
Drug resistance in targeted cancer therapies with RAF inhibitors

Abstract
Hyperactive RAS/RAF/MEK/ERK signaling has a well-defined role in cancer biology. Targeting this pathway results in complete or partial regression of most cancers. In recent years, cancer genomic studies have revealed that genetic alterations that aberrantly activate the RAS/RAF/MEK/ERK signaling mainly occur on RAF or upstream, which motivated the extensive development of RAF inhibitors for cancer therapy. Currently, the first-generation RAF inhibitors have been approved for treating late-stage cancers with BRAF(V600E) mutations. Although these inhibitors have achieved promising outcomes in clinical treatments, their efficacy is abolished by quick-rising drug resistance. Moreover, cancers with hyperactive RAS exhibit intrinsic resistance to these drugs. To resolve these problems, the second-generation RAF inhibitors have been designed and are undergoing clinical evaluations. Here, we summarize the recent findings from mechanistic studies on RAF inhibitor resistance and discuss the critical issues in the development of next-generation RAF inhibitors with better therapeutic index, which may provide insights for improving targeted cancer therapy with RAF inhibitors.
Keywords
INTRODUCTION
RAS/RAF/MEK/ERK signaling plays a determinant role in cell fate and is tightly regulated in normal cells[1-3]. Hyperactivation of RAS/RAF/MEK/ERK signaling by germline mutations induces developmental disorders termed as RASopathies[4,5], whereas that by somatic alterations causes human cancers[6,7]. In human cancers, the majority of genetic alterations that aberrantly activate this pathway occur on receptor tyrosine kinases (RTKs), NF1, RAS, and RAF[8-10]. Oncogenic RTK alterations can be effectively targeted by tyrosine kinase inhibitors (TKIs) or neutralizing antibodies[11-14], while active RAS mutations except ~10% G12C have been thought as “undruggable” targets[15,16]. As the first kinase of this signaling cascade, RAF is thus an ideal target for developing drugs against cancers with RAS or RAF mutations as well as loss-of-function of NF1 tumor suppressor[17-20]. Indeed, the first-generation RAF inhibitors have been developed and applied to clinical cancer therapy[21-23]. Although these inhibitors have no effect on cancers with active RAS, they have achieved promising outcomes on cancers with BRAF(V600E), a dominant mutation of RAF in cancer genomes. However, their efficacy is completely abrogated by acquired resistance in 6-7 months[24-27]. Mechanistic studies have suggested that both intrinsic and acquired drug resistance arises largely from the unexpected homo- or hetero-dimerization of RAF proteins[28-33]. These findings indicate that the regulation of RAS/RAF/MEK/ERK signaling is much more complicated than anticipated and its understanding will help us develop novel RAF inhibitors with long-term efficacy, broad coverage, and minor side-effects. In this review, we discuss key issues in targeted cancer therapy with RAF inhibitors and the development of next-generation RAF inhibitors, which may facilitate targeted therapy against cancers with RAS/RAF mutations.
THE RAS/RAF/MEK/ERK SIGNALING IN CELL BIOLOGY
The RAS/RAF/MEK/ERK pathway is fundamental to cellular proliferation, differentiation, and survival in response to growth signals from extracellular environment[3,34]. The discovery of this pathway dates back to the 1970s when studies of viral oncogenes from sarcoma viruses identified RAS small GTPases[35-38]. Shortly after the discovery of RAS, an N-terminal truncated version of CRAF (RAF1) was isolated from murine and avian retroviruses[39,40]. In 1984, it was uncovered that epidermal growth factor (EGF) activated RAS and that RAF functioned downstream of RAS[41-43]. Subsequently, the EGFR-RAS-RAF link was further confirmed by studies done in Drosophila and C. elegans[44,45]. Downstream of RAF, MEK and ERK were identified as cytoplasmic protein kinases activated by mitogens in the 1990s[46-50]. Thereafter, RAF was identified as the upstream kinase of MEK in 1992 as well as the direct effector of RAS in 1993, which delineates the whole RAS/RAF/MEK/ERK signaling pathway[51-55].
The properties of components in the RAS/RAF/MEK/ERK signaling pathway
The first component of the RAS/RAF/MEK/ERK signaling pathway, RAS small GTPase has four isoforms HRAS, NRAS, KRAS4A, and KRAS4B, which are encoded by three genes[2]. RAS isoforms are highly homologous except their carboxyl-terminal hypervariable regions that undergo various post-translational modifications and anchor individual isoforms to different intracellular compartments[56,57]. Similar to other small GTPases, the activity of RAS proteins is primarily regulated by the GTP/GDP cycle. Active RAS proteins associate with GTP and adopt an active conformation that allows them to bind to and activate effectors, while inactive RAS proteins bound with GDP have altered conformation that impedes such interactions. In the GTP/GDP cycle of RAS proteins, the dislodging of GDP for GTP on RAS proteins is catalyzed by guanine exchange factor (GEF) proteins (i.e., Sos), whereas the hydrolysis of GTP into GDP by RAS proteins is facilitated by RAS GTPase activating proteins (GAPs) (i.e., NF1). Thus, the relative activity of GEF proteins vs. GAP proteins in cells shifts the GTP/GDP cycle and determines the output of RAS activity. However, it has to be noted that different RAS isoforms have differential propensities in activity regulation as well as tissue expression patterns and effector preferences[58], which leads to their distinct physiological and pathological functions.
The direct effector of RAS, RAF/KSR family kinase includes three RAF isoforms (ARAF, BRAF, and CRAF) and two close pseudokinases (KSR1 and KSR2)[59]. All RAF proteins have highly homologous sequences and share three conserved regions (CR): CR1, CR2, and CR3. CR1 contains a RAS binding domain (RBD) and a Cys-rich domain[60,61], while CR2 is characterized by Ser/Thr-rich sequence. CR3 is a putative kinase domain flanked with an N-terminal acidic motif (NTA) and a C-terminal regulatory tail[62-64]. In the inactive state, CR1 docks on CR3 and thus inhibits its kinase activity[65,66]. The association of RAS-GTP with CR1 relieves this inhibitory interaction and drives the dimerization of CR3, which further triggers the catalytic activity of CR3. Despite having a similar regulatory mechanism, RAF isoforms exhibit differential activities with an order as: BRAF > CRAF > ARAF. This likely arises from the diversity in their NTA motif and APE motif[62,67]. Different from RAF proteins, KSR1 and KSR2 have a coiled-coil fused sterile α-motif and Pro-rich stretch at their N-terminus, which are responsible for their plasma membrane recruitment, and lack the catalytic lysine in VAIK motif of kinase domain. By virtue of their low kinase activity and association with MEK and ERK, KSR proteins had been thought of as scaffolds for a long time[68,69]. However, recent studies have revealed that KSR proteins function as allosteric activators to stimulate the catalytic activity of RAF proteins through dimerization[62,70,71]. The allosteric activity of RAF/KSR family kinase is indispensable for their function[72,73].
The second level of kinase in the RAS/RAF/MEK/ERK signaling pathway is MEK, a dual specific kinase, which includes two isoforms: MEK1 and MEK2[74]. These two kinases have a simple structure that consists of a short regulatory N-terminus and a canonical kinase domain. However, their regulatory mechanism(s) is not completely understood. At present, what we have learned about their regulation mainly includes: (1) the N-terminal regulatory region of MEK1/2 forms a helix that locks the kinase domain in an inactive conformation, and disrupting this inhibitory helix triggers the catalytic activity of MEK1/2[75]; (2) RAF/MEK and MEK/MEK interactions are required for MEK activation by RAF, in which MEK can be phosphorylated by RAF or itself[67,76]; and (3) even after activation by RAF, MEK needs to function as a dimer to phosphorylate downstream ERK[76].
The terminal kinase of RAS/RAF/MEK/ERK signaling, ERK has two highly homologous isoforms (ERK1 and ERK2) as the upstream activator MEK does[77]. ERK isoforms comprise a central kinase domain flanked by short N- and C-terminal tails that regulate their activity, subcellular location, and substrate specificity[78]. ERKs also have redundant functions albeit with different expression patterns as MEKs do[79]. However, unlike RAFs and MEKs that have only certain substrates, ERKs recognize and phosphorylate over 200 proteins including transcription factors, protein kinases and phosphatases, and other structural or enzymatic proteins, which regulate diverse cell functions as well as compose a negative feedback loop for turning off the RAS/RAF/MEK/ERK signaling pathway[80,81].
The regulation of RAS/RAF/MEK/ERK signaling under physiological conditions
The regulation of RAS/RAF/MEK/ERK signaling is very complex and involves complicated intermolecular interactions and phosphorylation events[20,82,83]. Although the framework of RAS/RAF/MEK/ERK signaling was discovered almost three decades ago, the approximate regulatory network for this signaling was uncovered only recently [Figure 1]. In quiescent cells, RAF proteins are immobilized in inactive conformation by 14-3-3 dimers through intramolecular association and form face-to-face heterodimers with MEKs[84,85]. The engagement of RTKs with their ligands triggers their kinase activity and results in the autophosphorylation of intracellular carboxyl-tails, which serve as docking sites for recruiting Grb2 and Sos1 to plasma membrane[86]. Further, active Sos1 proteins on the plasma membrane exert their GEF activity towards RAS, which in turn actives RAS proteins to recruit RAF/MEK heterodimers to the plasma membrane through the RBDs of RAF proteins[87-89]. Since active RAS proteins form oligomers or nanoclusters, RAF proteins are enriched on the plasma membrane, which promotes their side-to-side dimerization[90-93]. RAF dimerization on the plasma membrane can be further enhanced by the switching of intramolecular into intermolecular associations of 14-3-3 dimers with RAF proteins, which eventually turns on the catalytic activity of RAF proteins[64,94]. Active RAF dimers autophosphorylate their activation loops and, at the same time, loosen RAF/MEK interaction to facilitate the assembly of MEK homodimers on their surface[62,76]. Once MEK proteins form face-to-face homodimers that dock on RAF dimers, RAF proteins will phosphorylate their activation loops or trigger the cross-phosphorylation of two protomers in MEK homodimers, which ultimately activates MEK dimers[76]. The assembly of MEK dimers on the surface of RAF dimers also reversely stabilizes RAF dimers, which illustrates why active MEKs can transactivate RAFs[62,95]. Subsequently, active MEK dimers release from or continuously dock on RAF dimers to phosphorylate ERKs[76]. Thereafter, phospho-ERKs travel to different intracellular compartments and phosphorylate their substrates as monomers or dimers, which gives rise to diverse cell responses[3,76]. Under physiological condition, active ERKs also drive a negative feedback loop through inhibitory phosphorylating EGFR, Sos1, and RAF, as well as upregulating dual-specificity phosphatases and Sprouty proteins, which shuts off the whole signaling pathway and returns cells back to quiescent status[80,81,96]. It must be noted that this general regulatory network may be fine-tuned by the properties of components that constitute this signaling pathway. For instance, in contrast to BRAF, CRAF and ARAF do not form heterodimers with MEK in the inactive state[84], and how MEKs are delivered to these two RAF proteins for activation remains an intriguing question in current studies.
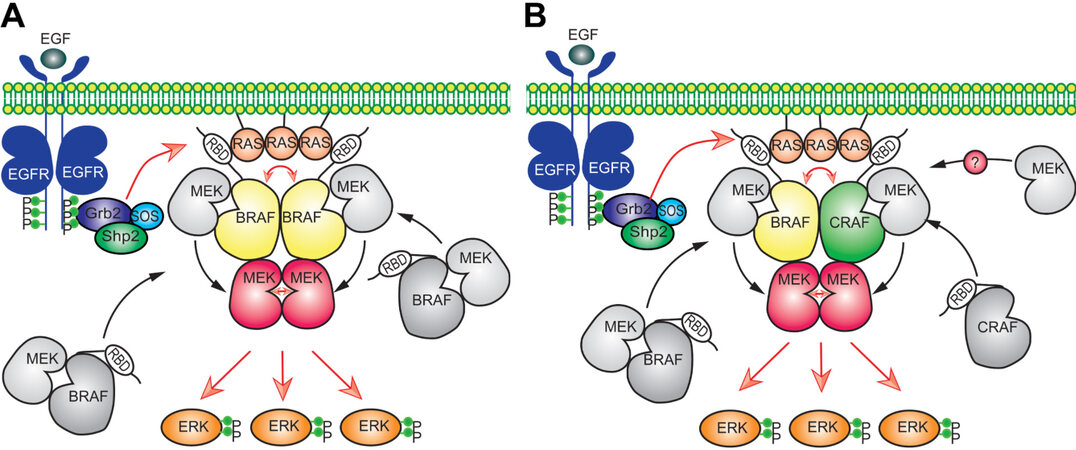
Figure 1. The regulatory mechanism of RAS/RAF/MEK/ERK signaling pathway. RAF and MEK exist as heterodimers in cytosol of quiescent cells (A). Upon Ras activation, these heterodimers are recruited to the plasma membrane where they form transient tetramers through side-to-side dimerization of RAF. Further, this side-to-side RAF dimerization activates RAF themselves and also loosens RAF/MEK heterodimers, which facilitate the MEK dimerization on the surface of RAF dimer. Subsequently, active RAF dimer phosphorylates MEK dimer docking on its surface or promotes intra-dimer MEK transphosphorylation. Thereafter, active MEK dimer docking on RAF dimer or released from RAF dimer phosphorylates and activates ERK. Since, unlike BRAF, CRAF and ARAF do not heterodimerize with MEK in inactive state, how MEK is delivered to CRAF and ARAF for activation remains unknown in current studies (B). EGF: Epidermal growth factor; RBD: RAS binding domain.
HYPERACTIVE RAS/RAF/MEK/ERK SIGNALING IN CANCERS
The pivotal role of RAS/RAF/MEK/ERK signaling in cell biology has been highlighted by studies in developmental disorders and cancers even before the delineation of the entire pathway. Aberrant activation of RAS/RAF/MEK/ERK signaling by germline mutations causes developmental disorders termed as RASopathies that include neurofibromatosis type 1 (NF1), Noonan syndrome, Castello syndrome, and cardio-facio-cutaneous syndrome[4]. In contrast, dysregulating this pathway by somatic mutations results in cancers[19,20,97]. In this review, we focus on somatic mutations that directly hyperactivate RAS/RAF/MEK/ERK signaling in cancers [Figure 2].
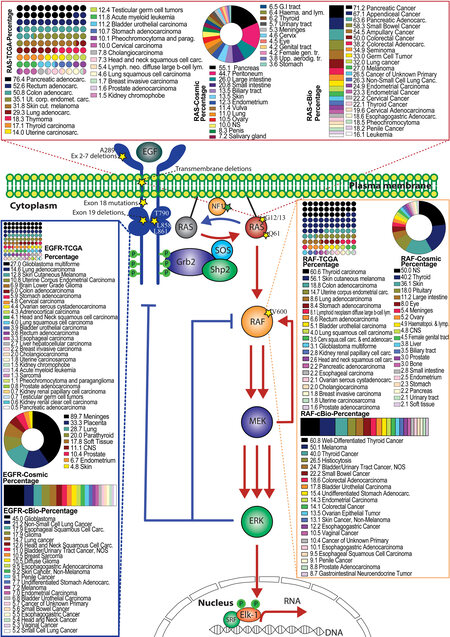
Figure 2. Genetic mutations that hyperactivate the RAS/RAF/MEK/ERK signaling pathway in human cancers. The RAS/RAF/MEK/ERK signaling is initiated on the plasma membrane where engagement of EGFR by its ligand activates Sos. In turn, Sos functions as a RAS-GEF to catalyze the GTP loading on RAS. Subsequently, RAS-GTP triggers the RAF/MEK/ERK kinase cascade and ultimately activates ERK. As the terminal kinase of this signaling cascade, active ERK phosphorylates numerous substrates and induces diverse cell responses. This signaling cascade is tightly regulated under physiological conditions through complicated mechanisms that include MEK-mediated positive feedback (red arrows) as well as NF1-driven GTP hydrolysis and ERK-mediated negative feedback (blue arrows). In cancer cells, this signaling cascade is mainly hyperactivated by genetic mutations on EGFR, NF1, RAS, and RAF. The hotspots for oncogenic mutations in these genes are labeled as yellow stars. The mutational prevalence of these genes in different type of cancers (calculated from TCGA, cBio, and COSMIC databases) are shown in boxes. TCGA data are shown in small circles forming square, while COSMIC data and cBio data are shown as a donut plot and a box graph, respectively. Numbers on the legends indicate the percentages of mutations in the particular cancer types. EGF: Epidermal growth factor.
RTK alterations
The first prominent target of oncogenic alterations that aberrantly activate RAS/RAF/MEK/ERK signaling is RTKs, particularly EGFRs. It is well established that EGFRs are activated through ligand engagement-driven dimerization of intracellular kinase domain[98-100]. Genetic alterations that trigger or bypass this process would hyperactivate EGFRs and thus drive tumorigenesis[101]. According to underlying activating mechanisms, oncogenic EGFR alterations can be classified as three groups: (1) overexpression of EGFRs that drives EGFR oligomerization independent of ligand association, which occurs in non-small cell lung cancer, glioblastoma, breast cancer, and pancreatic cancer[101-103]; (2) mutations in the extracellular ligand-binding domain (ECD), transmembrane domain (TMD), or juxtamembrane domain (JMD) of EGFRs that enhance the ligand affinity or bypass ligand binding, which are resident in gliomas, lung adenocarcinoma, breast cancer, and colorectal cancer[102,104]; and (3) constitutively active mutations in the kinase domain of EGFRs that mainly comprise β3-αC loop deletions (exon19del) and activation loop mutations (L858 in exon 21), which represent the majority of EGFR mutations in lung cancer and glioblastoma[105,106]. Targeting oncogenic EGFR alterations with neutralizing antibodies and TKIs has significantly changed the landscape of cancer treatment in recent years and has been comprehensively reviewed by many groups[107-109].
NF1 mutations
The second noticeable gene whose mutations hyperactivate RAS/RAF/MEK/ERK signaling in cancer is NF1 that encodes a RAS-GAP and functions as a tumor suppressor. NF1 was originally discovered in neurofibromatosis type 1, a common developmental disorder caused by germline mutations of NF1 gene. Somatic mutations of NF1 are responsible for ~10% of melanoma, lung cancer, glioblastoma, and bladder cancer cases[10]. Interestingly, NF1-mutated cancers have a heavier mutational burden and thus exhibit a better response to immunotherapy than those with RAS or RAF mutations[110,111].
RAS mutations
RAS is the most frequently mutated oncogene and has an impact on over 30% of cancers[112]. However, RAS mutations have isoform and tissue preferences, resulting in the enrichment of specific RAS mutations in certain cancers. For example, KRAS mutations are highly prevalent in pancreatic cancers (~90%), colorectal cancers (~35%), and lung cancers (~20%), while NRAS mutations exist in melanoma (~20%) and thyroid cancers (~20%)[113,114]. Furthermore, RAS mutations have two structural hotspots on G12/13 and Q61, which impairs RAS-GAP association or inhibits intrinsic GTPase activity of RAS proteins, and thus extends the half-life of RAS-GTP. Due to the extremely high affinity with GTP and the lack of a proper binding pocket for docking small molecular inhibitors, most RAS mutants are not druggable except G12C mutants that could be targeted with covalent small molecular inhibitors through docking in the switch II pocket and cross-linking with Cys12[115-119]. Therefore, targeting the downstream RAF/MEK/ERK kinase cascade has become a primary approach for treating cancers with non-G12C RAS mutations.
RAF mutations
Although the N-terminal truncated RAF (CRAF or RAF1) was first discovered as a retroviral oncogene in the early 1980s, oncogenic RAF mutations had not been observed in cancers until 2002[120]. Moreover, it is BRAF and not CRAF, ARAF, or KSR that is frequently mutated in cancer genomes[121]. A single point mutation (V600E) in the activation loop of BRAF represents > 90% of all cancer-related RAF mutations[17,20]. Mechanistic studies have suggested that oncogenic RAF mutations might be classified as three groups: (1) highly constitutively active mutants such as BRAF(V600E) that directly phosphorylate MEK and thus activate downstream signaling cascade; (2) kinase-dead or -impaired mutants such as BRAF(V471F) that function as allosteric activators and switch on downstream signaling through triggering the catalytic activity of wild-type RAFs; and (3) intermediate active mutants such as BRAF(G469A) that have enhanced dimer affinity and directly turn on downstream signaling as homodimers[122-124]. However, there are some unique RAF mutants such as BRAF(ΔNVTAP), BRAF(L505H), and BRAF(LLRins506) that do not fall into any of these categories[67,125-129], suggesting that oncogenic RAF mutations have diverse characters and complicated functional modes. Similar to RAS mutations, RAF mutations also have cancer-type preferences. Although RAF mutations exist in only ~7% of all cancers, they are highly prevalent in melanoma (~50%), thyroid cancer (~45%), and Langerhans cell histiocytosis (~50%)[17,20,130].
In contrast to dominant mutations of EGFR, NF1, RAS, and RAF, there are some rare activating mutations of Sos, MEK, and ERK in cancer genomes that constitutively turn on RAS/RAF/MEK/ERK signaling. The functional analysis of these rare mutations in cancer biology has been reviewed before[20,131], and we do not discuss them again here.
TARGET HYPERACTIVE RAS/RAF/MEK/ERK SIGNALING IN CANCERS WITH RAF INHIBITORS
Since oncogenic alterations that aberrantly activate RAS/RAF/MEK/ERK signaling mainly occur on RAFs, particularly BRAF or upstream, this family of kinases has been considered an ideal target for cancer drug development. Given the high prevalence of BRAF(V600E) in cancer genomes, Tsai et al.[21] resolved its high-resolution crystal structure and further utilized a structure-guided drug discovery approach to identify a potent and selective small molecule inhibitor of BRAF(V600E), PLX4720. This inhibitor exhibited a strong antitumor activity on BRAF(V600E)-harboring melanoma in vitro and in vivo[21], and its analog with more favorable pharmacokinetics, vemurafenib (PLX4032), was first approved for treating advance melanomas and then for other cancers with BRAF(V600E) mutation [Table 1][24,132,133]. In clinical therapy, vemurafenib exhibited an exceptional efficacy with manageable side effects on cancers with BRAF(V600E) mutation as a single agent or combined with cobimetinib (MEK inhibitor), although drug resistance occurs after 6-8 months of administration.
Small molecule RAF inhibitors that have been applied to clinical cancer therapy or are undergoing development
| Drug name/aliases | Structure | Remark | |
| First generation (Type I1/2) RAF inhibitor | Vemurafenib PLX4032 Zelboraf | 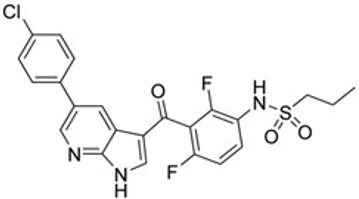 | Classification: Type I1/2 RAF inhibitor. PFS: 6.8 months (single agent), 9.9 months (combined with cobimetinib). ORR: 45% (single agent), 68% (combined with cobimetinib). OS: 15.9 months (median, single agent), 81% at 9 months (combined with cobimetinib). Side effects: new primary cutaneous malignancies (lower secondary cutaneous cancers in combination with MEKi). Clinically available (FDA approved in 2011 for single agent and in 2015 for combination with cobimetinib) |
| Dabrafenib GSK-2118436 TAFINLAR | 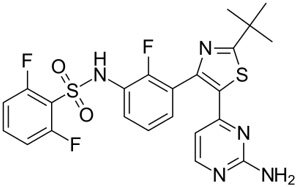 | Classification: Type I1/2 RAF inhibitor. PFS: 8.8 months (single agent), 11.0 months (combined with trametinib). ORR: 53% (single agent), 69% (combined with trametinib). OS (median): 18.7 months (single agent), 25.1 months (combined with trametinib). Side effects: new cutaneous and non-cutaneous malignancies (lower secondary cutaneous cancers in combination with MEKi). Clinically available (FDA approved in 2014 for single agent and in 2018 for combination with trametinib) (refer to trametinib) | |
| Encorafenib Lgx818 Braftovi |  | Classification: Type I RAF inhibitor. PFS: 9.6 months (single agent), 14.9 months (combined with binimetinib). ORR: 52% (single agent) (better than vemurafenib within the same cohort 41%), 64% (combined with binimetinib). OS (median): 23.5 months (single agent), 33.6 months (combined with binimetinib). Side effects: new primary malignancies, cutaneous and non-cutaneous. Clinically available (FDA approved in 2018 for combination with binimetinib and in 2020 for combination with cetuximab) | |
| Second generation (Type II) RAF Inhibitor | PLX8394 |  | Classification: RAF dimer breaker. Induces a conformation unfavorable for dimerization. Partial response rate: 22%. Side effects: diarrhea. Phase I/II is recruiting for advanced unresectable solid tumors (NCT02428712) |
| CCT3833/BAL3833 |  | Classification: Pan-RAF/SRC kinase inhibitor. Phase I for solid tumors was completed (NCT02437227) | |
| LY3009120 |  | Classification: Pan-RAF, RAF dimer inhibitor. Inhibits both RAF protomers. Side effects: fatigue, nausea, low appetite, and rash. Phase I for advanced or metastasized tumors was terminated (NCT02014116) | |
| BGB-283 Lifirafenib BEIGENE-283 | 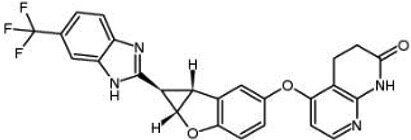 | Classification: Pan-RAF inhibitor. Side effects: hypertension and fatigue. Phase I is recruiting for advanced or refractory solid tumors (NCT03905148) | |
| Belvarafenib | 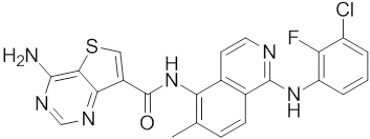 | Classification: Pan-RAF inhibitor. Side effects: rash, dermatitis, acneiform, and pyrexia. Phase I is recruiting for NRAS mutant advanced melanoma (NCT04835805) | |
| TAK-580 | 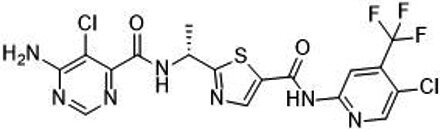 | Classification: Pan-RAF inhibitor. Side effects: rash and increased bilirubin in once-a-week treatment for ST or melanoma detected in Phase I. Phase I is recruiting for gliomas and other tumors (NCT03429803) | |
| Advanced RAF inhibitor | Trametinib Gsk1120212 | 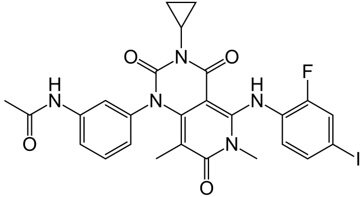 | Classification: RAF/MEK heterodimer breaker. Binds to the RAF/MEK interface. Prevents RAF/MEK association and MEK phosphorylation. PFS: 4.8 months (single agent), 11.0 months (combined with dabrafenib) ORR: 22% (single agent), 68% (combined with dabrafenib). OS (median): 15.6 months (single agent), 25.1 months (combined with dabrafenib). Side effects: hemorrhage, colitis, and venous thromboembolism. Clinically available (FDA approved in 2013 for single agent, also approved for combination therapy with dabrafenib in 2018) |
| RO5126766 |  | Classification: RAF/MEK complex inhibitor. Docks on RAF/MEK dimer interface and traps RAF/MEK complex in inactive state. Side effects: rash, elevated CPK, and diarrhea. Phase I is recruiting for solid tumors and multiple myeloma (NCT02407509) | |
| Trametiglue |  | Classification: RAF/MEK complex inhibitor. Derived from structure of trametinib. Docks on RAF/MEK dimer interface and traps RAF/MEK complex in inactive state. Clinical trial has not begun | |
Shortly after the discovery of vemurafenib, a second RAF inhibitor, dabrafenib (GSK2118436), was developed by the GSK group [Table 1][22]. Although dabrafenib is a reversible competitive inhibitor similar to vemurafenib, it is more selective for BRAF(V600E) over wild-type BRAF and other RAF kinases than vemurafenib[22]. Moreover, dabrafenib has a similar potency against BRAF(V600E) and BRAF(V600K)[134]. In clinical trials, dabrafenib exhibited a similar efficacy as well as side effects for treating advance melanoma with BRAF(V600E) mutation, but better activity on brain metastasis in contrast to vemurafenib[135-137], although no direct comparison between these two inhibitors has been carried out to reach a definitive conclusion. As for vemurafenib, the efficacy of dabrafenib was further extended when combined with a MEK inhibitor (trametinib)[138,139], which led to approval of dabrafenib plus trametinib, as a combinatorial agent for treating advance melanoma harboring BRAF(V600E/K) mutations in clinical therapy.
Although the combinatorial regimens of vemurafenib plus cobimetinib and dabrafenib plus trametinib have achieved promising outcomes at the initial therapeutic phase, drug resistance and adverse effects limit their effectiveness. To achieve better efficacy and tolerability of RAF inhibitors through optimizing their pharmacological properties, a third RAF inhibitor, encorafenib (LGX818), was developed by the Novartis group [Table 1][23]. In contrast to vemurafenib and dabrafenib, this inhibitor has over 10-fold slower off-rate and a much shorter serum half-life[23,140], which delays drug resistance and improves tolerability. Furthermore, encorafenib has much higher specificity than vemurafenib and dabrafenib, and it targets not only V600E but also V600K and V600D[141]. In clinical trials, encorafenib achieved significantly longer progression-free survival and fewer adverse effects on advance melanoma with V600 mutations as both monotherapy and combination therapy with the MEK inhibitor binimetinib, compared with vemurafenib[26,27,142]. In addition, encorafenib exhibited an excellent efficacy against metastatic colorectal cancers with BRAF(V600E) mutation when combined with cetuximab[143]. These trials led to the approval of encorafenib plus binimetinib or cetuximab for treating advance BRAF(V600E)-harboring cancers. Therefore, vemurafenib, dabrafenib, and encorafenib constitute the first-line agents against advance cancers with BRAF(V600E) mutation in current clinical therapy, which significantly changes the landscape of cancer therapy.
DRUG RESISTANCE IN TARGETED THERAPIES WITH RAF INHIBITORS
Although the first-generation RAF inhibitors achieved promising outcomes on treating BRAF(V600E)-harboring cancers, their efficacy is abrogated by quick-rising resistance within a median time of 6-7 months[24-27]. To understand molecular mechanisms that underlie RAF inhibitor resistance, extensive studies have been carried out. The overexpression/upregulation of RTKs such as PDGFRβ and EGRF was reported as the first type of alterations that led to RAF inhibitor resistance[144-146], which activates RAS and turns on CRAF-MEK-ERK signaling, and hence bypasses BRAF(V600E)-dependence for cancer cell growth. Downstream of RTKs, RAS mutations were identified as the second cause for RAF inhibitor resistance[144,147], which function similarly as RTK alterations. In other words, once cancer cells possess a high level of active Ras, the drug-loaded BRAF(V600E) will dimerize with CRAF and trigger its catalytic activity[28-30,32,33], which has been termed as the paradoxical effect of RAF inhibitors. Furthermore, N-terminal alternative splicing and overexpression of BRAF(V600E) were discovered in drug-resistant cancer patient samples[31,148,149]. This type of alterations enhances BRAF(V600E) homodimerization and impairs drug engagement. Unlike drug resistance that occurs in EGFR mutation-driven cancers or BCR-Abl-induced leukemia, secondary mutations such as gate-keeper mutations that cause drug resistance are rare in BRAF(V600E)-driven cancers. Thus far, only two cases (L505H and L514V) have been reported to confer drug resistance as secondary mutations in BRAF(V600E)-driven cancers[127,150]. As the direct effectors of RAF proteins, MEK also contributed to RAF inhibitor resistance, whose constitutively active mutations were identified in 3%-5% of cases[151,152]. Altogether, ~80% of drug resistant cases in BRAF(V600E)-harboring cancers arise from pathway reactivation of ERK signaling[151,152]. Although several mechanisms other than ERK reactivation have been suggested to be responsible for ~20% of drug resistance cases[153-156], none of them contributes to a significant portion of those cases.
Given the dominant role of RAF dimerization in the pathway reactivation of cancer cells upon RAF inhibitor treatment, a “monomer-dimer” hypothesis has been suggested to explain the majority of drug resistance cases in targeted therapy[31,157]. In this hypothesis, BRAF(V600E) was thought as a monomer to phosphorylate MEK, which has a high affinity for RAF inhibitors and hence can be inhibited by these drugs. In contrast, once BRAF(V600E) forms homodimers or heterodimers with other RAF proteins, its affinity for RAF inhibitors dramatically decreases and hence its catalytic activity cannot be blocked effectively by these drugs. This hypothesis was further strengthened by findings that BRAF mutants with in-frame deletions in
Recently, we and other groups identified a novel group of oncogenic BRAF mutants with in-frame insertions of LLR or VLR in the αC-β4 loop[129,161,162]. We found that these unique BRAF mutants had much lower dimer affinity than their wild-type counterpart or BRAF(V600E). Similar to other constitutively active BRAF mutants with low dimer affinity[67,163,164], they lost their catalytic activity upon purification from cells, which can be rescued by GST-fusion-enhanced dimerization. However, they exhibited a very strong resistance to all RAF inhibitors in clinical therapy or undergoing clinical trials in vitro and in vivo, indicating that the dimer affinity of RAF proteins is not directly correlated to RAF inhibitor resistance, although an enhanced dimerization could cause drug resistance. To uncover the molecular basis responsible for RAF inhibitor resistance, we determined the conformational alterations in these BRAF mutants through structure modeling and molecular dynamic simulation and found that the inserted LLR or VLR residues facilitated an assembly of large hydrophobic network that includes and stabilizes the regulatory spine (R-spine), a typical hydrophobic architecture of active protein kinases[165,166]. Since RAF inhibitor engagement broke the R-spine of BRAF[21,167,168], a stabilized R-spine would remarkably impair drug loading. Indeed, these BRAF mutants have much less affinity for RAF inhibitors, suggesting instead that a stabilized R-spine leads to drug resistance. Given that R-spine can be stabilized by different mutations via various manners, we then determined whether other BRAF mutants with a stabilized R-spine such as L485F and L505H[127,128] were resistant to RAF inhibitors, and found that all these mutants were resistant to RAF inhibitors with different extents related to their R-spine stability. The R-spine stability can also explain why BRAF(V600E) splicing variants or other mutants with high dimer affinity are resistant to RAF inhibitors: a strong dimerization would lead to a cooperative assembly of R-spine in two protomers of a dimer, and hence R-spine held in a tight dimer is more stable than that in a lax dimer, which impairs drug engagement. Altogether, our findings indicate that, instead of dimer affinity, R-spine stability is the root of RAF inhibitor resistance of RAF proteins.
DEVELOPING NEXT-GENERATION RAF INHIBITORS FOR OVERCOMING DRUG RESISTANCE
Since the majority of drug resistance in targeted therapies with first-generation RAF inhibitors was caused mainly by enhanced homo/heterodimerization of RAF proteins, two types of second-generation RAF inhibitors have been developed to resolve these issues: RAF dimer breakers (i.e., PLX8394) and pan-RAF inhibitors (i.e., BAL3833, LY3009120, BGB283, belvarafenib, and TAK-580) [Table 1]. RAF dimer breakers induce a conformation unfavorable for dimerization upon engagement with BRAF(V600E)[169] and hence impair homodimerization driven by N-terminal deletion or overexpression of BRAF(V600E). This type of inhibitors has exhibited promising efficacy in vivo against tumors that have alternative splicings or upregulation of BRAF(V600E) or other BRAF mutants with moderate dimer affinity [i.e., BRAF(G469A) and BRAF fusion mutants] and thus are resistant to first-generation RAF inhibitors[170,171]. Currently, this type of inhibitors is undergoing evaluation in stage I/II clinical trials. Unlike RAF dimer breakers, pan-RAF inhibitors are able to bind and block the catalytic activity of both protomers in RAF dimers[168,172,173]. This type of inhibitors may replace the Phe residue from DFG motif to form a much more stable hydrophobic network in RAF protomers upon engagement and is able to block the activity of RAF homo/heterodimers even with very high dimer affinity such as in BRAF(ΔNVTAP)[126]. Consistently, pan-RAF inhibitors have exhibited good efficacy against tumors that have active RAS mutations or high-affinity RAF dimer mutants in animal models, particularly when combined with MEK or ERK inhibitors[174-176]. However, since pan-RAF inhibitors target both wild-type and mutant RAF proteins, their therapeutic index should not be high as a single agent.
Although the second-generation RAF inhibitors might overcome resistance arising from RAF dimerization enhanced by BRAF alterations or active RAS, they can hardly block the activity of some emerging oncogenic BRAF mutants such as L505H and LLRins506[129]. As discussed above, both first- and second-generation RAF inhibitors disassemble the R-spine upon engagement and are unable to associate with RAF mutants possessing highly stabilized R-spines. Therefore, there is an unmet need for developing more effective RAF inhibitors that overcome drug resistance caused by these novel RAF alterations. Since the first two generations of RAF inhibitors belong to type I1/2 or II kinase inhibitors, the development of next-generation RAF inhibitors has been switched to type III and IV inhibitors that target the catalytic cleft or allosteric sites of RAF kinase domain, and thus may have much higher specificity and fewer off-target effects [Table 1]. In particular, given the unique activating and catalytic mechanisms of RAF family kinases, targeting RAF/RAF and RAF/MEK interactions has become an attractive strategy for designing novel RAF inhibitors[76]. Although no small molecular inhibitors that target RAF/RAF dimer interface have been developed at present, several groups have demonstrated that disrupting RAF/RAF dimers by using peptide inhibitors can effectively block hyperactive ERK signaling and thereby inhibit the growth of cancer cells harboring active RAF or RAS mutations[177-179], indicating that the RAF/RAF dimer interface is indeed a valid target for drug development. As for RAF/MEK interaction, it is dynamically altered in the process of MEK phosphorylation by RAF, as described above. Therefore, breaking the RAF/MEK complex or trapping the RAF/MEK complex in an inactive state would inhibit the catalytic activity of RAF. The first FDA-approved MEK inhibitor for cancer therapy, trametinib has been shown as a RAF/MEK heterodimer breaker, which binds to the interface of MEK and impairs RAF/MEK association as well as subsequent phosphorylation of MEK by RAF[76,180,181]. On the other hand, RO5126766, a RAF/MEK dual inhibitor that docks onto the RAF/MEK interface but prevents conformational alteration of the RAF/MEK complex, also blocks the MEK phosphorylation by RAF as trametinib does[182]. This allosteric inhibitor has exhibited promising activity against various cancers harboring RAS mutations in Phase I clinical trials[183-185]. Using a similar strategy, Khan et al.[181] developed a new RAF/MEK dual inhibitor, trametiglue, by modifying trametinib, which still docks onto the RAF/MEK interface and traps the RAF/MEK complex in an inactive state. In contrast to RO5126766, trametiglue has orders of magnitude higher potency and much slower off-rate kinetics, which results in better long-term inhibitory activity on both BRAF- and RAS-mutated tumors, although its clinical efficacy needs further evaluation. Altogether, these allosteric RAF inhibitors that target RAF/RAF or RAF/MEK interfaces without altering R-spine upon engagement could have higher efficacies against cancers harboring R-spine-stabilized RAF mutants, although they have not been tested on such cancers yet.
Besides these validated allosteric sites involved in RAF/RAF and RAF/MEK interactions, whether there are other allosteric cavities suitable for small molecule docking and hence developing advance RAF inhibitors remains unknown. Nevertheless, the high-resolution structures of RAF proteins and RAF/MEK complexes resolved in recent years[21,29,64,84,85,94,186] make possible in silico investigation of such allosteric sites and will accelerate the development of RAF inhibitors, particularly isoform-specific inhibitors through computation-aided molecular docking and optimization.
CONCLUSION
Targeting hyperactive RAS/RAF/MEK/ERK signaling with RAF inhibitors has achieved promising outcomes for treating cancers harboring BRAF(V600E) mutation. However, both intrinsic and acquired resistance have severely restricted their applications in cancer therapy. With a deeper understanding of regulatory mechanism(s) of RAF family kinases, next-generation RAF inhibitors that target allosteric sites on kinases and thus have longer efficacy, broader coverage, and fewer off-target effects are being developed, which sheds a light on overcoming emerging drug resistance in current cancer therapy.
DECLARATIONS
AcknowledgmentsWe thank all members of the Laboratory of Cancer Signaling (NCCS) for their helpful comments on this manuscript.
Authors’ contributionsConceptualization: Yap J, Hu J
Original draft writing: Degirmenci U, Yap J, Sim YRM, Qin S, Hu J
Review and editing: Hu J
Visualization: Degirmenci U, Hu J
Supervision: Hu J
Funding acquisition: Hu J
All authors have read and agreed to the published version of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was funded by Asia Fund for Cancer Research (AFCR-2017/2019-JH), SingHealth Foundation (AM/TP011/2018), National Medical Research Council (OFIRG18nov-0078), and Duke-NUS Medical School Khoo Foundation (Duke-NUS-KBrFA/2020/0036) to Jiancheng Hu.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta Mol Cell Res 2007;1773:1263-84.
2. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017;170:17-33.
3. Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol 2020;21:607-32.
5. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007;7:295-308.
8. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007;26:3291-310.
9. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007;26:3279-90.
10. Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics 2017;11:13.
11. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 2005;315:971-9.
12. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28-39.
13. Petit AM, Rak J, Hung MC, et al. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol 1997;151:1523-30.
14. Martinelli E, De Palma R, Orditura M, De Vita F, Ciardiello F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol 2009;158:1-9.
15. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014;13:828-51.
16. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov 2020;19:533-52.
17. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014;14:455-67.
18. Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011;30:3477-88.
19. Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol 2020;13:113.
20. Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 2020;9:198.
21. Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A 2008;105:3041-6.
22. Laquerre S, Arnone M, Moss K, et al. Abstract B88: a selective Raf kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-RafV600E mutation. Mol Cancer Ther 2009;8:B88.
23. Stuart DD, Li N, Poon DJ, et al. Abstract 3790: preclinical profile of LGX818: a potent and selective RAF kinase inhibitor. Cancer Res 2012;72:3790.
24. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010;467:596-9.
25. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30-9.
26. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2018;19:1315-27.
27. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19:603-15.
28. Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140:209-21.
29. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431-5.
30. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427-30.
31. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011;480:387-90.
32. Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol 2011;8:426-33.
33. Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KSM. Paradoxical oncogenesis-the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol 2013;10:390-9.
34. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 2004;5:875-85.
35. Scolnick EM, Rands E, Williams D, Parks WP. Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. J Virol 1973;12:458-63.
36. Hager GL, Chang EH, Chan HW, et al. Molecular cloning of the Harvey sarcoma virus closed circular DNA intermediates: initial structural and biological characterization. J Virol 1979;31:795-809.
37. Tsuchida N, Uesugi S. Structure and functions of the Kirsten murine sarcoma virus genome: molecular cloning of biologically active Kirsten murine sarcoma virus DNA. J Virol 1981;38:720-7.
38. Ellis RW, DeFeo D, Shih TY, et al. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 1981;292:506-11.
39. Rapp UR, Goldsborough MD, Mark GE, et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci U S A 1983;80:4218-22.
40. Sutrave P, Bonner TI, Rapp UR, Jansen HW, Patschinsky T, Bister K. Nucleotide sequence of avian retroviral oncogene v-mil: homologue of murine retroviral oncogene v-raf. Nature 1984;309:85-8.
41. Kamata T, Feramisco JR. Epidermal growth factor stimulates guanine nucleotide binding activity and phosphorylation of ras oncogene proteins. Nature 1984;310:147-50.
42. Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 1985;313:241-3.
43. Smith MR, DeGudicibus SJ, Stacey DW. Requirement for c-ras proteins during viral oncogene transformation. Nature 1986;320:540-3.
44. Ambrosio L, Mahowald AP, Perrimon N. Requirement of the Drosophila raf homologue for torso function. Nature 1989;342:288-91.
45. Han M, Golden A, Han Y, Sternberg PW. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 1993;363:133-40.
46. Ahn NG, Weiel JE, Chan CP, Krebs EG. Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J Biol Chem 1990;265:11487-94.
47. Boulton TG, Nye SH, Robbins DJ, et al. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991;65:663-75.
48. Crews CM, Erikson RL. Purification of a murine protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product: relationship to the fission yeast byr1 gene product. Proc Natl Acad Sci U S A 1992;89:8205-9.
49. Ray LB, Sturgill TW. Characterization of insulin-stimulated microtubule-associated protein kinase. Rapid isolation and stabilization of a novel serine/threonine kinase from 3T3-L1 cells. J Biol Chem 1988;263:12721-7.
50. Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc Natl Acad Sci U S A 1989;86:6940-3.
52. Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993;260:1658-61.
53. Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci U S A 1993;90:6213-7.
54. Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993;74:205-14.
55. Zhang XF, Settleman J, Kyriakis JM, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993;364:308-13.
56. Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 2003;4:373-84.
57. Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol 2011;13:39-51.
58. Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer 2011;2:216-31.
59. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015;16:281-98.
60. Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR. The ins and outs of Raf kinases. Trends Biochem Sci 1994;19:474-80.
61. Fischer A, Hekman M, Kuhlmann J, Rubio I, Wiese S, Rapp UR. B- and C-RAF display essential differences in their binding to Ras: the isotype-specific N terminus of B-RAF facilitates Ras binding. J Biol Chem 2007;282:26503-16.
62. Hu J, Stites EC, Yu H, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 2013;154:1036-46.
63. Dhillon AS, Yip YY, Grindlay GJ, et al. The C-terminus of Raf-1 acts as a 14-3-3-dependent activation switch. Cell Signal 2009;21:1645-51.
64. Kondo Y, Ognjenović J, Banerjee S, et al. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019;366:109-15.
65. Cutler REJ, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A 1998;95:9214-9.
66. Chong H, Guan KL. Regulation of Raf through phosphorylation and N terminus-C terminus interaction. J Biol Chem 2003;278:36269-76.
67. Yuan J, Ng WH, Lam PYP, et al. The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene 2018;37:5719-34.
68. Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 2002;16:427-38.
69. Nguyen A, Burack WR, Stock JL, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol 2002;22:3035-45.
70. Hu J, Yu H, Kornev AP, et al. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. P. roc Natl Acad Sci U S A 2011;108:6067-72.
71. Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009;461:542-5.
72. Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and Pseudokinases: lessons from RAF. Mol Cell Biol 2014;34:1538-46.
73. Taylor SS, Shaw A, Hu J, Meharena HS, Kornev A. Pseudokinases from a structural perspective. Biochem Soc Trans 2013;41:981-6.
74. Roskoski RJ. MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun 2012;417:5-10.
75. Mansour SJ, Candia JM, Matsuura JE, Manning MC, Ahn NG. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry 1996;35:15529-36.
76. Yuan J, Ng WH, Tian Z, et al. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal 2018;11:eaar6795.
77. Roskoski RJ. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 2012;66:105-43.
78. Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2011;2:195-209.
79. Buscà R, Pouysségur J, Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol 2016;4:53.
80. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 2007;1773:1213-26.
81. Lake D, Corrêa SAL, Müller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 2016;73:4397-413.
82. Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 2000;351:289-305.
83. Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein-protein interaction in the Raf/Mek/Erk pathway. FEBS Lett 2014;588:2398-406.
84. Haling JR, Sudhamsu J, Yen I, et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 2014;26:402-13.
85. Park E, Rawson S, Li K, et al. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2019;575:545-50.
88. Marshall M. Interactions between Ras and Raf: key regulatory proteins in cellular transformation. Mol Reprod Dev 1995;42:493-9.
89. Tran TH, Chan AH, Young LC, et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat Commun 2021;12:1176.
90. Lin WC, Iversen L, Tu HL, et al. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci U S A 2014;111:2996-3001.
91. Janosi L, Li Z, Hancock JF, Gorfe AA. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc Natl Acad Sci U S A 2012;109:8097-102.
92. Nan X, Collisson EA, Lewis S, et al. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci U S A 2013;110:18519-24.
93. Nan X, Tamgüney TM, Collisson EA, et al. Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proc Natl Acad Sci U S A 2015;112:7996-8001.
94. Liau NPD, Wendorff TJ, Quinn JG, et al. Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization. Nat Struct Mol Biol 2020;27:134-41.
95. Leicht DT, Balan V, Zhu J, et al. MEK-1 activates C-Raf through a Ras-independent mechanism. Biochim Biophys Acta 2013;1833:976-86.
96. Zhang Z, Kobayashi S, Borczuk AC, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 2010;31:577-86.
97. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer 2014;120:3446-56.
98. Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol 2005;25:7734-42.
99. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006;125:1137-49.
100. Jura N, Endres NF, Engel K, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009;137:1293-307.
101. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 2018;17:58.
102. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 2012;12:553-63.
103. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 2017;9:52.
104. Pahuja KB, Nguyen TT, Jaiswal BS, et al. Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 2018;34:792-806.e5.
105. Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J cancer 2006;118:257-62.
106. Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol 2008;26:1742-51.
107. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169-81.
108. Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol 2011;6:49-69.
109. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol 2018;12:3-20.
110. Cirenajwis H, Lauss M, Ekedahl H, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol 2017;11:438-451.
111. Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959-67.
112. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457-67.
113. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016;129:1287-92.
114. Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res 2020;80:2969-74.
115. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548-51.
116. Patricelli MP, Janes MR, Li LS, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 2016;6:316-29.
117. Janes MR, Zhang J, Li LS, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018;172:578-89.e17.
118. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217-23.
119. Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020;10:54-71.
120. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
122. Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855-67.
123. Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 2018;37:3183-99.
124. Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017;548:234-8.
125. Foster SA, Whalen DM, Özen A, et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 2016;29:477-93.
126. Chen SH, Zhang Y, van Horn RD, et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov 2016;6:300-15.
127. Wagenaar TR, Ma L, Roscoe B, Park SM, Bolon DN, Green MR. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res 2014;27:124-33.
128. Hu J, Ahuja LG, Meharena HS, et al. Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol 2015;35:264-76.
129. Yap J, Deepak RNVK, Tian Z, et al. The stability of R-spine defines RAF inhibitor resistance: a comprehensive analysis of oncogenic BRAF mutants with in-frame insertion of αC-β4 loop. Sci Adv 2021;7:eabg0390.
130. Zaman A, Wu W, Bivona TG. Targeting oncogenic BRAF: past, present, and future. Cancers (Basel) 2019;11:1197.
131. Baltanás FC, Zarich N, Rojas-Cabañeros JM, Santos E. SOS GEFs in health and disease. Biochim Biophys Acta Rev Cancer 2020;1874:188445.
132. Kim G, McKee AE, Ning YM, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation mutation. Clin Cancer Res 2014;20:4994-5000.
133. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726-36.
134. Trefzer U, Minor D, Ribas A, et al. BREAK-2: A phase IIA trial of the selective BRAF kinase inhibitor GSK2118436 in patients with BRAF mutation-positive (V600E/K) metastatic melanoma. Pigment Cell Melanoma Res 2011;24:1020.
135. Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012;379:1893-901.
136. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 2012;13:1087-95.
137. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358-65.
138. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol 2017;28:1631-9.
139. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367:1694-703.
140. Delord JP, Robert C, Nyakas M, et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin Cancer Res 2017;23:5339-48.
141. King AJ, Arnone MR, Bleam MR, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One 2013;8:e67583.
142. Goldinger SM, Graf NP, Galliker N, Conrad S, Dummer R. Hyperproliferative keratinocytic cutaneous adverse events and inflammatory palmoplantar erythrodysesthesia in melanoma patients treated with encorafenib compared to other BRAF inhibitors. J Clin Oncol 2017;35:9590.
143. Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 2019;381:1632-43.
144. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973-7.
145. Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF Inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010;18:683-95.
146. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-4.
147. Ahronian LG, Sennott EM, Van Allen EM, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov 2015;5:358-67.
148. Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies V600E B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 2012;3:724.
149. Saei A, Palafox M, Benoukraf T, et al. Loss of USP28-mediated BRAF degradation drives resistance to RAF cancer therapies. J Exp Med 2018;215:1913-28.
150. Wang J, Yao Z, Jonsson P, et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAFV600E-mutant brain tumor. Cancer Discov 2018;8:1130-41.
151. Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80-93.
152. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 2014;20:1965-77.
153. Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500-4.
154. Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014;508:118-22.
155. Lin L, Sabnis AJ, Chan E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015;47:250-6.
156. Lu H, Liu S, Zhang G, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017;550:133-6.
157. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401-9.
158. Röring M, Herr R, Fiala GJ, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J 2012;31:2629-47.
159. Diedrich B, Rigbolt KT, Röring M, et al. Discrete cytosolic macromolecular BRAF complexes exhibit distinct activities and composition. EMBO J 2017;36:646-63.
160. Cope N, Novak B, Candelora C, et al. Cover feature: biochemical characterization of full-length oncogenic BRAFV600E together with molecular dynamics simulations provide insight into the activation and inhibition mechanisms of RAF kinases (ChemBioChem 22/2019). ChemBioChem 2019;20:2802.[DOI: 10.1002/cbic.201900645].
161. Héritier S, Hélias-Rodzewicz Z, Chakraborty R, et al. New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer 2017;16:115.
162. Khater F, Langlois S, Cassart P, et al. Recurrent somatic BRAF insertion (p.V504_R506dup): a tumor marker and a potential therapeutic target in pilocytic astrocytoma. Oncogene 2019;38:2994-3002.
163. Yuan J, Ng WH, Yap J, et al. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho-Ser-621 in the C terminus of CRAF. J Biol Chem 2018;293:14276-84.
164. Yap J, Yuan J, Tee ZH, Huang X, Ng WH, Hu J. Characterize disease-related mutants of raf family kinases by using a set of practical and feasible methods. J Vis Exp 2019;2019:e59795.
165. Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci U S A 2008;105:14377-82.
166. Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 2011;36:65-77.
167. Waizenegger IC, Baum A, Steurer S, et al. A novel RAF kinase inhibitor with DFG-out-binding mode: high efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol Cancer Ther 2016;15:354-65.
168. Peng SB, Henry JR, Kaufman MD, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 2015;28:384-98.
169. Zhang C, Spevak W, Zhang Y, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015;526:583-6.
170. Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res 2014;27:479-84.
171. Yao Z, Gao Y, Su W, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 2019;25:284-91.
172. Tang Z, Yuan X, Du R, et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Ther 2015;14:2187-97.
173. Okaniwa M, Hirose M, Arita T, et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C -7-substituted 1,3-benzothiazole derivatives. J Med Chem 2013;56:6478-94.
174. Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA. Combined pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol Cancer Ther 2015;14:2700-11.
175. Ozkan-Dagliyan I, Diehl JN, George SD, et al. Low-dose vertical inhibition of the RAF-MEK-ERK cascade causes apoptotic death of KRAS mutant cancers. Cell Rep 2020;31:107764.
176. Yen I, Shanahan F, Lee J, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 2021;594:418-23.
177. Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 2013;49:751-8.
178. Beneker CM, Rovoli M, Kontopidis G, et al. Design and synthesis of type-IV inhibitors of BRAF kinase that block dimerization and overcome paradoxical MEK/ERK activation. J Med Chem 2019;62:3886-97.
179. Gunderwala AY, Nimbvikar AA, Cope NJ, Li Z, Wang Z. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol 2019;14:1471-80.
180. Lito P, Saborowski A, Yue J, et al. Disruption of CRAF-mediated MEK activation is required for effective mek inhibition in KRAS mutant tumors. Cancer Cell 2014;25:697-710.
181. Khan ZM, Real AM, Marsiglia WM, et al. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 2020;588:509-14.
182. Ishii N, Harada N, Joseph EW, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of raf activity. Cancer Res 2013;73:4050-60.
183. Martinez-Garcia M, Banerji U, Albanell J, et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res 2012;18:4806-19.
184. Chenard-Poirier M, Kaiser M, Boyd K, et al. Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. J Clin Oncol 2017;35:2506.
185. Sriskandarajah P, Boyd K, Xu W, et al. Update on clinical safety and efficacy of the novel oral dual RAF/MEK inhibitor RO5126766 (CH5127566) in RAS-mutant multiple myeloma. Blood 2018;132:323.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Degirmenci U, Yap J, Sim YRM, Qin S, Hu J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resist 2021;4:665-83. http://dx.doi.org/10.20517/cdr.2021.36
AMA Style
Degirmenci U, Yap J, Sim YRM, Qin S, Hu J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resistance. 2021; 4(3): 665-83. http://dx.doi.org/10.20517/cdr.2021.36
Chicago/Turabian Style
Degirmenci, Ufuk, Jiajun Yap, Yuen Rong M. Sim, Shiru Qin, Jiancheng Hu. 2021. "Drug resistance in targeted cancer therapies with RAF inhibitors" Cancer Drug Resistance. 4, no.3: 665-83. http://dx.doi.org/10.20517/cdr.2021.36
ACS Style
Degirmenci, U.; Yap J.; Sim YRM.; Qin S.; Hu J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resist. 2021, 4, 665-83. http://dx.doi.org/10.20517/cdr.2021.36
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 6
likes
Like This Article 6
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.