
Emerging RAS-directed therapies for cancer

 , ...
, ... Abstract
RAS oncogenes are the most commonly mutated oncogenes in human cancer, and RAS-mutant cancers represent a major burden of human disease. Though these oncogenes were discovered decades ago, recent years have seen major advances in understanding of their structure and function, including the therapeutic and prognostic significance of diverse isoforms. Targeting of these mutations has proven difficult, despite some successes with inhibition of RAS effector signalling. More recently, direct RAS inhibition has been achieved in a trial setting. While this has yet to be translated to everyday clinical practice, this development carries much promise. This review summarizes the diverse approaches that have been taken to RAS inhibition and then focuses on the most recent developments in direct inhibition of KRAS(G12C).
Keywords
INTRODUCTION
Since the discovery of RAS oncogenes as the transforming genes of oncogenic retroviruses[1,2] in the 1960s, great advances have been made in our understanding of their structure and role in human cancer. The three RAS genes, Kirsten rat sarcoma viral oncogene homolog (K-RAS), neuroblastoma RAS viral (v-ras) oncogene homolog (N-RAS) and Harvey rat sarcoma viral oncogene homolog (H-RAS), are the most commonly mutated oncogenes in human cancer with approximately one-third of all cancers driven by these oncoproteins, including 40%-50% of colorectal cancer and over 90% of pancreatic cancers[3,4]. The discovery of oncogenes and the elucidation of intracellular signalling pathways heralded an era of targeted therapy that has greatly improved the outlook for many cancers[5,6]. New agents targeting receptor kinases and their downstream mediators demonstrated the ability to stabilise and shrink tumours, with side effects that were frequently milder than those associated with standard cytotoxic chemotherapy. Throughout this enormous paradigm shift in cancer therapeutics, however, RAS stood apart, dominant and seemingly undruggable[4]. The development of direct RAS inhibitors proved very challenging. RAS has a high affinity towards GDP and GTP and a lack of deep hydrophobic pockets which would allow binding of small molecules[7]. Subtle differences in structure and variable activation of RAS proteins added greatly to the complexity and attention largely focused on downstream inhibition of the transduced signalling pathways[8]. In recent years, however, there have been some very promising developments in direct RAS targeting which would suggest that this has real potential as a therapeutic avenue. Here we aim to review current efforts at RAS inhibition in the context of both RAS family biology and the historical efforts which attempted, largely without success, to perturb its role as a major oncogenic driver.
RAS STRUCTURE AND FUNCTION
The RAS superfamily of genes has about 36 members, which encode for 39 proteins[9]. Three RAS genes, H-RAS, K-RAS and N-RAS, encode four protein isoforms: H-RAS, K-RAS4A, K-RAS4B and N-RAS. K-RAS4B is the predominant isoform and is referred to simply as K-RAS in this article. RAS proteins are small GTPases, of about 21kD molecular weight, and are monomeric proteins that have a central role in cell differentiation, adhesion, migration, proliferation and survival[10]. RAS proteins convey signals from growth factors and extracellular components, and are upstream of signalling pathways including the ERK pathway and the PI3K/mTOR survival pathway. As illustrated in Figure 1, they function as a membrane-bound molecular switch, alternating between an inactive GDP-bound state and an active GTP-bound state. This alternation is mediated by guanine nucleotide exchange factors (GEFs) and GTPase activating/accelerating proteins (GAPs). GEFs are activated by an upstream mitogenic signal, and they in turn cause an inactive RAS to shed its GDP and bind a GTP, which has a 10-fold higher cellular concentration than GDP, thereby becoming activated. This period of activity terminates when the intrinsic GTPase activity of RAS-GTP is enhanced by GAPs, leading to hydrolysis of the bound GTP. In normal cells, a tight equilibrium is maintained between the active and inactive states.
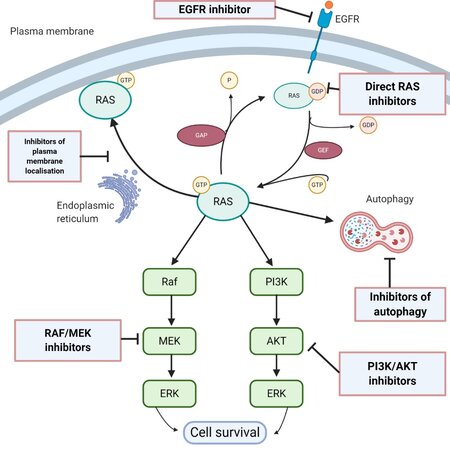
Figure 1. Scheme of the critical role of RAS in biological processes that regulate cell proliferation, survival and autophagy, and potential therapeutic targets (created with BioRender.com)
Pathogenesis
The normal function of RAS, as described above, can be deranged by mutations which unbalance this equilibrium. Single point missense mutations in codons G12 (most commonly), G13 or Q61 are responsible for converting proto-oncogenes to oncogenes. These mutations favour GTP binding and lead to constitutive activation of RAS, with reduction or loss of GTPase activity. These codons are implicated as their amino acid residues are found in the cavity where GTPase catalytic activity operates[10]. The consequence of this aberrantly activated RAS is prolonged oncogenic signalling rather than short, controlled bursts of activation[11]. There is subsequent activation of downstream signalling molecules such as PI3K, RAF and Rin1. By contrast, mutations in other RAS codons, or any nonsense mutations, are likely to inhibit rather than enhance the activity of RAS, and do not provide a survival advantage. As well as uncontrolled proliferation, the mutagenic RAS oncogene has been implicated in tumour immune resistance by causing intrinsic - as opposed to adaptive - upregulation of programmed death ligand 1[12].
RAS in cancer
Approximately 25%-30% of cancers contain mutations in one of the RAS isoforms, and they are considered an early genetic event in tumour progression[4]. For example, in pancreatic adenocarcinoma, where RAS mutations are virtually ever-present, precursor lesions - pancreatic intraepithelial neoplasia (PanIN) - contain RAS mutations which increase in frequency as they progress stepwise to malignancy. Though generally cooperative with other oncogenes during malignant transformation, these mutations are capable of neoplastic growth in the absence of further genetic abnormalities[13,14]. K-RAS is the isoform most frequently mutated and in addition to being almost inevitably mutated in pancreatic cancers, K-RAS mutations are present in approximately half of colorectal cancers and a third of lung cancers[3]. H-RAS mutations are found in salivary gland cancers (15% of salivary gland cancers), cervical cancers (9%) and urinary tract cancers (9%). N-RAS mutations are found in melanoma (17%), haematologic malignancies (10%) and thyroid cancers (7%)[15]. Not only are RAS mutations implicated in carcinogenesis, but also they are strongly associated with treatment resistance[16,17], and have been shown to be adverse prognostic markers in cancer[18]. RAS-mutated colorectal cancer will not benefit from EGFR-directed treatment with cetuximab or panitumumab. Similarly, in lung cancer RAS status has been shown to be an independent predictor for EGFR tyrosine kinase inhibition[19].
APPROACHES TO RAS INHIBITION
Given the central role of RAS both in carcinogenesis and tumour progression, the RAS oncoprotein is an important therapeutic target. Table 1 provides a summary of current trials of Ras inhibitors. Despite decades of efforts, however, it has proven extremely difficult to synthesise clinically effective direct inhibitors of RAS oncoproteins. This has been attributed to the high affinity of RAS towards GDP and GTP (in contrast to the low affinity of ATP for protein kinases, for example), and lack of deep hydrophobic pockets that would allow the binding of small molecules[7]. In addition, given variation in the frequency of isoform mutations within different cancer types, and in the specific mutations involved, there may not be a single effective RAS inhibitor for all RAS-mutated cancers[4]. Historical efforts to target RAS which have made it to the clinic could be broadly summarized as those focusing on RAS plasma membrane localisation and those attempting to indirectly block RAS by inhibiting the downstream effector signalling.
Ongoing RAS inhibitor trials
| Study/Phase | Drug | Disease | Biomarker | Clinicaltrials.gov registration no. | |
| Direct RAS inhibitors | |||||
| A phase I/II, study evaluating the safety, tolerability, PK, and efficacy of AMG 510 in subjects with solid tumours with a specific KRAS mutation (CodeBreak 100) | AMG510 | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT03600883 | |
| A Phase 1b, Protocol Evaluating the Safety, Tolerability, Pharmacokinetics, and Efficacy of AMG 510 (pINN) Sotorasib Monotherapy and in Combination with Other Anti-cancer Therapies in Subjects with Advanced Solid Tumours With KRAS p.G12C Mutation (CodeBreak 101) | AMG510 PD1 inhibitor MEK inhibitor SHP2 allosteric inhibitor Pan-ErbB TKI PD-L1 inhibitor EGFR inhibitor Chemotherapy | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT04185883 | |
| Phase I/II Study of MRTX849 in patients having a KRAS G12C Mutation KRYSTAL-1 | MRTX849 | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT03785249 | |
| First-in-Human Study of JNJ-74699157 in participants with Tumours Harboring the KRAS G12C Mutation | JNJ-74699157 (ARS-3248) | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT04006301 | |
| A Phase I/II study of LY3499446 administered to patients with advanced solid tumours with KRAS G12C mutation | LY3499446 Abemaciclib Cetuximab Erlotinib docetaxel | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT04165031 | |
| A Study to evaluate the safety, Pharmacokinetics, and activity of GDC-6036 in participants with advanced or metastatic solid tumours with a KRAS G12C mutation | GDC-6036 | Advanced/Metastatic solid tumours | KRASG12C Mutant | NCT04449874 | |
| Indirect Targeting of RAS | |||||
| SOS inhibitor | |||||
| A study to test different doses of BI-1701963 alone and in combined with trametinib in patients with different types of advanced cancer (solid tumours with KRAS mutation) | BI-1701963 BI-3406 trametinib | Advanced/Metastatic solid tumours | KRAS mutations | NCT04111458 | |
| SHP2 inhibitors | |||||
| Dose escalation of RMC-4630 monotherapy in relapsed/refractory solid tumours | RMC-4630 | Advanced/Metastatic solid tumours | Mutations that hyperactivate ERK pathway | NCT03634982 | |
| Dose finding study of TNO155 in adult patients with advanced solid tumours | TNO155 | Advanced/Metastatic solid tumours | EGFR or KRASG12C mutations | NCT03114319 | |
| Phase Ib study of TNO155 in combination with Spartalizumab or Ribociclib in selected malignancies | TNO155 Spartalizumab ribociclib | Advanced/Metastatic solid tumours | KRAS mutations | NCT04000529 | |
| First-in-Human Study of the SHP2 Inhibitor BBP-398 in patients with Advanced Solid Tumours | BBP-398 | Advanced/Metastatic solid tumours | MAPK-pathway alterations (excluding BRAF V600X) | NCT04528836 | |
| Farnesyltransferase inhibitors | |||||
| Phase II study of Tipifarnib in squamous head and neck cancer with HRAS mutations | Tipifarnib | Advanced head and neck squamous cell cancer | HRAS mutations | NCT02383927 | |
| RAF inhibitors | |||||
| Phase I study of LXH254 in patients with advanced solid tumours harbouring MAPK pathway alterations | LXH-254 Anti-PD-1 antibody | Advanced/Metastatic solid tumours | ERK pathway mutations | NCT02607813 | |
| Cobimetinib and HM95573 in Patients with Locally Advanced or Metastatic Solid Tumours | Belvarafenib cobimetinib | Advanced/Metastatic solid tumours | RAS or RAF mutation | NCT03284502 | |
| A Phase Ib Study of LXH254-centric Combinations in NSCLC or Melanoma | LXH-254 Trametinib LTT462 Ribociclib | Advanced/Metastatic solid tumours | KRAS or BRAF mutant NSCLC or NRAS mutant melanoma | NCT02974725 | |
| Study of the Safety and Pharmacokinetics of BGB-283 (Lifirafenib) and PD-0325901 (Mirdametinib) in Participants with Advanced or Refractory Solid Tumors | BGB-283 PD-0325901 | Advanced/Metastatic solid tumours | KRAS mutant NSCLC or endometrial cancer | NCT03905148 | |
| MEK inhibitors | |||||
| Trametinib and HDM201 in colorectal cancer patients with RAS/RAF mutant and TP53 wild-type advanced/metastatic colorectal cancer I | HDM201 (MDM2 inhibitor) Trametinib | Colorectal cancer | RAS mutant and TP53 wild type | NCT03714958 | |
| Atezolizumab and Cobimetinib in Treating Patients with Metastatic, Recurrent, or Refractory Non-small Cell Lung Cancer | Cobimetinib atezolizumab | Advanced/Metastatic solid tumours | KRAS mutation | NCT03600701 | |
| Study of MK-8353 + Selumetinib in Advanced/Metastatic Solid Tumors (MK-8353-014) | Selumetinib MK-8353 | Advanced/Metastatic solid tumours | none | NCT03745989 | |
| Trial of Trametinib and Ponatinib in Patients with KRAS Mutant Advanced Non-Small Cell Lung Cancer | Trametinib ponatinib | Advanced/Metastatic solid tumours | KRAS mutation | NCT03704688 | |
| Trametinib and Hydroxychloroquine in Treating Patients With Pancreatic Cancer | Trametinib hydroxychloroquine | Advanced pancreas cancer | none | NCT03825289 | |
| Trametinib and Docetaxel in Treating Patients with Recurrent or Stage IV KRAS Mutation Positive Non-Small Cell Lung Cancer | Trametinib Docetaxel | Metastatic NSCLC | KRAS mutation | NCT02642042 | |
| ERK inhibitors | |||||
| First-in-Human Study of KO-947 in Non-Hematological Malignancies | KO-947 | Advanced/Metastatic solid tumours | BRAF, KRAS, NRAS or HRAS mutation | NCT03051035 | |
| A Study of LY3214996 Administered Alone or in Combination with Other Agents in Participants with Advanced/Metastatic Cancer | LY-3214996 | Advanced melanoma or NSCLC | BRAF or NRAS mutations | NCT02857270 | |
| Adoptive Cell therapies | |||||
| Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12D Variant of Mutated RAS in HLA-A*11:01 Patients | Anti-RAS G12D mTCR | Advanced/Metastatic solid tumours | HLA-A11:01 RASG12D mutation | NCT03745326 | |
| Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12V Variant of Mutated RAS in HLA-A*11:01 Patients | Anti-RAS G12D mTCR | Advanced/Metastatic solid tumours | HLA-A11:01 RASG12D mutation | NCT03190941 | |
| Vaccine therapy | |||||
| A Study of mRNA-5671/V941 as Monotherapy and in Combination with Pembrolizumab (V941-001) | mRNA-5671 | Advanced NSCLC, non-MSI-high CRC, PDAC | HLA-A11:01 and/or HLA-C08:02; KRASG12C, KRASG12D, KRASG12V or KRASG13D mutation | NCT03948763 | |
| Immunotherapy | |||||
| A study of Avelumab, Binimetinib and Talazoparib in patients with locally advanced or metastatic RAS-mutant Solid Tumors I/II | Avelumab Binimetinib Talazoparib | Solid tumours | KRAS or NRAS mutant | NCT03637491 | |
| A study of Binimetinib + Nivolumab plus or minus Ipilimumab in patients with previously treated Microsatellite stable metastatic colorectal cancer with RAS mutation I/II | Binimetinib Nivolumab ipilimumab | Colorectal cancer | RAS mutations | NCT03271047 | |
RAS membrane localisation
In order to carry out their role, RAS proteins must become membrane-bound. This involves a complex series of post-translational modifications. Three enzymatic steps are necessary for RAS to associate with membranes - (1) prenylation of the CAAX box by farnesyltransferase (FTase); (2) cleavage of the terminal AAX residues by RAS converting enzymes RCE1; and (3) methylation of the cysteine residues of the CAAX box by isoprenylcysteine carboxyl methyltransferase ICMT. Farnesylation, the addition of farnesyl groups to RAS, is a critical step in creating a hydrophobic domain in RAS that allows the protein to associate with the plasma membrane, and therefore to be biologically active. Farnesyltransferases (FTases) are the enzymes responsible for this step, and they were an early target in efforts to inhibit RAS function[20]. Two farnesyltransferase inhibitors (FTIs), lonafarnib and tipifarnib, were investigated in Phase III trials either as monotherapy or in combination with chemotherapy in a number of different RAS-mutated tumours. Despite Phase I and II clinical trials showing some antitumour activity and low toxicity, no improvement in overall survival was reported in Phase III trials[21-23]. One reason for lack of “pan-RAS” efficacy for the FTI class is that K-RAS and N-RAS membrane localisation can be achieved in the absence of farnesyltransferases, via geranylgeranyl transferases. Attempts were made to target these enzymes with geranylgeranyltransferase inhibitors, but these were ineffective and associated with toxicity[24]. In contrast, H-RAS is not a substrate for geranylgeranyl transferase and therefore its membrane localization could be suppressed solely by FTIs[25]. Tipifarnib has demonstrated preclinical activity against a wide panel of H-RAS-mutated head and neck squamous cell carcinoma xenograft models and is undergoing clinical development in advanced head and neck cancers harbouring activating H-RAS mutations (NCT02383927)[26]. These efforts highlight the differences between RAS isoforms and the need for a tailored, isoform-specific approach to clinical trial design.
Salirasib is an S-trans, trans-farnesylthiosalicylic acid and a novel oral RAS inhibitor which competes with farnesylated RAS for binding sites on membranes. A recent trial demonstrated encouraging activity in patients with advanced solid tumours including a subset with K-RAS mutations[27]. A further target is isoprenylcysteine carboxyl methyltransferase (ICMT), an enzyme at the endoplasmic reticulum which increases RAS membrane affinity. It lacks homology with other protein methyltransferases, which adds to its specificity as a target. While agents that target ICMT have been isolated, and have demonstrated promising results in vitro against cancer cell lines, they have not been tested in the clinical setting[28]. Palmitoylation, the modification by the fatty acid palmitate, is necessary for the membrane interactions of H-RAS and N-RAS. Depalmitoylation inhibitors and palmitoyl acyltransferases have been described as having activity against RAS, but uncertainty regarding their specificity and concern about off-target effects, have impeded their further clinical development[4]. Many other proteins are modified by prenylation and farnesylation to ensure their correct subcellular localization, which makes off-target effects unavoidable even for highly specific inhibitors. Moreover, RAS can also signal from endomembranes (Golgi apparatus, endoplasmic reticulum) in addition to the plasma membrane[29,30]. It is unclear how localization inhibitors affect RAS signalling from different subcellular compartments.
Inhibitors of RAS effector signalling
Given the historic difficulties in directly targeting RAS, many efforts focused instead on inhibiting the downstream signal transduction pathways, either at a single point or as a combined approach targeting different nodal points. RAS effector families are involved in cancer initiation and maintenance, and it was hoped that inhibition of downstream proteins within these pathways could be an effective means of countering RAS-mediated oncogenesis[28]. These approaches have largely failed, perhaps mainly due to the inherent complexity and redundancy within these networks, but also due to a lack of specificity in the selection of inhibitors as well as isoform homogeneity within each subpopulation. In addition, the concept of linear pathways is misleading and has given way to that of signalling networks, whereby activated kinases interact via RAS (or other GTPases) with a large variety of signalling molecules resulting in highly interconnected networks.
EGFR: There is substantial crosstalk between EGFR tyrosine kinase and RAS[31]. With EGFR upstream of RAS, inactivation of these receptor tyrosine kinases can in theory reduce RAS activation, and this linear model explains the lack of clinical activity for EGFR inhibitors in colorectal cancer in the setting of K-RAS or N-RAS-mutant tumours[32,33]. This has also been demonstrated experimentally, through activation of the RAS signalling pathway by introduction of an activated K-RAS allele, confirming that as mutated RAS is constitutively active, disruption of signalling from EGFR impairs the therapeutic effect of anti-EGFR monoclonal antibodies[34]. Similarly in advanced non-small cell lung cancer (NSCLC), EGFR inhibition is insufficient to produce a response in the setting of RAS-mutant disease[35]. It is possible, however, that the specific RAS mutation plays a significant role in determining whether upstream inhibition of EGFR may be effective. We have seen in K-RAS G12D mutation-specific advanced solid tumours that a response can be achieved with pan-ERBB/EGFR inhibitors Afatinib and neratinib[36,37]. EGFR and pan-ERBB inhibitors have shown promise in preclinical studies in combination with both direct covalent RAS inhibitors and MEK inhibitors[37]. This synergistic effect between EGFR inhibitors and covalent RAS inhibitors appears to be the result of EGFR inhibition leading to a reduced amount of GTP-bound RAS, therefore leaving RAS in the unbound GDP state open to targeting by direct inhibition.
ERK pathway: Directly downstream of RAS signaling are the ERK and PI3K signaling pathways[38]. Active GTP-bound RAS results in RAF dimerization and phosphorylation, RAF kinase activity and, ultimately, phosphorylation of its substrates MEK1 and MEK2. The terminal kinases of this pathway, ERK1 and ERK2, act as growth promoting transcription factors. The RAS-RAF-MEK-ERK cascade is targeted with RAF kinase inhibitors, ERK inhibitors or MEK inhibitors. Eleven RAF kinase inhibitors have reached clinical evaluation and four are approved for use by the US Food and Drug Administration (FDA). Vemurafenib and dabrafenib are two ATP-competitive RAF inhibitors that are approved for use in BRAF-mutant metastatic melanoma. Further clinical evidence in both lung cancer and colorectal cancer has shown benefit for these agents when BRAF is mutated[39,40]. The current agents used in clinical practice, dabrafenib and vemurafenib, act on RAF monomers. However, in RAS-mutant cancer, RAF inhibition has been unsuccessful and the clinical experience has been very negative. The reason for this is that clinically used RAF inhibitors enhance RAF kinase homo-and heterodimerization, leading to the paradoxical activation of ERK signalling[41]. Homo- and heterodimerization of the RAF kinases BRAF and CRAF significantly increases their catalytic activities. The binding of RAF molecules to active RAS drives RAF dimerization by inducing conformational changes, dephosphorylation of inhibitory residues, and brings RAF molecules into proximity of each other[42]. Due to allosteric interactions between protomers in the RAF dimer, inhibitor binding to the first protomer in a dimer strongly decreases the affinity of the second protomer to the inhibitor. In this constellation the drug-bound RAF protomer allosterically activates the drug-free protomer causing paradoxical pathway activation and drug resistance[42]. As oncogenic RAS proteins are effective drivers of RAF kinase dimerization, RAS mutations lead to intrinsic or acquired resistance to RAF inhibitors. Overcoming dimerization-induced resistance to RAF inhibitors could lead to effective anti-RAS therapy[43].
Another interesting drawback to targeting wildtype RAF in RAS-mutant disease is that inhibition of wildtype RAF can paradoxically upregulate the ERK pathway in the setting of RAS mutations leading to downstream phosphorylation of MEK and ERK[44]. There are a number of newer agents that target RAF dimers rather than the monomer isoform all of which appear to demonstrate less paradoxical upregulation of the ERK pathway[45,46]. Belvarafenib and LXH-254 are pan-RAF inhibitors, effective against the monomer and dimer isoform, which are under clinical evaluation both as monotherapy and in combinations for RAS-mutant advanced solid tumours.
ERK upregulation through MEK appears to be the predominant method of resistance to BRAF directed monotherapy and we have seen both in preclinical and clinical studies that concurrent inhibition of BRAF and MEK can decrease acquired resistance and delay progression[47]. In RAS-mutant tumours, MEK inhibition as monotherapy has failed to demonstrate meaningful benefits largely due to the induction of feedback loops similar to those when RAF inhibitors are use in this setting[48]. Trametinib, Cobimetinib and Binimetinib are allosteric, non-ATP competitive inhibitors of MEK1 and MEK2, and are used in combination with RAF inhibitors in the treatment of melanoma. A number of trials have failed to show clinical benefit for MEK inhibition in advanced KRAS-mutant pancreatic, colorectal and non-small cell lung cancer[49-51]. One possible explanation for this is that inhibition of the ERK pathway induces autophagy, a process of cellular recycling that protects cells from the cytocidal effects of pathway inhibition[52]. Pancreatic cancer cells in particular utilize autophagy for growth and as a means of resistance to ERK inhibition[53]. Combining downstream MEK inhibition with hydroxychloroquine - an inhibitor of autophagy - displayed synergistic anti-proliferative effects against pancreatic ductal adenocarcinoma (PDA) cell lines and promoted striking regression of PDA xenografts from 2 patients with PDA which was superior to (standard of care) gemcitabine plus nab-paclitaxel chemotherapy. Highly encouraging clinical responses were also seen with this commercially available drug combination and clinical trials are ongoing [52,54].
ERK pathway: With ERK being the final kinase in the RAF-MEK-ERK pathway it would appear to be an attractive target to inhibit in RAS or RAF mutant tumours. Based on previous preclinical studies of the compound SCH-772984, a dual ERK1/2 inhibitor which demonstrated a reduction of phosphorylated ERK in a number of RAS-mutant cancer cell lines, an oral version MK-8353 was developed for clinical testing[55]. In a Phase I study, however, with 26 patients with advanced K-RAS or N-RAS mutated tumours, no objective responses were seen. It is now being tested in combination with a MEK inhibitor (selumetinib) and the anti-PD-1 inhibitor pembrolizumab in patients with RAS-mutant cancers. In K-RAS-mutant tumour models, ERK inhibitors such as GDC-0944 have shown efficacy in combination with the MEK inhibitor cobimetinib[56]. Phase I studies of the combination were stopped prematurely due to toxicity[57]. The ERK inhibitor was studied as monotherapy in a Phase I trial and appeared tolerable[58]. In this study, 14 patients had K-RAS-mutant advanced malignancies and of those 4 had stable disease and 10 had progression. ERK pathway suppression detected with NanoString gene expression was observed more commonly in those with BRAF-mutant tumours compared to those with KRAS-mutant tumours.
Ulixertinib is another ATP competitive ERK1/2 inhibitor that has shown clinical efficacy in N-RAS-mutant melanoma[59]. This in combination with nab-paclitaxel has been examined in a Phase 1 study in patients with advanced pancreas cancer and results are awaited (NCT02608229). Other studies are underway examining its role as monotherapy and in combination with other agents in patients with genetic alteration in the ERK pathway (NCT03698994, NCT04145297). LY-3214996 is a selective inhibitor of ERK1 and ERK2[60]. Unfortunately, however, in a Phase I study of this agent, only one patient with advanced RAS-mutant cancer had stable disease and the remaining patients had progressive disease[60]. KO-947, similarly a selective ERK1/2 inhibitor, demonstrated potent and sustained reduction in phosphorylated ERK in vitro in RAS-mutant cell lines[61]. This is now in clinical studies for patients with advanced RAS- or RAF-mutant tumours (NCT03051035).
PI3K pathway: The other major target of RAS effector signalling is the PI3K-AKT-mTOR pathway. PI3K is implicated in RAS-dependent cancer initiation and maintenance. While there are many inhibitors of this signalling pathway under investigation[62], they have demonstrated little activity as monotherapy in RAS-mutant cancers. KRAS and BRAF mutations are predictive of resistance to mTOR inhibition[63]. Moreover, inhibition of mTOR may lead to the upregulation and activation of MEK-ERK pathways. Therefore, the dual inhibition of the PI3K and the ERK pathway would appear to be a reasonable therapeutic target. With this in mind combination strategies have been explored in RAS mutant disease using both mTOR inhibitors along with RAF/MEK inhibitors[64]. Direct PI3K inhibitors have also been trialled with MEK inhibitors. However, in clinical trials, this combination was both toxic and only minimally efficacious[65].
AKT, which plays a key role in the activation of mTOR following its interaction with one of the 3 main PI3Ks, is amplified in the setting of RAS-mutant cancers[66]. Unfortunately no AKT inhibitors are approved for use in clinical practice. The evaluation of AKT inhibition with MEK inhibitors is under investigation, but similar challenging side effect profiles have been seen to that of combined mTOR and MEK inhibition[67].
Direct inhibitors of RAS
Attempts at direct RAS inhibition have been hampered by numerous challenges. Firstly, the activity of RAS is tightly governed by GEFs and GAPs, which control the high-affinity, picomolar interactions between GDP, GTP and RAS. Putative inhibitors struggle to overcome this affinity. Secondly, there are structural challenges. An effective inhibitor would typically require a deep hydrophobic pocket for binding. These pockets were long understood to be absent, although recent research demonstrates that they may arise dynamically as RAS goes through the GDP/GTP cycle[68,69]. Finally, there are selectivity and toxicity challenges. The “switch region”, which changes conformation upon GTP binding and recruits effector proteins, is highly conserved across other G-proteins in the body. Therefore, any agents which target this region in RAS, would be associated with risk of toxicity elsewhere.
A number of agents investigated were compounds that competed directly with GDP for the nucleotide binding site of RAS[70] and compounds that bind to RAS at the RAF binding site and inhibit RAS/RAF complex formation[71]. However, none were sufficiently potent to be considered for further investigation. More recently, however, direct targeting of RAS has been achieved for the G12C K-RAS mutation. This has been realised through the development of a small molecule inhibitor that binds covalently to the cysteine residue that results from the specific G12C mutation and has been shown to inhibit oncogenic RAS[72,73]. The inherently reactive nature of cysteine which is found at codon 12 of K-RAS(G12C) can be exploited for covalent small molecule inhibitors and the idea of targeting cysteine is one that is commonly exploited in drug discovery[74]. Another important feature of targeting this cysteine is that wild-type K-RAS lacks the cysteine in the active site unlike the mutant K-RAS(G12C).
Ostrem et al.[73] initially identified this novel allosteric binding pocket behind switch II referred to as the switch-II pocket, which led to the development of the first compounds to irreversibly target G12C. These compounds bind to K-RAS(G12C) in the GDP-bound inactive state, blocking SOS-catalysed nucleotide exchange and ultimately inhibiting K-RAS(G12C) association with RAF[73]. This switch II pocket is present in the GDP-bound inactive form of K-RAS only. Therefore, targeting of G12C needs to occur in the GDP-bound state. K-RAS(G12C) in its steady state is in the active GTP-bound state, but the presence of a high level of GTPase activity leaves it open to covalent attack[75]. The identification of this pocket led to a search for covalent inhibitors of K-RAS. The first was a molecule ARS-853[76], and its development led to the proof of concept that this specific isoform of K-RAS could be targeted using a covalent inhibitor.
A number of advances in the area ultimately led to the development of AMG510 (sotorasib)[73]. This particular G12C inhibitor has succeeded where others had not as its potency and selectivity was optimised through an interaction with a previously unexploited groove His95[77]. Preclinical studies have demonstrated responses and regression of K-RAS mutant tumours treated with AMG510. The results of the Phase I study of sotorasib (AMG510) demonstrated promising anticancer activity in patients with advanced solid tumours harbouring K-RAS(G12C) mutations. 129 patients were treated on the dose escalation study, 59 with NSCLC, 42 with colorectal cancer and 28 with other solid tumours. Sotorasib appeared to be well tolerated with 11.6% of grade 3 or 4 toxicity. 32.2% of the NSCLC had an objective response with a total of 88.1% having a response or stable disease. The median progression free survival was 6.3 months. In the colorectal cohort 7.1% had a confirmed response with 73.8% having a response or stable disease with a median PFS of 4 months[78]. This study represented the first clinical trial demonstrating objective response to direct KRAS inhibition. On December 8, 2020 the FDA granted Breakthrough Therapy designation for its investigational K-RAS(G12C) inhibitor, sotorasib, for the treatment of patients with locally advanced or metastatic NSCLC with K-RAS(G12C) mutations, as determined by an FDA-approved test, following at least one prior systemic therapy. Preclinical studies have also demonstrated that sotorasib was able to clear colon cancer from mice when given in combination with checkpoint inhibitors[79]. There is a Phase 1/2 study under way of sotorasib in solid tumours which will include a combination arm of sotorasib with an anti(PD-1/L1) (NCT03600883).
Adagrasib (MRTX849) is an additional agent under investigation in this field. It is a potent, highly selective inhibitor of KRAS(G12C)[80]. A Phase 1/2 study of adagrasib monotherapy in patients with pretreated NSCLC demonstrated an overall response rate of 45% and a high disease control rate[81]. This multiple-expansion-cohort trial is ongoing, investigating the use of adagrasib combined with pembrolizumab in NSCLC, afatinib in NSCLC or cetuximab in colorectal cancer. Research on this agent in nonclinical models has also demonstrated mechanisms of resistance, including KRAS nucleotide cycling and pathways that induce feedback reactivation or bypass KRAS dependence[80].
K-RAS(G12C) mutations only account for a small proportion of KRAS mutations that are found in cancer and are primarily found in lung cancer. As these irreversible allosteric inhibitors block RAS signalling by exclusively binding to the cysteine residue that results from the specific mutation, this limits their application to the particular allele they target. To target KRAS(G12D) and KRAS(G12V) different approaches are needed as these mutants lack the cysteines needed in the active state.
Attempting to design and develop drugs specifically targeting each individual RAS mutation would be extremely challenging and time-consuming, so direct targeting of ligand binding sites conserved on all RAS proteins (KRAS4A, 4B, NRAS and HRAS) has been thought to be one potential method of inhibiting RAS across all mutation and tumour types. In vitro studies have shown that Compound 3144, a molecule that binds a conserved residue Asp38 in switch-I, can block RAS effector binding[82]. This compound suppresses the growth of KRAS(G13D) tumours in vivo. A major concern, however, is that pan-inhibition of RAS potentially may lead to considerable toxicity as normal cellular function is reliant on RAS signalling in non-cancerous cells[83]. Models by which the deletion of all three RAS isoforms is carried out are not compatible with life, and therefore a pan inhibition of RAS in humans is likely to result in significant off-target toxicity. Nevertheless, it has been suggested that if such a compound were optimised for greater potency and specificity, this would be a viable approach[82].
Nucleotide exchange inhibition: In order to cycle between active GTP-bound and inactive GDP-bound states, RAS possesses intrinsic guanine nucleotide exchange and GTP hydrolysis activities. This cycling and exchange are accelerated by GEF and by GAPs, both of which change the activation state of RAS through covalent modifications. Upon activation of GEFs, nucleotide binding is destabilized and GDP is released. As GTP is much more prevalent than GDP in the cell, this loss of GDP leads to a transient formation of RAS-GTP, the active state[4]. GTP binding and activation of RAS leads to conformational changes in it, allowing it to bind effectors in RAS-binding domains. Mutations in RAS that are relevant to cancer usually lead to RAS permanently in the GTP bound active state. Efforts have been made to block this nucleotide exchange in an attempt to stop RAS transitioning to the active GTP bound form.
An alternative to blocking the direct nucleotide site is to inhibit the proteins that regulate the nucleotide exchange process. Normal RAS activation requires nucleotide exchange, processing, membrane localization and effector binding. Targeting any of these steps can be used to indirectly inhibit RAS. GEFs are responsible for releasing GDP from RAS allowing it to be replaced by GTP and ultimately leading to activation of RAS. In mammals, three families of RAS-specific GEFs exist: SOS, RASGRF1/Cdc25Mm and GRP/Cal-DAG-GEF[84]. Of these, SOS is the best known and studied of the RAS-specific GEFs and this has led to the development of a number of strategies and attempts to block it and ultimately inhibit RAS. SOS1 binds RAS at its catalytic binding site and thereby promotes exchange of GDP for GTP. RAS-GTP can also bind at an allosteric site on SOS1 to enhance GEF activity[85]. Genetic inactivation of SOS1 has been shown to decrease the survival of RAS-mutant tumor cells, but not in RAS wild type cells that are not reliant on RAS signaling[86]. Inhibition of SOS1 has been thought of as an attractive mechanism of RAS inhibition over direct RAS inhibitors as it does not appear to depend on targeting specific mutations.
BI-3406 is a potent and selective SOS1:K-RAS interaction inhibitor that potently decreased the formation of GTP-bound RAS and reduced cell proliferation of RAS-driven cancers both in vitro and in vivo. This orally bioavailable agent appears to reduce RAS-GTP level and inhibits ERK pathway signaling, thereby limiting the growth of tumor cells driven by RAS. Whilst most RAS variants appear to show reduction in cell proliferation when exposed to BI-3406, certain variants appear less sensitive. Mutations in codon 61 due to the resultant molecular conformation appear to have low intrinsic GTPase activity and are subsequently less sensitive to SOS1 inhibition[87]. Initial studies of SOS1 inhibitor BI-3406 suggest that it may benefit as many as 80%-90% of RAS-driven cancers[88].
SOS1 is phosphorylated by ERK, a kinase in the ERK pathway downstream of RAS, ultimately leading to the reduction of its GEF activity. It is thought that efforts to treat RAS-driven cancers with MEK inhibitors have failed in part as inhibition of MEK reduces the activity of ERK1/2, resulting in the release of a negative feedback loop, thus increasing the activity of SOS1-dependent formation of GTP-bound RAS. Combination therapy of a MEK inhibitor with BI-3406 blocks this negative feedback by reducing levels of phospho-MEK and phospho-ERK leading to sustained pathway inhibition and potentiating the benefit of SOS1 inhibition. A Phase I clinical trial of this combination to assess safety, tolerability and preliminary efficacy, as well as another SOS1 inhibitor, BI-1701963 in combination with Trametinib is ongoing (NCT040111458).
Although much less defined than SOS, another protein involved in the nucleotide exchange process is SHP2. SHP2 is a non-receptor protein tyrosine phosphatase that is required for the full activation of the ERK pathway[89]. Mutations in PTPN11, which encodes SHP2, cause “rasopathies” and are found in about 50% of patients with Noonan syndrome[90]. Although not fully defined, SHP2 appears to act as a scaffold protein, binding GRB2 and SOS1 in close proximity to RAS and ultimately thereby increasing RAS nucleotide exchange[91]. Research has explored the role of SHP2 inhibitors in the treatment of various cancers. In the preclinical setting, the allosteric SHP2 inhibitor SHP099 has been shown to inhibit myeloid leukaemia cell lines[92], and to elicit a response in colorectal cancer cell lines[93]. Of note, in the colorectal cancer cells, these responses seemed limited to cells that were sensitive to lapatinib, and therefore dependent on EGFR signalling. By contrast, RAS- or BRAF-mutant cells were generally resistant to SHP099.
CONCLUSION AND FUTURE DIRECTIONS
Effective and safe inhibition of RAS was considered a holy grail for cancer researchers decades ago, and it remains so today. While extraordinary advances have been made in our understanding of RAS and carcinogenesis - as well as methods of targeting its downstream effectors - the direct inhibition of RAS does not yet have a role in everyday practice. However, our deeper understanding of molecular pathogenesis is the foundation on which that future progress will be built, and effective treatments are now close.
The unmet need for proven therapies in this setting is clear, given the prevalence of RAS mutations in common cancers, many of which are highly fatal. Our recognition of the structure and clinical significance of diverse RAS isoforms represents a major step forward, of relevance to all future efforts at RAS targeting. Their importance is clear when considering candidate inhibitors of RAS plasma membrane localization, which are now reaching the clinic. Even individual isoforms, like K-RAS, have a range of possible mutations with different responses to targeted therapies[94]. Therefore, those designing clinical trials should carefully consider the need for isoform and mutation-specific approaches.
The development of inhibitors of RAS effector signalling, which are in widespread clinical practice, is an example of the concrete achievements of research in this area. This success has been tempered over time by failures, many of which can be attributed to misconceptions of RAS signalling pathways as unidirectional and linear. A more nuanced appreciation of these as delicately balanced signalling networks can help researchers refine their approach, and may warrant increased focus on drug combinations, as seen in trials involving inhibitors of MEK and autophagy[52].
Possibly showing greater promise than any of these initiatives, however, is efforts at direct targeting of RAS. The past 12 months has seen breakthroughs in this field that have been sought for years, vindicating decades of basic scientific endeavour. Successes here belie outdated dogma describing RAS as “undruggable”, demonstrating that drug exposure associated with preclinical efficacy can be achieved safely in patients[95]. However, these studies also raise questions - why did response rates to Sotorasib vary between cancers despite identical mutations, and what provoked early progression in some patients after an initial response? It is suggested that tumour heterogeneity or driver mutations alternative to K-RAS(G12C) may be the key to understanding this and that, as with inhibition of RAS effector signalling, drug combinations may be the next step[78]. In addition, optimizing monotherapy by developing inhibitors with higher affinity for inactive KRAS(G12C) or the use of pulsatile therapy has been suggested as approaches to maximise therapeutic effect[96].
In summary, a more textured understanding of RAS pathogenesis emerging from decades of basic scientific research has led to a more refined approach to RAS inhibition, which is now beginning to bear fruit. The coming months and years will hopefully take these efforts “over the line” to the routine use of effective therapeutics in the clinic setting, but it will be a long time before we have fully tapped the potential of RAS inhibition.
DECLARATIONS
Authors’ contributionsAll authors made substantial contributions to drafting the article, revising it and giving final approval prior to submission.
Availability of data and materialsNot applicable.
Financial support and sponsorshipFunding source: Work in Walter Kolch’s lab is funded by Science Foundation Ireland (SFI) under Grant Numbers 18/SPP/3522 and 14/IA/2395.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Harvey JJ. An unidentified virus which causes the rapid production of tumours in mice. Nature 1964;204:1104-5.
2. Kirsten WH, Mayer LA. . Morphologic responses to a murine erythroblastosis virus. J Natl Cancer Inst 1967;39:311-35.
3. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801-6.
4. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014;13:828-51.
5. Kwong LN, Davies MA. Targeted therapy for melanoma: rational combinatorial approaches. Oncogene 2014;33:1-9.
6. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther 2014;142:164-75.
7. Cromm PM, Spiegel J, Grossmann TN, Waldmann H. Direct Modulation of Small GTPase Activity and Function. Angew Chem Int Ed Engl 2015;54:13516-37.
8. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517-31.
10. Weinberg RA. . The biology of cancer. 2nd ed. New York, NY: Garland Science; 2014.
11. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011;11:761-74.
12. Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017;47:1083-1099.e6.
13. Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004;5:375-87.
14. Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 2003;4:111-20.
15. Murugan AK, Grieco M, Tsuchida N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol 2019;59:23-35.
16. Mengoli MC, Barbieri F, Bertolini F, Tiseo M, Rossi G. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: Report of two cases and review of the literature. Lung Cancer 2016;93:55-8.
17. Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: The role of MEK. Cancer Treat Rev 2017;53:61-9.
18. Marks JL, Broderick S, Zhou Q, et al. Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol 2008;3:111-6.
19. Cadranel J, Mauguen A, Faller M, et al. Impact of systematic EGFR and KRAS mutation evaluation on progression-free survival and overall survival in patients with advanced non-small-cell lung cancer treated by erlotinib in a French prospective cohort (ERMETIC project--part 2). J Thorac Oncol 2012;7:1490-502.
20. Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer 2011;11:775-91.
22. Wong NS, Meadows KL, Rosen LS, et al. A phase I multicenter study of continuous oral administration of lonafarnib (SCH 66336) and intravenous gemcitabine in patients with advanced cancer. Cancer Invest 2011;29:617-25.
23. Milojkovic Kerklaan B, Diéras V, Le Tourneau C, et al. Phase I study of lonafarnib (SCH66336) in combination with trastuzumab plus paclitaxel in Her2/neu overexpressing breast cancer: EORTC study 16023. Cancer Chemother Pharmacol 2013;71:53-62.
24. Lobell RB, Omer CA, Abrams MT, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res 2001;61:8758-68.
25. Whyte DB, Kirschmeier P, Hockenberry TN, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 1997;272:14459-64.
27. Furuse J, Kurata T, Okano N, et al. An early clinical trial of Salirasib, an oral RAS inhibitor, in Japanese patients with relapsed/refractory solid tumors. Cancer Chemother Pharmacol 2018;82:511-9.
28. Marín-Ramos NI, Ortega-Gutiérrez S, López-Rodríguez ML. Blocking Ras inhibition as an antitumor strategy. Semin Cancer Biol 2019;54:91-100.
29. Santra T, Herrero A, Rodriguez J, et al. An Integrated Global Analysis of Compartmentalized HRAS Signaling. Cell Rep 2019;26:3100-3115.e7.
30. Prior IA, Hancock JF. Ras trafficking, localization and compartmentalized signalling. Semin Cell Dev Biol 2012;23:145-53.
31. Volinsky N, Kholodenko BN. Complexity of receptor tyrosine kinase signal processing. Cold Spring Harb Perspect Biol 2013;5:a009043.
32. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757-65.
33. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626-34.
34. Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643-8.
35. Mao C, Qiu LX, Liao RY, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer 2010;69:272-8.
36. Moll HP, Pranz K, Musteanu M, et al. Afatinib restrains K-RAS-driven lung tumorigenesis. Sci Transl Med 2018;10:eaao2301.
37. Kruspig B, Monteverde T, Neidler S, et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Transl Med 2018;10:eaao2565.
38. Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011;2:261-74.
40. Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984-93.
41. Rauch N, Rukhlenko OS, Kolch W, Kholodenko BN. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr Opin Struct Biol 2016;41:151-8.
42. Jambrina PG, Rauch N, Pilkington R, et al. Phosphorylation of RAF Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angew Chem Int Ed Engl 2016;55:983-6.
43. Rukhlenko OS, Khorsand F, Krstic A, et al. Dissecting RAF Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic RAS Signaling. Cell Syst 2018;7:161-179.e14.
44. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431-5.
45. Peng SB, Henry JR, Kaufman MD, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015;28:384-98.
46. Nakamura A, Arita T, Tsuchiya S, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res 2013;73:7043-55.
47. Robert C, Grob JJ, Stroyakovskiy D, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019;381:626-36.
48. Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 2013;501:232-6.
49. Bodoky G, Timcheva C, Spigel DR, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs 2012;30:1216-23.
50. Bennouna J, Lang I, Valladares-Ayerbes M, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs 2011;29:1021-8.
51. Hainsworth JD, Cebotaru CL, Kanarev V, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol 2010;5:1630-6.
52. Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 2019;25:620-7.
53. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717-29.
54. Rahib L, Chen K, Ocean AJ, et al. Use of a real-world data approach to rapidly generate outcomes data following a case study of a novel treatment combination in pancreatic adenocarcinoma. J Clin Oncol 2020;38:e16735.
55. Moschos SJ, Sullivan RJ, Hwu WJ, et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018;3:92352.
56. Merchant M, Moffat J, Schaefer G, et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017;12:e0185862.
57. Weekes C, Lockhart A, LoRusso P, et al. A Phase Ib Study to Evaluate the MEK Inhibitor Cobimetinib in Combination with the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Oncologist 2020;25:833-e1438.
58. Varga A, Soria JC, Hollebecque A, et al. A First-in-Human Phase I Study to Evaluate the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Clin Cancer Res 2020;26:1229-36.
59. Sullivan RJ, Infante JR, Janku F, et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov 2018;8:184-95.
60. Bhagwat SV, McMillen WT, Cai S, et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther 2020;19:325-36.
61. Burrows F, Kessler L, Chen J, et al. Abstract 5168: KO-947, a potent ERK inhibitor with robust preclinical single agent activity in MAPK pathway dysregulated tumors. Cancer Research 2017;77 (13 Supplement):5168.
62. Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 2013;71:1395-409.
63. Belmont PJ, Jiang P, McKee TD, et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci Signal 2014;7:ra107.
64. Mohan S, Vander Broek R, Shah S, et al. MEK Inhibitor PD-0325901 Overcomes Resistance to PI3K/mTOR Inhibitor PF-5212384 and Potentiates Antitumor Effects in Human Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2015;21:3946-56.
65. Shapiro GI, LoRusso P, Kwak E, et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Invest New Drugs 2020;38:419-32.
66. Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci U S A 1998;95:14950-5.
67. Tolcher AW, Kurzrock R, Valero V, et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother Pharmacol 2020;85:673-83.
68. Kessler D, Bergner A, Böttcher J, et al. Drugging all RAS isoforms with one pocket. Future Med Chem 2020;12:1911-23.
69. Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 2019;116:15823-9.
70. Taveras A, Remiszewski S, Doll R, et al. Ras oncoprotein inhibitors: The discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorganic & Medicinal Chemistry 1997;5:125-33.
71. Karaguni IM, Herter P, Debruyne P, et al. The new sulindac derivative IND 12 reverses Ras-induced cell transformation. Cancer Res 2002;62:1718-23.
72. Lim SM, Westover KD, Ficarro SB, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl 2014;53:199-204.
73. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548-51.
74. Gehringer M, Laufer SA. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J Med Chem 2019;62:5673-724.
75. Janes MR, Zhang J, Li LS, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172:578-589.e17.
76. Patricelli MP, Janes MR, Li LS, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 2016;6:316-29.
77. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217-23.
78. Hong DS, Fakih MG, Strickler JH, et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383:1207-17.
79. Saiki AY, Gaida K, Rex K, et al. . Discovery and in vitro characterization of AMG 510-a potent and selective covalent small-molecule inhibitor of KRASG12C [abstract]. Proceedings of AACR Annual Meeting; 2019 Mar 29-Apr 3; Atlanta, GA; 2019.
80. Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10:54-71.
81. Jänne PA, Rybkin II, Spira A, et al. KRYSTAL-1: updated safety and efficacy data with Adagrasib (MRTX849) in NSCLC With KRASG12C mutation from a phase 1/2 study. Available from: https://www.mirati.com/wp-content/uploads/Janne-849-001_NSCLC-ENA-Presentation_25Oct2020_FINAL.pdf [Last accessed on 7 Apr 2021].
82. Welsch ME, Kaplan A, Chambers JM, et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017;168:878-889.e29.
83. Nakamura K, Ichise H, Nakao K, et al. Partial functional overlap of the three ras genes in mouse embryonic development. Oncogene 2008;27:2961-8.
84. Hennig A, Markwart R, Esparza-Franco MA, Ladds G, Rubio I. Ras activation revisited: role of GEF and GAP systems. Biol Chem 2015;396:831-48.
85. Freedman TS, Sondermann H, Friedland GD, et al. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci U S A 2006;103:16692-7.
86. Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun 2012;3:1168.
87. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 2015;13:1325-35.
88. Hofmann MH, Gmachl M, Ramharter J, et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov 2020; doi: 10.1158/2159-8290.CD-20-0142.
89. Shi ZQ, Yu DH, Park M, Marshall M, Feng GS. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol Cell Biol 2000;20:1526-36.
91. Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 2008;20:453-9.
92. Sun X, Ren Y, Gunawan S, et al. Selective inhibition of leukemia-associated SHP2E69K mutant by the allosteric SHP2 inhibitor SHP099. Leukemia 2018;32:1246-9.
93. Chen YN, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148-52.
94. De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304:1812-20.
95. LoRusso PM, Sebolt-Leopold JS. One Step at a Time - Clinical Evidence That KRAS Is Indeed Druggable. N Engl J Med 2020;383:1277-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Conroy M, Cowzer D, Kolch W, Duffy AG. Emerging RAS-directed therapies for cancer. Cancer Drug Resist 2021;4:543-58. http://dx.doi.org/10.20517/cdr.2021.07
AMA Style
Conroy M, Cowzer D, Kolch W, Duffy AG. Emerging RAS-directed therapies for cancer. Cancer Drug Resistance. 2021; 4(3): 543-58. http://dx.doi.org/10.20517/cdr.2021.07
Chicago/Turabian Style
Conroy, Michael, Darren Cowzer, Walter Kolch, Austin G. Duffy. 2021. "Emerging RAS-directed therapies for cancer" Cancer Drug Resistance. 4, no.3: 543-58. http://dx.doi.org/10.20517/cdr.2021.07
ACS Style
Conroy, M.; Cowzer D.; Kolch W.; Duffy AG. Emerging RAS-directed therapies for cancer. Cancer Drug Resist. 2021, 4, 543-58. http://dx.doi.org/10.20517/cdr.2021.07
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 57 clicks
Cite This Article 57 clicks

 Like This Article 38
likes
Like This Article 38
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.