
Teaching an old dog new tricks: reactivated developmental signaling pathways regulate ABCB1 and chemoresistance in cancer

Abstract
Oncogenic multidrug resistance (MDR) is a multifactorial phenotype intimately linked to deregulated expression of detoxification transporters. Drug efflux transporters, particularly the MDR P-glycoprotein ABCB1, represent a central mechanism by which not only chemotherapeutic drugs are extruded or sequestered to prevent drug delivery to their intracellular targets, but also for inhibiting apoptotic cell death cues, such as removal of proapoptotic signals. Several cell populations exhibiting the MDR phenotype co-exist within a tumor, such as cells forming the bulk tumor cell mass, cancer stem cells, and cancer persister cells. The key to regulation of ABCB1 expression is the cellular transcriptional machinery. Developmental signaling pathways (e.g, Hedgehog, Notch, Wnt/β-catenin, TGFβ, PITX2) are pivotal in governing cell proliferation, survival, differentiation and guiding cell migration during embryogenesis, and their reactivation during carcinogenesis, which is of particular significance for tumor initiation, progression, and metastasis, also leads to the upregulation of ABCB1. These pathways also drive and maintain cancer cell stemness, for which ABCB1 is used as a marker. In this review, the contribution of canonical and non-canonical developmental signaling pathways in transcriptional regulation of ABCB1 to confer MDR in cancer is delineated.
Keywords
Introduction
Bacterial resistance to antibiotic drugs was first described after the discovery that penicillin prompted bacteria to develop several important defense mechanisms, including the expression of efflux transporters in the outer cell wall. The broad range of substrates used by these transport proteins resulted in coining the term multidrug resistance (MDR) as pathogens can limit the accumulation of drugs targeted against them[1]. Widespread use of chemotherapeutic drugs in cancer therapy resulted in the evolution of an analogous program in mammalians, and the often-reported human form of MDR in tumor cells. According to patient records, the first human case of resistance to chemotherapy was observed in 1942 during the use of nitrogen mustard gas to treat a patient with recurrent lymphosarcoma[2].
The discovery of a molecular link strongly associated with the MDR phenotype: P-glycoprotein (“altered drug permeability glycoprotein”), abbreviated as P-gp, gp170, P170 or, according to current nomenclature, ABCB1 (ATP-binding cassette sub-family B member 1) was achieved by exposure of non-drug resistant cell lines to increasing concentrations of cytotoxins[3]. In colchicine-resistant Chinese hamster ovary cells, a membrane glycoprotein absent in parental cells was associated with an ostensible “permeability barrier” to colchicine toxicity[4]. This glycoprotein was further characterized and designated as P-gp based on its apparent ability to control drug permeation by modulating properties of hydrophobic membrane regions in drug-resistant cells[5]. In the first instance, the druggability of ABCB1 was promising because it represented a molecular link to MDR. Drug development has spawned three generations of ABCB1 inhibitors, which are usually effective in reversing chemoresistance in cell lines but are hampered in in vivo models or human clinical trials by physiological ABCB1 expression at barrier and excretion sites, culminating in adverse side effects. Development of the “fourth generation” ABCB1 modulators is focused on already available natural compounds[6], yet it is becoming increasingly apparent that MDR is a multifactorial phenomenon that will require targeting of a mechanism underlying several MDR-contributing factors[7,8].
ABCB1-dependent MDR commonly involves regulation of expression, such as gene transcription, epigenetics, or post-translational modifications[7,9]. Due to ABCB1’s indispensable roles in tissue protection and detoxification, its expression follows developmental patterns in the fetus as the key sites for ABCB1 begin to develop. These include endothelium of the blood-brain-barrier, proximal tubule of the kidney, intestinal epithelium, and liver hepatocytes. Little is known about ABCB1 expression during gestation; it is presumably under the control of transcription factors belonging to developmental signaling pathways that orchestrate the intricate and highly choreographed process of cell growth and patterning[10,11].
With exception of some specialized tissues, developmental signaling pathways such as Hedgehog, Notch, and Wnt, are switched off and remain dormant in adult life. However, they can be aberrantly reactivated during carcinogenesis[12-14], wherein cellular proliferation and migration becomes uncontrolled. Upregulation of ABCB1 through these signaling pathways poses a major hurdle because not only is it pivotal to tumor MDR, but it is also a major contributor to the survival of self-renewing tumor cells that are capable of regenerating a tumor after treatment, and thus a marker of cancer cell stemness.
In this review, the resurgence of signaling activity of pathways classically associated with development of oncogenic ABCB1-dependent MDR will be discussed. Also, the discovery of the HOX gene and homeodomain protein PITX2 (Paired Like Homeodomain 2) as a positive ABCB1 regulator will be presented.
ABCB1
Physiological expression of ATP-binding cassette (ABC) transporters, such as multidrug resistance (MDR) P-glycoprotein MDR1/ABCB1, multidrug resistance related protein MRP1/ABCC1, and breast cancer resistance protein 2 BRCP2/ABCG2, at barrier sites, including colon, liver, kidney, and blood-brain barrier, serve to protect against xenobiotics and toxic metabolites[15]. The importance of ABCB1 in protection from damaging agents is highlighted by an anecdote associated with the generation of the Abcb1a-/- mouse. Though the mouse was viable and showed no functional deficiency, it was far more sensitive to chemical challenges as exemplified by an accidental observation where the centrally-neurotoxic pesticide, ivermectin, proved lethal to Abcb1a-/-, but not Abcb1+/+, mice due to compromised blood-brain barrier function[16].
The canonical MDR protein, ABCB1, belongs to the evolutionary-conserved ABC superfamily, a large group of transmembrane (TM) transporters that utilize energy to translocate various substrates across membranes. At the plasma membrane, ABCB1 extrudes a broad range of structurally unrelated substrates directed towards the extracellular space. Substrate suitability for ABCB1 is determined and limited by size, charge, and hydrophobicity; neutral or cationic compounds ranging from 300-4000 Da and compounds with high hydrophobicity are favored, though amphiphilic compounds are also translocated[17-20]. Moreover, ABCB1 is a lipid translocase with broad specificity[21], extruding simple phospholipids and sphingolipids[22,23].
Structure and function of ABCB1
ABCB1 structure
Full length human ABCB1 was cloned in 1986[24,25], but it required more than a decade to visualize the tertiary structure of mouse ABCB1 by X-ray crystallography[26]. Functional ABC transporters typically consist of at least two TM domains (TMDs) and two nucleotide binding domains (NBDs)[17]. In humans, ABCB1 protein is synthesized from the ABCB1 gene (previously MDR1) generating a single polypeptide chain that is folded to form two homologous parts, each consisting of six a-helical TMDs and an NBD, joined by a cytoplasmic linker region. The α-helical TMDs of each homologous half create a bundle that comes together to form a large internal cavity, which can easily accommodate two compounds[27]. In the resting state, ABCB1 is found in a triangular conformation where the extracellular bundle “heads” are close to each other. The substrate-binding pocket faces inward to accept compounds only from intracellular origin, that is, the cytoplasm and inner membrane leaflet (IML) of the lipid bilayer, but not from the extracellular compartment or outer membrane leaflet (OML)[28]. The predicted substrate-binding pocket contains mostly hydrophobic and aromatic residues, and each substrate has a defined amino acid subset for its recognition[29].
ABCB1 transport function
In the catalytic cycle, substrate binding stimulates ATP binding to the NBDs, leading to their dimerization, which in turn, drives the large structural change of ABCB1 from an inward to outward-facing conformation. The substrate is then released on the opposite side of the membrane through decreased binding affinity, facilitated by ATP hydrolysis or by the law of mass action. Following hydrolysis of ATP, NBD dimerization is disrupted and ABCB1 returns to the pre-transport state[26].
Structural and mutational studies reveal potential mechanisms by which ABCB1 may extrude a wide variety of structurally unrelated substrates[17]. The “vacuum cleaner” model describes recognition, extraction, and extrusion of hydrophobic cationic substrates partitioned in the hydrophobic portion of the IML[28,30,31]. However, this model is increasingly challenged because ABCB1 transport kinetics do not follow Michaelis-Menten kinetics. An alternative “oscillation” hypothesis has been proposed: continuous interchange between inward and outward conformations results in the stochastic extrusion of substrates translocated to the IML by flippase activity[32] or passive distribution via lateral diffusion[33]. According to this model, effective ABCB1 inhibitors likely lock ABCB1 in the outward conformation and thereby hinder substrate extrusion.
Garnering evidence supports both the vacuum cleaner and oscillation models[34,35]. The partition coefficient of a substrate into the lipid bilayer, dependent on its chemistry, is an important determinant of ABCB1 affinity and thus transport[36]. Interestingly, in addition to the hydrophobic core of the membrane, ABCB1 potentially binds substrates that partition at the cytoplasmic surface of the membrane[34,37,38]. For extrusion, the molecule laterally diffuses to the substrate-binding chamber of ABCB1 driven by its hydrophobic residues, and without direct involvement of the hydrophobic core. Taken together, efflux of substrates by ABCB1 is a multistep process involving (1) initial substrate binding to membrane lipids; (2) entry; partitioning into the lipid bilayer/interfacial region or flippase-driven movement into the IML (3) recognition by ABCB1; (4) uptake from the hydrophobic bilayer core or interfacial region, and binding pocket entry; and finally (5) transport to the opposite/extracellular compartment through a conformational change of ABCB1 facilitated by ATP hydrolysis. Thus, an apparent combination of lipid binding and ABCB1 substrate recognition forms the basis of ABCB1 efflux function.
ABCB1 upregulation in chemoresistant cancer cells
Ineffective chemotherapy treatment is one of the greatest challenges posed by modern cancer treatment, which in part, results from upregulation of broad range transporters such as ABCB1, in many cancer cells. High expression of ABCB1 in tumors is associated with increased cell survival due to efficient extrusion of chemotherapeutic drugs, evasion of apoptosis, and increased metastatic potential, resulting in poor prognosis[9,39]. As detailed below, levels of ABCB1 are predominantly regulated by transcription factors[40] such as developmental transcription factors TCF4 (T-cell factor 4)[41] or PITX2[42,43] that have become oncogenic.
Intracellular localization of ABCB1
Drug resistance is not only determined by the expression of ABCB1 in the plasma membrane, wherein ATP-dependent drug efflux to the extracellular space prevents its accumulation in the cell, but also by the intracellular expression of functional ABCB1[44-46] that further restricts drugs from entering the nucleus where drug targets are commonly localized. Cytoplasmic ABCB1 expression was identified in drug resistant gastric carcinoma cells with immunohistocytochemistry[47]. Using pulse chase experiments and tracking of autofluorescent daunorubicin, an anthracycline chemotherapeutic drug, nucleus-derived fluorescent vesicles appeared to be trafficked to the cell periphery followed by their exocytosis[47]. Similar findings were reported in LLC-PK1 renal proximal tubule cells[48], and the acidic nature of the ABCB1 expressing or drug-accumulating vesicles was revealed[45,48,49]. The presence of functional ABCB1 in vesicular structures was confirmed using immunostaining, which ruled out endocytic vesicles and fluorescent substrate accumulation[44]. The origin of ABCB1/drug-accumulating vesicles is still unclear. An acidic vesicular pool that accumulates anthracycline drugs is well-defined and accumulating evidence points towards lysosomes as a potential target, wherein ABCB1 is localized as part of its degradation/turnover cycle[50] and internalized into after tumor micro-environment stress signals, such as hypoxia or nutrient starvation[51]. Moreover, lysosome biogenesis was found to be induced, facilitating mislocalization of doxorubicin to lysosomes and consequent drug resistance[51,52].
A further line of defense against nuclear drug accumulation has been evidenced directly in the nuclear envelope[46,53,54]. A different glycoside chain could explain the targeted trafficking of ABCB1 to the nucleus[54]. Kinetic studies have suggested that vesicles bud from the nuclear envelope to expel drugs from the cell through exocytosis[47], whereas direct nuclear-to-cytosol efflux activity of ABCB1 might be affected by local ATP concentrations and lipid composition of nuclear membranes.
Lipid microenvironment in ABCB1 functionalization
Integral membrane proteins often exhibit lipid binding specificity and are dependent on their lipid microenvironment for correct function. Functionalization of ABCB1 is determined not only by the composition of lipids in its immediate vicinity but also by fluidity, phospholipid headgroup, and fatty acyl chain length (reviewed in[55]). Small, specialized membrane domains (< 100 nm), also known as lipid rafts (LRs), are highly enriched in cholesterol, glycosphingolipids, and phospholipids with saturated fatty acids, particularly SM, and provide an optimal lipid microenvironment for integral membrane protein function (reviewed in[56,57]). ABCB1 localizes to LRs[58,59], possibly due to its capacity to bind cholesterol[60] or SM[8]. Cholesterol depletion reduces ABCB1 transport activity, which is associated with migration of ABCB1 out of LRs, indicating ABCB1 activity dependence on LR localization[61,62]. In support, cholesterol repletion enhances ABCB1 activity[62] and movement of substrates across a lipid membrane is more efficient when cholesterol and SM are present[63].
Transcriptional regulation of ABCB1
ABCB1 expression is predominantly ruled at the transcriptional level. The eukaryotic transcriptional machinery is a large multi-protein complex, comprised of (co-)activators, (co-)repressors, polymerases, and more[64,65]. DNA accessibility and responsiveness are additional crucial factors for the transcriptional machinery to bind to target DNA sequences and initiate or repress gene expression.
The promoter region of ABCB1 contains multiple sites for activating transcription factors, including myc, Sp1, AP-1, nuclear factor kappa B (NF-κB), TCF (reviewed in[40]), and the more recently-discovered, Sp3[66], octamer-binding transcription factor 4 (OCT-4)/POU5F1[67] and PITX2[42,43]. Repression of ABCB1 expression can occur through the binding of oncogenic chimeric proteins[68,69], DNA methylation[70,71] or a combination of transcription factors, for example, NF-κB with c-Fos[72]. Furthermore, recent findings in drug resistant ovarian and breast cancers showed evidence of multiple aberrant transcriptional fusions of ABCB1, placing it under control of alternative promoter regions from genes in chromosomal proximity to ABCB1[73]. These data point towards non-canonical mechanisms of transcriptional regulation that broaden the range of ABCB1 regulators and thus increasing disposition to MDR progression. The full impact of upstream signaling pathways in regulating ABCB1 transcriptional fusions is yet to be clarified. Thus, elevated ABCB1 levels can be controlled through several ways, including increased activator activity, decreased repressor activity, chromatin remodeling[74], DNA priming[75], and transcriptional fusions[76].
Since many of the aforementioned transcription factors are altered by stressors, ABCB1 is highly susceptible to upregulation during a stress response. ABCB1 expression can also be modified by single nucleotide polymorphisms (SNPs)[77,78], which play an important role in cancer therapy, gene rearrangements[79], mutations[17,80], epigenetic regulation[81,82], and post-translational mechanisms[83].
ABCB1 expression in chemoresistant cancer stem cells
Cancer stem cells
Intratumor heterogeneity stems from various intratumor cell populations[84] and/or spatial heterogeneity between intratumor structures and the tumor microenvironment[85]. Tumor initiating or cancer stem (-like) cells (CSCs) possess self-renewing capacity, ability to differentiate into multiple tumor cell types, and display enhanced resistance to chemotherapy and apoptosis cues[86-89]. In solid tumors, they were first identified, isolated, and characterized by presence of the stem cell marker, CD133, in the brain[90]. During therapy, some tumor cell populations are incapable of defending themselves and undergo cell death, whereas CSCs are well equipped with defense mechanisms such as altered drug transporter expression, which render them impervious to the death cues proffered by anticancer drugs[91]. Due to the small number of CSCs in a tumor, they are often below detectable levels and may remain dormant for a significant time (i.e, years to decades). The existence of CSCs has been much debated[92] and could be limited to certain hemopoietic cancers and some solid tumors. Several hypotheses exist for the origin of CSCs, such as cell fusion, de-differentiation of tumor cells, or genomic instability (reviewed in[93]).
Since their discovery, a multitude of CSC markers have emerged. Aside from CD133+, which is also found in persister cancer cells[94], CD24-, CD44+, ABCB1+, ABCG2+ and ALDH+ are common phenotypic markers for CSCs. Because of the challenges in identifying expression patterns of cell surface CSC-specific markers across all CSCs from different cancer tissues, current methodologies take advantage of the ABC transporter substrate Hoechst-33342 (Hoechst) combined with flow cytometry analysis to isolate the so-called side population. Since CSCs express high ABC transporter levels, there is less accumulation of Hoechst dye in the cells, which can be detected through cellular fluorescence intensity[95].
ABCB1 in cancer stem cells
The challenge of eradicating MDR cancers lies not only in tumor cells that do not respond to chemotherapeutic therapy and continue to proliferate, but also encompasses the tenacity of CSCs[96] that are drug-tolerant and contribute to tumor repopulation. CSCs undergo genetic changes such as increased expression of antiapoptotic Bcl-2 and multiple ABC drug transporters, causing acquisition of apoptosis resistance and chemoresistance, respectively[89,97,98].
ABCB1+ CSCs have been isolated from non-small cell lung cancer[99], ovarian cancer[100-102], colorectal cancer[103-105], pancreatic cancer[106], oral squamous cell carcinoma[107] and glioblastoma[108,109]. Similar to tumor cells and epithelial cells, ABCB1 expression (mRNA and protein) in CSCs is largely governed by transcriptional regulation, which has been demonstrated for Kelch-like ECH-associated protein 1 (KEAP) with nuclear factor erythroid 2-related factor 2 (NRF2)[110], OCT-4/POU5F1[67,111], NANOG/STAT3[112], Twist[113], and Wnt/β-catenin[114]. Intriguingly, expression or activation of surface markers CD133+[115] or CD44+[112], respectively, engages ABCB1-elevating signal transduction, indicating that stemness is inherently linked to chemoresistance.
Developmental signaling pathways
A complicated, highly choreographed series of molecular events orchestrate cell lineage commitment, cell expansion, cell differentiation, organ asymmetry, positioning, patterning, and organism sculpturing during embryonic development[116-118]. Developmental signaling pathways usually remain dormant in adult life but are frequently reactivated during carcinogenesis, contributing to cell survival, proliferation, resistance to apoptosis and MDR[12-14]. With a limited combination of receptors and ligands, the pleiotropic effects of gene transcription targeted by developmental signaling need to be adequately communicated. Aside from correct timings, receptor-ligand interactions are finely tuned and highly discriminating, such that even slight variations in the associated interaction dynamics and kinetics have a profound effect on signal transduction. These communication codes encompass differences in signal amplitude, frequency, duration, fold change, ligand combinations, ligand concentrations, and kinetics of ligand binding (reviewed in[119]).
Classical developmental signaling pathways in ABCB1 regulation in cancer
Notch signaling
In contrast to other developmental signaling pathways, which are engaged following docking of a secreted ligand to cell surface receptors, the Notch pathway is activated through ligand-receptor binding of contiguous cells. Notch signaling is active in several developmental programs, mostly in the ones that determine cell differentiation, but also in the cell proliferation and stem cell maintenance programs in tissues such as the heart, central nervous system, and pancreas (reviewed in[120]).
Canonical Notch signaling
Four receptors (Notch1-4) and multiple ligands (Delta, Jagged) have been identified in mammalians. Notch receptors are type I membrane proteins with a single transmembrane pass. Upon docking of the ligand onto the Notch receptor, a conformational change permits a successive two-step cleavage event in a process termed regulated intramembrane proteolysis[121-124]. The first and rate-limiting step involves matrix metalloproteases (ADAMs, A Disintegrin And Metalloproteinases), which dissociate the extracellular and intracellular domains of Notch receptor at the S2 cleavage site. The Notch extracellular domain (NECD) is shed from the membrane and the cleavage product Notch Extracellular Truncation (NEXT) remains membrane bound. The second step requires scission in the transmembrane domain by the γ-secretase complex[125,126] at the S3/4 cleavage sites, releasing the Notch intracellular domain (NICD) from the membrane into the cytoplasm and permitting its translocation to the nucleus. Endocytosis of the Notch receptor occurs and is crucial for signal transmission. Moreover, Notch signals can be conveyed differentially through the generation of different products that have varying signal duration and downstream targets[127], allowing for the communication of distinct ligand-receptor combinations[119].
Another level of regulation exists in the cytoplasm. There, NICD turnover is controlled by modification of the C-terminal PEST domain, which is rich in proline (P), glutamic acid (E), serine (S), and threonine (T). Generally, phosphorylation and ubiquitination affect degradation, whereas hydroxylation and acetylation affect the half-life of NCID[127]. Once NICD reaches the nucleus, it binds to CSL/RBPjK (CBF-1, Suppressor of Hairless, Lag-2/Recombination signal Binding Protein for immunoglobulin Kappa J region) and regulates gene transcription. Pertinent to Notch’s multifaceted role in embryonic development, not one set of genes can be assigned to Notch that is regulated in the same way in every cell or by activation mode, adding to Notch’s complexity and diversity. Common gene signatures include the HESR (Hairy and Enhancer of Split-Related) genes, which encode basic helix-loop-helix-type transcriptional repressors, c-myc, cyclin D1, and Snail[127].
Non-canonical Notch signaling
Notch signaling independent of CSL in both ligand-dependent and independent manners are considered to be non-canonical and is often associated with disease[128]. Increase in NICD (cytoplasmic or membrane tethered) is usually prerequisite and rather than directly altering gene transcription, NICD interacts with components of other signaling pathways such as Akt/PKB (protein kinase B), Yin-Yang-1 (YY1), NF-κB, β-catenin or hypoxia-inducible factor 1-α (HIF-1α), to control transcription (reviewed in[128]). The interaction between Notch and β-catenin has been best described - both NCID and uncleaved Notch receptor can directly interact with the active form of β-catenin, repressing its activity, either by sequestration or by targeting β-catenin to the endo-/lysosomal compartment by endocytosis, respectively (reviewed in[129]).
Regulation of ABCB1 by Notch
Notch signaling has been positively linked to ABCB1 expression in ovarian cancer[130] and cholangiocarcinoma[131]. In recurrent ovarian cancer, Notch3 signaling was overactive. Immunostaining for Notch3 evidenced elevated nuclear levels (up to ~4-fold) in recurrent ovarian serous carcinoma tissues compared to primary carcinoma tissues from the same patients. Furthermore, aberrant activation of Notch3 was associated with poor prognosis[130]. Overexpression of NCID3 in a normal ovarian epithelial cell line (IOSE-80pc) or a low-grade serous ovarian carcinoma cell line (MPSC1) increased stemness, as characterized through mRNA expression of stem cell markers as well as ABCB1 mRNA expression, which correlated with increased resistance to carboplatin[130]. Expansion of a stem cell-like population of cells as well as a ABCB1+ cell population was also reported in pancreatic cancer, in which Notch was activated by the adipocyte hormone leptin, though co-staining for stem cell markers and ABCB1 was not performed[132]. In cell line models for hepatic cholangiocarcinoma, downregulation of Notch1 culminated in decreased ABCB1 expression and sensitization to the fluoropyrimidine 5-fluorouracil[131], an antimetabolite anticancer drug.
Interestingly, studies targeting upstream regulators of the Notch receptor, either through Notch ligand Jagged[133] or receptor regulation[134], did not observe changes in ABCB1 or ABCB1, respectively, pointing to the involvement of the non-canonical Notch signaling pathway. Downregulation of Jagged1 in drug resistant ovarian cancer cell line SKOV3TRip2 did not alter ABCB1 mRNA, but resulted in diminished GLI2 (Glioma-associated oncogene homolog 2), although not GLI1[133]. Similarly, GLI2 downregulation resulted in lowered Jagged1 levels, suggesting bidirectional regulation, and increased sensitivity to the cytotoxic drug, docetaxel. In cisplatin-resistant ovarian cancer cells (SKOV3, A2780), downregulation of caveolin-1 had no impact on total ABCB1 protein expression despite the increased apoptosis to cisplatin[134], a DNA damaging agent. Caveolin-1 can indirectly upregulate Notch receptors via mitogen-associated protein kinase (MAPK) signaling and transcriptional upregulation of POFUT1, a fucosyltransferase, which in turn, activates Notch signaling, as demonstrated by increased NCID, HEY1 (Hairy Ears, Y-Linked 1), and HES1 by immunoblotting and immunofluorescence in hepatocellular carcinoma[135]. In addition, caveolin-1 negates γ-secretase activity, as evidenced by cleavage of amyloid-β-precursor protein and Notch[136]. Loss of caveolin-1 culminated in the distribution of γ-secretase to clathrin-coated non-caveolar endocytic vesicles suggesting that caveolin-1 regulates γ-secretase activity by modulating its spatial distribution, thereby impacting Notch activation.
Collectively, therefore, ABCB1 appears to be independent of ligand-Notch receptor interaction as well as Notch receptor cleavage. The transcriptional regulation of ABCB1 by Notch involves engaging non-canonical Notch signaling, which modulates other signaling networks to alter gene expression. Known non-canonical Notch targets include YY-1[137], NF-κB[138,139], β-catenin[140], and HIF1α[141], all of which are well-evidenced regulators of the ABCB1 gene, but data are lacking for their regulation by Notch signaling in chemoresistant cancer associated with ABCB1. The lack of requirement of Notch receptor cleavage would imply the involvement of active β-catenin, which has been shown to interact with intact membrane-bound Notch[129], and is strongly linked to carcinogenesis and ABCB1. Notch signaling could be engaged through active β-catenin to further strengthen the MDR phenotype and/or cell stemness.
Hedgehog/GLI
The Hedgehog (HH) family of secreted signaling proteins were first discovered in Drosophila where it was found to function in segment polarity of larvae[142,143]. The vertebrate hedgehog genes, encoding three isoforms Sonic Hedgehog (SHH), Desert Hedgehog (DHH), and Indian Hedgehog (IHH), were soon identified and had similar polarizing activity[144-146].
In the absence of HH, the Hedgehog pathway is maintained in an inactive state through the interaction of tumor suppressor Patched 1 (PTCH1) and the proto-oncogene Smoothened (SMO) at the plasma membrane. In ciliated cells, this occurs at the primary cilium. PTCH1 is the primary receptor for HH ligand and in the unbound state exports cholesterol from the lipid bilayer to prevent the activation of SMO, a G-protein coupled receptor belonging to the Frizzled (Fzd) family, although without direct interaction[147], and keeps SMO internalized. When secreted, HH binds to two PTCH1 receptors[148], disrupts cholesterol extrusion, relieves inhibition of SMO and permits changes in HH target genes[149].
Central to the HH response is the nuclear translocation of glioma-associated oncogene homolog (GLI) transcription factors, which are zinc-finger proteins and whose turnover in the cytoplasm is determined by several mechanisms. Stabilization of Suppressor of fused (SUFU) through dual phosphorylation, mediated by protein kinase A (PKA) and glycogen synthase kinase3-β (GSK-3β)[150], results in cleavage by the proteasome of GLI transcription factors generating repressor forms (GLI-R), which translocate to the nucleus and suppress transcriptional activation of HH/GLI target genes, such as elements of the HH signaling pathway (HH, GLI)[151,152]. SUFU can also sequester GLIs by direct binding[153] or promote proteasome processing of the repressor form of GLI3 through the recruitment of GSK3β[154]. GLI transcription factors are directly phosphorylated by PKA[155], MAPK[156], casein kinase 1 (CK1)[157] or Fused family kinases[158] to affect HH signaling.
ABCB1 regulation by HH/GLI
Initial studies applied SHH ligand to LnCAP prostate cancer and Seg-1 oesophagus cell lines, wherein ABCB1 was upregulated and endogenous ABCB1 could be downregulated by GLI siRNA[159]. In chemoresistant Lucena-1 myeloid leukemia cells, PTCH1 and SMO were elevated and GLI1 was more present in the nuclei compared to their chemosensitive K562 counterparts[160]. Inhibition of the HH signaling pathway using cyclopamine and vitamin D3, which both bind to SMO, resulted in decreased expression of PTCH1, SMO, and ABCB1 as well as decreased resistance to the drugs, vincristine (antimicrotubule agent), doxorubicin and mitoxantrone (anthracenedione derivative). Similar effects were observed with GLI inhibitor, Gant61, indicating regulation of ABCB1 by HH/GLI signaling, Interestingly, cyclopamine did not increase chemosensitivity in several further cancer cell lines, including ACHN renal cancer, Jurkat T-lymphocytes, and PC3 prostate cancer cells, suggesting that either these cells do not have active HH/GLI signaling or chemoresistance is governed by other transcription factors[160]. A comparative study for PTCH1, SMO, and nuclear GLI expression between the cell lines was not performed.
Recent studies have identified canonical and non-canonical GLI consensus sequences in multiple ABC transporters, including ABCB1, in chemoresistant cancer cells. In agreement with Queiroz et al.[160], high GLI expression was observed in Colo205 that could be increased by treatment with 5-fluorouracil and oxaliplatin (DNA damaging agent) in a Gant61-sensitive manner[161]. Chromatin immunoprecipitation (ChIP) assays in Colo205 transfected with GLI shRNA or in GLI-overexpressing HCT115 cells, which have low GLI levels, evidenced active regulation of ABCB1 and ABCB1 by GLI, as previously described for electromobility shift assay (EMSA) assays in ovarian cancer cells[162]. In the presence of GLI shRNA, decreased GLI binding to the ABCB1 promoter region was observed, which was in line with decreased transcriptional activity, assessed by pull-down with anti-acetyl-H3 antibody. Analogous to GLI, ABCB1 was elevated by 5-fluorouracil and oxaliplatin, and basal levels could be attenuated by Gant61. In overexpression studies, increased GLI promoter binding was correlated with augmented transcriptional activity, and increased mRNA and protein expression of ABCB1. In silico analysis of patient-derived mRNA expression profiles from three different databases proved inconclusive for ABCB1: ABCB1 was lower in two datasets and higher in the third . Moreover, ABCB1 was linked to poorer prognosis lower survival in two out of the four datasets analyzed[161]. The significance of HH/GLI signaling for ABCB1 expression is further exemplified in the effectiveness of HH pathway inhibitors in reducing ABCB1 protein expression and/or activity[163-165], as well as reducing tumors and prolonging survival in a mouse medulloblastoma model[166].
The β-1,4-galactosyltransferase (B4GALT) family of enzymes is responsible for the synthesis of complex N-linked oligosaccharides present in many glycoproteins as well as for the generation of glycolipids. B4GALT has also been reported to affect the HH signaling pathway, leading to increased MDR in both drug resistant leukemia cell lines and patient samples from acute and chronic myeloid leukemias[167,168]. Overexpression of B4GALT1 or B4GALT5 in HL60 cells resulted in increased expression of key HH signaling components (PTCH1, SMO, GLI-1) as well as ABCB1 and ABCC1, in a cyclopamine-sensitive manner. Furthermore, B4GALT1 and B4GALT5 were elevated by more than 2-fold in > 50% of patient samples exhibiting MDR[167]. With their key role in glycosylation, B4GALT enzymes might either directly affect PTCH1 or SMO synthesis or activity through increased glycosylation. Alternatively, elevated glycolipids could aid in the clustering of cholesterol and impact PTCH1-SMO signaling in the membrane to favor signal transmission.
Sustained HH signaling was found to be the consequence of lowered PTCH1 levels, regulated by microRNA, in glioblastoma multiforme. Repression of PTCH1 in glioblastoma cells exhibiting resistance to temozolomide (a DNA alkylating agent) could be attributed to the microRNA (miRNA), hsa-mir-9-(1-3), which targeted PTCH1 mRNA, and was confirmed in human tissue samples using in silico analysis[169]. In contrast, in multiple myeloma cells and flank mouse models, deregulated HH signaling with downstream increase in proliferation, downturned spontaneous apoptosis and increased drug resistance were a result of augmented autocrine signaling of HH ligands, which are increased in CD138+ multiple myeloma cells[170]. The authors did not observe an increase in ABCB1, but did not test ABCC1 or ABCG2. Rather, the drug resistance acquired was explained by increased expression of the antiapoptotic protein, Bcl-2.
Wnt/β-catenin
The Wnt pathways
The Wnt signaling pathway controls cell proliferation and body patterning throughout the development of both vertebrates and invertebrates. It plays a key role in body axis formation (reviewed in[171-173]). Several branches of the Wnt-mediated signaling cascade have been described[41,174-176]. In mammals, the most prominent is the canonical Wnt pathway that mediates activation of the β-catenin/TCF/lymphoid enhancer factor (TCF/LEF) transcriptional machinery[41,177]. There are three non-canonical pathways: the JNK/planar cell polarity (PCP) pathway, a Ca2+ releasing pathway for cell motility and adhesion, and a PKA-dependent pathway for myogenesis[178].
The canonical Wnt pathway
Wnts are secreted cysteine rich glycoproteins that are essential for a wide array of developmental and physiological processes. Currently, 19 human Wnt genes are identified[179]. The Wnt proteins signal across the plasma membrane by interacting with Wnt receptors, which consist of a heterodimeric complex of Fzd receptors and members of the low-density-lipoprotein-related protein (LRP) family, such as LRP5/6[180]. This trimeric complex formation is a prerequisite for Wnt signaling.
Inhibitors of Wnt signaling belong to small protein families, including soluble Fzd related proteins (sFRP), Dickkopf (Dkk), and Wnt inhibitory factor (WIF) (reviewed in[41,181,182]). Their common feature is to antagonize Wnt signaling by preventing ligand-receptor interactions or Wnt receptor maturation. Conversely, the Wnt activators, R-spondin and Norrin, promote Wnt signaling by binding to Wnt receptors or releasing a Wnt-inhibitory step. Recent studies have uncovered the Lgr5 family, whose members bind R-spondins with high affinity to potently enhance Wnt signals, in adults (reviewed in[183,184]) as well as CSCs[185,186].
In the absence of Wnts, cytoplasmic β-catenin is tagged for degradation by a multi-protein degradation (“destruction”) complex orchestrated by the tumor suppressor protein, Axin. Axin acts as a scaffold for this complex by directly interacting with its other components, adenomatous polyposis coli (APC) and the kinases, GSK3β and CK1, which constitutively phosphorylate β-catenin to promote subsequent ubiquitylation and continuous elimination by the ubiquitin-proteasome pathway (reviewed by[187]).
When Wnt ligands bind to the Fzd-LRP receptor complex, the cytoplasmic tail of LRP5/6 is phosphorylated by GSK3β and CK1, resulting in the binding of Axin[188,189]. In a process that involves activation of the cytoplasmic protein, Dishevelled (Dvl), this leads to disruption of the destruction complex, promoting stabilization, accumulation, and nuclear translocation of the co-activator β-catenin, where it triggers transcription of target genes by associating with transcription factors TCF/LEF[190-192] (reviewed in[171,193]). In the absence of Wnt signaling, TCF/LEF repress target genes, helped by transcriptional co-repressors such as transducin-like enhancer protein (TLE)/groucho(Gro) to silence Wnt responsive genes[194,195]. Upon Wnt signaling, nuclear β-catenin displaces Gro from TCF/LEF and recruits transcriptional coactivators and histone modifiers, Bcl9, Pygopus and CREB-binding protein (CBP)/p300[196-198], which form a multimeric complex with TCF/LEF to drive expression of genes, such as c-myc, cyclin D1, and ABCB1[199-202]. More Wnt target genes can be found on the Wnt homepage[203].
Wnt signaling and ABCB1-dependent MDR in cancer
Since the initial landmark study on APC mutations in the development of colorectal carcinogenesis[204] (reviewed in[205,206]), aberrant Wnt signaling has been shown to affect many cancer tissues (reviewed in[176-178,207-211]). Because Wnt signaling is a key driver of most types of tissue stem cells[212], aberrant Wnt signaling plays an important role in the induction and maintenance of cancer stemness[209].
As a rule of thumb, Wnt contributes to carcinogenesis after genetic mutations and epigenetic mechanisms affecting pathway components, resulting in altered expression of Wnt relevant genes, including ABCB1[208,209,213]. Both mechanisms either positively or negatively interfere with the Wnt pathway, depending on whether stimulatory or inhibitory regulators of Wnt signaling are targeted. Cooperativity between the Wnt signaling and various other developmental signaling pathways has also been implicated in promoting or even potentiating carcinogenesis, such as NF-κB (see[208] and below).
Various epigenetic control mechanisms affect Wnt signaling to alter ABCB1 target gene expression and MDR [Table 1]. CpG island hypermethylation is important for gene inactivation in cancer cells and has been described in almost every type of tumor (reviewed in[214,215]). It affects various gene loci of the Wnt/β-catenin pathway, thereby modulating ABCB1 expression and includes APC (see[216-219]), upstream modulators such as cyclooxygenase 2 (PTGS2)[216,219] or SFRP5[220], and/or target genes including the ABCB1 gene locus[216-219] in various malignancies, e.g, prostate adenocarcinoma, non-small cell lung cancer, or leukemia cells.
Regulation of Wnt signaling and target gene ABCB1 by ncRNAs in malignant tissues and cells
| ID* | Expression in malignant tissue/cell | Malignant tissue/cell | Effect on ABCB1 expression | Target gene | Effect on target gene expression | Mechanism responsible for ↑ Wnt signaling (Wnt) and ABCB1
(direct/indirect) | Ref. |
|---|---|---|---|---|---|---|---|
| miRNAs | |||||||
| hsa-mir-451a | ↓ | Colorectal CSC | ↑ | MIF (Macrophage migration inhibitory factor) | ↓ (mRNA + protein) | MIF↑→COX2↑→Wnt↑→ABCB1↑ | [336] |
| hsa-mir-27a | ↓ | Hepatocellular carcinoma | ↑ | FZD7 (Frizzled 7) | ↓ (protein) | Fzd7↑→Wnt↑→ABCB1↑ | [337] |
| hsa-mir-33a | ↓ | Pancreatic ductal adenocarcinoma | ↑ | CTNNB1 (β-catenin) | ↓ (mRNA + protein) | β-catenin ↑→Wnt↑→ABCB1↑ | [338] |
| hsa-mir-134 | ↓ | Oral squamous CSC | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [339] |
| hsa-mir-506 | ↓ | Colorectal carcinoma | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [340] |
| hsa-mir-122 | ↓ | Hepatocellular carcinoma | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [341] |
| lncRNAs | |||||||
| CCAL | ↑ | Colorectal carcinoma | ↑ | TFP2A (Activating Enhancer Binding Protein 2 Alpha) | ↓ (protein) | TFP2A↓→Wnt↑→ABCB1↑ | [231] |
| PVT1/HSA-LNCG007059 | ↑ | Bladder urothelial carcinoma | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [342] |
| HOTAIR/HSA-LNCG003959 | ↑ | Non-small cell lung carcinoma | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [343] |
| GAS5/HSA-LNCG004395 | ↓ | Breast cancer | ↑ | hsa-mir-221-3p | ↓ (miRNA) | hsa-mir-221-3p↓→DKK2↓→ Wnt↑→ABCB1↑ | [232] |
| CRNDE/HSA-LNCG006310 | ↑ | AML | ↑ | n.d. | n.d. | Wnt↑→ABCB1↑ | [344] |
Acetylation and deacetylation are counteracting, post-translational epigenetic modifications that affect various histone and non-histone proteins[221], whereby acetylation by histone acetyl transferases (HATs) increases transcriptional activation and deacetylation by histone deacetylases (HDACs) is associated with transcriptional deactivation[222]. In a study performed on breast cancer cells, binding of hyaluronan to the CSC marker, CD44+, a target gene of Wnt signaling[223], upregulated HAT CBP/p300, thus promoting acetylation of β-catenin and the inflammatory transcription factor, NF-κB-p65, leading to activation of TCF/LEF and NF-κB-specific transcription. This resulted in upregulation of ABCB1 and the anti-apoptotic gene, Bcl-xL (BCL2L1), and promoted chemoresistance in MCF-7 cells[224]. Conversely, activation of the HDAC Sirtuin 1, by the antioxidant resveratrol, prevented these effects with consequent chemosensitivity and caspase-3 mediated apoptosis.
Among non-coding RNAs (ncRNAs)[225,226], miRNAs are ~22 nucleotide RNAs that function by direct RNA silencing and post-transcriptional regulation of mRNA targets[227]. miRNAs function via complementary base-pairing with sequences within mRNA molecules, which results in repression of protein synthesis. When they are complexed with Argonaute protein, miRNAs use seed sequences near their 5’ end to base pair with a target mRNA to induce deadenylation and decay or translational regulation[227,228]. In various MDR cancers, lower levels of miRNAs targeting mRNAs of Wnt/β-catenin signaling components were associated with increased ABCB1 expression [Table 1].
Long non-coding RNAs (lncRNAs) (> 200 nucleotides) are autonomously transcribed RNAs found in the nucleus, cytoplasm, or both, that do not encode proteins and their specific functions are still under investigation[229,230]. Nevertheless, several studies have investigated the role of various lncRNAs on Wnt/β-catenin signaling and ABCB1 expression in cancer [Table 1]. In most of these reports, increased levels of lncRNAs resulted in activation of the Wnt pathway, increased ABCB1 expression, and MDR. In some instances, the complexity of lncRNA functions became apparent by indirect effects on Wnt signaling, e.g, via interactions with epigenetic or miRNA regulation. Upregulation of the lncRNA CCAL induced by decreased CpG island methylation and increased acetylation of the CCAL promoter region enhanced the development of colorectal cancer by increased proteasomal degradation of transcription factor AP-2a, which derepresses Wnt/β-catenin signaling and ABCB1 expression[231]. In adriamycin resistant MCF-7 cells and breast cancer tissues, decreased levels of the lncRNA GAS5 suppressed the expression of miRNA hsa-mir-221-3p, which relieves expression of the Wnt inhibitor DKK2, resulting in increased Wnt signaling and ABCB1 upregulation[232].
Genetic alterations of the Wnt pathway components are found in most cancer studies on ABCB1 expression and MDR, which are under the control of Wnt signaling. They comprise mutations, deletions or amplifications, resulting in enhancement or reduction/loss of activity of ligands, receptors, and its cytosolic or nuclear components [Table 2] (reviewed in[176,208,209]).
Genetic and expression changes of Wnt signaling in malignant tissues and cells and their impact on target gene ABCB1
| Affected Wnt signaling gene | Expression/Function in malignant tissue/cell | Malignant tissue/cell | Effect on ABCB1 expression | Mechanism responsible for ↑ Wnt signaling (Wnt) and ABCB1 (direct or indirect) | Ref. |
|---|---|---|---|---|---|
| WNT3a | ↑ | Glioblastoma | ↑ | Wnt↑→ABCB1↑ | [114] |
| WNT5a | ↑ | Uterus sarcoma & breast cancer | ↑ | Hypomethylation WNT5A→PKA↑→CRE/TCF↑→
ABCB1↑ | [233] |
| FZD1 | ↑ | Neuroblastoma
AML Breast cancer | ↑ | Wnt↑→ABCB1↑ | [345-347] |
| FZD7 | ↑ | Esophageal squamous cell Hepatocellular | ↑ | Wnt↑→ABCB1↑ | [348,349] |
| LGR5 | ↑ | Colorectal CSC | ↑ | Wnt↑→ABCB1↑ | [105] |
| DVL1-3 | ↑ | Colorectal | ↑ | Nuclear Tcf4/β-catenin complex↑→ABCB1↑ | [350] |
| APC | ↓* | Colorectal CSC | ↑ | Wnt↑→ABCB1↑ | [351] |
| CTNBB1 | ↑
↑** ↑ | Colorectal CSC
Colorectal CML | ↑ | Wnt ↑→ABCB1↑
Wnt ↑→ABCB1↑ Nuclear Tcf4/β-catenin complex↑→ABCB1↑ | [352-354] |
| CREBBP | ± | Uterus sarcoma
Erythroleukemia Colorectal adenocarcinoma | ↑ | MEK1/2/ERK1/2↑→Nuclear Tcf4/β-catenin/CBP complex↑→ABCB1↑ | [355] |
| PYGO2 | ↑ | Breast cancer
Brain glioma | ↑ | Nuclear Tcf4/β-catenin/PYGO2 complex↑→ABCB1↑ | [356,357] |
Examples of other Wnt dependent mechanisms of increased ABCB1 expression are described as follows. Wnt5a is upregulated in MDR uterus sarcoma and breast cancer cells, and is associated with hypomethylation of CpG islands of a Wnt5a intron sequence[233]. Wnt5a increases cAMP response elements and TCF/LEF transcriptional activity, ABCB1 and chemoresistance in MDR cancer cells, suggesting that PKA dependent non-canonical Wnt signaling also regulates ABCB1 expression[233]. Chronic inflammation and oxidative stress are common and co-substantial pathological processes accompanying and contributing to cancers[234]. The pro-inflammatory transcription factor, NF-κB-p65, upregulates PTGS2[235] and ABCB1[139,236]. Coincidently, both PTGS2 and ABCB1 are also target genes of Wnt/β-catenin [202,237,238]. Gutkind et al.[239] first demonstrated that prostaglandin E2 (PGE2), the product of cyclooxygenase 2, enhances colon cancer progression by binding with the G protein-coupled receptor, EP2, by a signaling route that involves the activation of phosphoinositide-3-kinase (PI3K) and AKT/PKB, leading to the inactivation of Axin and release of GSK3β from its complex with Axin. This process relieves the inhibitory phosphorylation of β-catenin and consequently upregulates PTGS2 and ABCB1[239]. Although initially described for colorectal cancer progression, this mechanism has been described in other cancer types[240-243]. This positive feedback loop (via upregulation of PTGS2) would further enhance aberrant Wnt signaling in affected cancer tissues, and possibly ABCB1 upregulation. Interestingly, this cooperative inflammatory and carcinogenic signaling mechanism has led to promising therapeutic anti-cancer concepts with nonsteroidal anti-inflammatory drugs (reviewed in[244-246]).
Other developmental signaling pathways
Additional transcription factors or aberrant epigenetic misregulation have been reported to regulate ABCB1 expression and/or activity.
Bone morphogenetic proteins & transforming growth factor β
The TGFβ superfamily comprises multiple ligands, such as BMPs, TGFβ, Activins as well as Nodal and Lefty, and their corresponding receptors at the cell surface[247]. In embryonic development, they are required for axis formation, left-right-patterning, organ asymmetry, gastrulation, and organogenesis (reviewed in[248]). Upon ligand binding, a heteromultimeric receptor complex transduces signals to the intracellular milieu via receptor-activated Smad proteins, which are phosphorylated through the acquired serine/threonine kinase receptor activity[249,250]. TGFβ, Nodal, and Activin receptors generally activate Smad2/3, whereas BMP receptors, which do not exhibit cooperative assembly, use Smad1/5/8, though other combinations of receptors and Smads have also been observed[251,252]. Cytosolic co-Smad4 is recruited to the phosphorylated Smads forming a complex that is exported out of the nucleus at a slower rate than monomeric Smads, leading to their accumulation in the nucleus[253] and subsequently to the elevation of transcriptionally regulating genes associated with cell proliferation, cell cycle, apoptosis, and cell differentiation. Signaling to the nucleus can also occur through Smad-independent pathways, such as MAPK and PI3K/AKT.
Deregulation of TGFβ signaling is well documented in cancer playing a role, not only in tumor cell growth and survival but also in determining tumor heterogeneity and self-renewal of CSCs (reviewed in[254]). Exogenous application of TGFβ induces spontaneous neoplastic transformation of hepatocytes, correlating with augmented ABCB1[255], as well as increasing the side population, ABCB1 mRNA, TGFβ receptor mRNA, and MAPK signaling in lymphoma cell lines[256]. In A549 lung cancer cells, antisense oligonucleotides targeting hsa-mir-10a reduced phosphorylated Smad2, survival proteins (Bcl-2, Survivin), and ABC drug transporters (ABCB1, ABCC1) with increased sensitivity to cisplatin[257].
Fibroblast growth factor
The fibroblast growth factors (FGF) are a family of 23 known secreted growth factors recognized by five FGF receptor (FGFR) tyrosine kinases[258]. Ligand binding induces FGFR dimerization and can initiate several phosphorylation cascades, such as PKC, STAT (Signal Transducer And Activator Of Transcription), MAPK, and PI3K/AKT. Transcriptional regulation of genes associated with proliferation, differentiation, and growth depends on STAT1/3/5, FOXO1 (Forkhead Box O1) or ETS (V-Ets Avian Erythroblastosis Virus E26 Oncogene Homolog) transcription factor activity. In cancer, FGF signaling contributes to cell growth and survival, chemoresistance, and neoangiogenesis[259].
In paclitaxel-resistant prostate cancer PC3 cells, ETS1 silencing inhibited the activity of the ABCB1 promoter, which contains ETS binding sites, reduced ABCB1 protein, and reversed resistance to paclitaxel[260], an antimicrotubule drug. ETS mediated regulation of ABCB1 was verified in a gastric cancer cell line exhibiting enhanced ABCB1 expression and vincristine resistance using ETS2 overexpression combined with ChIP assays[261]. At the level of FGF, immunohistochemical studies of bladder cancer tissue from patients showed correlation between basic FGF and ABCB1 signals[262].
Hippo/YAP
Growth factors, signaled through cell surface receptors, and cell density, signaled through cell-cell contacts, are the major regulators of the Hippo/YAP pathway, which have multiple roles in the course of development, including growth control and morphogenesis[263]. When a growth signal is triggered, the transcription factor Yes-associated protein (YAP) escapes cytosolic degradation and translocates to the nucleus to drive gene transcription. When growth should be inhibited, YAP is phosphorylated[264,265], retained in the cytosol by binding to 14-3-3, or degraded by the proteasome. Though there are indications in the literature that ABCB1 mRNA and protein are driven by Hippo/YAP signaling in both tumor cells[266,267] and ovarian CSCs[268], further experimental evidence is required to ascertain direct or indirect as well as positive or negative transcriptional regulation.
YBX1
Y-box binding protein 1 (YBX1 or YB-1) is a transcription factor, expressed at various stages of development and in early hematopoiesis[269,270], but is also active in tumor cells[271-273]. Deregulation of YBX-1 in carcinogenesis seems to be attributed to a combination of epigenetic alterations and MAPK signaling[274-276]. Furthermore, YBX1 binds to the promoter region of ABC transporters, including ABCB1, and predicts poor outcome (reviewed in[277]). Moreover, YBX1 transcriptionally regulates genes involved in cell proliferation[271], cell cycle[278] and metabolism[273], which can, in turn, impact ABCB1 expression and activity, e.g., GSK3β[271] or NF-κB[279].
Snail
The Snail transcription factors (SNAI1-3) are well evidenced in epithelial-mesenchymal transition (EMT) whereby metastatic tumor cells acquire the ability to detach from the tumor mass[280,281]. In embryonic development, Snail is intimately involved in early patterning[282], most likely through transcriptional repression[283] and conferring resistance to cell death by cell cycle inhibition[284]. Epigenetic alterations, such as changes in non-coding mRNA or DNA methylation status, are the major contributors to the deregulation of Snail in cancer (reviewed in[285]).
In hepatocellular carcinoma cell lines (MHCCLM3, SMMC-7721), MDR was increased by augmented levels of ABCB1, ABCB1 and ABCG2 when SLUG (SNAI2) was downregulated. ABCB1 promoter luciferase assay and promoter sequence analysis predicted SNAI2 promoter binding[286]. Though these observations do not align with the EMT role of Snail family members, the authors suggest a tumor suppressor role for SNAI2 that is tumor-type specific[286]. Conversely, in colorectal cancer, elevated Snail correlated positively with tumor size and metastatic nature in patient tissue samples. Snail overexpression in HCT116 and SW480 cells resulted in upregulation of ABCB1 mRNA and protein without significant effects on other relevant MDR ABC transporters (ABCCs, ABCG2). ABCB1 promoter luciferase and ChIP assays evidenced direct binding of Snail to the ABCB1 promoter[287], indicating that Snail and ABCB1 promote EMT. The transcription repressor function of Snail does not align with the positive transcription regulation of ABCB1. However, rather than simply preventing transcription through blockade of the RNA polymerase, repressors can also modify DNA looping by binding at multiple DNA sites, priming a transcription site to modulate cellular responsiveness or fine-tuning the transcriptional response[288].
A novel developmental transcriptional regulator of ABCB1 in cancer: PITX2
The paired-like homeodomain (PITX) subfamily of bicoid class homeodomain proteins consists of three paralogues, PITX1, PITX2, PITX3, which have essential roles in embryonic development, including organ asymmetry[289], through their function as transcriptional regulators. The PITX proteins harbor a highly homologous homeobox domain (97% homology), in which a crucial lysine residue is expressed at residue 50, which is determinant of DNA binding specificity[289]. A high degree of similarity is also seen in the C-terminus (55%-70%) which is required for protein-protein interactions, such as dimerization with other PITX proteins. The N-terminus shows fewer common amino acids with homology varying from 18%-31%[289]. PITX2 undergoes alternative splicing and the use of different promoter regions results in four isoforms (PITX2A, PITX2B, PITX2C, PITX2D)[290,291], which exhibit isoform transcriptional specificity, sometimes working in synergism, for target genes in embryonic development and cellular functions[292]. For example, PITX2B regulates heart asymmetry[293], while PITX2A alters cytoskeleton and migration properties[294], and PITX2C preferentially activates atrial natriuretic factor expression in cardiogenesis[295]. PITX2D does not harbor transcriptional activity since the homeodomain is non-functional. Rather, PITX2D suppresses transcriptional activity of other PITX2 isoforms through interaction and formation of heterodimeric complexes[292]. PITX2 isoforms can also work synergistically depending on the targeted promoter[292].
PITX1 governs the development of anterior structures and the brain, including the pituitary gland; PITX2 determines organ asymmetry and pituitary gland development whereas PITX3 is involved in lens formation and maintenance of midbrain dopaminergic neurons[289,296]. Pitx2 knockout mice are embryonically lethal due to developmental deficits in multiple organs[297] whereas Pitx1 knockout mice die after birth[298]. In adults, PITX2 activity is restricted to selective tissues, appearing to be important in cardiac injury recovery[299] and maintenance of mature pituitary function[300]. The increase in cell death occurrence during development in Pitx2 knockout mice serves as an indication of its potentially pivotal role in governing cell life and death[300]. Mutations in PITX2 manifest in autosomal dominant Axenfeld-Rieger syndrome, a group of diseases affecting the development of the anterior segment of the eye[301,302].
PITX2 in cancer
Though PITX1 has been linked to cancer, it has anti-tumorigenic properties[303-305] and a role for PITX3 has not been well-established. In contrast, PITX2 was identified as a target gene of human acute leukemia ALL1 in ALL1-/- embryonic stem cells. Furthermore, its downregulation in leukemia cell lines with ALL1-inactivating chromosomal rearrangements was indicative of the oncogenic property of PITX2/ARP1 in leukemia[306], possibly in conjunction with hypermethylation of its promoter, observed in acute myeloid leukemia[307]. Early studies linked PITX2 to Wnt/β-catenin signaling, which is frequently deregulated in tumorigenesis, either by acting as a β-catenin target gene[308] or working synergistically with β-catenin to regulate promoter activity and hence gene expression[309]. Subsequently, PITX2 mediated regulation of cyclin A1[310], cyclin D2/CCND2[308,311], cyclin D1/CCND1, and c-Myc/MYC[312] has been observed. Though increased PITX2 has been associated with cancer progression in various tumors[313-315], the subset of genes regulated by PITX2 appears to be cancer specific. The presence of PITX2 in sites outside of the primary tumor suggests its role in invasion and metastatic potential[316].
Several cellular factors could promote aberrant PITX2 expression in cancer. In silico analysis found the highest PITX2 RNA changes in human colon adenocarcinomas[43] and could be attributed to APC, a β-catenin suppressor frequently mutated in colon cancer, thus driving β-catenin excess, nuclear translocation, and transcriptional upregulation of PITX2[308]. As mentioned above, DNA methylation patterns are tumor-specific and impact cellular architecture. Hypermethylation of the PITX2 promoter, resulting in decreased PITX2 expression, has predicted survival[317,318], but has also been associated with poor prognosis[307,319], and is a prospective prognostic biomarker for prostate cancer[320]. Recent evidence points to miRNA-mediated regulation of PITX2. PITX2 is negatively regulated by miRNAs[321], but PITX2 can also regulate miRNAs to effectuate its downstream effects, such as augmented cell proliferation[322]. Potential additional malfunctioning mechanisms involved in aberrant oncogenic PITX2 include mRNA export, translation initiation factors, protein folding, and proteolysis in the cytosol and nucleus.
Upregulation of PITX2 appears to be an early event in tumorigenesis observed in tumor tissue sample at early stages[43], suggesting that PITX2 could increase susceptibility or permissibility to manifestation of mutation events or adaptive mechanisms by upregulating genes favoring tumor progression such as cell cycle control genes[312,314,322], proto-oncogenes[312,314], EMT markers[315], and MDR mediating factors[42]. Moreover, PITX2 could transcriptionally repress tumor counteracting/suppressor genes or epigenetically alter gene expression[323,324]. Regulation of PITX2 cytoplasmic-nuclear shuttling remains uninvestigated. Evidence for its multifunctional C-terminal tail, which has protein interaction capabilities[325], a potential nuclear localization sequence[326], and PKC phosphorylation sites[327] suggests that PITX2 could be regulated analogously to other related HOX transcription factors, namely through cytosolic levels.
PITX2 regulation of ABCB1
In renal cell carcinoma (RCC) and colon cancer cell lines exhibiting MDR and augmented ABCB1 expression, we have identified PITX2 as a contributor to chemotherapeutic drug resistance through transcriptional upregulation of ABCB1[42] as well as ABCC1, ABCG2, and the drug uptake transporter, SLC22A3[43]. The PITX2 consensus sequence, TAATCC, was found at -7,626 bp and -14,510 bp in the ABCB1 promoter region[42]. Using ChIP assays by pulling down overexpressed myc-tagged PITX2C[42] or endogenous PITX2[43], PITX2 was evidenced to bind to both sites in chemoresistant colon and renal cancer cell lines and was supported by RNAi studies wherein PITX2 downregulation resulted in attenuated ABCB1 mRNA and ABCB1 protein as well as chemoresistance[42,43]. The action of PITX2 on ABCB1 is independent of β-catenin since PITX2 overexpression in β-catenin-deficient mouse keratinocytes increased ABCB1 expression, cell survival, and chemoresistance[42] and the GSK3β inhibitor, SB216763 (which would augment β-catenin stabilization), decreased PITX2 promoter activity[43].
Though all PITX2 isoforms can upregulate ABCB1, synergism of PITX2 isoforms on drug transporters was not evident; PITX2C had the greatest effect on ABCB1 upregulation and resistance to vincristine in RCC[43]. The effectivity of PITX2C was confirmed in Caki-1 cells, derived from a metastatic skin site of a primary renal tumor, where PITX2C maximally increased promoter activity of cyclin D1/CCND1 and mRNA expression of select PITX2 target genes (Aquisap, A., Zarbock, R. Lee, W.K., unpublished data). A recent report detailing a new role for PITX2C in gastrulation during embryonic development could underpin PITX2C’s apparent enhanced oncogenicity and/or metastatic potential. During gastrulation, cells migrate into the three primary germ layers (mesoderm, ectoderm, endoderm), a process that is driven by pitx2c-mediated expression of the chemokine cxc112b in zebrafish[328]. These findings reiterate the concept that specific gene subsets are regulated by each PITX2 isoform[292]. This is further exemplified in a study of ectopic PITX2A, PITX2B, or PITX2C overexpression in ovarian cancer cell lines, wherein TGFβ signaling promoted invasion and EMT was activated, but with PITX2 isoform gene activation selectivity[315]. Moreover, even when all PITX2 isoforms increased prostate cancer cell mobility, only PITX2A conferred a specific mobility advantage in the presence of Wnt5a stimulation[329].
The molecular identification of the reactivated PITX2 transcriptional complex in cancer is currently unknown; however, it appears that PITX2 requires additional co-factors or interaction partners to transcriptionally regulate its target genes[330-332], as commonly seen in developmental PITX2 signaling with, for example, SOX2 (sex determining region Y-box 2) and LEF1[333] or FOXC1 (Forkhead box C1)[334]. As mentioned above, the Wnt/β-catenin pathway is not a pre-requisite for PITX2, however, it could be envisaged that co-activation of Wnt/β-catenin is likely to enhance and strengthen PITX2-mediated transcription of ABCB1 in a self-propagating cycle.
From PITX2’s role in embryonic development and reactivation in cancer, it is reasonable to assume its influence on CSCs, which, as detailed above, harbor strong defense mechanisms against chemotherapeutics. Only a single study has reported increased mutations of PITX2 in CSCs derived from patient bladder cancer tissue samples[335]. Using cell surface markers and flow cytometry, single cell bladder CSCs were distinguished from cancerous non-stem cells and bladder epithelial cells. Single cell sequencing identified somatic nonsynonymous mutations in PITX2 in bladder cancer tissue obtained through transurethral resection. These mutations were found exclusively in bladder CSCs, although their role in bladder CSC self-renewal and tumor propagation was not further investigated[335].
Conclusions
Chemoresistance develops in multiple cell populations within the heterogenic tumor. Malignant cells may display inherent or acquired MDR, depending on the tumor tissue origin and use of chemotherapeutic drugs, respectively. However, CSCs or persister cells use enhanced chemoresistance as part of their defense mechanisms to counteract cytotoxic cues, permitting their survival and regeneration of tumor tissue. Mounting evidence points to the regulation of the MDR transporter, ABCB1, by several reactivated developmental signaling pathways, as delineated in this work [Figure 1]. There are still many unanswered questions. For example, do these pathways interact in a feed-forward loop? Is the timing of pathway activation crucial to ABCB1 regulation analogous to pathway co-ordination in embryonic development? Do these pathways regulate directed cell movement within the tumor and does ABCB1 contribute to this potential mechanism? With advancing technology and model systems, it is conceivable that our improved molecular understanding of MDR phenotype regulation will help increase the effectiveness of cancer therapy.
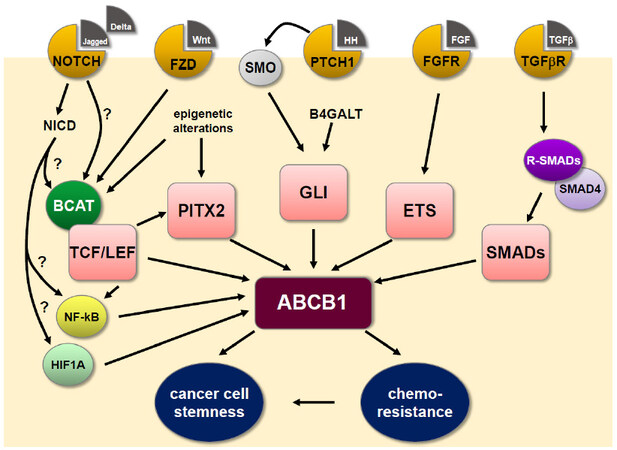
Figure 1. Summary of ABCB1 regulation by developmental signaling pathways in cancer. The MDR P-glycoprotein ABCB1 is central to conferring oncogenic chemoresistance and is also a phenotypical marker for cancer cell stemness. Alterations in the transcription of ABCB1 represent the major regulatory pathway and the ABCB1 promoter region harbors consensus binding sequences for numerous transcription factors, including those primarily involved in embryonic development, which can resurge during carcinogenesis. See main text for further details. B4GALT: β-1,4-galactosyltransferase; BCAT: β-catenin; ETS: erythroblast transformation specific; FGF: fibroblast growth factor; FGFR: fibroblast growth factor receptor; Fzd: frizzled; GLI: glioma-associated oncogenes; HH: hedgehog; HIF1A: hypoxia-inducible factor 1α; NF-κB: nuclear factor kappa B; PTCH1: patched 1; PITX2: paired-like homeodomain transcription factor 2; R-SMADs: receptor-activated SMADs; SMO: smoothened; TCF/LEF: T-cell factor/lymphoid enhancer factor; TGFβ: transforming growth factor β; TGFβR: transforming growth factor β receptor; Wnt: wingless-INT.
Declarations
AcknowledgementsWe would like to thank Aaron Aquisap and Dr. Ralf Zarbock for their valuable contributions through experimental observations and discussions.
Authors’ contributionsConceptualized the manuscript: Lee WK
Wrote and edited the manuscript: Lee WK, Thévenod F
Availability of data and materialsNot applicable.
Financial support and sponsorshipW.K.L has received funding from the Intramural Research Program at Witten/Herdecke University and Westmann-Westerdorp Foundation. The research of F.T. on ABCB1 was funded by DFG (German Research Foundation) Grants TH345 and the Centre for Biomedical Education and Research (ZBAF) at Witten/Herdecke University.
Conflicts of interestThe authors declare no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationAll authors have approved the final manuscript.
Copyright© The Author(s) 2021.
REFERENCES
1. Hyde SC, Emsley P, Hartshorn MJ, et al. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 1990;346:362-5.
2. Christakis P. The birth of chemotherapy at Yale. Bicentennial lecture series: Surgery Grand Round. Yale J Biol Med 2011;84:169-72.
3. Biedler JL, Riehm H. Cellular resistance to actinomycin D in Chinese hamster cells in vitro: cross-resistance, radioautographic, and cytogenetic studies. Cancer Res 1970;30:1174-84.
4. Ling V. Drug resistance and membrane alteration in mutants of mammalian cells. Can J Genet Cytol 1975;17:503-15.
5. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152-62.
6. Karthikeyan S, Hoti SL. Development of Fourth Generation ABC Inhibitors from Natural Products: A Novel Approach to Overcome Cancer Multidrug Resistance. Anticancer Agents Med Chem 2015;15:605-15.
7. Genovese I, Ilari A, Assaraf YG, Fazi F, Colotti G. Not only P-glycoprotein: Amplification of the ABCB1-containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins. Drug Resist Updat 2017;32:23-46.
8. Lee WK, Kolesnick RN. Sphingolipid abnormalities in cancer multidrug resistance: Chicken or egg? Cell Signal 2017;38:134-45.
9. Nobili S, Lapucci A, Landini I, Coronnello M, Roviello G, Mini E. Role of ATP-binding cassette transporters in cancer initiation and progression. Semin Cancer Biol 2020;60:72-95.
10. Han LW, Gao C, Mao Q. An update on expression and function of P-gp/ABCB1 and BCRP/ABCG2 in the placenta and fetus. Expert Opin Drug Metab Toxicol 2018;14:817-29.
11. Huang QT, Shynlova O, Kibschull M, et al. P-glycoprotein expression and localization in the rat uterus throughout gestation and labor. Reproduction 2016;152:195-204.
12. Dempke WCM, Fenchel K, Uciechowski P, Chevassut T. Targeting Developmental Pathways: The Achilles Heel of Cancer? Oncology 2017;93:213-23.
13. Franco C, Hess S. Recent proteomic advances in developmental, regeneration, and cancer governing signaling pathways. Proteomics 2015;15:1014-25.
14. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445-64.
15. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48-58.
16. Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest 2013;123:4131-3.
17. Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene 2003;22:7468-85.
18. Chen Y, Simon SM. In situ biochemical demonstration that P-glycoprotein is a drug efflux pump with broad specificity. J Cell Biol 2000;148:863-70.
19. Shapiro AB, Ling V. The mechanism of ATP-dependent multidrug transport by P-glycoprotein. Acta Physiol Scand Suppl 1998;643:227-34.
20. Alvarez M, Paull K, Monks A, et al. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J Clin Invest 1995;95:2205-14.
21. van Helvoort A, Smith AJ, Sprong H, et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 1996;87:507-17.
22. Eckford PD, Sharom FJ. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem J 2005;389:517-26.
23. Lee WK, Torchalski B, Kohistani N, Thévenod F. ABCB1 protects kidney proximal tubule cells against cadmium-induced apoptosis: roles of cadmium and ceramide transport. Toxicol Sci 2011;121:343-56.
24. Chen CJ, Chin JE, Ueda K, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986;47:381-9.
25. Gros P, Croop J, Housman D. Mammalian multidrug resistance gene: complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell 1986;47:371-80.
26. Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009;323:1718-22.
27. Loo TW, Bartlett MC, Clarke DM. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J Biol Chem 2003;278:39706-10.
28. Chen Y, Pant AC, Simon SM. P-glycoprotein does not reduce substrate concentration from the extracellular leaflet of the plasma membrane in living cells. Cancer Res 2001;61:7763-9.
29. Loo TW, Clarke DM. Functional consequences of phenylalanine mutations in the predicted transmembrane domain of P-glycoprotein. J Biol Chem 1993;268:19965-72.
30. Raviv Y, Pollard HB, Bruggemann EP, Pastan I, Gottesman MM. Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J Biol Chem 1990;265:3975-80.
31. Qu Q, Sharom FJ. Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry 2002;41:4744-52.
32. Eytan GD. Mechanism of multidrug resistance in relation to passive membrane permeation. Biomed Pharmacother 2005;59:90-7.
33. Rauch C. The “multi” of drug resistance explained by oscillating drug transporters, drug-membrane physical interactions and spatial dimensionality. Cell Biochem Biophys 2011;61:103-13.
34. Omote H, Al-Shawi MK. Interaction of transported drugs with the lipid bilayer and P-glycoprotein through a solvation exchange mechanism. Biophys J 2006;90:4046-59.
35. Clay AT, Sharom FJ. Lipid bilayer properties control membrane partitioning, binding, and transport of p-glycoprotein substrates. Biochemistry 2013;52:343-54.
36. Seelig A, Landwojtowicz E. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur J Pharm Sci 2000;12:31-40.
37. Shapiro AB, Ling V. Extraction of Hoechst 33342 from the cytoplasmic leaflet of the plasma membrane by P-glycoprotein. Eur J Biochem 1997;250:122-9.
38. Shapiro AB, Ling V. Transport of LDS-751 from the cytoplasmic leaflet of the plasma membrane by the rhodamine-123-selective site of P-glycoprotein. Eur J Biochem 1998;254:181-8.
39. Breier A, Barancik M, Sulova Z, Uhrik B. P-glycoprotein--implications of metabolism of neoplastic cells and cancer therapy. Curr Cancer Drug Targets 2005;5:457-68.
42. Lee WK, Chakraborty PK, Thévenod F. Pituitary homeobox 2 (PITX2) protects renal cancer cell lines against doxorubicin toxicity by transcriptional activation of the multidrug transporter ABCB1. Int J Cancer 2013;133:556-67.
43. Lee WK, Thévenod F. Oncogenic PITX2 facilitates tumor cell drug resistance by inverse regulation of hOCT3/SLC22A3 and ABC drug transporters in colon and kidney cancers. Cancer Lett 2019;449:237-51.
44. Shapiro AB, Fox K, Lee P, Yang YD, Ling V. Functional intracellular P-glycoprotein. Int J Cancer 1998;76:857-64.
45. Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem 2013;288:31761-71.
46. Calcabrini A, Meschini S, Stringaro A, Cianfriglia M, Arancia G, Molinari A. Detection of P-glycoprotein in the nuclear envelope of multidrug resistant cells. Histochem J 2000;32:599-606.
47. Seidel A, Hasmann M, Loser R, et al. Intracellular localization, vesicular accumulation and kinetics of daunorubicin in sensitive and multidrug-resistant gastric carcinoma EPG85-257 cells. Virchows Arch 1995;426:249-56.
48. Crivellato E, Candussio L, Rosati AM, Decorti G, Klugmann FB, Mallardi F. Kinetics of doxorubicin handling in the LLC-PK1 kidney epithelial cell line is mediated by both vesicle formation and P-glycoprotein drug transport. Histochem J 1999;31:635-43.
49. Bobichon H, Colin M, Depierreux C, Liautaud-Roger F, Jardillier JC. Ultrastructural changes related to multidrug resistance in CEM cells: role of cytoplasmic vesicles in drug exclusion. J Exp Ther Oncol 1996;1:49-61.
50. Katayama K, Kapoor K, Ohnuma S, et al. Revealing the fate of cell surface human P-glycoprotein (ABCB1): The lysosomal degradation pathway. Biochim Biophys Acta 2015;1853:2361-70.
51. Al-Akra L, Bae DH, Sahni S, et al. Tumor stressors induce two mechanisms of intracellular P-glycoprotein-mediated resistance that are overcome by lysosomal-targeted thiosemicarbazones. J Biol Chem 2018;293:3562-87.
52. Stefan SM, Jansson PJ, Kalinowski DS, Anjum R, Dharmasivam M, Richardson DR. The growing evidence for targeting P-glycoprotein in lysosomes to overcome resistance. Future Med Chem 2020;12:473-7.
53. Molinari A, Calcabrini A, Meschini S, et al. Subcellular detection and localization of the drug transporter P-glycoprotein in cultured tumor cells. Curr Protein Pept Sci 2002;3:653-70.
54. Szaflarski W, Sujka-Kordowska P, Januchowski R, et al. Nuclear localization of P-glycoprotein is responsible for protection of the nucleus from doxorubicin in the resistant LoVo cell line. Biomed Pharmacother 2013;67:497-502.
55. Sharom FJ. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front Oncol 2014;4:41.
56. Brown DA. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology (Bethesda) 2006;21:430-9.
57. Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol 2011;3:a004697.
58. Lavie Y, Fiucci G, Liscovitch M. Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J Biol Chem 1998;273:32380-3.
59. Klappe K, Hummel I, Hoekstra D, Kok JW. Lipid dependence of ABC transporter localization and function. Chem Phys Lipids 2009;161:57-64.
60. Wang E, Casciano CN, Clement RP, Johnson WW. Cholesterol interaction with the daunorubicin binding site of P-glycoprotein. Biochem Biophys Res Commun 2000;276:909-16.
61. Fenyvesi F, Fenyvesi E, Szente L, et al. P-glycoprotein inhibition by membrane cholesterol modulation. Eur J Pharm Sci 2008;34:236-42.
62. Troost J, Lindenmaier H, Haefeli WE, Weiss J. Modulation of cellular cholesterol alters P-glycoprotein activity in multidrug-resistant cells. Mol Pharmacol 2004;66:1332-9.
63. Modok S, Heyward C, Callaghan R. P-glycoprotein retains function when reconstituted into a sphingolipid- and cholesterol-rich environment. J Lipid Res 2004;45:1910-8.
64. Deng B, Melnik S, Cook PR. Transcription factories, chromatin loops, and the dysregulation of gene expression in malignancy. Semin Cancer Biol 2013;23:65-71.
65. Martinez E. Multi-protein complexes in eukaryotic gene transcription. Plant Mol Biol 2002;50:925-47.
66. Gromnicova R, Romero I, Male D. Transcriptional control of the multi-drug transporter ABCB1 by transcription factor Sp3 in different human tissues. PLoS One 2012;7:e48189.
67. Lettnin AP, Wagner EF, Carrett-Dias M, et al. Silencing the OCT4-PG1 pseudogene reduces OCT-4 protein levels and changes characteristics of the multidrug resistance phenotype in chronic myeloid leukemia. Mol Biol Rep 2019;46:1873-84.
68. Asakura K, Uchida H, Miyachi H, et al. TEL/AML1 overcomes drug resistance through transcriptional repression of multidrug resistance-1 gene expression. Mol Cancer Res 2004;2:339-47.
69. Lutterbach B, Sun D, Schuetz J, Hiebert SW. The MYND motif is required for repression of basal transcription from the multidrug resistance 1 promoter by the t(8;21) fusion protein. Mol Cell Biol 1998;18:3604-11.
70. Ando T, Nishimura M, Oka Y. Decitabine (5-Aza-2’-deoxycytidine) decreased DNA methylation and expression of MDR-1 gene in K562/ADM cells. Leukemia 2000;14:1915-20.
71. El-Osta A, Baker EK, Wolffe AP. Profiling methyl-CpG specific determinants on transcriptionally silent chromatin. Mol Biol Rep 2001;28:209-15.
72. Ogretmen B, Safa AR. Negative regulation of MDR1 promoter activity in MCF-7, but not in multidrug resistant MCF-7/Adr, cells by cross-coupled NF-kappa B/p65 and c-Fos transcription factors and their interaction with the CAAT region. Biochemistry 1999;38:2189-99.
73. Christie EL, Pattnaik S, Beach J, et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat Commun 2019;10:1295.
74. Kumar R, Li DQ, Muller S, Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016;35:4423-36.
75. Moro H, Hattori N, Nakamura Y, et al. Epigenetic priming sensitizes gastric cancer cells to irinotecan and cisplatin by restoring multiple pathways. Gastric Cancer 2020;23:105-15.
76. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489-94.
77. Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1). J Bioenerg Biomembr 2007;39:481-7.
78. Wolking S, Schaeffeler E, Lerche H, Schwab M, Nies AT. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin Pharmacokinet 2015;54:709-35.
79. Huff LM, Lee JS, Robey RW, Fojo T. Characterization of gene rearrangements leading to activation of MDR-1. J Biol Chem 2006;281:36501-9.
80. Rund D, Azar I, Shperling O. A mutation in the promoter of the multidrug resistance gene (MDR1) in human hematological malignancies may contribute to the pathogenesis of resistant disease. Adv Exp Med Biol 1999;457:71-5.
81. Reed K, Hembruff SL, Sprowl JA, Parissenti AM. The temporal relationship between ABCB1 promoter hypomethylation, ABCB1 expression and acquisition of drug resistance. Pharmacogenomics J 2010;10:489-504.
82. Huo H, Magro PG, Pietsch EC, Patel BB, Scotto KW. Histone methyltransferase MLL1 regulates MDR1 transcription and chemoresistance. Cancer Res 2010;70:8726-35.
83. Yague E, Armesilla AL, Harrison G, et al. P-glycoprotein (MDR1) expression in leukemic cells is regulated at two distinct steps, mRNA stabilization and translational initiation. J Biol Chem 2003;278:10344-52.
84. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017;168:613-28.
85. Yuan Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb Perspect Med 2016;6.
86. Bakhshinyan D, Adile AA, Qazi MA, et al. Introduction to Cancer Stem Cells: Past, Present, and Future. Methods Mol Biol 2018;1692:1-16.
87. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, ME LL. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol 2020;60:166-80.
88. Koren E, Fuchs Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist Updat 2016;28:1-12.
89. Wang YH, Scadden DT. Harnessing the apoptotic programs in cancer stem-like cells. EMBO Rep 2015;16:1084-98.
90. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821-8.
91. Cho Y, Kim YK. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front Oncol 2020;10:764.
92. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009;15:1010-2.
94. Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551:247-50.
95. Boesch M, Hoflehner E, Wolf D, Gastl G, Sopper S. Harnessing the DNA Dye-triggered Side Population Phenotype to Detect and Purify Cancer Stem Cells from Biological Samples. J Vis Exp 2017; doi: 10.3791/55634.
97. McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor-initiating cells. Expert Opin Drug Metab Toxicol 2016;12:633-44.
98. Begicevic RR, Falasca M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int J Mol Sci 2017;18:2362.
99. Hashida S, Yamamoto H, Shien K, et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci 2015;106:1377-84.
100. Eyre R, Harvey I, Stemke-Hale K, Lennard TW, Tyson-Capper A, Meeson AP. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumour Biol 2014;35:9879-92.
101. Kobayashi Y, Seino K, Hosonuma S, et al. Side population is increased in paclitaxel-resistant ovarian cancer cell lines regardless of resistance to cisplatin. Gynecol Oncol 2011;121:390-4.
102. Yamamoto M, Suzuki S, Togashi K, et al. AS602801 Sensitizes Ovarian Cancer Stem Cells to Paclitaxel by Down-regulating MDR1. Anticancer Res 2019;39:609-17.
103. Emmink BL, Van Houdt WJ, Vries RG, et al. Differentiated human colorectal cancer cells protect tumor-initiating cells from irinotecan. Gastroenterology 2011;141:269-78.
104. Goto S, Kawabata T, Li TS. Enhanced Expression of ABCB1 and Nrf2 in CD133-Positive Cancer Stem Cells Associates with Doxorubicin Resistance. Stem Cells Int 2020;2020:8868849.
105. Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ. Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomed Pharmacother 2013;67:791-9.
106. Zhou J, Wang CY, Liu T, et al. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J Gastroenterol 2008;14:925-30.
107. Naik PP, Mukhopadhyay S, Panda PK, et al. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif 2018;51:e12411.
108. Nakai E, Park K, Yawata T, et al. Enhanced MDR1 expression and chemoresistance of cancer stem cells derived from glioblastoma. Cancer Invest 2009;27:901-8.
109. Shervington A, Lu C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Invest 2008;26:535-42.
110. Sadeghi MR, Jeddi F, Soozangar N, et al. Nrf2/P-glycoprotein axis is associated with clinicopathological characteristics in colorectal cancer. Biomed Pharmacother 2018;104:458-64.
111. Marques DS, Sandrini JZ, Boyle RT, Marins LF, Trindade GS. Relationships between multidrug resistance (MDR) and stem cell markers in human chronic myeloid leukemia cell lines. Leuk Res 2010;34:757-62.
112. Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem 2008;283:17635-51.
113. Deng JJ, Zhang W, Xu XM, Zhang F, Tao WP, Ye JJ, Ge W. Twist mediates an aggressive phenotype in human colorectal cancer cells. Int J Oncol 2016;48:1117-24.
114. Riganti C, Salaroglio IC, Caldera V, et al. Temozolomide downregulates P-glycoprotein expression in glioblastoma stem cells by interfering with the Wnt3a/glycogen synthase-3 kinase/beta-catenin pathway. Neuro Oncol 2013;15:1502-17.
115. Angelastro JM, Lame MW. Overexpression of CD133 promotes drug resistance in C6 glioma cells. Mol Cancer Res 2010;8:1105-15.
116. White MD, Zenker J, Bissiere S, Plachta N. Instructions for Assembling the Early Mammalian Embryo. Dev Cell 2018;45:667-79.
117. Chazaud C, Yamanaka Y. Lineage specification in the mouse preimplantation embryo. Development 2016;143:1063-74.
118. Zhang HT, Hiiragi T. Symmetry Breaking in the Mammalian Embryo. Annu Rev Cell Dev Biol 2018;34:405-26.
119. Li P, Elowitz MB. Communication codes in developmental signaling pathways. Development 2019;146.
120. Siebel C, Lendahl U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev 2017;97:1235-94.
122. Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A 1996;93:1683-8.
123. Lal M, Caplan M. Regulated intramembrane proteolysis: signaling pathways and biological functions. Physiology (Bethesda) 2011;26:34-44.
124. Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998;393:382-6.
125. De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003;38:9-12.
126. Zhang X, Li Y, Xu H, Zhang YW. The gamma-secretase complex: from structure to function. Front Cell Neurosci 2014;8:427.
127. Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development 2011;138:3593-612.
128. Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol 2014;4:345.
129. Andersen P, Uosaki H, Shenje LT, Kwon C. Non-canonical Notch signaling: emerging role and mechanism. Trends Cell Biol 2012;22:257-65.
130. Park JT, Chen X, Trope CG, Davidson B, Shih Ie M, Wang TL. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am J Pathol 2010;177:1087-94.
131. Wu WR, Zhang R, Shi XD, et al. Notch1 is overexpressed in human intrahepatic cholangiocarcinoma and is associated with its proliferation, invasiveness and sensitivity to 5-fluorouracil in vitro. Oncol Rep 2014;31:2515-24.
132. Harbuzariu A, Rampoldi A, Daley-Brown DS, et al. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017;8:7740-52.
133. Steg AD, Katre AA, Goodman B, et al. Targeting the notch ligand JAGGED1 in both tumor cells and stroma in ovarian cancer. Clin Cancer Res 2011;17:5674-85.
134. Zou W, Ma X, Hua W, Chen B, Cai G. Caveolin-1 mediates chemoresistance in cisplatin-resistant ovarian cancer cells by targeting apoptosis through the Notch-1/Akt/NF-kappaB pathway. Oncol Rep 2015;34:3256-63.
135. Zhang C, Huang H, Zhang J, et al. Caveolin-1 promotes invasion and metastasis by upregulating Pofut1 expression in mouse hepatocellular carcinoma. Cell Death Dis 2019;10:477.
136. Kapoor A, Hsu WM, Wang BJ, et al. Caveolin-1 regulates gamma-secretase-mediated AbetaPP processing by modulating spatial distribution of gamma-secretase in membrane. J Alzheimers Dis 2010;22:423-42.
137. Antonio-Andres G, Rangel-Santiago J, Tirado-Rodriguez B, Jimenez-Hernandez E, Torres Nava J, Medina-Sanson A, Huerta-Yepez S, et al. Role of Yin Yang-1 (YY1) in the transcription regulation of the multi-drug resistance (MDR1) gene. Leuk Lymphoma 2018;59:2628-38.
138. Bentires-Alj M, Barbu V, Fillet M, et al. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003;22:90-7.
139. Thévenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem 2000;275:1887-96.
140. Yamada T, Mori Y, Hayashi R, et al. Suppression of intestinal polyposis in Mdr1-deficient ApcMin/+ mice. Cancer Res 2003;63:895-901.
141. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002;62:3387-94.
142. Lee JJ, von Kessler DP, Parks S, Beachy PA. Secretion and localized transcription suggest a role in positional signaling for products of the segmentation gene hedgehog. Cell 1992;71:33-50.
143. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature 1980;287:795-801.
144. Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993;75:1401-16.
145. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001;15:3059-87.
146. Johnson RL, Riddle RD, Laufer E, Tabin C. Sonic hedgehog: a key mediator of anterior-posterior patterning of the limb and dorso-ventral patterning of axial embryonic structures. Biochem Soc Trans 1994;22:569-74.
147. Stone DM, Hynes M, Armanini M, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996;384:129-34.
148. Qi X, Schmiege P, Coutavas E, Wang J, Li X. Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 2018;560:128-32.
150. Chen Y, Yue S, Xie L, Pu XH, Jin T, Cheng SY. Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. J Biol Chem 2011;286:13502-11.
152. Pak E, Segal RA. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev Cell 2016;38:333-44.
153. Merchant M, Vajdos FF, Ultsch M, et al. Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol Cell Biol 2004;24:8627-41.
154. Kise Y, Morinaka A, Teglund S, Miki H. Sufu recruits GSK3beta for efficient processing of Gli3. Biochem Biophys Res Commun 2009;387:569-74.
155. Niewiadomski P, Kong JH, Ahrends R, et al. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep 2014;6:168-81.
156. Antonucci L, Di Magno L, D’Amico D, et al. Mitogen-activated kinase kinase kinase 1 inhibits hedgehog signaling and medulloblastoma growth through GLI1 phosphorylation. Int J Oncol 2019;54:505-14.
157. Shi Q, Li S, Li S, Jiang A, Chen Y, Jiang J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc Natl Acad Sci U S A 2014;111:E5651-60.
158. Han Y, Wang B, Cho YS, Zhu J, Wu J, Chen Y, Jiang J. Phosphorylation of Ci/Gli by Fused Family Kinases Promotes Hedgehog Signaling. Dev Cell 2019;50:610-26 e4.
159. Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007;26:5674-9.
160. Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010;29:6314-22.
161. Po A, Citarella A, Catanzaro G, et al. Hedgehog-GLI signalling promotes chemoresistance through the regulation of ABC transporters in colorectal cancer cells. Sci Rep 2020;10:13988.
162. Chen Y, Bieber MM, Teng NN. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol Carcinog 2014;53:625-34.
163. Chen YJ, Kuo CD, Chen SH, et al. Small-molecule synthetic compound norcantharidin reverses multi-drug resistance by regulating Sonic hedgehog signaling in human breast cancer cells. PLoS One 2012;7:e37006.
164. Lu YL, Ma YB, Feng C, et al. Co-delivery of Cyclopamine and Doxorubicin Mediated by Bovine Serum Albumin Nanoparticles Reverses Doxorubicin Resistance in Breast Cancer by Down-regulating P-glycoprotein Expression. J Cancer 2019;10:2357-68.
165. Zhang Y, Laterra J, Pomper MG. Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/Pgp. Neoplasia 2009;11:96-101.
166. Lee MJ, Hatton BA, Villavicencio EH, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci U S A 2012;109:7859-64.
167. Zhou H, Ma H, Wei W, et al. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis 2013;4:e654.
168. Zhou H, Zhang Z, Liu C, et al. B4GALT1 gene knockdown inhibits the hedgehog pathway and reverses multidrug resistance in the human leukemia K562/adriamycin-resistant cell line. IUBMB Life 2012;64:889-900.
169. Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, Ligon KL, Greco SJ, Rameshwar P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015;6:1190-201.
170. Liu Z, Xu J, He J, et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014;124:2061-71.
172. Petersen CP, Reddien PW. Wnt signaling and the polarity of the primary body axis. Cell 2009;139:1056-68.
173. Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development 2018;145.
175. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17:9-26.
176. Wiese KE, Nusse R, van Amerongen R. Wnt signalling: conquering complexity. Development 2018;145:dev165902.
177. Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017;169:985-99.
178. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013;13:11-26.
179. Mikels AJ, Nusse R. Wnts as ligands: processing, secretion and reception. Oncogene 2006;25:7461-8.
180. MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb Perspect Biol 2012;4:a007880.
181. Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol 2013;5:a015081.
182. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci 2003;116:2627-34.
183. de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev 2014;28:305-16.
184. Morgan RG, Mortensson E, Williams AC. Targeting LGR5 in Colorectal Cancer: therapeutic gold or too plastic? Br J Cancer 2018;118:1410-8.
185. de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017;543:676-80.
186. Shimokawa M, Ohta Y, Nishikori S, et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017;545:187-92.
187. Stamos JL, Weis WI. The beta-catenin destruction complex. Cold Spring Harb Perspect Biol 2013;5:a007898.
188. Tamai K, Zeng X, Liu C, et al. A mechanism for Wnt coreceptor activation. Mol Cell 2004;13:149-56.
189. Zeng X, Tamai K, Doble B, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005;438:873-7.
190. Behrens J, von Kries JP, Kuhl M, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996;382:638-42.
191. Brunner E, Peter O, Schweizer L, Basler K. pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. Nature 1997;385:829-33.
192. Molenaar M, van de Wetering M, Oosterwegel M, et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996;86:391-9.
194. Fang M, Li J, Blauwkamp T, Bhambhani C, Campbell N, Cadigan KM. C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J 2006;25:2735-45.
195. Levanon D, Goldstein RE, Bernstein Y, et al. Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proc Natl Acad Sci U S A 1998;95:11590-5.
196. Brack AS, Murphy-Seiler F, Hanifi J, et al. BCL9 is an essential component of canonical Wnt signaling that mediates the differentiation of myogenic progenitors during muscle regeneration. Dev Biol 2009;335:93-105.
197. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J 2000;19:1839-50.
198. Schwab KR, Patterson LT, Hartman HA, et al. Pygo1 and Pygo2 roles in Wnt signaling in mammalian kidney development. BMC Biol 2007;5:15.
199. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509-12.
200. Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 1999;96:5522-7.
201. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999;398:422-6.
202. Yamada T, Takaoka AS, Naishiro Y, et al. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res 2000;60:4761-6.
203. Nusse R. .
204. Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235-7.
206. Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta 1997;1332:F127-47.
207. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008;8:387-98.
210. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401-4.
211. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1.
212. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014;346:1248012.
213. Ghandadi M, Valadan R, Mohammadi H, Akhtari J, Khodashenas S, Ashari S. Wnt-beta-catenin Signaling Pathway, the Achilles’ Heels of Cancer Multidrug Resistance. Curr Pharm Des 2019;25:4192-207.
214. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 2002;21:5427-40.
216. Cho NY, Kim BH, Choi M, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 2007;211:269-77.
217. Enokida H, Shiina H, Urakami S, et al. Multigene methylation analysis for detection and staging of prostate cancer. Clin Cancer Res 2005;11:6582-8.
218. Lin Q, Geng J, Ma K, et al. RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non-small cell lung cancer in China. J Cancer Res Clin Oncol 2009;135:1675-84.
219. Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004;64:1975-86.
220. Wang H, Wang X, Hu R, et al. Methylation of SFRP5 is related to multidrug resistance in leukemia cells. Cancer Gene Ther 2014;21:83-9.
221. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 2019;20:156-74.
222. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998;20:615-26.
223. Wielenga VJ, Smits R, Korinek V, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol 1999;154:515-23.
224. Bourguignon LY, Xia W, Wong G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J Biol Chem 2009;284:2657-71.
225. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014;157:77-94.
228. Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012;336:237-40.
229. Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018;172:393-407.
230. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 2016;17:47-62.
231. Ma Y, Yang Y, Wang F, et al. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/beta-catenin signalling pathway via suppression of activator protein 2alpha. Gut 2016;65:1494-504.
232. Chen Z, Pan T, Jiang D, et al. The lncRNA-GAS5/miR-221-3p/DKK2 Axis Modulates ABCB1-Mediated Adriamycin Resistance of Breast Cancer via the Wnt/beta-Catenin Signaling Pathway. Mol Ther Nucleic Acids 2020;19:1434-48.
233. Hung TH, Hsu SC, Cheng CY, et al. Wnt5A regulates ABCB1 expression in multidrug-resistant cancer cells through activation of the non-canonical PKA/beta-catenin pathway. Oncotarget 2014;5:12273-90.
235. Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem 1995;270:31315-20.
236. Zhou G, Kuo MT. NF-kappaB-mediated induction of mdr1b expression by insulin in rat hepatoma cells. J Biol Chem 1997;272:15174-83.
237. Haertel-Wiesmann M, Liang Y, Fantl WJ, Williams LT. Regulation of cyclooxygenase-2 and periostin by Wnt-3 in mouse mammary epithelial cells. J Biol Chem 2000;275:32046-51.
238. Howe LR, Subbaramaiah K, Chung WJ, Dannenberg AJ, Brown AM. Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res 1999;59:1572-7.
239. Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 2005;310:1504-10.
240. Lim K, Han C, Xu L, Isse K, Demetris AJ, Wu T. Cyclooxygenase-2-derived prostaglandin E2 activates beta-catenin in human cholangiocarcinoma cells: evidence for inhibition of these signaling pathways by omega 3 polyunsaturated fatty acids. Cancer Res 2008;68:553-60.
241. Liu XH, Kirschenbaum A, Weinstein BM, Zaidi M, Yao S, Levine AC. Prostaglandin E2 modulates components of the Wnt signaling system in bone and prostate cancer cells. Biochem Biophys Res Commun 2010;394:715-20.
242. Nunez F, Bravo S, Cruzat F, Montecino M, De Ferrari GV. Wnt/beta-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS One 2011;6:e18562.
243. Wu M, Guan J, Li C, et al. Aberrantly activated Cox-2 and Wnt signaling interact to maintain cancer stem cells in glioblastoma. Oncotarget 2017;8:82217-30.
244. Gala MK, Chan AT. Molecular pathways: aspirin and Wnt signaling-a molecularly targeted approach to cancer prevention and treatment. Clin Cancer Res 2015;21:1543-8.
245. Vallee A, Lecarpentier Y, Vallee JN. Targeting the Canonical WNT/beta-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs. Cells 2019;8:726.
246. Zimmerman ZF, Moon RT, Chien AJ. Targeting Wnt pathways in disease. Cold Spring Harb Perspect Biol 2012;4.
247. Martinez-Hackert E, Sundan A, Holien T. Receptor binding competition: A paradigm for regulating TGF-beta family action. Cytokine Growth Factor Rev 2021;57:39-54.
248. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 2009;16:329-43.
249. Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-beta pathway for cancer therapy. Clin Cancer Res 2012;18:4514-21.
251. Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol 2008;28:6889-902.
252. Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF. TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J 2009;28:88-98.
253. Schmierer B, Tournier AL, Bates PA, Hill CS. Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc Natl Acad Sci U S A 2008;105:6608-13.
254. Derynck R, Turley SJ, Akhurst RJ. TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 2021;18:9-34.
255. Zhang X, Wang T, Batist G, Tsao MS. Transforming growth factor beta 1 promotes spontaneous transformation of cultured rat liver epithelial cells. Cancer Res 1994;54:6122-8.
256. Tomiyasu H, Watanabe M, Goto-Koshino Y. Regulation of expression of ABCB1 and LRP genes by mitogen-activated protein kinase/extracellular signal-regulated kinase pathway and its role in generation of side population cells in canine lymphoma cell lines. Leuk Lymphoma 2013;54:1309-15.
257. Sun W, Ma Y, Chen P, Wang D. MicroRNA-10a silencing reverses cisplatin resistance in the A549/cisplatin human lung cancer cell line via the transforming growth factor-beta/Smad2/STAT3/STAT5 pathway. Mol Med Rep 2015;11:3854-9.
258. Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015;4:215-66.
259. Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR Signaling in Cancer. Clin Cancer Res 2015;21:2684-94.
260. Kato T, Fujita Y, Nakane K, et al. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem Biophys Res Commun 2012;417:966-71.
261. Wang J, Zeng X, Luo T, Jin W, Chen S. Involvement of V-Ets erythroblastosis virus E26 oncogene homolog 2 in regulation of transcription activity of MDR1 gene. Acta Biochim Biophys Sin (Shanghai) 2012;44:752-8.
262. Gan Y, Wientjes MG, Au JL. Expression of basic fibroblast growth factor correlates with resistance to paclitaxel in human patient tumors. Pharm Res 2006;23:1324-31.
264. Liu AM, Wong KF, Jiang X, Qiao Y, Luk JM. Regulators of mammalian Hippo pathway in cancer. Biochim Biophys Acta 2012;1826:357-64.
266. Suemura S, Kodama T, Myojin Y, et al. CRISPR Loss-of-Function Screen Identifies the Hippo Signaling Pathway as the Mediator of Regorafenib Efficacy in Hepatocellular Carcinoma. Cancers (Basel) 2019;11:1362.
267. Nguyen CDK, Yi C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019;5:283-96.
268. Xia Y, Zhang YL, Yu C, Chang T, Fan HY. YAP/TEAD co-activator regulated pluripotency and chemoresistance in ovarian cancer initiated cells. PLoS One 2014;9:e109575.
269. Bhullar J, Sollars VE. YBX1 expression and function in early hematopoiesis and leukemic cells. Immunogenetics 2011;63:337-50.
270. Evans MK, Matsui Y, Xu B, et al. Ybx1 fine-tunes PRC2 activities to control embryonic brain development. Nat Commun 2020;11:4060.
271. Liu Z, Li Y, Li X, et al. Overexpression of YBX1 Promotes Pancreatic Ductal Adenocarcinoma Growth via the GSK3B/Cyclin D1/Cyclin E1 Pathway. Mol Ther Oncolytics 2020;17:21-30.
272. Shibata T, Tokunaga E, Hattori S, et al. Y-box binding protein YBX1 and its correlated genes as biomarkers for poor outcomes in patients with breast cancer. Oncotarget 2018;9:37216-28.
273. Xu L, Li H, Wu L, Huang S. YBX1 promotes tumor growth by elevating glycolysis in human bladder cancer. Oncotarget 2017;8:65946-56.
274. Hartley AV, Wang B, Mundade R, et al. PRMT5-mediated methylation of YBX1 regulates NF-kappaB activity in colorectal cancer. Sci Rep 2020;10:15934.
275. Jurchott K, Kuban RJ, Krech T, et al. Identification of Y-box binding protein 1 as a core regulator of MEK/ERK pathway-dependent gene signatures in colorectal cancer cells. PLoS Genet 2010;6:e1001231.
276. Zhao P, Deng Y, Wu Y, et al. Long noncoding RNA SNHG6 promotes carcinogenesis by enhancing YBX1-mediated translation of HIF1alpha in clear cell renal cell carcinoma. FASEB J 2021;35:e21160.
277. Kuwano M, Shibata T, Watari K, Ono M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci 2019;110:1536-43.
278. Kotake Y, Arikawa N, Tahara K, Maru H, Naemura M. Y-box Binding Protein 1 Is Involved in Regulating the G2/M Phase of the Cell Cycle. Anticancer Res 2017;37:1603-8.
279. Martin M, Hua L, Wang B, et al. Novel Serine 176 Phosphorylation of YBX1 Activates NF-kappaB in Colon Cancer. J Biol Chem 2017;292:3433-44.
280. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20:69-84.
281. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178-96.
282. Murray SA, Gridley T. Snail1 gene function during early embryo patterning in mice. Cell Cycle 2006;5:2566-70.
283. De Craene B, van Roy F, Berx G. Unraveling signalling cascades for the Snail family of transcription factors. Cell Signal 2005;17:535-47.
284. Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 2004;18:1131-43.
285. Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des 2014;20:1698-705.
286. Zhao XY, Li L, Wang XB, et al. Inhibition of Snail Family Transcriptional Repressor 2 (SNAI2) Enhances Multidrug Resistance of Hepatocellular Carcinoma Cells. PLoS One 2016;11:e0164752.
287. Wang H, Li JM, Wei W, et al. Regulation of ATP-binding cassette subfamily B member 1 by Snail contributes to chemoresistance in colorectal cancer. Cancer Sci 2020;111:84-97.
288. Reynolds N, O’Shaughnessy A, Hendrich B. Transcriptional repressors: multifaceted regulators of gene expression. Development 2013;140:505-12.
289. Gage PJ, Suh H, Camper SA. The bicoid-related Pitx gene family in development. Mamm Genome 1999;10:197-200.
290. Gage PJ, Camper SA. Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet 1997;6:457-64.
291. Lamba P, Hjalt TA, Bernard DJ. Novel forms of Paired-like homeodomain transcription factor 2 (PITX2): generation by alternative translation initiation and mRNA splicing. BMC Mol Biol 2008;9:31.
292. Cox CJ, Espinoza HM, McWilliams B, et al. Differential regulation of gene expression by PITX2 isoforms. J Biol Chem 2002;277:25001-10.
293. Hernandez-Torres F, Franco D, Aranega AE, Navarro F. Expression patterns and immunohistochemical localization of PITX2B transcription factor in the developing mouse heart. Int J Dev Biol 2015;59:247-54.
294. Wei Q, Adelstein RS. Pitx2a expression alters actin-myosin cytoskeleton and migration of HeLa cells through Rho GTPase signaling. Mol Biol Cell 2002;13:683-97.
295. Ganga M, Espinoza HM, Cox CJ, et al. PITX2 isoform-specific regulation of atrial natriuretic factor expression: synergism and repression with Nkx2.5. J Biol Chem 2003;278:22437-45.
296. Semina EV, Reiter RS, Murray JC. Isolation of a new homeobox gene belonging to the Pitx/Rieg family: expression during lens development and mapping to the aphakia region on mouse chromosome 19. Hum Mol Genet 1997;6:2109-16.
297. Lin CR, Kioussi C, O’Connell S, et al. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 1999;401:279-82.
298. Szeto DP, Rodriguez-Esteban C, Ryan AK, et al. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev 1999;13:484-94.
299. Tao G, Kahr PC, Morikawa Y, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016;534:119-23.
300. Charles MA, Suh H, Hjalt TA, Drouin J, Camper SA, Gage PJ. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol 2005;19:1893-903.
301. Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med 2005;7:1-17.
303. Kolfschoten IG, van Leeuwen B, Berns K, et al. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 2005;121:849-58.
304. Mudie S, Bandarra D, Batie M, et al. PITX1, a specificity determinant in the HIF-1alpha-mediated transcriptional response to hypoxia. Cell Cycle 2014;13:3878-91.
305. Qi DL, Ohhira T, Fujisaki C, et al. Identification of PITX1 as a TERT suppressor gene located on human chromosome 5. Mol Cell Biol 2011;31:1624-36.
306. Arakawa H, Nakamura T, Zhadanov AB, et al. Identification and characterization of the ARP1 gene, a target for the human acute leukemia ALL1 gene. Proc Natl Acad Sci U S A 1998;95:4573-8.
307. Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood 2001;97:2823-9.
308. Kioussi C, Briata P, Baek SH, et al. Identification of a Wnt/Dvl/beta-Catenin --> Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 2002;111:673-85.
309. Vadlamudi U, Espinoza HM, Ganga M, et al. PITX2, beta-catenin and LEF-1 interact to synergistically regulate the LEF-1 promoter. J Cell Sci 2005;118:1129-37.
310. Liu Y, Huang Y, Zhu GZ. Cyclin A1 is a transcriptional target of PITX2 and overexpressed in papillary thyroid carcinoma. Mol Cell Biochem 2013;384:221-7.
311. Huang Y, Guigon CJ, Fan J, Cheng SY, Zhu GZ. Pituitary homeobox 2 (PITX2) promotes thyroid carcinogenesis by activation of cyclin D2. Cell Cycle 2010;9:1333-41.
312. Baek SH, Kioussi C, Briata P, et al. Regulated subset of G1 growth-control genes in response to derepression by the Wnt pathway. Proc Natl Acad Sci U S A 2003;100:3245-50.
313. Hirose H, Ishii H, Mimori K, et al. The significance of PITX2 overexpression in human colorectal cancer. Ann Surg Oncol 2011;18:3005-12.
314. Fung FK, Chan DW, Liu VW, Leung TH, Cheung AN, Ngan HY. Increased Expression of PITX2 Transcription Factor Contributes to Ovarian Cancer Progression. PLoS One 2012;7:e37076.
315. Basu M, Bhattacharya R, Ray U, Mukhopadhyay S, Chatterjee U, Roy SS. Invasion of ovarian cancer cells is induced by PITX2-mediated activation of TGF-beta and Activin-A. Mol Cancer 2015;14:162.
316. Pillai SG, Dasgupta N, Siddappa CM, et al. Paired-like Homeodomain Transcription factor 2 expression by breast cancer bone marrow disseminated tumor cells is associated with early recurrent disease development. Breast Cancer Res Treat 2015;153:507-17.
317. Dietrich D, Hasinger O, Liebenberg V, Field JK, Kristiansen G, Soltermann A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn Mol Pathol 2012;21:93-104.
318. Lopez JI, Angulo JC, Martin A, et al. A DNA hypermethylation profile reveals new potential biomarkers for the evaluation of prognosis in urothelial bladder cancer. APMIS 2017;125:787-96.
319. Nimmrich I, Sieuwerts AM, Meijer-van Gelder ME, et al. DNA hypermethylation of PITX2 is a marker of poor prognosis in untreated lymph node-negative hormone receptor-positive breast cancer patients. Breast Cancer Res Treat 2008;111:429-37.
320. Dietrich D, Hasinger O, Banez LL, et al. Development and clinical validation of a real-time PCR assay for PITX2 DNA methylation to predict prostate-specific antigen recurrence in prostate cancer patients following radical prostatectomy. J Mol Diagn 2013;15:270-9.
321. Zhang JX, Chen ZH, Xu Y, et al. Downregulation of MicroRNA-644a Promotes Esophageal Squamous Cell Carcinoma Aggressiveness and Stem Cell-like Phenotype via Dysregulation of PITX2. Clin Cancer Res 2017;23:298-310.
322. Lozano-Velasco E, Vallejo D, Esteban FJ, et al. A Pitx2-MicroRNA Pathway Modulates Cell Proliferation in Myoblasts and Skeletal-Muscle Satellite Cells and Promotes Their Commitment to a Myogenic Cell Fate. Mol Cell Biol 2015;35:2892-909.
323. Hilton T, Gross MK, Kioussi C. Pitx2-dependent occupancy by histone deacetylases is associated with T-box gene regulation in mammalian abdominal tissue. J Biol Chem 2010;285:11129-42.
324. Mikeska T, Bock C, Do H, Dobrovic A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Rev Mol Diagn 2012;12:473-87.
325. Amendt BA, Sutherland LB, Russo AF. Multifunctional role of the Pitx2 homeodomain protein C-terminal tail. Mol Cell Biol 1999;19:7001-10.
326. Kozlowski K, Walter MA. Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet 2000;9:2131-9.
327. Espinoza HM, Ganga M, Vadlamudi U, et al. Protein kinase C phosphorylation modulates N- and C-terminal regulatory activities of the PITX2 homeodomain protein. Biochemistry 2005;44:3942-54.
328. Collins MM, Maischein HM, Dufourcq P, Charpentier M, Blader P, Stainier DY. Pitx2c orchestrates embryonic axis extension via mesendodermal cell migration. Elife 2018;7.
329. Vela I, Morrissey C, Zhang X, et al. PITX2 and non-canonical Wnt pathway interaction in metastatic prostate cancer. Clin Exp Metastasis 2014;31:199-211.
330. Huang Y, Huang K, Boskovic G, et al. Proteomic and genomic analysis of PITX2 interacting and regulating networks. FEBS Lett 2009;583:638-42.
331. Liu Y, Huang Y, Fan J, Zhu GZ. PITX2 associates with PTIP-containing histone H3 lysine 4 methyltransferase complex. Biochem Biophys Res Commun 2014;444:634-7.
332. Simard A, Di Giorgio L, Amen M, Westwood A, Amendt BA, Ryan AK. The Pitx2c N-terminal domain is a critical interaction domain required for asymmetric morphogenesis. Dev Dyn 2009;238:2459-70.
333. Yu W, Sun Z, Sweat Y, et al. Pitx2-Sox2-Lef1 interactions specify progenitor oral/dental epithelial cell signaling centers. Development 2020;147.
334. Berry FB, Lines MA, Oas JM, et al. Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum Mol Genet 2006;15:905-19.
335. Yang Z, Li C, Fan Z, et al. Single-cell Sequencing Reveals Variants in ARID1A, GPRC5A and MLL2 Driving Self-renewal of Human Bladder Cancer Stem Cells. Eur Urol 2017;71:8-12.
336. Bitarte N, Bandres E, Boni V, et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells 2011;29:1661-71.
337. Chen Z, Ma T, Huang C, et al. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/beta-catenin pathway in hepatocellular carcinoma cells. Cell Signal 2013;25:2693-701.
338. Liang C, Wang Z, Li YY, Yu BH, Zhang F, Li HY. miR-33a suppresses the nuclear translocation of beta-catenin to enhance gemcitabine sensitivity in human pancreatic cancer cells. Tumour Biol 2015;36:9395-403.
339. Ghosh RD, Ghuwalewala S, Das P, et al. MicroRNA profiling of cisplatin-resistant oral squamous cell carcinoma cell lines enriched with cancer-stem-cell-like and epithelial-mesenchymal transition-type features. Sci Rep 2016;6:23932.
340. Zhou H, Lin C, Zhang Y, et al. miR-506 enhances the sensitivity of human colorectal cancer cells to oxaliplatin by suppressing MDR1/P-gp expression. Cell Prolif 2017;50.
341. Cao F, Yin LX. miR-122 enhances sensitivity of hepatocellular carcinoma to oxaliplatin via inhibiting MDR1 by targeting Wnt/beta-catenin pathway. Exp Mol Pathol 2019;106:34-43.
342. Liu Z, Zhang H. LncRNA plasmacytoma variant translocation 1 is an oncogene in bladder urothelial carcinoma. Oncotarget 2017;8:64273-82.
343. Guo F, Cao Z, Guo H, Li S. The action mechanism of lncRNA-HOTAIR on the drug resistance of non-small cell lung cancer by regulating Wnt signaling pathway. Exp Ther Med 2018;15:4885-9.
344. Kang Y, Zhang S, Cao W, Wan D, Sun L. Knockdown of LncRNA CRNDE suppresses proliferation and P-glycoprotein-mediated multidrug resistance in acute myelocytic leukemia through the Wnt/beta-catenin pathway. Biosci Rep 2020;40.
345. Flahaut M, Meier R, Coulon A, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene 2009;28:2245-56.
346. Wang YH, Imai Y, Shiseki M, Tanaka J, Motoji T. Knockdown of the Wnt receptor Frizzled-1 (FZD1) reduces MDR1/P-glycoprotein expression in multidrug resistant leukemic cells and inhibits leukemic cell proliferation. Leuk Res 2018;67:99-108.
347. Zhang H, Zhang X, Wu X, et al. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/beta-catenin pathway. Cancer Lett 2012;323:106-13.
348. Liu X, Yan Y, Ma W, Wu S. Knockdown of frizzled-7 inhibits cell growth and metastasis and promotes chemosensitivity of esophageal squamous cell carcinoma cells by inhibiting Wnt signaling. Biochem Biophys Res Commun 2017;490:1112-8.
349. Chen Z, Huang C, Ma T, et al. Reversal effect of quercetin on multidrug resistance via FZD7/beta-catenin pathway in hepatocellular carcinoma cells. Phytomedicine 2018;43:37-45.
350. Zhang K, Li M, Huang H, et al. Dishevelled1-3 contribute to multidrug resistance in colorectal cancer via activating Wnt/beta-catenin signaling. Oncotarget 2017;8:115803-16.
351. Ku JL, Shin YK, Kim DW, et al. Establishment and characterization of 13 human colorectal carcinoma cell lines: mutations of genes and expressions of drug-sensitivity genes and cancer stem cell markers. Carcinogenesis 2010;31:1003-9.
352. Chikazawa N, Tanaka H, Tasaka T, et al. Inhibition of Wnt signaling pathway decreases chemotherapy-resistant side-population colon cancer cells. Anticancer Res 2010;30:2041-8.
353. Correa S, Binato R, Du Rocher B, Castelo-Branco MT, Pizzatti L, Abdelhay E. Wnt/beta-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012;12:303.
354. Stein U, Fleuter C, Siegel F, et al. Impact of mutant beta-catenin on ABCB1 expression and therapy response in colon cancer cells. Br J Cancer 2012;106:1395-405.
355. Xia Z, Guo M, Liu H, et al. CBP-dependent Wnt/beta-catenin signaling is crucial in regulation of MDR1 transcription. Curr Cancer Drug Targets 2015;15:519-32.
356. Zhang ZM, Wu JF, Luo QC, et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/beta-catenin pathway. Oncogene 2016;35:4787-97.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lee WK, Frank T. Teaching an old dog new tricks: reactivated developmental signaling pathways regulate ABCB1 and chemoresistance in cancer. Cancer Drug Resist 2021;4:424-52. http://dx.doi.org/10.20517/cdr.2020.114
AMA Style
Lee WK, Frank T. Teaching an old dog new tricks: reactivated developmental signaling pathways regulate ABCB1 and chemoresistance in cancer. Cancer Drug Resistance. 2021; 4(2): 424-52. http://dx.doi.org/10.20517/cdr.2020.114
Chicago/Turabian Style
Lee, Wing-Kee, Thévenod Frank. 2021. "Teaching an old dog new tricks: reactivated developmental signaling pathways regulate ABCB1 and chemoresistance in cancer" Cancer Drug Resistance. 4, no.2: 424-52. http://dx.doi.org/10.20517/cdr.2020.114
ACS Style
Lee, W.K.; Frank T. Teaching an old dog new tricks: reactivated developmental signaling pathways regulate ABCB1 and chemoresistance in cancer. Cancer Drug Resist. 2021, 4, 424-52. http://dx.doi.org/10.20517/cdr.2020.114
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 24
likes
Like This Article 24
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.