
Proteasomal adaptations to FDA-approved proteasome inhibitors: a potential mechanism for drug resistance?
 0
0Abstract
With proteasome inhibitors (PIs) becoming clinically available since 2003, outcomes for patients with multiple myeloma (MM) have dramatically changed, improving quality of life and survival. Despite the impressive treatment success, however, almost all MM patients who initially respond to these PIs eventually develop resistance. Furthermore, a portion of MM patients is inherently unresponsive to the PIs. Extensive mechanistic investigations identified several non-proteasomal signaling pathways suspected to be linked to the PI resistance, for which several excellent reviews are currently available. On the other hand, it is still unclear how cancer cells under high PI environments adapt to spare proteasome activity essential for survival and proliferation regardless of cancer evolution stages. This review outlines current progress towards understanding the proteasomal adaptations of cells in response to PI treatment to maintain necessary proteasome activity. A better understanding of cellular proteasomal changes in response to the PIs could provide a rationale to develop new therapeutics that could be used to overcome resistance to existing PI drugs.
Keywords
INTRODUCTION
One of the most fundamental processes in mammalian cells is the ubiquitin-proteasome system (UPS)-mediated protein degradation. In addition to disposing of misfolded or damaged proteins, the UPS is also responsible for the degradation of many signaling molecules in a highly controlled manner, regulating critical biological processes, such as cell cycle, inflammation, and DNA repair[1]. As such, the UPS is firmly believed to be a crucial component of several traditional cancer hallmarks, including angiogenesis, unlimited replication, and sustained proliferation[2]. The UPS’s fundamental importance to cancer cells is further endorsed by suggesting to include proteotoxic stress, which is delicately controlled by the UPS, as an additional cancer hallmark by Luo et al.[3]. More importantly, the UPS is also thought to be vital for cancer cells during their evolutionary processes to select clones conferring resistance to anticancer therapies, including proteasome inhibitor (PI)-based therapies[4].
The multi-protease complex 26S proteasome is a final executioner in the UPS, recognizing and deubiquitinating polyubiquitinated proteins and breaking them down into smaller peptide fragments [Figure 1A]. The 26S proteasome’s 20S core consists of 4-stacked rings comprising two identical outer α-rings and two identical inner β-rings, each containing 7 subunits [Figure 1B]. There are two main types of proteasomes in mammalian cells: the constitutive proteasome (cP) and immunoproteasome (iP). The cP has three catalytic subunits (β1, β2, β5) on each β-ring displaying three distinct substrate preferences: referred to as caspase-like (C-L), trypsin-like (T-L), and chymotrypsin-like (CT-L) activities, respectively. In immune cells, the constitutive subunits β1, β2, and β5, which are also commonly referred to as Y, Z, and X, are replaced by three immunosubunits β1i, β2i, and β5i, respectively, to form the iP. The three immunosubunits β1i, β2i, and β5i are also conveniently referred to as LMP2, MECL1, and LMP7. In non-immune cells, the iP can also be induced by pro-inflammatory cytokines such as interferon (IFN)-γ [Figure 1B]. Although the iP is shown to play a significant role in antigen presentation, its exact function in non-immune cells remains to be investigated.
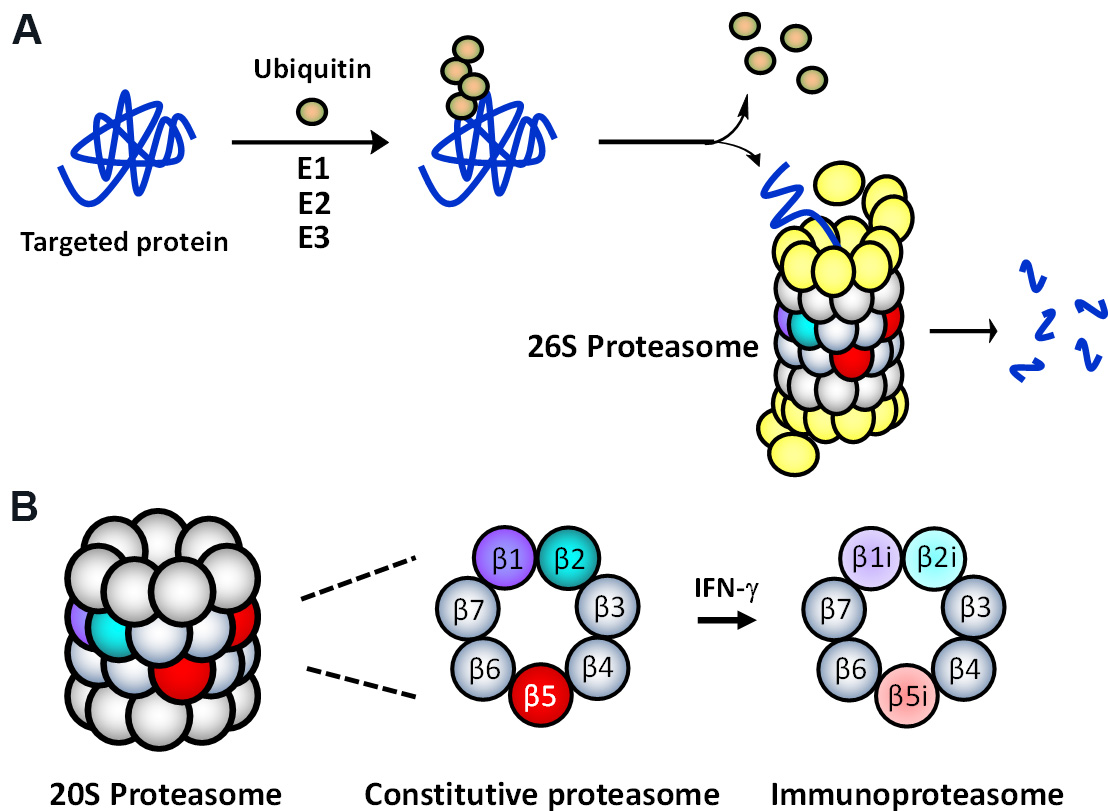
Figure 1. (A) The ubiquitin-mediated protein degradation pathway. Proteins, polyubiquitinated by an enzyme complex containing a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin-protein ligase (E3), are recognized and degraded by the 26S proteasome. (B) Two main (or standard) 20S proteasome subtypes present in mammalian cells: the constitutive proteasome (cP) and immunoproteasome (iP).
Due to increased proteotoxic stress and high metabolism in rapidly proliferating cancer cells, the demand for the UPS (and proteasome) activity is much higher than in resting normal cells, ultimately leading to the development of FDA-approved anticancer proteasome inhibitors (PIs)[1]. With several PIs becoming clinically available since 2003 [Figure 2], PI drugs, together with immunomodulatory agents, have become the mainstays of treatment regimens for multiple myeloma (MM) patients. The dipeptides bortezomib and its oral counterpart ixazomib share a boronic acid pharmacophore and bind to β5 via a tight non-covalent interaction between the boron atom and the nucleophilic oxygen lone pair of Thr01 within the β5 active site. On the other hand, the covalent inhibitor carfilzomib, a tetrapeptide with C-terminal α',β'-epoxyketone, forms a seven-membered, 1,4-oxazepano adduct between the epoxyketone pharmacophore and the catalytic Thr01 within the β5 active site[5,6]. As a result, the covalent interaction of carfilzomib leads to less recovery of CT-L activity in cells than bortezomib[7]. Regardless, the prognosis of patients with MM has substantially improved over the years, increasing the 5-year survival rate from ~35% to ~54%[8-10]. Despite the treatment success, however, those who respond to PIs inevitably relapse[11], and a portion of the MM patient population is also inherently unresponsive to the PIs[12-14]. Once relapsed on a PI drug, most patients are likely resistant to another PI drug, posing a major clinical hurdle. Clinical trials revealed that more than 75% of patients who relapsed on bortezomib-based therapies display cross-resistance to carfilzomib[15], significantly restricting carfilzomib-based treatments.
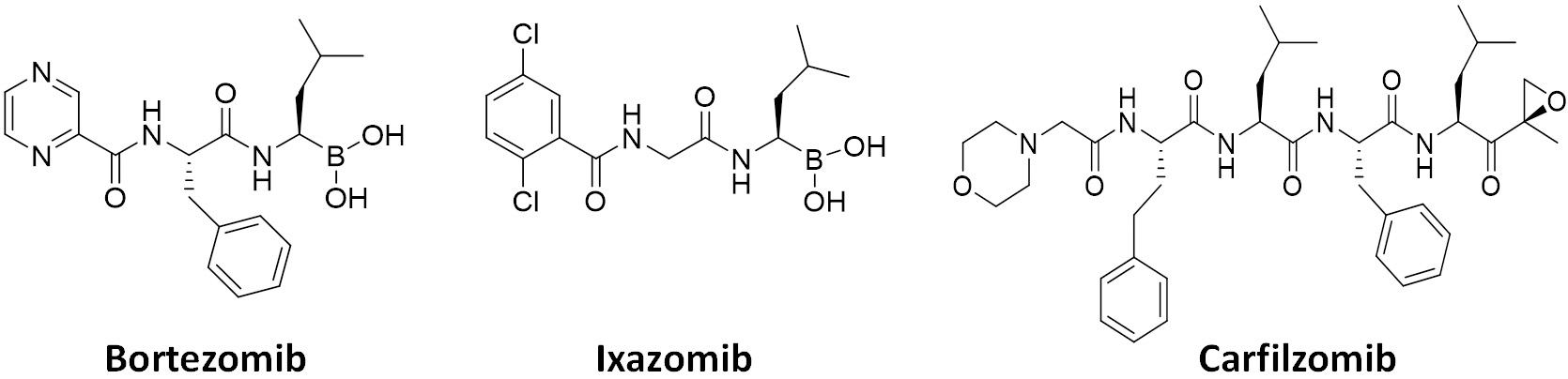
Figure 2. FDA-approved proteasome inhibitors. Bortezomib and ixazomib contain a boronic acid pharmacophore, while a tetrapeptide carfilzomib has an epoxyketone pharmacophore.
Extensive mechanistic investigations identified several non-proteasomal PI resistance mechanisms, such as those involving dysregulations of unfolded protein response, autophagy, aggresomes, MARCKS proteins, or efflux transporters[16-23]. These mechanistic studies were performed primarily using established cell lines or small sample sizes of primary tissues, needing further verification for their clinical relevance and potential applicability to the development of new therapeutics to overcome PI resistance. On the other hand, it has been proposed that when MM cells in patients are exposed and adapted to therapeutic doses of PIs, the proteasome activity in MM cells might be sufficiently salvaged to allow for cell survival and proliferation[24]. This premise has been further bolstered by findings that cells with acquired or intrinsic PI resistance remain sensitive to proteasome activity disruption[25-29], supporting that the proteasome activity is essential even to the PI-resistant cells. However, to date, it remains a controversial topic as to how cancer cells evade the inhibitory activity of PI to spare proteasome activity necessary for cell survival. Of note, it has been shown that PI-resistant cells are unlikely to have enhanced metabolic activity to inactivate PIs[29]. With this in mind, this review outlines the current understanding of proteasomal adaptations in response to PI treatment in cancer cells. It should be noted that the information summarized here is mainly from studies performed using cell line models and a limited number of primary samples. We expect that a better understanding of proteasomal adaptations to PIs in current clinical use could provide a strategy to develop new drugs that can be used to overcome clinical PI resistance.
PROTEASOMAL ADAPTATIONS
Point mutations in the PI-targeted proteasome subunits
Earlier studies performed using cell line models revealed that specific mutations occur primarily on a particular proteasome β-subunit, β5, which is responsible for the CT-L activity and a major target of all three FDA-approved PIs (bortezomib, carfilzomib, and ixazomib). For example, researchers observed a high level of acquired resistance to bortezomib when human myelomonocytic THP1 cells were exposed to increasing concentrations of bortezomib[30]. Further studies found a specific point mutation (Ala49Thr) residing in a highly conserved active site in the catalytic β5 subunit of the cP[31]. Given that Ala49 resides within the substrate-binding pocket, the Ala-to-Thr substitution is thought to impact bortezomib binding negatively. Similarly, using Jurkat cells cultured with increasing concentrations of bortezomib, another research group isolated two additional bortezomib-resistant clones containing β5 point mutation: Ala49Val and a conjoined mutant (Ala49Thr and Ala50Val), in addition to the previously reported Ala49Thr clone [Table 1]. These three mutants were ~22-67-fold more resistant to bortezomib compared to parental Jurkat cells. Modeling of β5 complexed with bortezomib suggests that both Ala49 and Ala50 are involved in interactions with bortezomib, verified by the X-ray study result[32].
β5 mutations found in cells with acquired resistance to bortezomib
| Cell lines | β5 | Fold difference (cell viability) | Ref. | |
| THP-1 | Ala49Thr | ~45-129 | Oerlemans et al.[30] | |
| Jurkat | Ala49Thr | Selected at 500 nM | Lu et al.[31] | |
| Jurkat | Ala49Thr Ala49Val Ala49Thr & Ala50Val | ~22 ~39 ~67 | Lü et al.[70] | |
| HT-29 | Cys63Phe | ~30 | Suzuki et al.[7] | |
| CEM | Cys52Phe Ala49Val & Cys52Phe Ala49Thr | ~170 | Franke et al.[33] | |
| RPMI 8266 | Ala49Thr | ~40 | ||
| THP-1 | Ala49Thr | Selected at 100 nM | ||
| Ala49Thr & Met45Ile | Selected at 500 nM | |||
| Met45Ile | Selected at 100 nM (2nd independent panel) | |||
| Yeast | Met45Ile Cys63Phe | 21.4 0.8 | Huber et al.[41] | |
| KBM7 | Met45Ile | Exposed to 18 nM bortezomib and 700 nM MG132 | Tsvetkov et al.[42] | |
| RPMI 6226 KMS11R FR4R XG-1R | Met104Val | At least 5-fold | Shi et al.[35] | |
| MM1.SR | Thr80Ala | |||
| KMS-18 | Thr21Ala | ~2 | Allmeroth et al.[34] | |
| KMS-27 | Ala49Val | |||
| L363 MM.1S | Ala50Val | 9 | Against ixazomib | Brünnert et al.[36] |
| Thr21Ala | 13 | |||
| Primary MM* | Ala20Thr Ala27Pro Met45Ile Cys63Tyr | From an MM patient refractory to bortezomib | Barrio et al.[40] | |
| Tyr42Cys | One out of 1,241 newly diagnosed patients with MM | |||
Multiple additional mutations were also identified in bortezomib-resistant blood cancer cell lines (AML, ALL, and MM), displaying ~40-170-fold resistance[33]. In these cell lines, mutations were clustered in the bortezomib binding region or its proximity. To distinguish between an expansion of a pre-existing β5 subunit mutant subclone and the de novo acquisition of a mutation and its subsequent outgrowth, researchers generated an additional independent panel of bortezomib-resistant cell lines. Identification of different mutations in these cell lines verified that the observed bortezomib resistance is due to de novo acquisition of multiple mutations in the β5 gene (PSMB5)[33].
Allmeroth et al.[34] also reported one previously known (Ala49Val) mutation and a new one (Thr21Ala) in KMS cell lines treated with increasing bortezomib concentrations [Table 1]. Interestingly, bortezomib-resistant KMS cells with Thr21Ala substitution were shown to be hypersensitive to carfilzomib and oprozomib. It is presumed that the replacement of Thr with Ala (having a smaller side chain) creates a bigger binding pocket, affording a tighter binding for carfilzomib and oprozomib, which possess a bulkier pharmacophore (epoxyketone) than bortezomib (boronic acid). In contrast, Ala49 to Val (having a bulkier side chain) mutation provided a cross-resistance to carfilzomib, presumably due to reduced accessibility of carfilzomib and oprozomib to the binding site within β5. Additionally, they employed an unbiased forward genetic approach in haploid cells using N-ethyl-N-nitrosourea mutagenesis to screen mutations that can impact the effectiveness of bortezomib, selecting several clones showing at least 2-fold bortezomib resistance. By analyzing these clones, they identified two new β5 mutation sites within the substrate pocket that can impact the effectiveness of bortezomib binding (Ser130, Tyr169). On the other hand, a systemic screening approach for bortezomib-resistant MM cell lines yielded 5 MM cell lines acquiring previously unidentified point mutations (Met104Val and Thr80Ala) [Table 1][35]. Although these mutant-screening data may offer essential insights into the potential direction of second-generation PI treatment for patients who have developed resistance to initial PI treatment, their clinical relevance needs to be established.
For PIs other than bortezomib, Brünnert et al.[36] reported the generation of MM cell lines that display a ~10-fold higher resistance to ixazomib than their parental cell lines. In two ixazomib-resistant MM cell lines, they found different β5 mutations (Thr21Ala in MM.1S and Ala50Val in L363 cell lines), whereas no mutations were found in the ixazomib-resistant AMO1 cell line. These ixazomib-resistant MM cell lines also exhibited high resistance to bortezomib and carfilzomib, indicating a common mechanical mode of resistance between the three PIs.
Until now, multiple large-scale screening efforts to identify somatic mutations of PSMB5 from primary MM samples have failed to verify PSMB5 mutations as a clinically relevant mechanism of PI resistance[37-39]. In a recent screening study with 1241 newly diagnosed MM patients, Barrio et al.[40] identified a Tyr42Cys mutation of β5 from a single patient sample [Table 1]. Currently, the importance of the Tyr42 mutation in clinical PI resistance remains unclear. They also reported four somatic PSMB5 mutations from primary samples collected from a single MM patient refractory to bortezomib [Table 1]. Three of them (Ala20Thr, Ala27Pro, Met45Ile) reside within the substrate-binding site of β5, while Cys63Tyr is located near the S1 pocket area of the β5[40]. The Met45Ile mutant was previously reported in established cell lines with acquired bortezomib resistance[41,42], and all the four mutations were shown to induce PI resistance, indicating that they are also involved with bortezomib binding as shown in structural and modeling studies[7,41,42]. While Ala20Thr and Ala27Pro mutations effectively abolished the effectiveness of bortezomib, both mutants remained sensitive to carfilzomib. Although a single patient sample was used to identify and verify these β5 mutants as a potential cause of PI resistance, the results suggest that PI resistance caused by specific β5 mutations can be overcome by alternative PIs that allow for stronger binding to the mutants. Ultimately, a larger scale of β5 mutation analysis using clinical samples may be needed to offer a helpful treatment direction in the future. There have been no reports showing β5 mutations as a potential cause of carfilzomib resistance in cell lines or primary tissue samples. Instead, several studies demonstrated that Pgp (P-glycoprotein) upregulation is a major mechanism of carfilzomib resistance in established cell lines[19,43].
Upregulation of β5 subunit, a primary target of proteasome inhibitor drugs
A few reports showed that PSMB5 mRNA and β5 protein were overexpressed in response to chronic exposure to PIs. When Jurkat cells were repeatedly exposed to bortezomib over 6 months, researchers found increased expression of PSMB5 mRNA, which was also coincident with increased CT-L activity [Table 2][44]. In the same Jurkat cells, a considerable decrease in IκB-α levels was observed, indicating that the bortezomib-mediated upregulation of NF-κB activity may contribute to survival and proliferation of bortezomib-resistant cells. THP1 cells exposed to bortezomib over 6 months also led to bortezomib-resistant cells showing ~60-fold drug resistance, with a dramatic overexpression of β5 mutant (Ala49Thr) but not other subunits[30]. While increased β5 mutant expression led to no marked changes in the CT-L activity (measured using Suc-LLVY-AMC) compared to parental THP1 cells, siRNA-mediated PSMB5 silencing restored bortezomib sensitivity, indicating that the β5 mutant contributes to bortezomib resistance.
β5 overexpression found in cancer cells in response to bortezomib treatment
| Cell lines | Overexpression | Fold difference | Ref. | |
| Jurkat | PSMB5 mRNA (CT-L activity increased) | Exposed to bortezomib for 6 months | Lu et al.[44] | |
| THP1 | β5 | ~50 (β5 activity) | Oerlemans et al.[30] | |
| HepG2 | β5, β1 | ~15 (cell viability) | Wu et al.[45] | |
| HuH7 | ~39 (cell viability) | |||
| U266 JJN3 | β5 (& β6) | ~2-8 (cell viability) | Shi et al.[35] | |
| Primary MM | β5, β6, β6 | |||
| L363 MM.1S AMO1 | β5, β1 | 9 | Against ixazomib | Brünnert et al.[36] |
| 13 | ||||
| 11 | ||||
| Primary MM | PSMB5 mRNA | ~5 (mRNA) | Shuqing et al.[46] | |
| Primary TNBC | PSMB5 mRNA CT-L activity | Not determined (PSMB5 is indicative of poor prognosis) | Wei et al.[47] | |
PSMB5 mRNA overexpression has also been observed in bortezomib-resistant hepatocellular carcinoma (HCC) cell lines (HepG2 and Huh7) treated with stepwise increasing concentrations of bortezomib over ~6 months[45]. Bortezomib-resistant HepG2 cells displayed ~2.8 and ~6-fold higher CT-L and caspase-like (C-L) activities, respectively, relative to the parental cells, consistent with increased expression of β5 and β1 subunits in the drug-resistant cell lines [Table 2]. This observation suggests that the increased expression of β5 may be one of the mechanisms underlying bortezomib resistance regardless of cancer types. In addition, β5 was also highly upregulated in all three ixazomib-resistant MM cell lines examined (L363, MM.1S, AMO1), while β1 upregulation was found only in the ixazomib-resistant AMO1 cells[36].
The overexpression of the PSMB5 gene is also reported in primary MM cells collected from a refractory patient to bortezomib-based treatment[46]. Specifically, mRNA levels of PSMB5 in MM cells collected from patients who underwent 6-cycle of intensive bortezomib-based treatment were about 5-fold higher relative to before bortezomib treatment, with no PSMB5 mutation [Table 2]. In a separate study using 10 MM patient samples collected before and after bortezomib treatment, researchers found that in 8 of those matched patient samples, PSMB5, 6, and 7 mRNA levels were significantly higher in bortezomib-treated primary samples than untreated counterparts[35]. It is currently unknown whether these upregulations observed in MM primary samples are linked to bortezomib resistance in these patients. Furthermore, PSMB5 mRNA was also highly expressed in primary triple-negative breast cancer (TNBC) tissue compared to normal tissues and non-TNBC breast cancer samples[47]. Not surprisingly, the CT-L activity in these primary TNBC tissues was considerably higher than in non-TNBC tissue breast cancer samples. Further analysis of survival data revealed that TNBC patients with relatively lower PSMB5 levels had much longer progression-free and overall survival than the high PSMB5 patients, suggesting the status of PSMB5 expression as an important indicator of TNBC prognosis. Currently, it is unclear whether there is a correlation between PSMB5 overexpression and drug resistance in TNBC. Mechanically, the overexpression of proteasome catalytic subunits in response to PIs is proposed to be related to a gain-of-function via increased assembly of 20S proteasome in response to PIs[48].
Paradoxically, researchers found that reduced expression of 19S proteasome regulatory subunits is directly associated with intrinsic bortezomib resistance in cell line studies[42,49]. Furthermore, they also observed that reduced 19S subunit mRNA expression is correlated with bortezomib resistance and poor outcome for myeloma patients treated with bortezomib[49]. Similarly, Acosta-Alvear et al.[50] identified several 19S subunits that induce resistance to carfilzomib in cancer cells using a systematic RNAi screening approach. In both cases, reduced expression of the 19S subunits is shown to cause significant changes in the spectrum of proteasome substrate and the remodeling of the transcriptome, indicating a potential mechanism of PI resistance. Interestingly, by analyzing gene expression data sets from 170 newly diagnosed, uniformly treated MM patients, Song et al.[51] reported that the 19S subunit Rpn11, a deubiquitinating enzyme (DUB), is more highly expressed in patient MM cells than in normal plasma cells and its expression is directly correlated with poor patient survival. Although researchers show that inhibition of Rpn11 can overcome resistance to bortezomib, whether overexpression of Rpn11 is directly correlated with resistance to bortezomib is currently unknown.
Aberrant expression of immunoproteasome and constitutive proteasome subunits
It has been shown that established MM cells and primary MM samples express iP subunits (β1i, β2i, or β5i)[29,52], even though the presence of the fully assembled standard iP containing β1i-β2i-β5i in those cells has not been verified. Busse et al.[53] also reported that three bortezomib-resistant cell lines, selected from 12 hematopoietic cancer cell lines by incubating with increasing concentrations of PS-341 (bortezomib), express commonly lower levels of both β2 and β1i than bortezomib-sensitive hematopoietic cancer cell lines examined. Specifically, bortezomib-resistant DG75 and KARPAS442 cell lines expressed much lower levels of β2i than bortezomib-resistant RAJI cells [Table 3], suggesting the impaired assembly of the standard 20S iP. They also found that solid cancer cell lines with low expression levels of β2 and iP subunits tend to show low bortezomib sensitivity[53]. In another study, Niewerth et al.[23] reported that bortezomib-resistant sublines obtained from human leukemia cell lines (RPMI 8226, CCRF-CEM, and THP1) contain a significantly lower amount of iP subunits (in particular, β5i) while increased cP subunits (including β5 mutant) compared to their parental cell lines, perhaps promoting β5 mutant incorporation into the fully assembled 20S proteasome in place of β5i. Based on this finding, they suggested that downregulation of β5i expression may be a major determinant in acquiring bortezomib resistance. Similarly, bortezomib-adapted Namalwa (human Burkitt’s lymphoma) cells displayed increased expression of cP subunits (β1, β2, and β5) but a completely down-regulated expression of iP subunits (β1i, β2i, and β5i). Despite significantly increased expression of cP subunits at the expense of iP subunits, the proteasomal proteolytic activities were only slightly increased compared to the parental Namalwa cells.
Aberrant expression of iP and cP subunits
| Cell lines | Expression | Fold difference (cell viability) | Ref. |
| RAJI DG75 KARPAS442 | ↓ β2, β1i ↓ β2i (KARPAS442 & DG75 only) | Selected at 10-40 nM bortezomib | Busse et al.[53] |
| RPMI 8226 CCRF-CEM THP1 | ↓ β1i, β2i, β5i ↑ β1, β2, β5 | ~40-150 | Niewerth et al.[23] |
| Namalwa (MES-SA) | ↓ β1i, β2i, β5i ↑ β1, β2, β5 | Growing at 12.5 nM of bortezomib | Fuchs et al.[71] |
| Primary bone marrow plasma cells | ↑ β5i | After carfilzomib therapy | Woodle et al.[55] |
There have been many clinical trials to evaluate the effectiveness of PIs in combination with other agents for the treatment of acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL), despite several setbacks showing only modest single-agent activity. Using samples from patients with relapsed and refractory acute leukemia, Niewerth et al.[54] investigated the iP/cP expression ratios related to bortezomib-containing therapy responses. They found that AML patients who achieved complete remission (CR) have higher pre-treatment iP/cP ratios than patients who did not achieve CR. Similarly, ALL patients with higher iP/cP ratios showed an excellent initial response to the bortezomib-containing therapy; however, the result was not statistically significant due to insufficient sample sizes. Taken together, the result further supports that the iP/cP expression ratios may contribute to the bortezomib-containing therapy response in these disease types. Moving forward, further clinical studies may be needed to direct the course of the PI-containing therapy for the treatment of the diseases.
One of the major immunologic barriers in transplantation is donor-specific antibodies (DSAs), which have a deleterious effect on allografts. Bone marrow plasma cells (BMPCs) are a well-known source of long-term antibody production, causing severe transplantation rejection. In recent years, PIs, including carfilzomib, have been actively investigated as a promising strategy to suppress BMPC-mediated antibody production but suffer from acquired drug resistance of these BMPCs. Woodle et al.[55] reported that BMPCs with acquired carfilzomib resistance display reduced sensitivity to other PIs such as bortezomib and ixazomib but enhanced sensitivity to the iP subunit β5i-selective inhibitor (ONX-0914). Using patient bone marrow biopsies, they confirmed that catalytically active iPs are upregulated in BMPCs collected after carfilzomib therapy[55]. Specifically, expression of β5i (and the proteasome activator PA28) was significantly higher in BMPC lysates collected after carfilzomib treatment than in those collected before. Expression levels of other proteasomal catalytic subunits (β1, β1i, β2, β2i, and β5) remained unchanged before and after carfilzomib treatment. The induction of β5i after carfilzomib therapy was also verified ex vivo by incubating carfilzomib in CD138+ BMPCs isolated from 8 individual HLA-sensitized patients. The incorporation of β5i into the catalytically active 20S proteasome complex was verified using a fluorescently labeled β5i-selective covalent probe, which fluorescently labels β5i in live cells. The data collectively support that upregulation of β51 contributes to acquired drug resistance of these BMPCs. However, it needs to be determined whether β5i upregulation leads to the assembly of the standard 20S iP containing β1i-β2i-β5i or non-standard 20S proteasome subtypes such as β1-β2-β5i or β1i-β2-β5i.
Formation of non-standard 20S proteasomes
When the standard proteasome subtypes are present in cells, an equivalent expression of the standard sets of three catalytic subunits (β1-β2-β5 or β1i-β2i-β5i) is expected to be simultaneously detected. However, the evidence demonstrated that different tissue cells, including cancer, express inequivalent sets of standard catalytic subunits[52,56-65], indicating the presence of non-standard types of 20S proteasomes containing mixed assortments of cP and iP catalytic subunits, such as β1-β2i-β5i or β1i-β2-β5. Parlati et al.[66] reported that normal and malignant hematopoietic cells express high levels of β5 and β5i, while expression levels of β2i and β1i are significantly lower relative to β5i. Based on the subunit expression information, they proposed that non-standard 20S proteasomes containing β1-β2-β5i or β1i-β2-β5i are likely to present in the hematopoietically derived cells [Figure 3][66]. While non-standard proteasome subtypes’ identity remains to be determined, non-standard proteasomes appear to display different sensitivities to PIs than cP and iP[57,62,67,68]. However, it is unclear whether the presence of non-standard proteasomes is linked to PI resistance.
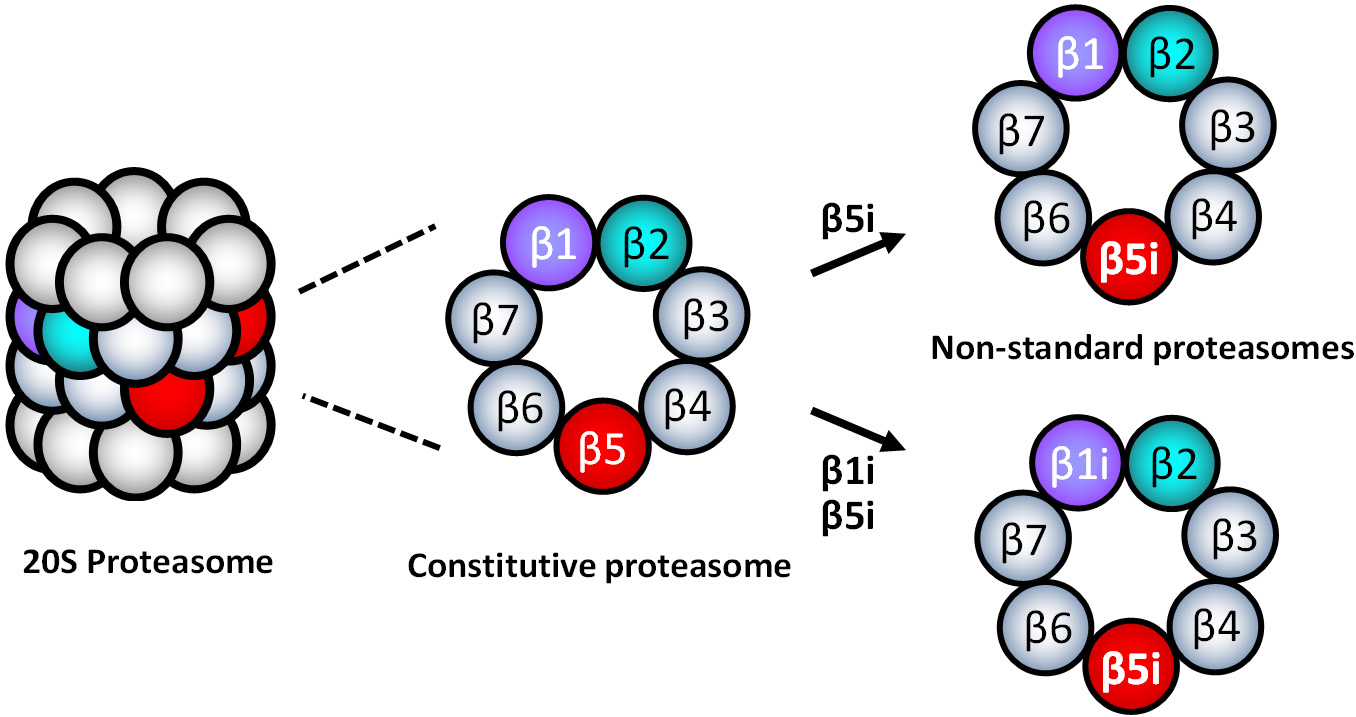
Figure 3. Examples of non-standard 20S proteasomes containing β5i-β1i-β2 or β5i-β1-β2 composition.
In this regard, Lee et al.[29] recently reported that intrinsically carfilzomib-resistant H727 cells express most of the iP and cP catalytic subunits while lacking β1i, suggesting the presence of non-standard proteasome subtypes such as β1-β2i-β5i, β1-β2i-β5, or β1-β2-β5i. Despite the de novo carfilzomib resistance, it has been shown that the proteasome activity in H727 cells remained vital for cell survival and proliferation. When incubated with IFN- to induce the assembly of the standard iP, H727 cells became sensitive to carfilzomib, suggesting that non-standard proteasome subtypes are involved with de novo carfilzomib resistance in H727 cells. It was also shown that alternative PIs with different structural features could overcome the de novo carfilzomib resistance in H727 cells[69]. Taken together, they suggested that non-standard proteasomes present in H727 cells may contribute to the intrinsic carfilzomib resistance, having a different drug binding affinity than the standard proteasome types.
CONCLUSION
The proteasome-mediated protein degradation remains vital to cancer cells regardless of PI resistance status. Proteasomal adaptations in response to PI treatment are also noted in surviving and proliferating tumor cells. Point mutations or overexpression of proteasome catalytic β-subunits have been frequently observed in PI-resistant cell lines and limited numbers of primary tumor samples. Studies also suggest a potential role of non-standard 20S proteasome subtypes in PI resistance. However, the lack of extensive clinical evidence supporting the proteasomal contribution to acquired or intrinsic PI resistance indicates that there remains much room for further investigations of clinical samples. Moving forward, a better understanding of the proteasomal adaptation to PI treatment will help to determine a course of PI-based therapy. In summary, proteasomal adaptations appear to be partially responsible for PI resistance, either acquired or de novo, implying that the proteasome-targeting strategy may remain a valid therapeutic strategy for MM patients who relapsed on or refractory to PIs in current clinical use.
DECLARATIONS
AcknowledgmentsThe author wants to thank the former and current lab members for their hard work and dedication and the National Institutes of Health (R01 CA188354) for the proteasome inhibitor development project financially.
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNational Institutes of Health (R01 CA188354).
Conflicts of interestThe author declared that there is no conflict of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Park JE, Miller Z, Jun Y, Lee W, Kim KB. Next-generation proteasome inhibitors for cancer therapy. Transl Res 2018;198:1-16.
3. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136:823-37.
4. Furukawa Y, Kikuchi J. Molecular basis of clonal evolution in multiple myeloma. Int J Hematol 2020;111:496-511.
5. Schrader J, Henneberg F, Mata RA, et al. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016;353:594-8.
6. Carmony K, Lee W, Kim KB. High-resolution snapshots of proteasome inhibitors in action revise inhibition paradigms and inspire next-generation inhibitor design. Chembiochem 2016;17:2115-7.
7. Suzuki E, Demo S, Deu E, et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS One 2011;6:e27996.
8. Yang Y, Li Y, Gu H, Dong M, Cai Z. Emerging agents and regimens for multiple myeloma. J Hematol Oncol 2020;13:150.
9. Liu J, Liu W, Mi L, et al. Union for China Lymphoma Investigators of the Chinese Society of Clinical Oncology. Incidence and mortality of multiple myeloma in China, 2006-2016: an analysis of the Global Burden of Disease Study 2016. J Hematol Oncol 2019;12:136.
10. Bergsagel PL. Where we were, where we are, where we are going: progress in multiple myeloma. Am Soc Clin Oncol Educ Book 2014:199-203.
11. Kumar S, Rajkumar SV. Many facets of bortezomib resistance/susceptibility. Blood 2008;112:2177-8.
12. Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003;348:2609-17.
13. Herndon TM, Deisseroth A, Kaminskas E, et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res 2013;19:4559-63.
14. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142-52.
15. Vij R, Wang M, Kaufman JL, et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012;119:5661-70.
16. Verbrugge SE, Al M, Assaraf YG, et al. Overcoming bortezomib resistance in human B cells by anti-CD20/rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Exp Hematol Oncol 2013;2:2.
17. Verbrugge SE, Assaraf YG, Dijkmans BA, et al. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 2012;341:174-82.
18. McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 2008;11:164-79.
19. Ao L, Wu Y, Kim D, et al. Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm 2012;9:2197-205.
20. Leung-Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013;24:289-304.
21. Nikesitch N, Ling SC. Molecular mechanisms in multiple myeloma drug resistance. J Clin Pathol 2016;69:97-101.
22. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 2015;18:18-35.
23. Niewerth D, Kaspers GJ, Assaraf YG, et al. Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J Hematol Oncol 2014;7:7.
24. Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol 2014;12:94.
25. Kraus M, Bader J, Geurink PP, et al. The novel β2-selective proteasome inhibitor LU-102 synergizes with bortezomib and carfilzomib to overcome proteasome inhibitor resistance of myeloma cells. Haematologica 2015;100:1350-60.
26. Mirabella AC, Pletnev AA, Downey SL, et al. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem Biol 2011;18:608-18.
27. Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009;113:4667-76.
28. Li X, Wood TE, Sprangers R, et al. Effect of noncompetitive proteasome inhibition on bortezomib resistance. J Natl Cancer Inst 2010;102:1069-82.
29. Lee MJ, Miller Z, Park JE, Bhattarai D, Lee W, Kim KB. H727 cells are inherently resistant to the proteasome inhibitor carfilzomib, yet require proteasome activity for cell survival and growth. Sci Rep 2019;9:4089.
30. Oerlemans R, Franke NE, Assaraf YG, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008;112:2489-99.
31. Lü S, Yang J, Song X, et al. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J Pharmacol Exp Ther 2008;326:423-31.
32. Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006;14:451-6.
33. Franke NE, Niewerth D, Assaraf YG, et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012;26:757-68.
34. Allmeroth K, Horn M, Kroef V, Miethe S, Müller RU, Denzel MS. Bortezomib resistance mutations in PSMB5 determine response to second-generation proteasome inhibitors in multiple myeloma. Leukemia 2021;35:887-92.
35. Shi CX, Zhu YX, Bruins LA, et al. Proteasome subunits differentially control myeloma cell viability and proteasome inhibitor sensitivity. Mol Cancer Res 2020;18:1453-64.
36. Brünnert D, Kraus M, Stühmer T, et al. Novel cell line models to study mechanisms and overcoming strategies of proteasome inhibitor resistance in multiple myeloma. Biochim Biophys Acta Mol Basis Dis 2019;1865:1666-76.
37. Kortuem KM, Braggio E, Bruins L, et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J 2016;6:e397.
38. Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol 2015;33:3911-20.
39. Egan JB, Shi CX, Tembe W, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012;120:1060-6.
40. Barrio S, Stühmer T, Da-Viá M, et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019;33:447-56.
41. Huber EM, Heinemeyer W, Groll M. Bortezomib-resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015;23:407-17.
42. Tsvetkov P, Mendillo ML, Zhao J, et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. Elife 2015;4:e08467.
43. Besse A, Stolze SC, Rasche L, et al. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018;32:391-401.
44. Lü S, Chen Z, Yang J, et al. Overexpression of the PSMB5 gene contributes to bortezomib resistance in T-lymphoblastic lymphoma/leukemia cells derived from Jurkat line. Exp Hematol 2008;36:1278-84.
45. Wu YX, Yang JH, Saitsu H. Bortezomib-resistance is associated with increased levels of proteasome subunits and apoptosis-avoidance. Oncotarget 2016;7:77622-34.
46. Shuqing L, Jianmin Y, Chongmei H, Hui C, Wang J. Upregulated expression of the PSMB5 gene may contribute to drug resistance in patient with multiple myeloma when treated with bortezomib-based regimen. Exp Hematol 2011;39:1117-8.
47. Wei W, Zou Y, Jiang Q, et al. PSMB5 is associated with proliferation and drug resistance in triple-negative breast cancer. Int J Biol Markers 2018;33:102-8.
48. Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem 2005;280:11840-50.
49. Tsvetkov P, Sokol E, Jin D, et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc Natl Acad Sci U S A 2017;114:382-7.
50. Acosta-Alvear D, Cho MY, Wild T, et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife 2015;4:e08153.
51. Song Y, Li S, Ray A, et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene 2017;36:5631-8.
52. Carmony K, Sharma LK, Lee DM, Park JE, Lee W, Kim KB. Elucidating the catalytic subunit composition of distinct proteasome subtypes: a crosslinking approach employing bifunctional activity-based probes. Chembiochem 2015;16:284-92.
53. Busse A, Kraus M, Na IK, et al. Sensitivity of tumor cells to proteasome inhibitors is associated with expression levels and composition of proteasome subunits. Cancer 2008;112:659-70.
54. Niewerth D, Kaspers GJ, Jansen G, et al. Proteasome subunit expression analysis and chemosensitivity in relapsed paediatric acute leukaemia patients receiving bortezomib-containing chemotherapy. J Hematol Oncol 2016;9:82.
55. Woodle ES, Tremblay S, Brailey P, et al. Proteasomal adaptations underlying carfilzomib-resistance in human bone marrow plasma cells. Am J Transplant 2020;20:399-410.
56. Klare N, Seeger M, Janek K, Jungblut PR, Dahlmann B. Intermediate-type 20 S proteasomes in HeLa cells: "asymmetric" subunit composition, diversity and adaptation. J Mol Biol 2007;373:1-10.
57. Guillaume B, Chapiro J, Stroobant V, et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc Natl Acad Sci U S A 2010;107:18599-604.
58. Fabre B, Lambour T, Delobel J, et al. Subcellular distribution and dynamics of active proteasome complexes unraveled by a workflow combining in vivo complex cross-linking and quantitative proteomics. Mol Cell Proteomics 2013;12:687-99.
59. Gohlke S, Kloss A, Tsokos M. Adult human liver contains intermediate-type proteasomes with different enzymatic properties. Ann Hepatol 2014;13:429-38.
60. Drews O, Wildgruber R, Zong C, et al. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics 2007;6:2021-31.
61. Gomes AV, Young GW, Wang Y, et al. Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol Cell Proteomics 2009;8:302-15.
62. Kloss A, Meiners S, Ludwig A, Dahlmann B. Multiple cardiac proteasome subtypes differ in their susceptibility to proteasome inhibitors. Cardiovasc Res 2010;85:367-75.
63. Pelletier S, Schuurman KG, Berkers CR, Ovaa H, Heck AJ, Raijmakers R. Quantifying cross-tissue diversity in proteasome complexes by mass spectrometry. Mol Biosyst 2010;6:1450-3.
64. Zheng J, Dasgupta A, Bizzozero OA. Changes in 20S subunit composition are largely responsible for altered proteasomal activities in experimental autoimmune encephalomyelitis. J Neurochem 2012;121:486-94.
65. Gohlke S, Mishto M, Textoris-Taube K, et al. Molecular alterations in proteasomes of rat liver during aging result in altered proteolytic activities. Age (Dordr) 2014;36:57-72.
66. Parlati F, Lee SJ, Aujay M, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009;114:3439-47.
67. Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J Mol Biol 2000;303:643-53.
68. Dahlmann B, Ruppert T, Kloetzel PM, Kuehn L. Subtypes of 20S proteasomes from skeletal muscle. Biochimie 2001;83:295-9.
69. Lee MJ, Bhattarai D, Yoo J, et al. Development of novel epoxyketone-based proteasome inhibitors as a strategy to overcome cancer resistance to Carfilzomib and Bortezomib. J Med Chem 2019;62:4444-55.
70. Lü S, Yang J, Chen Z, et al. Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Exp Hematol 2009;37:831-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kim KB. Proteasomal adaptations to FDA-approved proteasome inhibitors: a potential mechanism for drug resistance?. Cancer Drug Resist 2021;4:634-45. http://dx.doi.org/10.20517/cdr.2021.27
AMA Style
Kim KB. Proteasomal adaptations to FDA-approved proteasome inhibitors: a potential mechanism for drug resistance?. Cancer Drug Resistance. 2021; 4(3): 634-45. http://dx.doi.org/10.20517/cdr.2021.27
Chicago/Turabian Style
Kim, Kyung Bo. 2021. "Proteasomal adaptations to FDA-approved proteasome inhibitors: a potential mechanism for drug resistance?" Cancer Drug Resistance. 4, no.3: 634-45. http://dx.doi.org/10.20517/cdr.2021.27
ACS Style
Kim, KB. Proteasomal adaptations to FDA-approved proteasome inhibitors: a potential mechanism for drug resistance?. Cancer Drug Resist. 2021, 4, 634-45. http://dx.doi.org/10.20517/cdr.2021.27
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 30
likes
Like This Article 30
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.