
DNA direct reversal repair and alkylating agent drug resistance

Abstract
DNA direct reversal repair (DRR) is unique in that no DNA synthesis is required to correct the error and therefore repair via such mechanisms are error-free. In humans, DRR is carried out by two different pathways: the O6-methylguanine-DNA methyltransferase (MGMT) and the alkylated DNA repair protein B (AlkB) homologs. The use of alkylating agents is the standard of care for many cancers. However, the use of those drugs is usually halted when resistance develops. This review will examine repair of alkylating agent damage mediated by DRR, resistance mechanisms and potential ways to overcome such resistance.
Keywords
Introduction
DNA alkylating agents are still used for the treatment of many cancers. However, continued treatment with alkylating agents, even in drug cocktails, generally results in drug resistance. Increasing the utility of those drugs requires the understanding of the sources that oppose the therapeutic effects of alkylating agents. One of the principal mechanisms that contributes to alkylating agent resistance is DNA repair.
DNA alkylation damage occurs at all bases. The level of DNA damage at the individual bases does not correlate with the biological impact of the damage. Major damage sites at N7 of guanine does not generally cause mutation or cell death associated with therapeutic impact. More minor sites of alkylation damage, including O6 of guanine, N1 of adenine, or N3 of cytosine are more closely aligned with therapeutic responses of alkylating agents[1,2].
The number of alkylating agents used in chemotherapy is too long to cite in this brief review. To provide some examples, there are monofunctional methylating agents (temozolomide or TMZ), larger monofunctional alkylating agents (cyclohexyl chloroethyl nitrosourea, CCNU, or lomustine) [Figure 1]. There are bifunctional agents that interact and can cause inter- or intrastrand crosslinks (nitrogen mustards or busulfan) [Figure 1]. SN1 alkylating agents (e.g., temozolomide, lomustine in Figure 1) form reactive intermediates that then react with DNA according to kinetics dependent only on the concentration of alkylating agent, whereas SN2 alkylating agents (e.g., nitrogen mustard, busulfan in Figure 1) react directly with DNA and manifest kinetics that depend on the concentration of both the alkylating agent and the target. DNA that is damaged by these agents cause is restored to normal by mechanisms of DNA repair.
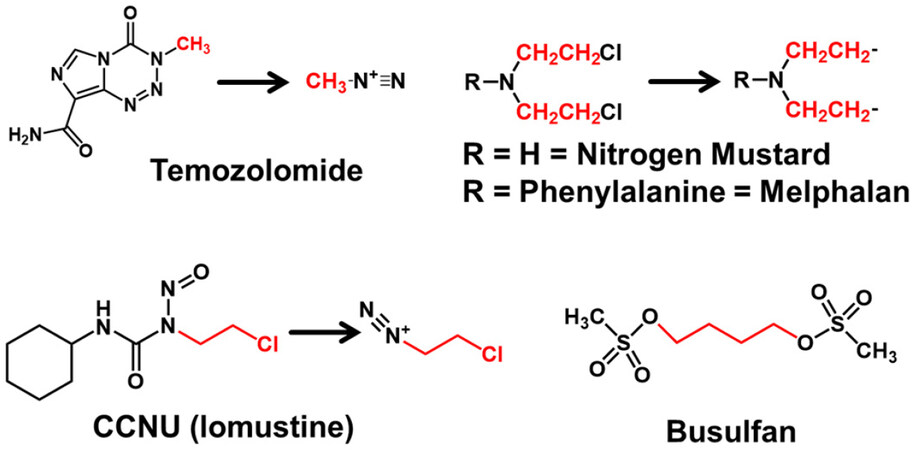
Figure 1. Selected chemotherapeutic alkylating agents. DNA damage is monofunctional (temozolomide, CCNU) or bifunctional (nitrogen mustard, melphalan, busulfan). Alkylating agents use SN1 (temozolomide, CCNU) or SN2 mechanisms (nitrogen mustard, melphalan, busulfan). The red portions of the structure are the moieties that alkylate DNA.
DNA repair is often collectively referenced, but DNA repair consists of numerous pathways that can restore genomic integrity and depend on the type of damage inflicted by an agent. Thus, detailing the ways that tumors develop resistance ultimately will require understanding how the ensemble of these pathways function together to protect cells [Figure 2]. Modification of DNA bases can lead to the activation of these pathways, depending on how the initial insult is addressed. Base or nucleotide excision repair can directly eliminate the damage. If damage levels are too high, cells can undergo apoptosis. Another possibility is the formation of double-strand breaks that can cause cell death. The simplest type of repair that will be described in this review is direct reversal repair (DRR), which does not require any DNA synthesis and is therefore error-free. DRR is conducted by O6-methylguanine-DNA methyltransferase (MGMT) and the alkylated DNA repair protein B (AlkB) homologs ALKBH2 and ALKBH3. Failure to repair prior to replication can result in an alternative DNA base that can lead to mutations. If the alternative base pairs are maintained there is a possibility that mismatch repair (MMR) will play a role in cell death. However, if replication is not continued through the mispairs, arrest of the replication fork can lead to a double-strand break that would be lethal if left unrepaired. To limit the scope of this review, we will concentrate on resistance associated with one type of DNA repair, DRR.
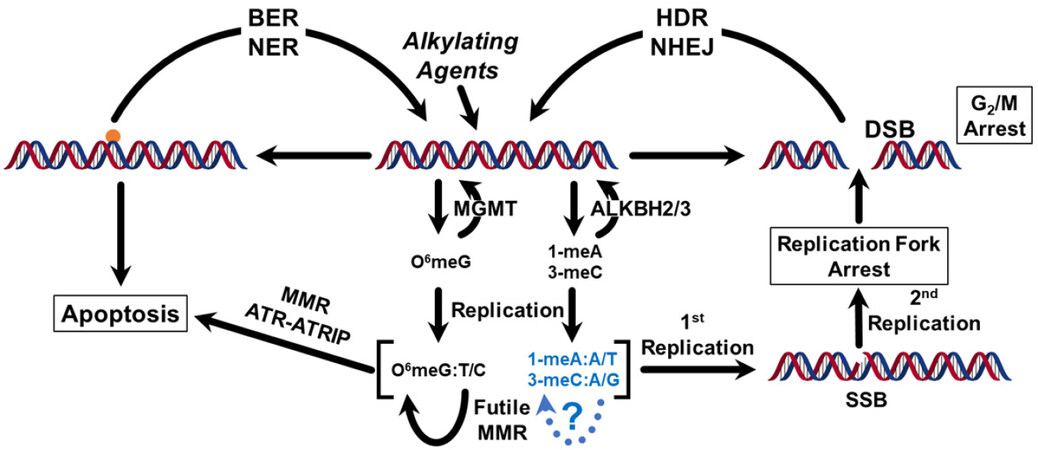
Figure 2. General DNA repair pathways implicated in the elimination of DNA damage from alkylating agents. The blue sections and dotted lines have not yet been described in the literature. The possible base pairs for O6meG, 1-meA, and 3-meC are indicated. If cells complete the first round of replication, following a second round of replication, a mutation is fixed in 50% of the cells. If replication is arrested and a second round of replication occurs, a DSB can form. Modeled after Reference[3]. BER: Base excision repair; NER: nucleotide excision repair; MMR: mismatch repair; DSB: double-strand break; HDR: homology-directed repair; NHEJ: non-homologous end-joining; SSB: single strand break; ATR: ataxia telangiectasia and Rad3 related; ATRIP: ATR interacting protein; O6meG: O6methylguanine; 1-meA: 1-methyladenine; 3-meC: 3-methylcytosine.
O6-methylguanine-DNA methyltransferase pathway
The DRR MGMT pathway is found in all cells. The reaction mechanism is a direct transfer of the alkylated base to the cysteine-145 receptor in MGMT in the conserved 5-amino acid active site PCHRV [Figure 3][4,5]. That transfer is accompanied by ubiquitination of MGMT and subsequent degradation of the protein in the 26S proteasome[6]. In this repair, a single MGMT protein repairs a single O6-methylguanine (O6-meG), which represents a substantial energy requirement to remove a single damage from a base[7].
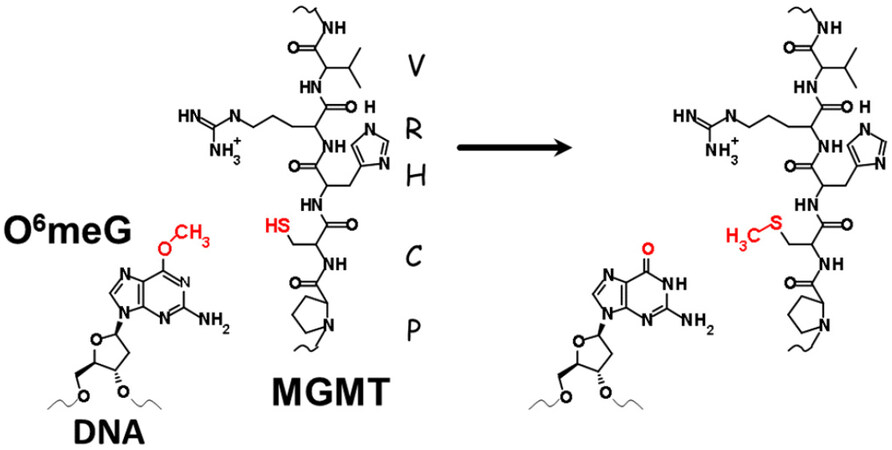
Figure 3. Transfer of methyl group from O6meG to the cysteine residue of MGMT. Conserved amino acids around the active site are shown. MGMT: O6-Methylguanine-DNA Methyltransferase; O6meG: O6methylguanine.
Persistence of O6meG during replication, can lead to the incorporation of thymine opposite O6meG. If no repair occurs and replication is completed the O6meG:T base pairing can form [Figure 4]. A subsequent round of replication leads to a G→A mutation.
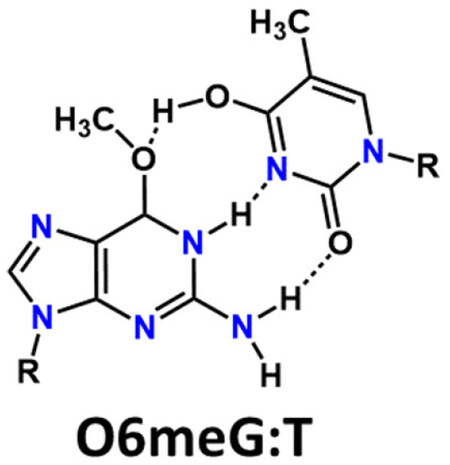
Figure 4. O6methylguanine:thymine base pairing that leads to G→A mutation. The pairing is based on data from the crystal structure of the pair in oligodeoxyribonucleotides[8]. Note that the thymine tautomer formed is not the standard tautomer drawn for thymine in Watson-Crick base pairs.
Persistent O6methylguanine lesions and cell death
When replication is finished, the base pair depicted in Figure 4 can be recognized by an active MMR pathway. If the MMR pathway removes the thymine, another thymine could be reinserted in a futile repair process. Repeated futile repair cycles eventually result in the cell undergoing apoptosis [Figure 2].
Although MGMT inhibitors can deplete MGMT activity, another way to reduce the MGMT levels is to take advantage of the variations in the levels of MGMT found in different cells. In many tumors, MGMT deficiency is linked to epigenetic methylation of the MGMT promoter region to produce 5-methylcytosines that silence expression[9-11]. MGMT promoter silencing is frequently found in tumors[12,13]. Tumors with low MGMT levels usually produce positive patient outcomes.
Alternatives to silencing MGMT expression in tumors have used drugs, such as O6-benzylguanine, which deplete MGMT protein levels by inactivating the protein. These MGMT inhibitors inactivate the protein and are still undergoing clinical evaluation. One potential consideration for the poor outcomes using MGMT inactivation is that drugs that deplete MGMT could also require other DNA repair systems, as noted for the futile repair cycles.
Resistance to alkylating agent damage by MGMT
The levels of MGMT in tissues and in tumors vary greatly[14,15]. Tumors with low or MGMT-deficiency respond better to dacarbazine or temozolomide therapy than do tumors with MGMT levels similar to those found in normal cells[16-21]. Therefore, tumors with MGMT levels approximating those of normal cells are more resistant to alkylating agent treatment. Conversely, tumors with low MGMT levels respond more favorably to treatment as compared to tumors with normal MGMT levels[17,22-24]. Thus, improved outcomes for patients with lower MGMT levels indicate that normal MGMT levels provide resistance to dacarbazine or temozolomide treatment.
Resistance to alkylating agent damage by MGMT in concert with mismatch repair
MGMT protects against alkylating agent damage, and the discovery that tumors and cell lines can spontaneously have the MGMT promoter silenced, certainly suggested that there could be cumulative effects that would result in further sensitization of tumors to alkylating agent treatments. The MMR system is another major DNA repair pathway that is often defective in tumors[25]. Hereditary nonpolyposis colon cancer has defective MMR genes that result in genomic instability[26-28], but other tumor types can also evolve MMR deficiencies by mutations, although also by epigenetic silencing, particularly of MLH1[29-34]. The sensitivity of MMR-proficient cells in the absence of MGMT is due to the futile MMR cycles[35,36] that are induced by O6meG:T mispairing [Figures 2 and 3]. The role of MMR was demonstrated using Msh2-deficient mouse embryonic stem cells and O6-benzylguanine that inactivates MGMT[37]. Further mouse model work demonstrated the resistance to alkylating agents that arises with the simultaneous inactivation of both the Mgmt and Mlh1 and the increase in mutations upon treatment as compared to either of the genes inactivated independently[38,39].
AlkB homolog pathway
The AlkB homolog (ALKBH) pathway in this review refers to both ALKBH2 and ALKBH3 (ALKBH), which reversibly remove numerous modified bases but principally 1-methyladenine (1-meA) and 3-methylcytosine (3-meC)[40-42]. There are 9 proteins among the AlkB homologs, but the functions for most are diverse and include removal of RNA modifications[43]. Numerous protein groups use similar reaction mechanisms, which include proline hydroxylation and ten-eleven translocation[44-47]. The ALKBH reaction mechanism uses an oxidative demethylation with a Fe(II) that is coordinated to amino acids in the proteins, along with α-ketoglutarate. The removal of the methyl group involves the conversion of α-ketoglutarate to succinate with the release of carbon dioxide and formaldehyde [Figure 5].
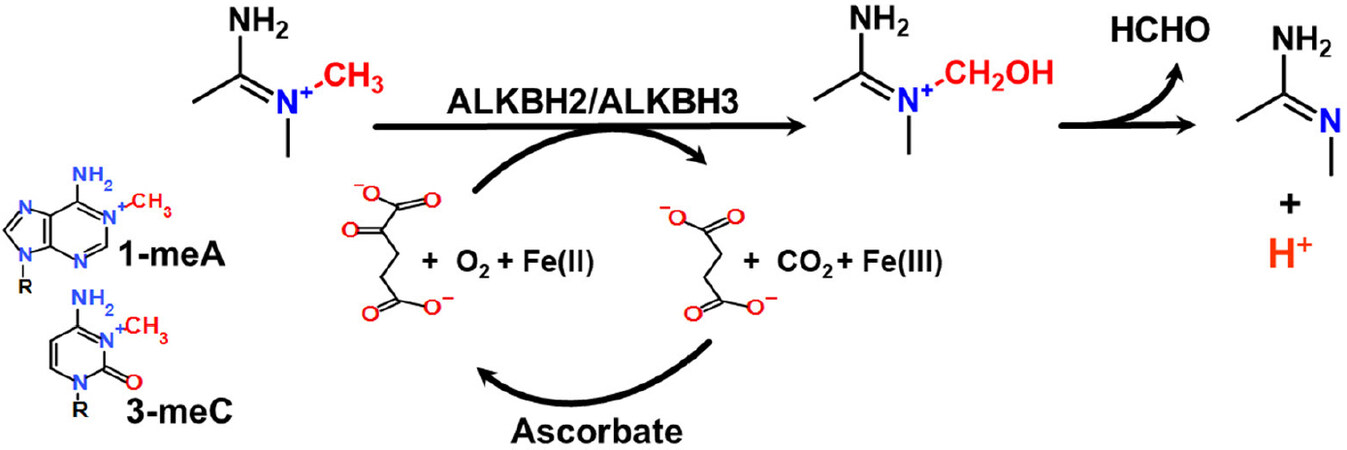
Figure 5. Repair of 1-methyladenine and 3-methylcyotsine. The reaction converts α-ketoglutarate to succinate with CO2 release and requires oxygen and Fe(II). There is also a release of formaldehyde to regenerate the normal base. 1-meA: 1-methyladenine; 3-meC: 3-methylcytosine.
Persistence of 1-meA and 3-meC can lead to mutations that are reported to be A→T transversions or C→T transitions[48], which involve 1-meA pairing with T or possibly bypass of the 3-meC, because only a single base pair can form with the 1-meC modification [Figure 6]. The bypass of C by translesion DNA polymerases inserting A opposite to produce a C→T transition mutation is suggested by an in vitro steady state kinetics study that showed a substrate preference for insertion of dATP, based on the kcat/KM ratio[49].
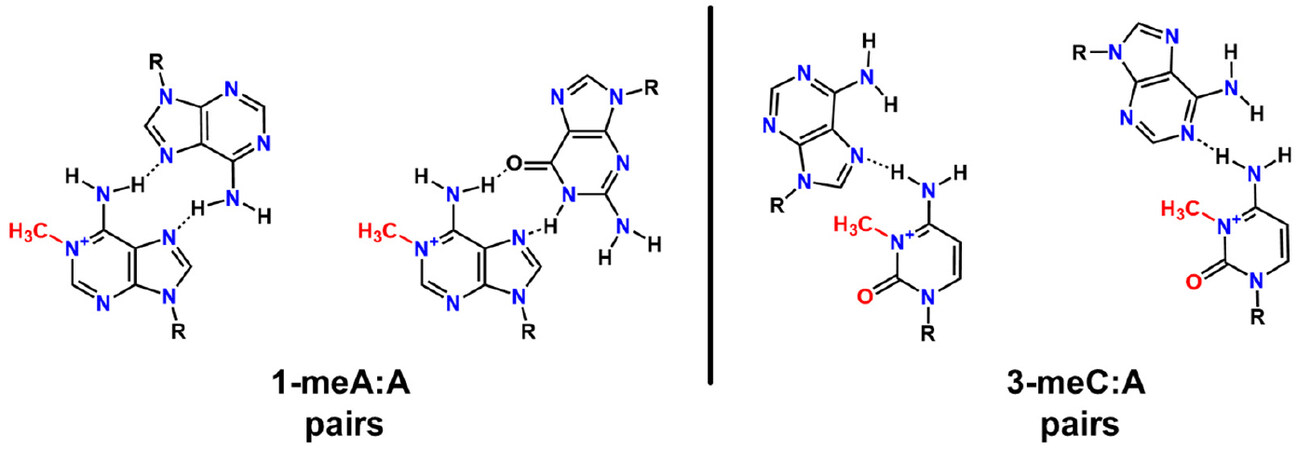
Figure 6. 1-methyladenine:adenine and 3-methylcytosine:adenine base pairings, which lead to A→T and C→T mutations, respectively. These possible base pairing schemes are derived from the observation of mutations in a mouse model system with Alkbh2 or Alkbh3 deleted[48]. The 3-methylcytosine (3-meC) modified base would most probably use a translesion DNA polymerase for insertion opposite 3-meC due to the single-base pairing available with the modification at the N3 position. Note that the left 1-meA:A pair would be in a parallel helix, whereas the right structure would be in an antiparallel helix.
Persistent 1-methyladenine and 3-methylcytosine lesions and cell death
Currently, the consequences of the completion of replication with the base pair structures in Figure 6 is unknown. On the basis of the link between MGMT and MMR, it is possible that mispairing occurs that leads to cell death [Figure 2], but that is not reported. Moreover, understanding the links between the MGMT and ALKBH pathways is important to the understanding of how tumor cells protect themselves against alkylating agent chemotherapy.
Resistance to alkylating agent damage by ALKBH
Expression of genes in the ALKBH pathway in tissues varies[50], as does the level in tumors[51-54]. The increased expression of ALKBH3 in some pancreatic cancers is associated with poor outcomes and reduced survival in patients, but it is unclear if high ALKBH3 levels are also associated with resistance to treatment with alkylating agents in those patients[52]. In breast cancer cell lines, there are also differences in the level of ALKBH3, with certain lines (e.g., MCF-7) having relatively high levels of ALKBH3 and other cell lines (e.g., Bt-474) having relatively low levels of ALKBH3[53]. Despite those differences in ALKBH3 levels, the contribution to alkylating agent resistance has not been evaluated. However, the accumulation of 3-meC suggests that cells with lower 3-meC are more resistant to alkylating agent treatment. Despite the varied levels of ALKBH3 in breast tumor cell lines, the survival of cells treated with methylating agents was not evaluated[53]. In 265 primary breast cancer samples, 72 patients showed epigenetic silencing[53]. ALKBH3 is also linked to prostate cancer and an alternate name is prostate cancer-1 (PCA-1)[55,56]. Thus, targeting ALKBH3 is a potential way to use alkylating agent therapy for prostate cancer[55].
ALKBH pathway resistance can be overcome by reducing the activity of the ALKBH proteins. One way that ALKBH activities can be reduced is by the presence of mutated isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2) [Figure 7]. Both those proteins are responsible for conversion of isocitrate to α-ketoglutarate. Mutated IDH1 or IDH2 is found in many cancers[57-59]. Mutated IDH1 or 2 leads to the production of (R)-2-hydroxyglutarate, which disrupts the activity of ALKBH[60]. This loss of activity renders cells more susceptible to alkylating agent treatment. Another way to restore the sensitivity of cells to alkylating agents by reducing IDH1 or IDH2 levels is to limit glutamine levels using glutaminase inhibitors[61]. Glutamine is a precursor of α-ketoglutarate, and inhibiting glutaminase levels can limit α-ketoglutarate levels, making cells sensitive to alkylating agents by lowering ALKBH activities. Both these methods can reduce resistance to alkylating agents in tumor cells.
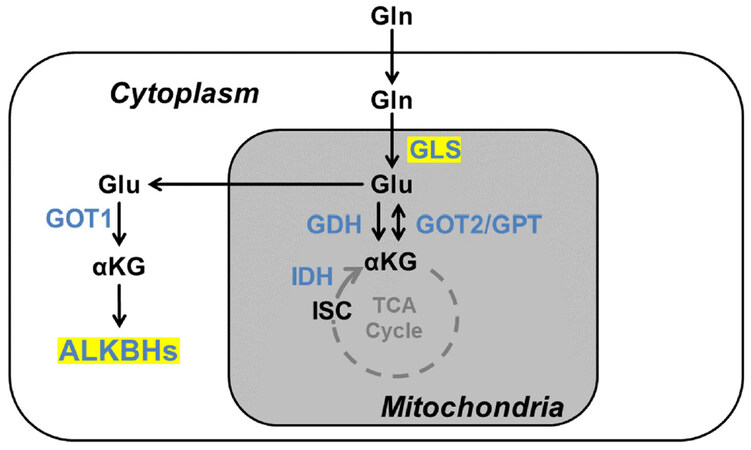
Figure 7. Glutamine metabolism producing 2-oxoglutarate. Enzymes are indicated in blue. ALKBHs represents ALKBH2 and ALKBH3 proteins that require αKG as a cofactor in this review. Gln: Glutamine; Glu: glutamate; αKG: α-ketoglutarate, ISC: isocitrate; TCA: tricarboxylic acid cycle; GDH: glutamate dehydrogenase; GLS: glutaminase; GOT: glutamate oxaloacetate transaminase; IDH: isocitrate dehydrogenases.
Possible resistance to alkylating agent damage by ALKBH proteins in concert with mismatch repair
Resistance is manifested when both MGMT and MMR are not functional[39]. O6-meG can block or impede DNA synthesis[62]. Both 1-meA and 3-meC can also arrest DNA synthesis, which suggests that elimination of both ALKBH pathways. Such and MMR pathways could have similar outcomes. The existence of such resistance would diminish the therapeutic outcomes for alkylating agent chemotherapy. Therefore, examining the link between the ALKBH and MMR pathways in alkylating agent damage should be a priority, because resistance to alkylating agents is often linked to MMR gene silencing.
Interaction between the MGMT and ALKBH repair pathways
Resistance provided by MGMT and ALKBH pathways is not necessarily independent but could be cumulative or even synergistic, even though the lesions repaired by both pathways are different. Such a link could provide answers concerning resistance observed for both MGMT and ALKBH pathways separately. Investigating potential contributions of these 2 DRR mechanisms would help to understand DNA repair resistance mechanisms, which could lead to improved alkylating agent treatment efficacy.
Conclusion
DRR is the simplest form of DNA repair, but still there are questions that are left unanswered. MGMT provides resistance to alkylation damage for O6-guanine modifications. Absence of MGMT is a positive indicator of response to alkylating agent treatment. However, the MMR pathway is needed for chemotherapy to be successful. The ALKBH pathway is less studied as compared to the MGMT pathway. Thus, we still need to address the role of MMR in ALKBH pathway repair and how the two DRR pathways work to resist alkylating agent treatments. Therefore, despite the simplicity of direct repair pathways biochemically, significant questions remain about how these pathways function to resist alkylating agent chemotherapy.
Declarations
Authors’ contributionsMade substantial contributions to conception and design of the review and writing: Gutierrez R, O’Connor TR.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by California Tobacco-Related Disease Research Program Grant: Combined genotoxic effects of alternative tobacco products and alcohol use (No. 28IR-0050, TRO) and Irell & Manella Graduate School of Biological Sciences: Payson and Chu Fellowship.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012;12:104-20.
2. Ahmad A, Nay SL, O’Connor TR. Direct Reversal Repair in Mammalian Cells. In: Clark Chen, editor. Advances in DNA Repair. Rijeka, Croatia: InTech; 2015. pp. 95-128.
3. Kondo N, Takahashi A, Ono K, Ohnishi T. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids 2010;2010:543531.
4. Tano K, Shiota S, Collier J, Foote RS, Mitra S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc Natl Acad Sci U S A 1990;87:686-90.
5. Hayakawa H, Koike G, Sekiguchi M. Expression and cloning of complementary DNA for a human enzyme that repairs O6-methylguanine in DNA. J Mol Biol 1990;213:739-47.
6. Xu-Welliver M, Pegg AE. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002;23:823-30.
7. Lindahl T, Demple B, Robins P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J 1982;1:1359-63.
8. Warren JJ, Forsberg LJ, Beese LS. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc Natl Acad Sci U S A 2006;103:19701-6.
9. Costello JF, Futscher BW, Kroes RA, Pieper RO. Methylation-related chromatin structure is associated with exclusion of transcription factors from and suppressed expression of the O-6-methylguanine DNA methyltransferase gene in human glioma cell lines. Mol Cell Biol 1994;14:6515-21.
10. Costello JF, Futscher BW, Tano K, Graunke DM, Pieper RO. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J Biol Chem 1994;269:17228-37.
11. Harris LC, Remack JS, Brent TP. In vitro methylation of the human O6-methylguanine-DNA methyltransferase promoter reduces transcription. Biochim Biophys Acta 1994;1217:141-6.
12. Esteller M, Toyota M, Sanchez-Cespedes M, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res 2000;60:2368-71.
13. Qi Z, Tan H. Association between MGMT status and response to alkylating agents in patients with neuroendocrine neoplasms: a systematic review and meta-analysis. Biosci Rep 2020;40:BSR20194127.
14. Mitra S, Kaina B. Regulation of repair of alkylation damage in mammalian genomes. Prog Nucleic Acid Res Mol Biol 1993;44:109-42.
15. Christmann M, Verbeek B, Roos WP, Kaina B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta 2011;1816:179-90.
16. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997-1003.
17. Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 2009;15:338-45.
18. Boeckmann L, Nickel AC, Kuschal C, et al. Temozolomide chemoresistance heterogeneity in melanoma with different treatment regimens: DNA damage accumulation contribution. Melanoma Res 2011;21:206-16.
19. Cen L, Carlson BL, Pokorny JL, et al. Efficacy of protracted temozolomide dosing is limited in MGMT unmethylated GBM xenograft models. Neuro Oncol 2013;15:735-46.
20. Fan CH, Liu WL, Cao H, Wen C, Chen L, Jiang G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis 2013;4:e876.
21. Bengtsson D, Schrøder HD, Andersen M, et al. Long-term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab 2015;100:1689-98.
22. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000;343:1350-4.
23. Middlemas DS, Stewart CF, Kirstein MN, et al. Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin Cancer Res 2000;6:998-1007.
24. Bruheim S, Bruland OS, Breistol K, Maelandsmo GM, Fodstad O. Human osteosarcoma xenografts and their sensitivity to chemotherapy. Pathol Oncol Res 2004;10:133-41.
25. Pećina-Šlaus N, Kafka A, Salamon I, Bukovac A. Mismatch repair pathway, genome stability and cancer. Front Mol Biosci 2020;7:122.
26. Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027-38.
27. Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993;75:1215-25.
28. Young J, Leggett B, Gustafson C, et al. Genomic instability occurs in colorectal carcinomas but not in adenomas. Hum Mutat 1993;2:351-4.
29. Felsberg J, Thon N, Eigenbrod S, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 2011;129:659-70.
30. von Bueren AO, Bacolod MD, Hagel C, et al. Mismatch repair deficiency: a temozolomide resistance factor in medulloblastoma cell lines that is uncommon in primary medulloblastoma tumours. Br J Cancer 2012;107:1399-408.
31. Shinsato Y, Furukawa T, Yunoue S, et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 2013;4:2261-70.
32. Taverna P, Liu L, Hanson AJ, Monks A, Gerson SL. Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemother Pharmacol 2000;46:507-16.
33. Helleman J, van Staveren IL, Dinjens WN, et al. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer 2006;6:201.
34. Gee ME, Faraahi Z, McCormick A, Edmondson RJ. DNA damage repair in ovarian cancer: unlocking the heterogeneity. J Ovarian Res 2018;11:50.
35. Karran P, Bignami M. Self-destruction and tolerance in resistance of mammalian cells to alkylation damage. Nucleic Acids Res 1992;20:2933-40.
36. Day RS 3rd, Babich MA, Yarosh DB, Scudiero DA. The role of O6-methylguanine in human cell killing, sister chromatid exchange induction and mutagenesis: a review. J Cell Sci Suppl 1987;6:333-53.
37. de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995;82:321-30.
38. Kawate H, Sakumi K, Tsuzuki T, et al. Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc Natl Acad Sci U S A 1998;95:5116-20.
39. Takagi Y, Takahashi M, Sanada M, Ito R, Yamaizumi M, Sekiguchi M. Roles of MGMT and MLH1 proteins in alkylation-induced apoptosis and mutagenesis. DNA Repair (Amst) 2003;2:1135-46.
40. Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A 2002;99:16660-5.
41. Koivisto P, Robins P, Lindahl T, Sedgwick B. Demethylation of 3-methylthymine in DNA by bacterial and human DNA dioxygenases. J Biol Chem 2004;279:40470-4.
42. Aas PA, Otterlei M, Falnes PO, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003;421:859-63.
43. Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem 2014;83:585-614.
44. Ciesielski P, Jóźwiak P, Krześlak A. TET proteins and epigenetic modifications in cancers. Postepy Hig Med Dosw (Online) 2015;69:1371-83.
45. Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J Biol Chem 2015;290:20734-42.
46. Guengerich FP. Introduction: Metals in Biology: alpha-Ketoglutarate/Iron-Dependent Dioxygenases. J Biol Chem 2015;290:20700-1.
47. Hashimoto H, Zhang X, Vertino PM, Cheng X. The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations. J Biol Chem 2015;290:20723-33.
48. Nay SL, Lee DH, Bates SE, O’Connor TR. Alkbh2 protects against lethality and mutation in primary mouse embryonic fibroblasts. DNA Repair (Amst) 2012;11:502-10.
49. Furrer A, van Loon B. Handling the 3-methylcytosine lesion by six human DNA polymerases members of the B-, X- and Y-families. Nucleic Acids Res 2014;42:553-66.
50. Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O’Connor TR. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem 2005;280:39448-59.
51. Pilžys T, Marcinkowski M, Kukwa W, et al. ALKBH overexpression in head and neck cancer: potential target for novel anticancer therapy. Sci Rep 2019;9:13249.
52. Yamato I, Sho M, Shimada K, et al. PCA-1/ALKBH3 contributes to pancreatic cancer by supporting apoptotic resistance and angiogenesis. Cancer Res 2012;72:4829-39.
53. Stefansson OA, Hermanowicz S, van der Horst J, et al. CpG promoter methylation of the ALKBH3 alkylation repair gene in breast cancer. BMC Cancer 2017;17:469.
54. Johannessen TC, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol 2013;15:269-78.
55. Konishi N, Nakamura M, Ishida E, et al. High expression of a new marker PCA-1 in human prostate carcinoma. Clin Cancer Res 2005;11:5090-7.
56. Liu BQ, Wu YD, Li PH, Wei JX, Zhang T, Liu RL. Prostate cancer antigen-1 as a potential novel marker for prostate cancer. Asian J Androl 2007;9:821-6.
57. Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091-100.
58. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 2012;18:5562-71.
59. Schaap FG, French PJ, Bovée JV. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv Anat Pathol 2013;20:32-8.
60. Wang P, Wu J, Ma S, et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep 2015;13:2353-61.
61. Tran TQ, Ishak Gabra MB, Lowman XH, et al. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol 2017;15:e2002810.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Gutierrez R, O’Connor TR. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist 2021;4:414-23. http://dx.doi.org/10.20517/cdr.2020.113
AMA Style
Gutierrez R, O’Connor TR. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resistance. 2021; 4(2): 414-23. http://dx.doi.org/10.20517/cdr.2020.113
Chicago/Turabian Style
Gutierrez, Roberto, Timothy R. O’Connor. 2021. "DNA direct reversal repair and alkylating agent drug resistance" Cancer Drug Resistance. 4, no.2: 414-23. http://dx.doi.org/10.20517/cdr.2020.113
ACS Style
Gutierrez, R.; O’Connor TR. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 2021, 4, 414-23. http://dx.doi.org/10.20517/cdr.2020.113
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 17 clicks
Cite This Article 17 clicks

 Like This Article 1
likes
Like This Article 1
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.