1)- the mediated mechanism and was required for the clearance of DNA breaks, thereby inducing resistance to chemotherapy and radiotherapy[135]. Overexpression of Zeb1 activated ATM kinase by recruiting the transcriptional coactivators p300/PCAF to the ATM promoter, thus rendering chemoresistance in breast cancer cells[136]. In addition, the activated ATM was previously described to phosphorylate and stabilize Zeb1, indicating a positive feedback loop between Zeb1 and ATM regulation[136,137]. Activation of YAP and FOXM1 axis induced EMT-associated EGFR-TKI resistance in lung cancer by dysregulating mitosis. This was indicated by an increased abundance of spindle assembly checkpoint components including polo-like kinase 1, aurora kinases, survivin, and kinesin spindle protein[80]. FOXM1 was suggested as a potential biomarker for resistant lung cancers and its presence predicted a worse clinical outcome[80].
EMT is associated with activation of alternative pathways in acquired resistance to TKIs
Besides the resistance to conventional chemotherapeutics, EMT also contributes to the resistance of EGFR-targeted agents in many types of cancer cells[138]. Other than metabolic changes such as ROS or environmental cues like hypoxia, EMT can be induced by the activation of alternative pathways involved in acquired resistance to EGFR-TKIs, and the induced EMT further exaggerates the resistance. Poh et al.[139] reported a case of EMT causatively inducing acquired resistance to the second-generation EGFR-TKI, afatinib in a patient with advanced non-small cell lung cancer (NSCLC), but negative for a secondary EGFR mutation. AXL is a receptor tyrosine kinase and its correlation with EMT has been demonstrated in NSCLC, breast cancer, and pancreatic cancer[140]. Acquisition of EMT phenotypes and AXL kinase activation were reported in the osimertinib-resistant cell line[140]. The insulin-like growth factor (IGF) signaling is crucial in growth, development and apoptosis. Upregulation of IGF1 receptor (IGF1R) was found to correlate with EMT and EGFR-TKIs resistance[141]. Notably, either silencing of IGF1R or direct inhibition of EMT by overexpression of E-cadherin substantially reduced EMT and resistance[141]. The mechanistic study suggested the activation of MAPK/ERK pathway downstream IGF1R induced EMT, which is indicated by increased nuclear β-catenin and Snail; the induction of EMT is associated with promoted resistance to EGFR-TKIs[141]. Sato et al.[142] reported a feedback loop between MEK/ERK and PI3K/Akt pathways in EGFR-TKI-resistant NSCLC cell lines, suggesting resistance to EGFR inhibition may be a result and also a cause of the activation of PI3K/Akt-induced EMT. In advanced hepatocellular carcinoma, the sorafenib-activated Akt is thought to account for EMT and resistance to sorafenib via EMT-related upregulation of P-gp[107]. Treatment with a combined inhibition of MEK and PI3K pathways reversed EMT to MET and restored sensitivity to EGFR-TKIs[128,142].
Inhibiting EMT to reduce drug resistance can be achieved by targeting characteristics associated with EMT, including TGF-β signaling, mesenchymal markers (vimentin, Slug, and Snail), AXL, EGFR[143,144], and IGF signaling[141,145]. The development of therapeutics has been intensively reviewed[143,144]. In our recent study aiming at understanding the mechanistic roles of eATP in promoting EMT, we compared the gene expression changes induced by eATP and TGF-β, which would identify potential therapeutic targets (e.g., Stanniocalcin-1) involved in EMT and drug resistance. Table 2 is included below to summarize factors/pathways described in this section.
Major factors and pathways involved in EMT-related drug resistance in cancer
| Involved factor | Normal biological function | Functions in cancer drug resistance | Major Ref. | Additional remarks |
| CSC induction | Not applicable | Promoting stemness | [52,70,118] | |
| TGF-β | Inducing EMT and metastasis | Inducing drug resistant state of EMT and CSC | [120-126] | A master inducer of EMT and CSC |
| EMT-associated transcription factors | Expression of EMT-related genes | Upregulating drug transporters, anti-apoptosis Enhancing DNA damage repair | [127-131] [132,133] [134-137] | Snail, Slug, Twist, Zeb, FOXM1 and FOXC2 |
| Alternative RTK signaling | Cell growth, proliferation and survival | Activating EMT and related resistance | [128,140-142] | AXL, IGF, MEK/ERK and PI3K/Akt |
PATHWAYS CONNNECTING EMT AND CSC AND THE IMPLICATION IN DRUG RESISTANCE
Many recent studies suggest that EMT and CSC are intimately interconnected and their involvements in drug resistance represent different manifestations of a combined EMT/CSC phenotype[11,70,146]. One possible mechanism is that the activation of EMT and induction of CSC during tumor development are associated with activation of numerous signaling pathways that control CSC functions including self-renewal, differentiation, survival and metastatic potential. Among them, the Akt, Notch, Hh, Wnt/β-catenin, and NF-κB signaling pathways are responsible for inducing drug resistance. A better understanding of the mechanistic linkage between EMT, CSC and drug resistance would smooth the path for the identification of anticancer targets and promote therapeutic efficacy.
Akt
Akt promotes cell survival, cell proliferation, induction of EMT and maintenance of stemness, it is considered as the master regulator of drug resistance[147]. Dysregulation of PI3K/Akt signaling prevails in human cancer and Akt often interacts with other signaling pathways involved in EMT/CSC to confer drug resistance. For instance, hypoxia synergistically enhanced gemcitabine-induced interaction between Akt and notch1, which in turn promoted cell stemness and chemoresistance[148]. Akt/glycogen synthase kinase 3-β activated Wnt signaling and promoted stemness and cisplatin resistance[149]. Akt cross talks with MAPK and NF-κB inducing overexpression of P-gp and MDR in cancer cells with EMT/CSC phenotypes[128,150], as well as activating programmed death-ligand 1 expression which leads to immunosuppression[147]. Under hypoxia, PI3K/Akt coupled with mTOR and activated Hif-1α and Hif-2α, which promoted quiescence and chemoresistance via ABC transporters[79]. Combined therapy using PI3K inhibitor with inhibitors of other pathways that interact with AKT, like MAPK, might be a potent therapeutic strategy against drug resistance[142].
Notch
Notch signaling plays a crucial role in cell to cell communications, cell fate, angiogenesis, and CSCs of several tumors[151,152]. Notch signaling consists of two types of membrane-bound ligands: Delta-like ligands, and Jagged ligands. The binding of Notch ligands with four trans-membrane notch receptors (notch 1-4) leads to coordinated communication between adjacent cells[151]. Firstly, Notch cross talks with EMT: Notch induces EMT by upregulating EMT-TFs such as Twist, Snail, Slug, and Zeb; while Notch can be regulated by growth factors relevant to EMT such as fibroblast growth factor (FGF) and PDGF[138]. Further, Notch is an important mediator of CSC-related chemoresistance. For instance, CD133+/CD24+ high renal cell carcinoma CSCs showed enhanced notch signaling, and blockage of Notch-1 or notch-2 reversed the stemness, invasiveness, migratory potential, and chemoresistance to cisplatin and sorafenib[153]. Also, Notch directly activates the transcription of the anti-apoptotic survivin, which serves as a Notch effector and contributes to CSC maintenance[154]. Moreover, activation of Notch signaling enhances the efflux activity of ABC transporters and promotes MDR. In NSCLC, Notch-1 contributes to chemoresistance via a Notch-1/AP-1/miR-451/MDR-1 axis, and inhibition of Notch-1 led to increased miR-451 and sensitized tumors to taxane-based treatment[155]. Patients of Stage II CRC with overexpression of hairy and enhancer of split-1 (HES1), a downstream transcriptional factor of Notch, showed higher recurrence rates after chemotherapy[156]. The over-expressed HES1 was found to correlate with increased MRP1, MRP2 and P-gp expression and chemoresistance in CRC cells[156]. Finally, Notch mediates resistance to EGFR-targeted agents by enhancing DNA damage repair and/or activating pro-survival signaling pathways. For instance, a combined blockade of EGFR and Notch-2/3 prevented acquired resistance to EGFR inhibitors and radiation, by reducing EGFR-TKI-induced ALDH+ and radiation-induced CD133+ stem cell subpopulations and expression of EMT and DNA repair genes[157]. In another example, Notch1 was necessary to induce trastuzumab resistance in breast cancer in vitro and in vivo[158]. Notch1 suppressed PTEN expression to activate ERK1/2 signaling, leading to breast CSC survival and bulk cell proliferation[158]. High Notch-1 protein expression was suggested as a marker to predict poorer survival in women with HER2+ breast cancer[158]. In gastric cancer cells, long-term treatment with trastuzumab resulted in upregulation of Notch ligand Jagged-1, leading to acquired resistance with EMT and CSC phenotypes through an IL-6/STAT3/Jagged-1/Notch positive feedback loop[159]. STAT3 is a well-known major cell survival factor and its activation in this axis contributed to the enhanced survival signaling and resistance to trastuzumab[159].
Hh
Hh signaling is central to embryogenesis, hyperactivation of this pathway has been found to be tumorigenic in many cancers[151]. Binding of Hh ligand to its receptor, Patched induces the activation of Smoothened (SMO) and Gli1; the SMO-mediated translocation of Gli1 into the cell nucleus drives the transcription of Hh target genes[151]. Hh signaling plays a key role in the maintenance of CSCs and chemo-resistance. In a study with an organoid culture model from patient-derived colorectal tumors, treatment with Hh inhibitors reduced expression of stem cell markers c-Myc, CD44, and Nanog, together with their transcription factor Gli1[160]. Combined treatment of Hh inhibitors with chemotherapeutics decreased the cell viability of organoids compared with the treatment of a single drug[160]. Finally, Hh signaling promotes drug resistance by inducing overexpression of P-gp and BCRP[161]. Approved SMO inhibitors are capable of reversing chemoresistance in gastric cancer and basal cell carcinomas[161]. Likewise in ovarian cancer cells, Hh signaling enhanced resistance to cisplatin, which was probably mediated by Hh transcription factor Gli2 which activated gene expression of P-gp[162]. Besides, BCRP was reported to mediate drug resistance in hepatoma cells under the transcriptional control of Gli1/2, and the inhibition of Gli1 or ABCB2 gene expression ameliorated resistance to various chemotherapies in hepatoma cells[163]. Other ABC transporters were also found to confer chemoresistance as transcriptional targets of Hh/Gli signaling in CRC cells, including ABCA2, P-gp, ABCB4, ABCB7, ABCC2 and ABCG1[164]. These findings suggest a connecting role of Hh signaling in CSC, EMT and chemo-resistance.
Wnt
Wnt signaling helps determine cell fate during embryogenesis and regulates the homeostasis of many tissues in adults[151]. Aside from its function in normal cells, Wnt also plays a major role in maintaining CSCs in various cancer types. The binding of Wnt to frizzled receptors initiates two distinct signaling cascades, termed canonical or noncanonical. The activation of the canonical pathway results in translocation of β-catenin into the nucleus which transcriptionally controls a set of genes involved in cell migration [matrix metallopeptidases (MMPs)], survival (Myc) and proliferation (peroxisome proliferator-activated
NF-κB
NF-κB is a well-documented mediator of the inflammatory response, during which the activation of NF-κB via toll-like receptors stimulation promotes stemness[178], providing a link between inflammation and CSC phenotypes. Under hypoxic conditions, Hif-1α and Hif-2α induce the CSC features likely via NF-κB signaling pathway activation[179,180]. NF-κB is also suggested as a direct regulator of EMT-TFs and is associated with metastatic potential[181]. Resistance to radiotherapy correlates with increased EMT/CSC phenotypes induced via NF-κB[182]. Inhibition of NF-κB activity by metformin was associated with reduction of stemness and synergistically sensitized resistant lung cancer cells to EGFR-TKIs[183]. All the major pathways discussed in this section are summarized in Table 3.
Major pathways connecting EMT with CSC in drug resistance in cancer
| Contributing factor | Normal biological function | Function in cancer drug resistance | Major Ref. | Additional remarks |
| Akt | Cell growth and cell survival | Promoting stemness and related resistance | [79,128,147-149] | Crosstalk with other signaling pathways |
| Notch | Embryogenesis and tissue homeostasis | Inducing CSC and EMT, anti-apoptosis, promoting MDR | [138,153,157,159] [153] [155,156] | |
| Hedgehog | Embryogenesis and tissue homeostasis | CSC maintenance, promoting MDR | [160] [161-164] | |
| Wnt | Embryogenesis and tissue homeostasis | CSC maintenance, anti-apoptosis, enhancing DNA damage repair, promoting MDR | [176] [165,167] [168,169] [166] | Alternative pathway in resistance to EGFR inhibition |
| NF-κB | Gene expression, cytokine production Immune regulation | Promoting stemness, EMT and related resistance | [178-183] | Master inducer and regulator |
TME regulation on EMT, CSC and drug resistance
Drug resistance can be induced by non-cancer cells present within the TME. Environmental stress due to anticancer therapies can initiate cancer cell secretion of chemoattractants, which recruit various cells like macrophages into the TME and assist in the differentiation of the latter in a paracrine manner[184]. The newly differentiating non-cancer cells, such as cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), provide feedback to the cancer cells to promote stemness and resistance. CAF secretion of either exosome containing miRNA, Wnt proteins[185], Interleukin (IL)-1B, TNFα or fibrillar collagen can activate EMT of cancer cells[186]. For example, exosome transfer of miR-92 from CAFs releases the inhibition of the Wnt/β-catenin pathway, which promotes stemness, EMT and chemoresistance[187]. CAFs can be activated by Hh ligand produced by tumor cells, leading to the expression of FGF5 and production of fibrillar collagen, which forms a supportive niche to facilitate cancer cells to acquire EMT/CSC phenotype and chemoresistance[15]. Combination of SMO inhibitor and chemotherapy improved response for 3 of 12 patients in a clinical trial[15]. TAMs can induce EMT and stemness of cancer cells and related treatment resistance by the secretion of Wnt proteins[188], TGF-β, IL-6, IL-10 and TNFα[189]. In addition, The higher proportion of M2-like TAMS to M1-like TAMs creates an immunosuppressive TME possibly by releasing immunosuppressive cytokines, IL-10 and TGF-β, and activating induced regulatory T cells which inhibit the cytotoxic function of effector T and NK cells[190,191].
All these discussed drug resistance mechanisms are schematically shown in Figure 1.
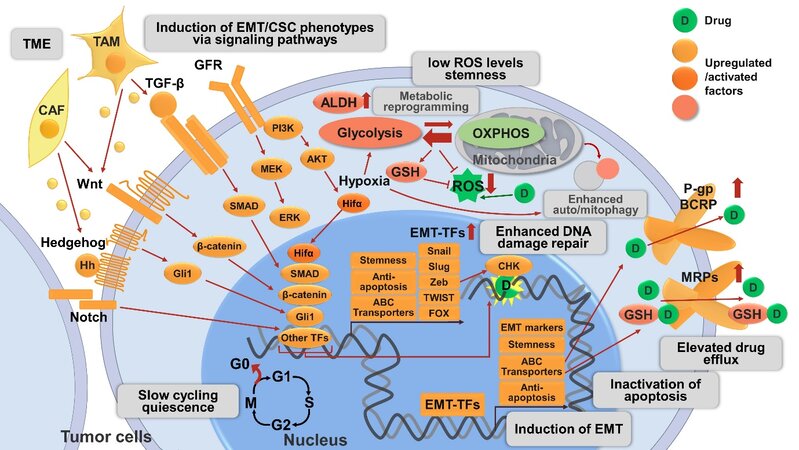
Figure 1. Mechanisms related to CSC and EMT involved in drug resistance in cancer described in this review. The CSC phenotype is a plastic state and can be adopted by most cancer cells. CSCs are slow-cycling or quiescent, and circumvent therapies targeting rapidly-dividing cells. CSCs exhibit increased activities of detoxifying proteins including ALDH and ABC transporters (P-gp, BCRP). CSCs maintain low levels of ROS by metabolic reprogramming and/or auto/mitophagy to protect from radio- and chemo-therapies. Hypoxia and Hifα are crucial regulators of metabolic reprogramming primarily by increasing flux to glycolysis and antioxidant production (e.g., GSH) and inducing autophagy. Hypoxia and Hifα are also emerging inducers of EMT/CSC phenotype, one of the mechanisms is via NF-κB signaling pathway activation (not shown in the figure). EMT is a crucial regulator of and tightly interconnected with CSC, their involvement in drug resistance may represent different manifestations of the EMT/CSC phenotype. Activation of diverse signaling pathways are involved in the induction of EMT/CSC phenotype, including developmental pathways (e.g., Wnt/β-catenin, Hh/Gli1, Notch), cell survival pathways (e.g., GFR), and EMT-related pathways (e.g., TGF-β/SMAD signaling). The above pathways act independently or cross talk with each other to induce EMT/CSC phenotypes, leading to elevated drug resistance by various mechanisms: (1) The activation of these pathways allows the maintenance of CSC properties, including enhanced drug resistance. (2) These pathways converge on EMT-TFs (e.g., Snail, Slug, Zeb, Twist, FOX, etc.) to alter the expression of EMT markers (e.g., increase in N-cadherin, vimentin; decrease in E-cadherin) and induce EMT; the downstream transcription factor of the above pathways, as well as EMT-TFs, can upregulate ABC transporters, leading to enhanced drug efflux. EMT-TFs also enhance stemness and anti-apoptotic signaling. (3) The activation of these pathways is associated with enhanced anti-apoptotic machinery and thereby promotes tumor cell survival. (4) Certain pathways and EMT-TFs like Zeb1 can enhance ATM and CHK1/2-mediated DNA-damage repair and promote resistance to genotoxic therapies. (5) Non-cancer cells such as CAFs and TAMs in the TME can also activate these signaling pathways by secreted proteins and thus promote drug resistance. CSC: Cancer stem cell; EMT: epithelial-mesenchymal transition; ALDH: aldehyde dehydrogenase; ABC transporters: ATP-binding cassette (ABC) transporters; P-gp: P-glycoprotein; BCRP: breast cancer resistance protein; ROS: reactive oxygen species; Hifα: hypoxia-inducible factors α; GSH: glutathione; NF-κB: nuclear factor-κB; Hh: Hedgehog; GFR: growth factor receptor; TGF-β: transforming growth factor-β; EMT-TFs: EMT-inducing transcriptional factors; Zeb: zinc finger E-box binding homeobox; FOX: forkhead box; ATM: ataxia telangiectasia mutated; CHK: checkpoint kinase; CAF: cancer-associated fibroblasts; TAM: tumor-associated macrophages; TME: tumor microenvironment.
ATP AND ATP-MEDIATED DRUG RESISTANCE
Intracellular ATP promotes drug resistance
Adenosine 5’-triphosphate (ATP) is a multifunctional molecule participating in a myriad of cellular processes. ATP, as an energy supplier, signaling transducer, extracellular messenger, has been proven to be a significant player in tumor growth, survival and resistance[192-195]. Cancer cells are known to have higher levels of intracellular ATP (iATP) compared with their non-cancerous counterparts, likely due to the elevated glucose transport and aerobic glycolysis, namely the Warburg effect[196,197]. Notably, resistant cancer cell lines are found to have even higher iATP levels than their parental cancer cells, and the increased iATP levels have been demonstrated to contribute to the establishment and maintenance of resistance[198]. In colon cancer, direct delivery of ATP into cancer cells induced conversion of drug-sensitive cancer cells to drug-resistant cancer cells, while ATP depletion by glycolysis inhibition restored their sensitivity to chemotherapy[198]. This suggests iATP levels are a core determinant of chemoresistance[198].
Accordingly, depleting iATP levels emerges as a promising strategy to restore drug sensitivity. Metformin, an AMPK activator and a prescription drug used to treat type 2 diabetes, significantly hampered the production of iATP, leading to a critical energy crisis which impaired the capability of the breast CSCs to repair chemotherapy-induced DNA damage in vitro. The addition of eATP completely abrogated such synergistic effects of metformin on the sensitivity to chemotherapeutics[199]. In one attempt, a covalent conjugate of nucleobases and peptides, or nucleopeptides, was engineered, which can selectively sequester ATP over ADP. This nucleopeptide slowed down efflux pumps in multidrug resistance cancer cells and boosted the efficacy of doxorubicin[200]. A recently described estrogen receptor α biomodulator, BHPI, is capable of inducing toxic hyperactivation of the endoplasmic reticulum stress sensor, the unfolded protein response. By this action, BHPI depleted iATP and nearly blocked P-gp-mediated drug efflux, which restored doxorubicin and paclitaxel sensitivity in ovarian cancer cells[201]. A novel nanoparticle was described, which independently encapsulated doxorubicin in the core and glucose oxidase (GOx) in the shell[202]. The fast release of GOx by acid-sensitive degradation of the shell consumed glucose and deprived ATP, which suppressed ATP-dependent drug efflux and facilitated the sequential accumulation of doxorubicin in breast cancer cells and dramatically improved the efficacy of anticancer drugs for MDR cells[202].
Extracellular ATP induces drug resistance
eATP levels of various cancer types are 103 to 104 times higher than those in their corresponding normal tissues[1]. The abnormally high concentration of eATP is majorly released from dying tumor cells, which can be induced by cellular stress, hypoxia, inflammation, platelet aggregation and anticancer therapies[203]. The functions of high eATP levels have been primarily revealed by our study. We investigated the role of eATP related to resistance: eight anticancer drugs, including both targeted and chemotherapeutic drugs, were tested in five cancer cell lines of five different organ origins. We found that eATP can promote the increase of iATP and cancer cell survival in most cases[10]. When studying promoted drug resistance to sunitinib due to eATP in NSCLC A549 cells, we saw that eATP can be internalized by cancer cells through clathrin- and caveolae-mediated endocytosis, but mainly by macropinocytosis indicated by colocalization of fluorescent-ATP with a macropinocytosis tracer (high molecular weight Dextran), resulting in substantially elevated iATP levels from 150% to 200% of the original iATP concentrations[8-10]. ATP levels increased by eATP is not a transient effect. The level elevation persists as long as the eATP is present[9]. Activation of macropinocytosis is a hallmark of some cancers, particularly for those harboring oncogenic Ras mutations[204]. We inhibited macropinocytosis by either siRNA knockdown of macropinocytosis-essential enzyme PAK1 or a macropinocytosis inhibitor IPA3, and the inhibition resulted in the reduction of iATP levels and survival of eATP and sunitinib treated A549 cells[10]. Thus, the elevated iATP level from macropinocytosis and other endocytosis-mediated eATP internalization is responsible, at least in part, for the observed drug resistance. One of the possible drug resistance mechanisms induced by the internalization of eATP is that more abundant iATP molecules compete with tyrosine kinase inhibitors, which are ATP competitors for the ATP binding site located on RTKs, leading to increased phosphorylation and activation of downstream signaling pathways[10]. Besides, the enhanced iATP is found to affect cancer cell metabolism, particularly those related to energy metabolism (unpublished observation).
In the five cell lines studied, increased iATP was found to correlate with drug resistance status when the ABC transporters expressed by the cell line matched those required for the efflux of a given drug[10]. Indeed, our results showed that the eATP molecules being internalized served as an energy supplement and enhanced efflux activity of ABC transporters in two cancer cell lines, resulting in increased cell survival[10]. In addition to directly providing energy, eATP was also shown to regulate expression levels of ABC transporters[10]. These results indicated the profound effects of eATP on modulating ABC transporter activity to potentiate efflux of anticancer drugs and enhancing drug resistance. Wang et al.[205] reported a lipid membrane-coated silica carbon nanoparticle engineered to produce ROS in mitochondria under near-infrared laser irradiation. The introduced ROS species reduced iATP by oxidizing available NADH for ATP synthesis, and thereby suppressed the efflux of chemotherapeutics as well as reduced the expression of P-gp and its distribution on the plasma membrane, which is consistent with our findings[205].
Additionally, our recent study demonstrated that eATP induced cancer cell migration and invasion. When exposing lung cancer cells to an ATP concentration equivalent to that in intratumoral extracellular space, we found accelerated detachment, EMT, migration, and invasion of lung cancer cells[12]. Mechanistically, we detected an increase in expression levels of MMPs, mesenchymal-phenotype molecules, EMT-TFs (vimentin, Snail and Slug) and decreased levels of epithelial phenotype markers (E-cadherin, β-catenin and ZO-1)[12]. Notably, these effects did not require TGF-β and were semi-independent of the activation of purinergic signaling (P2X7)[12]. Moreover, the knockout of a key macropinocytosis-associated gene, sorting nexin 5, significantly reduced micropinocytosis and the resulted iATP levels, cell growth, migration/invasion rates in vitro, and slowed down tumor formation and growth in nude mice[12]. As summarized in the previous content, EMT is intimately associated with the acquisition of resistance, and thus these results may reveal new mechanisms of the eATP-induced drug resistance. Overall, our studies suggested the multifunctional roles of ATP as an energy supplier facilitating drug efflux and a signaling transducer activating signaling pathways related to cell survival and resistance, drug efflux and tumor migration/invasion.
eATP can also contribute to cancer drug resistance as an extracellular messenger through P2 purinergic signaling, which is composed of nucleotides/nucleosides (mainly ATP and adenosine) acting as signaling ligands with their corresponding membrane receptors and modulating diverse signaling pathways[206]. There are two P2 receptor families: P2X receptors which are ATP-gated ion channels and P2Y receptors which are mainly activated by ATP or ADP. P2X and P2Y family members emerge as players in resistance to chemotherapies[207,208]. Ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, or CD39) eATPase activity, which converts eATP to AMP, coordinately activated mitochondrial stress response via the downstream P2RY13/cAMP/PKA axis and promoted cytarabine resistance by enhancing mitochondrial OXPHOS activity in acute myeloid leukemia (AML). With such evidence, eATP was proposed as a key factor in chemoresistance of AML and CD39 as a new marker associated with a poor response to chemotherapy[207]. In the extracellular compartment, tumor cells can induce platelet activation and aggregation. Activated platelets release ADP and ATP, which activated ADP purinergic receptor P2Y12 expressed on pancreatic ductal adenocarcinoma and hence promoted expression of gemcitabine resistance markers Slug and cytidine deaminase (CDD)[209]. Exogenous ADP and ATP (100 µM) also increased the expression levels of Slug and CDD while P2Y12 inhibition completely blocked the survival signals initiated in cancer cells by platelet-derived ADP and ATP[209]. P2X7 receptor behaves as a bi-functional receptor. Tumor cell killing by mmol/L ATP concentration is in most cases mediated by the overstimulation of P2X7 and large pores opening on the plasma membrane. In contrast, low level of P2X7 stimulation often promotes cell survival and proliferation. P2X7 can potentiate OXPHOS, aerobic glycolysis and biosynthesis, which endows P2X7-expressing cells with a striking proliferative advantage[210]. A recent study described a distinct conformational form of P2X7, termed non-pore functional P2X7 (nfP2X7), which is unable to form a pore upon eATP stimulation. The exposure to a high ATP concentration (0.5-1 mM) drove nfP2X7 expression and was essential for tumor cell survival[211]. These results suggest a role of P2X7 in resistance to apoptosis-inducing chemotherapies. Besides, P2X7 expression is intimately associated with cancer cell metastatic potential and invasiveness[210,212]. In addition to P2X7, Some other purinergic receptors (PRs) are likely to be involved in drug resistance as well. These PRs have different EC50s compared with P2X7, they respond to different levels of extracellular ATP and mediates presently-poorly defined actions in drug resistance.
Nevertheless, several chemotherapeutic agents have the capacity to induce immunogenic cell death (ICD) which can lead to the generation of immunological memory and improve relapse-free survival[213]. In this scenario, the release of eATP from stressed or dying tumor cells during ICD is recognized as a damage-associated molecular pattern and is indispensable for the ICD[213]. For example, released eATP can ligate P2RX7 on dendritic cells to drive its recruitment and activation, which then generate adaptive anticancer immunity by the process of tumor-derived antigens and secretion of interleukin-1β[214]. eATP also executes immune-activating functions by communicating with other immune cells such as monocytes, macrophages, T cells, eosinophils, neutrophils and B cells[203]. Importantly, ICD depends not only on the release of eATP but also on its stability in the TME[215]. CD39 and cell surface purinergic enzyme ecto-5’-nucleotidase (NT5E, or CD73) can ultimately convert eATP to adenosine (ADO). While eATP promotes antitumor immunity, ADO attenuates immune response against tumors. Adenosine induced signaling would upregulate anti-inflammatory molecules, decrease anti-tumor function of immune cells (e.g., T cells, B cells, dendritic cells, mast cells, natural killer, macrophages) and activate immunoregulatory cells (e.g., regulatory T cells), shaping an immunosuppressive environment and diminishing the effect of immunotherapies[203,216]. In addition, autophagy of tumor cells is required for chemotherapy-induced ICD, in which it facilitates the release of eATP over the generation of adenosine[217].
Therefore, inhibition of purinergic signaling might improve the response to anticancer drugs. Among purinergic receptors, P2X7 receptor is a potential candidate, as the application of its antagonists was suggested in the inhibition of tumor growth and migration[203]. To limit the conversion from eATP to adenosine, CD39 and CD73 emerge as targets for anticancer immunotherapy[213]. One study reported CD39 blockade augmented eATP/P2X7-mediated proinflammatory antitumor responses and the ultimate release of IL18, which facilitated the significant expansion and infiltration of intratumor effector T cells, and reversed anti-PD-1 resistance[208]. Furthermore, depletion of eATP may also reduce drug resistance. One novel approach described nanoparticles that can simultaneously release Fe3+ and doxorubicin at the TME upon near infrared laser irradiation, and Fe3+ can deplete eATP through metal ion-triphosphate coordination, sensitizing chemotherapy[218]. The potential inhibitory approaches may include pH-dependent activation of ATPase introduced in a prodrug form to reduce TME eATP levels or by inhibiting pathways or factors downstream from eATP’s purinergic receptor signaling. The former approach is based on the observation that the TME is more acidic compared to the normal tissues while the latter strategy relies on the identification and testing of key players in the eATP induced EMT and CSC. Finally, inhibition of macropinocytosis can reduce cell uptake of eATP and thus iATP levels, which might alleviate the drug resistance and EMT promoted by eATP.
All these discussed ATP-related drug resistance mechanisms are shown in Figure 2.
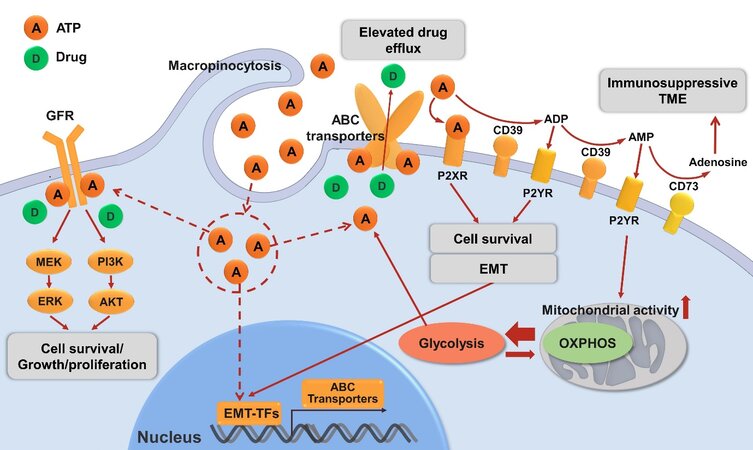
Figure 2. Mechanisms of drug resistance in cancer related to ATP described in this review. eATP can be degraded to ADP, AMP by ecto-nucleotidase CD39 or sequentially to immunosuppressive adenosine by ecto-nucleotidase CD73. eATP acts as messengers outside of cancer cells through purinergic signaling including P2X receptor (P2XR, e.g., P2X7) while ADP/AMP act through P2Y receptor (P2XR, e.g., P2Y12, P2Y13) to promote cell survival signals, energy generation or/and EMT, contributing to drug resistance. eATP is also internalized by cancer cells via macropinocytosis, which results in significantly elevated iATP levels. The increased iATP molecules become more competitive against ATP analog anticancer drugs for the intracellular ATP binding domain of RTKs of GFRs on cancer cell plasma membrane, and thereby reduce RTK phosphorylation and downstream signaling involved in cell growth, proliferation and survival. Elevated iATP levels also enhance the efflux activity of ABC transporters for out-pumping anticancer drugs from cancer cells. Additionally, our recent study identified a role of iATP directly inducing EMT, and EMT confers drug resistance by numbers of mechanisms including upregulating ABC transporters. All these mechanisms work together to promote drug resistance by increasing cell survival signaling, reducing intracellular drug concentration, and inducing EMT. Further studies are needed for the final validation of ATP-mediated mechanisms of drug resistance. eATP: Intratumoral extracellular ATP; ENTPD1: ectonucleoside triphosphate diphosphohydrolase 1 (CD39); NT5E: ecto-5’-nucleotidase (CD73); EMT: epithelial-mesenchymal transition; iATP: intracellular ATP; RTK: receptor tyrosine kinase; GFR: growth factor receptor; ABC transporters: ATP-binding cassette (ABC) transporters.
CONCLUSIONS
Through this review, we show that eATP, which has been found to be present in TME with concentrations in hundreds of micromolar, is an emerging inducer and regulator of drug resistance in cancer. eATP has been shown by us and others to be an inducer and regulator for EMT, which is closely associated with the formation of increasing CSC subpopulations in tumors. EMT/CSC phenotypes work together to increase therapy resistance by various mechanisms discussed above. Thus, eATP in TME is a factor, along with TGF-β, working through inducing EMT and CSC, and leading to a drug resistance state in cells and tumors. To reduce drug resistance and enhance anticancer therapeutic effects, inhibiting and blocking eATP is likely to be a promising approach[219].
DECLARATIONS
Authors’ contributionsPerformed major literature search, manuscript writing, figure drawing: Zhang H
Performed literature search, wrote a section and edited manuscript: Steed A
Performed literature search, wrote a section and edited manuscript: Co M
Conceptualized the topic and manuscript content, wrote part of the manuscript, supervised and provided funding for the project: Chen X
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported in part by a NIH grant R15 CA242177-01 to Chen X.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 2008;3:e2599.
2. Wilhelm K, Ganesan J, Müller T, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 2010;16:1434-8.
3. Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011;334:1573-7.
4. Falzoni S, Donvito G, Di Virgilio F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013;3:20120101.
5. Morciano G, Sarti AC, Marchi S, et al. Use of luciferase probes to measure ATP in living cells and animals. Nat Protoc 2017;12:1542-62.
6. Liu Y, Zhang W, Cao Y, Liu Y, Bergmeier S, Chen X. Small compound inhibitors of basal glucose transport inhibit cell proliferation and induce apoptosis in cancer cells via glucose-deprivation-like mechanisms. Cancer Lett 2010;298:176-85.
7. Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 2012;11:1672-82.
8. Qian Y, Wang X, Liu Y, et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett 2014;351:242-51.
9. Qian Y, Wang X, Li Y, Cao Y, Chen X. Extracellular ATP a new player in cancer metabolism: NSCLC cells internalize ATP in vitro and in vivo using multiple endocytic mechanisms. Mol Cancer Res 2016;14:1087-96.
10. Wang X, Li Y, Qian Y, et al. Extracellular ATP, as an energy and phosphorylating molecule, induces different types of drug resistances in cancer cells through ATP internalization and intracellular ATP level increase. Oncotarget 2017;8:87860-77.
11. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019;2:141-60.
12. Cao Y, Wang X, Li Y, Evers M, Zhang H, Chen X. Extracellular and macropinocytosis internalized ATP work together to induce epithelial-mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int 2019;19:254.
13. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci 2019;234:116781.
14. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci 2017;18:2362.
15. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun 2018;9:2897.
16. Mondal S, Bhattacharya K, Mandal C. Nutritional stress reprograms dedifferention in glioblastoma multiforme driven by PTEN/Wnt/Hedgehog axis: a stochastic model of cancer stem cells. Cell Death Discov 2018;4:110.
17. Filipponi D, Emelyanov A, Muller J, Molina C, Nichols J, Bulavin DV. DNA damage signaling-induced cancer cell reprogramming as a driver of tumor relapse. Mol Cell 2019;74:651-63.e8.
18. D'Alterio C, Scala S, Sozzi G, Roz L, Bertolini G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin Cancer Biol 2020;60:351-61.
19. Lytle NK, Ferguson LP, Rajbhandari N, et al. A multiscale map of the stem cell state in pancreatic adenocarcinoma. Cell 2019;177:572-86.e22.
20. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008;8:755-68.
21. Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila) 2010;3:1361-4.
22. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013;501:328-37.
23. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008;27:1749-58.
24. Mueller MT, Hermann PC, Witthauer J, et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009;137:1102-13.
26. Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011;146:633-44.
27. Dirkse A, Golebiewska A, Buder T, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun 2019;10:1787.
29. Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 2001;42:1007-17.
30. Li YJ, Lei YH, Yao N, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer 2017;36:52.
31. Kadioglu O, Saeed MEM, Munder M, Spuller A, Greten HJ, Efferth T. Effect of ABC transporter expression and mutational status on survival rates of cancer patients. Biomed Pharmacother 2020;131:110718.
32. Hu J, Li J, Yue X, et al. Expression of the cancer stem cell markers ABCG2 and OCT-4 in right-sided colon cancer predicts recurrence and poor outcomes. Oncotarget 2017;8:28463-70.
33. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer 2018;18:452-64.
34. Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res 2005;65:6207-19.
35. Ling X, Wu W, Fan C, et al. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J Exp Clin Cancer Res 2018;37:240.
36. Keshet GI, Goldstein I, Itzhaki O, et al. MDR1 expression identifies human melanoma stem cells. Biochem Biophys Res Commun 2008;368:930-6.
37. Gong F, Dong D, Zhang T, Xu W. Long non-coding RNA FENDRR attenuates the stemness of non-small cell lung cancer cells via decreasing multidrug resistance gene 1 (MDR1) expression through competitively binding with RNA binding protein HuR. Eur J Pharmacol 2019;853:345-52.
38. Xu H, Liu T, Li W, Yao Q. SMAR1 attenuates the stemness of osteosarcoma cells via through suppressing ABCG2 transcriptional activity. Environ Toxicol 2021;36:1090-8.
39. Stefan SM. Multi-target ABC transporter modulators: what next and where to go? Future Med Chem 2019;11:2353-8.
40. Chefetz I, Grimley E, Yang K, et al. A pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep 2019;26:3061-75.e6.
41. Mazor G, Levin L, Picard D, et al. The lncRNA TP73-AS1 is linked to aggressiveness in glioblastoma and promotes temozolomide resistance in glioblastoma cancer stem cells. Cell Death Dis 2019;10:246.
42. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015;31:16-27.
43. Li Y, Chen T, Zhu J, Zhang H, Jiang H, Sun H. High ALDH activity defines ovarian cancer stem-like cells with enhanced invasiveness and EMT progress which are responsible for tumor invasion. Biochem Biophys Res Commun 2018;495:1081-8.
44. Singh S, Brocker C, Koppaka V, et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med 2013;56:89-101.
45. Kim D, Choi BH, Ryoo IG, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis 2018;9:896.
46. Harati M, Rodemann HP, Toulany M. Nanog signaling mediates radioresistance in ALDH-positive breast cancer cells. Int J Mol Sci 2019;20:1151.
47. Muralikrishnan V, Hurley TD, Nephew KP. Targeting aldehyde dehydrogenases to eliminate cancer stem cells in gynecologic malignancies. Cancers (Basel) 2020;12:961.
48. Chatterjee R, Chatterjee J. ROS and oncogenesis with special reference to EMT and stemness. Eur J Cell Biol 2020;99:151073.
49. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458:780-3.
50. Qian X, Nie X, Yao W, et al. Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin Cancer Biol 2018;53:248-57.
51. Park HK, Hong JH, Oh YT, et al. Interplay between TRAP1 and Sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res 2019;79:1369-82.
52. Wu MJ, Chen YS, Kim MR, et al. Epithelial-mesenchymal transition directs stem cell polarity via regulation of mitofusin. Cell Metab 2019;29:993-1002.e6.
53. Huang H, Aladelokun O, Ideta T, Giardina C, Ellis LM, Rosenberg DW. Inhibition of PGE2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci Rep 2019;9:4954.
54. Meng Q, Shi S, Liang C, et al. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018;37:5843-57.
55. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016;1863:2977-92.
56. Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003;22:7369-75.
57. Lu H, Samanta D, Xiang L, et al. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl Acad Sci U S A 2015;112:E4600-9.
58. Lan D, Wang L, He R, Ma J, Bin Y, Chi X, et al. Exogenous glutathione contributes to cisplatin resistance in lung cancer A549 cells. Am J Transl Res 2018;10:1295-309.
59. Akan I, Akan S, Akca H, Savas B, Ozben T. Multidrug resistance-associated protein 1 (MRP1) mediated vincristine resistance: effects of N-acetylcysteine and Buthionine sulfoximine. Cancer Cell Int 2005;5:22.
60. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 2018;217:2291-8.
61. Boswell-Casteel RC, Fukuda Y, Schuetz JD. ABCB6, an ABC transporter impacting drug response and disease. AAPS J 2017;20:8.
62. Tong X, Zhao J, Zhang Y, Mu P, Wang X. Expression levels of MRP1, GST-π, and GSK3β in ovarian cancer and the relationship with drug resistance and prognosis of patients. Oncol Lett 2019;18:22-8.
63. Wang B, Shen C, Li Y, et al. Oridonin overcomes the gemcitabine resistant PANC-1/Gem cells by regulating GST pi and LRP/1 ERK/JNK signalling. Onco Targets Ther 2019;12:5751-65.
64. El-Readi MZ, Eid S, Abdelghany AA, Al-Amoudi HS, Efferth T, Wink M. Resveratrol mediated cancer cell apoptosis, and modulation of multidrug resistance proteins and metabolic enzymes. Phytomedicine 2019;55:269-81.
65. Chatterjee A, Gupta S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett 2018;433:33-42.
66. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res 2015;34:111.
67. El-Sahli S, Wang L. Cancer stem cell-associated pathways in the metabolic reprogramming of breast cancer. Int J Mol Sci 2020;21:9125.
68. Fonseca NA, Cruz AF, Moura V, Simões S, Moreira JN. The cancer stem cell phenotype as a determinant factor of the heterotypic nature of breast tumors. Crit Rev Oncol Hematol 2017;113:111-21.
69. Luo M, Shang L, Brooks MD, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab 2018;28:69-86.e6.
70. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611-29.
71. Dhawan A, Madani Tonekaboni SA, Taube JH, et al. Mathematical modelling of phenotypic plasticity and conversion to a stem-cell state under hypoxia. Sci Rep 2016;6:18074.
72. Emami Nejad A, Najafgholian S, Rostami A, et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int 2021;21:62.
73. Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J 2018;475:1611-34.
74. Zhao H, Duan Q, Zhang Z, et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med 2017;21:2055-67.
75. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J 2017;36:252-9.
76. Qiang L, Wu T, Zhang HW, et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ 2012;19:284-94.
77. Yeo CD, Kang N, Choi SY, et al. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: a possible link to epigenetic regulation. Korean J Intern Med 2017;32:589-99.
78. Zhang Z, Fang E, Rong Y, Han H, Gong Q, Xiao Y, et al. Hypoxia-induced lncRNA CASC9 enhances glycolysis and the epithelial-mesenchymal transition of pancreatic cancer by a positive feedback loop with AKT/HIF-1α signaling. Am J Cancer Res 2021;11:123-37.
79. Schöning JP, Monteiro M, Gu W. Drug resistance and cancer stem cells: the shared but distinct roles of hypoxia-inducible factors HIF1α and Hif-2α. Clin Exp Pharmacol Physiol 2017;44:153-61.
80. Nilsson MB, Sun H, Robichaux J, et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med 2020;12:eaaz4589.
81. Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010;2:823-42.
82. Folmes CD, Martinez-Fernandez A, Faustino RS, et al. Nuclear reprogramming with c-Myc potentiates glycolytic capacity of derived induced pluripotent stem cells. J Cardiovasc Transl Res 2013;6:10-21.
83. Das PK, Pillai S, Rakib MA, et al. Plasticity of cancer stem cell: origin and role in disease progression and therapy resistance. Stem Cell Rev Rep 2020;16:397-412.
84. Hirpara J, Eu JQ, Tan JKM, et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol 2019;25:101076.
85. Dar S, Chhina J, Mert I, et al. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci Rep 2017;7:8760.
86. Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med 2009;13:2780-6.
87. Zhou Y, Zhou Y, Shingu T, et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem 2011;286:32843-53.
88. Schornack PA, Gillies RJ. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 2003;5:135-45.
89. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer 2017;17:577-93.
90. Riemann A, Rauschner M, Gießelmann M, Reime S, Haupt V, Thews O. Extracellular acidosis modulates the expression of Epithelial-Mesenchymal Transition (EMT) markers and adhesion of epithelial and tumor cells. Neoplasia 2019;21:450-8.
91. Linden C, Corbet C. Therapeutic targeting of cancer stem cells: integrating and exploiting the acidic niche. Front Oncol 2019;9:159.
92. Andreucci E, Peppicelli S, Ruzzolini J, et al. The acidic tumor microenvironment drives a stem-like phenotype in melanoma cells. J Mol Med (Berl) 2020;98:1431-46.
93. Ruzzolini J, Peppicelli S, Andreucci E, et al. Everolimus selectively targets vemurafenib resistant BRAFV600E melanoma cells adapted to low pH. Cancer Lett 2017;408:43-54.
94. Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009;15:501-13.
95. Khacho M, Tarabay M, Patten D, et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun 2014;5:3550.
96. Corbet C, Bastien E, Santiago de Jesus JP, et al. TGF-β2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun 2020;11:454.
97. Li J, Condello S, Thomes-Pepin J, et al. Lipid Desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017;20:303-314.e5.
98. Thews O, Nowak M, Sauvant C, Gekle M. Hypoxia-induced extracellular acidosis increases p-glycoprotein activity and chemoresistance in tumors in vivo via p38 signaling pathway. Adv Exp Med Biol 2011;701:115-22.
99. Cheng GM, To KK. Adverse cell culture conditions mimicking the tumor microenvironment upregulate ABCG2 to mediate multidrug resistance and a more malignant phenotype. ISRN Oncol 2012;2012:746025.
100. Smith AG, Macleod KF. Autophagy, cancer stem cells and drug resistance. J Pathol 2019;247:708-18.
101. Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016;85:152-67.
102. Corbet C, Ragelle H, Pourcelle V, et al. Delivery of siRNA targeting tumor metabolism using non-covalent PEGylated chitosan nanoparticles: Identification of an optimal combination of ligand structure, linker and grafting method. J Control Release 2016;223:53-63.
103. Fitzwalter BE, Towers CG, Sullivan KD, et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell 2018;44:555-65.e3.
104. Liang C, Dong Z, Cai X, et al. Hypoxia induces sorafenib resistance mediated by autophagy via activating FOXO3a in hepatocellular carcinoma. Cell Death Dis 2020;11:1017.
105. Ho CJ, Gorski SM. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers (Basel) 2019;11:1775.
106. Ryoo IG, Choi BH, Ku SK, Kwak MK. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol 2018;17:246-58.
107. Jang JE, Eom JI, Jeung HK, et al. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J Exp Clin Cancer Res 2020;39:66.
108. Yao N, Wang C, Hu N, et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis 2019;10:232.
109. Yan C, Li T-S. Dual role of mitophagy in cancer drug resistance. Anticancer Res 2018;38:617-21.
110. Liu K, Lee J, Kim JY, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell 2017;68:281-92.e5.
111. Yan C, Luo L, Guo CY, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett 2017;388:34-42.
112. Tam SY, Wu VWC, Law HKW. Hypoxia-induced epithelial-mesenchymal transition in cancers: HIF-1α and beyond. Front Oncol 2020;10:486.
113. Gjorevski N, Boghaert E, Nelson CM. Regulation of epithelial-mesenchymal transition by transmission of mechanical stress through epithelial tissues. Cancer Microenviron 2012;5:29-38.
114. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol 2017;11:805-23.
115. Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 2016;21:965.
116. Jolly MK, Somarelli JA, Sheth M, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther 2019;194:161-84.
117. Bocci F, Gearhart-Serna L, Boareto M, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Natl Acad Sci U S A 2019;116:148-57.
118. Wilson MM, Weinberg RA, Lees JA, Guen VJ. Emerging mechanisms by which EMT programs control stemness. Trends Cancer 2020;6:775-80.
119. Wang Z, Sun H, Provaznik J, Hackert T, Zöller M. Pancreatic cancer-initiating cell exosome message transfer into noncancer-initiating cells: the importance of CD44v6 in reprogramming. J Exp Clin Cancer Res 2019;38:132.
120. Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res 2002;307:1-14.
121. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741-51.
122. Tripathi V, Shin JH, Stuelten CH, Zhang YE. TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 2019;38:3185-200.
123. Xu X, Zhang L, He X, et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun 2018;502:160-5.
124. Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013;154:61-74.
125. Oshimori N, Oristian D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015;160:963-76.
126. Zhu X, Chen L, Liu L, Niu X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front Oncol 2019;9:1044.
127. Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2011;2:e179.
128. Dong J, Zhai B, Sun W, Hu F, Cheng H, Xu J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS One 2017;12:e0185088.
129. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr 2015;9:317-24.
130. Karaosmanoğlu O, Banerjee S, Sivas H. Identification of biomarkers associated with partial epithelial to mesenchymal transition in the secretome of slug over-expressing hepatocellular carcinoma cells. Cell Oncol (Dordr) 2018;41:439-53.
131. Roh YG, Mun MH, Jeong MS, et al. Drug resistance of bladder cancer cells through activation of ABCG2 by FOXM1. BMB Rep 2018;51:98-103.
132. Yochum ZA, Cades J, Wang H, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019;38:656-70.
133. Paranjape AN, Soundararajan R, Werden SJ, et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene 2016;35:5963-76.
134. Sun S, Yang X, Qin X, Zhao Y. TCF4 promotes colorectal cancer drug resistance and stemness via regulating ZEB1/ZEB2 expression. Protoplasma 2020;257:921-30.
135. Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015;14:481-7.
136. Zhang X, Zhang Z, Zhang Q, et al. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis 2018;9:57.
137. Zhang P, Wei Y, Wang L, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol 2014;16:864-75.
138. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett 2013;341:41-5.
139. Poh ME, Liam CK, Rajadurai P, Chai CS. Epithelial-to-mesenchymal transition (EMT) causing acquired resistance to afatinib in a patient with epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma. J Thorac Dis 2018;10:E560-3.
140. Namba K, Shien K, Takahashi Y, et al. Activation of AXL as a preclinical acquired resistance mechanism against osimertinib treatment in EGFR-mutant non-small cell lung cancer cells. Mol Cancer Res 2019;17:499-507.
141. Zhou J, Wang J, Zeng Y, Zhang X, Hu Q, Zheng J, et al. Implication of epithelial-mesenchymal transition in IGF1R-induced resistance to EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget 2015;6:44332-45.
142. Sato H, Yamamoto H, Sakaguchi M, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci 2018;109:3183-96.
143. Otsuki Y, Saya H, Arima Y. Prospects for new lung cancer treatments that target EMT signaling. Dev Dyn 2018;247:462-72.
144. Cho ES, Kang HE, Kim NH, Yook JI. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch Pharm Res 2019;42:14-24.
145. Wu X, Wu Q, Zhou X, Huang J. SphK1 functions downstream of IGF-1 to modulate IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells: A preliminary in vitro study. J Cancer 2019;10:4264-9.
146. May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011;13:202.
147. Jabbarzadeh Kaboli P, Salimian F, Aghapour S, et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer - a comprehensive review from chemotherapy to immunotherapy. Pharmacol Res 2020;156:104806.
148. Zhang Z, Han H, Rong Y, et al. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J Exp Clin Cancer Res 2018;37:291.
149. Mohapatra P, Shriwas O, Mohanty S, et al. CMTM6 drives cisplatin resistance by regulating Wnt signaling through the ENO-1/AKT/GSK3β axis. JCI Insight 2021;6:143643.
150. Yuan Z, Liang X, Zhan Y, et al. Targeting CD133 reverses drug-resistance via the AKT/NF-κB/MDR1 pathway in colorectal cancer. Br J Cancer 2020;122:1342-53.
151. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011;8:97-106.
152. Gu JW, Rizzo P, Pannuti A, Golde T, Osborne B, Miele L. Notch signals in the endothelium and cancer "stem-like" cells: opportunities for cancer therapy. Vasc Cell 2012;4:7.
153. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res 2017;36:41.
154. Lee CW, Raskett CM, Prudovsky I, Altieri DC. Molecular dependence of estrogen receptor-negative breast cancer on a notch-survivin signaling axis. Cancer Res 2008;68:5273-81.
155. Huang J, Chen Y, Li J, et al. Notch-1 Confers Chemoresistance in lung adenocarcinoma to taxanes through AP-1/microRNA-451 mediated regulation of MDR-1. Mol Ther Nucleic Acids 2016;5:e375.
156. Sun L, Ke J, He Z, et al. HES1 Promotes colorectal cancer cell resistance to 5-Fu by inducing of EMT and ABC transporter proteins. J Cancer 2017;8:2802-8.
157. Hu S, Fu W, Li T, et al. Antagonism of EGFR and Notch limits resistance to EGFR inhibitors and radiation by decreasing tumor-initiating cell frequency. Sci Transl Med 2017;9:eaag0339.
158. Baker A, Wyatt D, Bocchetta M, et al. Notch-1-PTEN-ERK1/2 signaling axis promotes HER2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018;37:4489-504.
159. Yang Z, Guo L, Liu D, Sun L, Chen H, Deng Q, et al. Acquisition of resistance to trastuzumab in gastric cancer cells is associated with activation of IL-6/STAT3/Jagged-1/Notch positive feedback loop. Oncotarget 2014;6:5072-87.
160. Usui T, Sakurai M, Umata K, et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int J Mol Sci 2018;19:1098.
161. Cho Y, Kim YK. Cancer stem cells as a potential target to overcome multidrug resistance. Front Oncol 2020;10:764.
162. Zhang H, Hu L, Cheng M, Wang Q, Hu X, Chen Q. The Hedgehog signaling pathway promotes chemotherapy resistance via multidrug resistance protein 1 in ovarian cancer. Oncol Rep 2020;44:2610-20.
163. Zhou XT, Ding J, Li HY, et al. Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J Cell Mol Med 2020;24:4298-311.
164. Po A, Citarella A, Catanzaro G, et al. Hedgehog-GLI signalling promotes chemoresistance through the regulation of ABC transporters in colorectal cancer cells. Sci Rep 2020;10:13988.
165. Periplocin from Cortex periplocae inhibits cell growth and down-regulates survivin and c-myc expression in colon cancer in vitro and in vivo via β-catenin/TCF signaling. Oncol Rep 2010:24.
166. Vesel M, Rapp J, Feller D, et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir Res 2017;18:52.
167. He L, Zhu H, Zhou S, et al. Wnt pathway is involved in 5-FU drug resistance of colorectal cancer cells. Exp Mol Med 2018;50:1-12.
168. Fukumoto T, Zhu H, Nacarelli T, et al. N6-Methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res 2019;79:2812-20.
169. Yamamoto TM, McMellen A, Watson ZL, et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol Carcinog 2019;58:1770-82.
171. Chen G, Gao C, Gao X, et al. Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther 2018;17:806-13.
172. Solberg NT, Waaler J, Lund K, Mygland L, Olsen PA, Krauss S. TANKYRASE inhibition enhances the antiproliferative effect of PI3K and EGFR inhibition, mutually affecting β-CATENIN and AKT signaling in colorectal cancer. Mol Cancer Res 2018;16:543-53.
173. Park YL, Kim HP, Cho YW, et al. Activation of WNT/β-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int J Cancer 2019;144:389-401.
174. Lee E, Ha S, Logan SK. Divergent androgen receptor and beta-catenin signaling in prostate cancer cells. PLoS One 2015;10:e0141589.
175. Zhang Z, Cheng L, Li J, et al. Inhibition of the Wnt/β-Catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res 2018;78:3147-62.
176. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445-64.
177. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther 2021;6:62.
178. Yeh DW, Huang LR, Chen YW, Huang CF, Chuang TH. Interplay between inflammation and stemness in cancer cells: the role of toll-like receptor signaling. J Immunol Res 2016;2016:4368101.
179. Qin J, Liu Y, Lu Y, et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep 2017;7:10592.
180. Mohiuddin IS, Wei SJ, Kang MH. Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim Biophys Acta Mol Basis Dis 2020;1866:165432.
181. Pires BR, Mencalha AL, Ferreira GM, et al. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS One 2017;12:e0169622.
182. Zou K, Li Z, Zhang Y, et al. β-Elemene enhances radiosensitivity in non-small-cell lung cancer by inhibiting epithelial-mesenchymal transition and cancer stem cell traits via Prx-1/NF-kB/iNOS signaling pathway. Aging (Albany NY) 2020;13:2575-92.
183. Li L, Wang T, Hu M, Zhang Y, Chen H, Xu L. Metformin overcomes acquired resistance to EGFR TKIs in EGFR-Mutant lung cancer via AMPK/ERK/NF-κB signaling pathway. Front Oncol 2020;10:1605.
184. Unver N. Macrophage chemoattractants secreted by cancer cells: Sculptors of the tumor microenvironment and another crucial piece of the cancer secretome as a therapeutic target. Cytokine Growth Factor Rev 2019;50:13-8.
185. Hu YB, Yan C, Mu L, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019;38:1951-65.
186. Bu L, Baba H, Yasuda T, Uchihara T, Ishimoto T. Functional diversity of cancer-associated fibroblasts in modulating drug resistance. Cancer Sci 2020;111:3468-77.
187. Hu JL, Wang W, Lan XL, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer 2019;18:91.
188. Lv J, Feng ZP, Chen FK, et al. M2-like tumor-associated macrophages-secreted Wnt1 and Wnt3a promotes dedifferentiation and metastasis via activating β-catenin pathway in thyroid cancer. Mol Carcinog 2021;60:25-37.
189. Chen Y, Tan W, Wang C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco Targets Ther 2018;11:3817-26.
190. Wang N, Liu W, Zheng Y, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-κB/SOX4 signaling. Cell Death Dis 2018;9:880.
191. Zhou K, Cheng T, Zhan J, et al. Targeting tumor-associated macrophages in the tumor microenvironment. Oncol Lett 2020;20:234.
192. Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int 2005;5:30.
193. Nieminen AI, Eskelinen VM, Haikala HM, et al. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A 2013;110:E1839-48.
194. Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med 2015;5:a006098.
196. Pecqueur C, Oliver L, Oizel K, Lalier L, Vallette FM. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol 2013;2013:805975.
197. Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029-33.
198. Zhou Y, Tozzi F, Chen J, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res 2012;72:304-14.
199. Soo JS, Ng CH, Tan SH, et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis 2015;20:1373-87.
200. Wang H, Feng Z, Qin Y, Wang J, Xu B. Nucleopeptide assemblies selectively sequester ATP in cancer cells to increase the efficacy of doxorubicin. Angew Chem 2018;130:5025-9.
201. Zheng X, Andruska N, Lambrecht MJ, et al. Targeting multidrug-resistant ovarian cancer through estrogen receptor α dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget 2018;9:14741-53.
202. Liu G, Wang L, Liu J, et al. Engineering of a core-shell nanoplatform to overcome multidrug resistance via ATP deprivation. Adv Healthc Mater 2020;9:e2000432.
203. Feng LL, Cai YQ, Zhu MC, Xing LJ, Wang X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int 2020;20:110.
204. Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633-7.
205. Wang H, Gao Z, Liu X, et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat Commun 2018;9:562.
206. Campos-Contreras ADR, Díaz-Muñoz M, Vázquez-Cuevas FG. Purinergic signaling in the hallmarks of cancer. Cells 2020;9:1612.
207. Aroua N, Boet E, Ghisi M, et al. Extracellular ATP and CD39 activate cAMP-mediated mitochondrial stress response to promote cytarabine resistance in acute myeloid leukemia. Cancer Discov 2020;10:1544-65.
208. Li XY, Moesta AK, Xiao C, et al. Targeting CD39 in cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov 2019;9:1754-73.
209. Elaskalani O, Falasca M, Moran N, Berndt MC, Metharom P. The role of platelet-derived ADP and ATP in promoting pancreatic cancer cell survival and gemcitabine resistance. Cancers (Basel) 2017;9:142.
210. Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017;36:293-303.
211. Gilbert SM, Oliphant CJ, Hassan S, et al. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019;38:194-208.
212. Arnaud-Sampaio VF, Rabelo ILA, Ulrich H, Lameu C. The P2X7 receptor in the maintenance of cancer stem cells, chemoresistance and metastasis. Stem Cell Rev Rep 2020;16:288-300.
213. Kepp O, Loos F, Liu P, Kroemer G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol Rev 2017;280:83-92.
214. Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009;15:1170-8.
215. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97-111.
216. Ohta A. A Metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 2016;7:109.
217. Pietrocola F, Pol J, Kroemer G. Fasting improves anticancer immunosurveillance via autophagy induction in malignant cells. Cell Cycle 2016;15:3327-8.
218. Luo Y, Qiao B, Zhang P, et al. TME-activatable theranostic nanoplatform with ATP burning capability for tumor sensitization and synergistic therapy. Theranostics 2020;10:6987-7001.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zhang H, Steed A, Co M, Chen X. Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer. Cancer Drug Resist 2021;4:684-709. http://dx.doi.org/10.20517/cdr.2021.32
AMA Style
Zhang H, Steed A, Co M, Chen X. Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer. Cancer Drug Resistance. 2021; 4(3): 684-709. http://dx.doi.org/10.20517/cdr.2021.32
Chicago/Turabian Style
Zhang, Haiyun, Alexander Steed, Milo Co, Xiaozhuo Chen. 2021. "Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer" Cancer Drug Resistance. 4, no.3: 684-709. http://dx.doi.org/10.20517/cdr.2021.32
ACS Style
Zhang, H.; Steed A.; Co M.; Chen X. Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer. Cancer Drug Resist. 2021, 4, 684-709. http://dx.doi.org/10.20517/cdr.2021.32
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 27
likes
Like This Article 27
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.