
Key genes and drug delivery systems to improve the efficiency of chemotherapy

Abstract
Cancer cells can develop resistance to anticancer drugs, thereby becoming tolerant to treatment through different mechanisms. The biological mechanisms leading to the generation of anticancer treatment resistance include alterations in transmembrane proteins, DNA damage and repair mechanisms, alterations in target molecules, and genetic responses, among others. The most common anti-cancer drugs reported to develop resistance to cancer cells include cisplatin, doxorubicin, paclitaxel, and fluorouracil. These anticancer drugs have different mechanisms of action, and specific cancer types can be affected by different genes. The development of drug resistance is a cellular response which uses differential gene expression, to enable adaptation and survival of the cell to diverse threatening environmental agents. In this review, we briefly look at the key regulatory genes, their expression, as well as the responses and regulation of cancer cells when exposed to anticancer drugs, along with the incorporation of alternative nanocarriers as treatments to overcome anticancer drug resistance.
Keywords
Introduction
Drug resistance and inefficient cancer therapy accounts for up to 90% of cancer-related deaths[1]. Resistance occurs when a cancer cell develops the ability to prevent chemotherapeutic drugs from entering inside the cell, or reduces the amount of the drug that can enter inside it to non-damaging levels. Current cancer management programs include surgery, radiation therapy, immunotherapy and/or chemotherapy (including toxic, non-targeted, and targeted therapy). The problem of drug resistance is highly sustained by intrinsic and extrinsic factors. Although many cancers initially respond successfully to chemotherapy, the development of drug resistance occurs in most patients[2]. The initial solution to the problem of resistance to single-agent chemotherapy is the combined administration of agents that have non-overlapping mechanisms of action.
Cancer cells have many growth mechanisms; they release proliferative signals while avoiding growth suppressor molecules to resist cell death. Anticancer agents cause DNA damage by targeting cellular replication as well as the growth signaling molecules of rapidly dividing cells[3]. Usually, cytotoxic drugs are cell-cycle specific and target a phase of the process. In this way, they normally induce mitochondria-mediated caspase-dependent apoptosis. For example, tamoxifen hormone targets the G1-phase, antimetabolites target the S-phase, podophyllotoxins target the G2-phase, and taxanes target the M-phase of the cell cycle[4]. In contrast, there are anticancer agents which modulate cellular process that are cell cycle-independent[5]. For example, alkylating and platinum-based agents can disrupt DNA at any stage of the cell cycle, and anthracyclines, as doxorubicin, interfere with DNA replication and mainly generates reactive oxygen species (ROS)[5,6]. However, anticancer drugs affect healthy cells and produce severe side effects. Depending on their mechanism, anticancer drugs can be divided into (1) alkylating agents, (2) antimetabolites, (3) mitotic spindle inhibitors, (4) topoisomerase inhibitors, (5) anthracyclines, and others[7][Table 1].
Common classes of anticancer drugs
| Classification | Mechanism of action | Type of drug | Examples of drugs | Cancer type | Ref. |
|---|---|---|---|---|---|
| Alkylating agents | Add alkyl groups to guanine on DNA; create cross links within the DNA | Platinum-based agents | Cisplatin
Carboplatin Oxaliplatin | Breast, Leukemia, Lymphoma, Multiple Myeloma, Sarcoma, Brain Cancer, Ovary, Lung | [8-10] |
| Nitrogen mustards | Chlorambucil
Cyclophosphamide | ||||
| Alkylsulfonates | Busulfan | ||||
| Antimetabolites | Interfere with vital metabolic pathways by acting as a false substrate during cell cycle synthesis phase | Pyrimidine antagonists | 5-Fluorouracil
Gemcitabine | Leukemia, Breast, Ovary, Intestinal Tract, Pancreatic, Colorectal | [11-14] |
| Purine antagonists | Fludarabine | ||||
| Purine analogs | 6-Mercaptopurine | ||||
| antifolates | Methotrexate | ||||
| Ribonucleotide reductase inhibitors | Hydroxyurea | ||||
| Mitotic spindle inhibitors | Inhibit microtubule polymerization causing disruption of mitotic spindle formation | Taxanes | Paclitaxel
Docetaxel | ALL, Burkitt lymphoma, Hodgkin lymphoma, Neuroblastoma, Rhabdomyosarcoma, Wilms tumor, NSCLC, Ovarian, Head and neck | [14,15] |
| Vinca alkaloids | Vincristine
Vinblastine | ||||
| Topoisomerase inhibitors | Prevents resealing of DNA breaks | Topoisomerase inhibitors I and II | Topotecan
Etoposide | Leukemia,
Lung, ovarian, gastrointestinal, and other cancers | [14,16] |
| anti-tumor antibiotics | Different mechanisms (free radical formation, lipid peroxidation, direct membrane effects, and enzyme interactions) | Anthracyclines | Doxorubicin | ALL, AML, Hodgkin’s and non-Hodgkin’s lymphoma, Bladder, Breast, Metastatic cancers, Esophageal | [14,17,18] |
| Daunorubicin | |||||
| Tyrosine kinase inhibitors | Blocks the action of tyrosine kinases | Small molecules inhibitors | Erlotinib
Lapatinib Ripretinib | Breast, CML, NSCLC, Lung, Renal, Hepatocellular, Prostate, Renal, Colorectal, ALL, GIST | [19-22] |
In chemotherapy, cytotoxic agents target different metabolic pathways, mainly that of the apoptotic machinery. Cancer cells can become resistant to cytotoxic agents and several cancer treatments due to the dysregulation of cell signaling pathways, as is described in several reviews[1,7,23-28]. One of the principal causes of drug resistance is the patient’s genetic predisposition, where the individual inherits genetic characteristics of an ineffective drug response. Another case of tumor drug resistance is the growth of cancer cells which adapt and survive after drug exposure. In this last scenario, exposure of tumor cells to the drug leads to resistance because of a genetic adaptation to the tumor microenvironment. Epigenetic modifications and an imbalance of random mutations influencing signaling processes also support carcinogenesis[7,26,29].
Table 2 presents a list of the most well-known anticancer drugs used in chemotherapy for more than 20 years. Unfortunately, these anticancer drugs are not entirely effective and lead to the development of drug resistance to cancer treatment. Most genes presented in this review have developed resistance to one or more of these drugs.
Common chemotherapeutic drugs associated with cancer cell resistance#
| Chemotherapeutics | Cell cycle phase disrupted | Cellular pathways affected | Main resistance-related genes | Ref. |
|---|---|---|---|---|
| Cisplatin | DNA repair /
Any phase of the cell cycle | -DNA damage
DNA-platinum adducts leading to apoptosis | BRCA (1 and 2)- DNA damage repair | [30-33] |
| Doxorubicin | DNA replication and DNA repair | -DNA structure changes --Formation of free radicals and oxidative damage | BCL-2- Cardiotoxicity to non-cancer cells | [34-37] |
| Paclitaxel | Mitosis | -Cytoplasmic microtubule-assembling disruptor
-Cell replication inhibitor | MDR1- Overexpression of P-gp, drug target alteration | [7,38,39] |
| 5-Fluorouracil | DNA synthesis (DS) | -DS inhibition through thymidylate synthase targeting
-Cell growth inhibition leading to apoptosis -DNA and RNA damage | BCL-2, Bcl-XL and p53 overexpression
-drug inactivation -drug target alteration | [40-42] |
Cancer-stem cells play an essential role in resistance to cancer treatment by promoting uncontrolled cell growth and generating tumors. Cancer-stem cells can self-renew and differentiate into multiple cell types. Such cells can persist in tumors as a distinct population, and can cause relapse and metastasis by giving rise to new tumors[43,44]. Furthermore, the tumor microenvironment can contribute to anticancer drug resistance, which decreases therapy effectiveness[45,46].
Drug resistance can develop due to intrinsic genetic causes or can be acquired upon exposure to chemotherapeutic drugs[47]. Tumors with intrinsic resistance exhibit cell heterogeneity and inherent decreased responsiveness to chemotherapy[1]. On the other hand, acquired resistance is generated by most cancer patients under chemotherapy as a gradual decrease in drug efficiency[47]. Furthermore, in this type of resistance, mutations can affect the expression level of the drug target, affecting the structure of the protein (mostly receptors) and the target of the therapy. Mutations can also affect other proteins within the cancer cells, which can become an oncogene, also known as a second proto-oncogenesis.
Cancer stem cells are also a result of mutations that turn them into a subset of cells within the tumor with a potential for self-renewal, differentiation, and tumorigenicity, making the tumor resistant to chemotherapy. Finally, chemotherapeutic drugs can also cause DNA damage in cancer cells and might increase the probability of the emergence of new mutations, including, for example, the activation of cell growth factors and cell defense systems[1].
The multidrug resistance (MDR) syndrome impedes the efficiency of cancer treatments, and it can occur during or after the cancer treatment. MDR can result from a difference in the structure or mechanism of anticancer drugs. MDR’s principal causes include increases in the efflux activity of drug pumps and a decrease in drug transporters within the membrane[48]. MDR is common in cancers such as ovarian, breast, cervical, lung, prostate, and melanoma[49]. Development of MDR is the main cause for failure of the most widely used chemotherapeutic drugs (paclitaxel, cisplatin, docetaxel, vincristine, epirubicin, 5-fluorouracil, and oxaliplatin), and leads to cancer recurrence after one or more years of treatment[50,51].
Some of the most well-studied cancer drug resistance mechanisms include drug inactivation, alteration of drug target, efflux pump, DNA damage repair, cell death inhibition, cancer cell heterogeneity, and epigenetics (explained in Table 3).
Mechanisms of anticancer drug resistance
| Mechanism | Short description | Ref. |
|---|---|---|
| Drug inactivation | Cancer cells generate an alternative mechanism that inactivates the drug that is inside the cell, contributing to modification, degradation, or complex formation. This inactivation decreases the drug’s toxicity levels, and reduces the damage and activity of the drug in cancer cells | [26] |
| Alteration of drug target | Altered or unrecognized protein structure in the drug’s transporter protein due to accumulated mutations can prevent proper attachment of the drug on its binding site. As a consequence, cancer cells become unable to internalize the cytotoxic drug, leading to their survival | [7,25] |
| Enhanced efflux pumps | The anticancer drug is pumped out of the cell through a transmembrane protein (efflux pump), preventing the accumulation of the effective drug concentration from causing toxicity in the cell, sabotaging the therapy | [26,29] |
| DNA-damage repair | Cancer cells may gain the ability to repair the DNA damage/breakage caused by anticancer drugs as a response to promote cell survival | [7,29] |
| Cell death inhibition | When proteins that induce cell death pathways (apoptosis, necrosis, or autophagy) are mutated or altered, they are unable to induce cell death | [52] |
| Tumor cell heterogeneity | Cancer cells multiply at an uncontrolled rate, accumulating genetic mutations and epigenetic changes, which lead to resistance and affect their sensitivity to cancer drugs. The generation of cell heterogeneity leads to the development of stem cell-like properties on the new growing cells. The stemness effect is common in cancer cells that are in circulation | [53] |
| Genetic factors | Include gene mutations, amplifications, and epigenetic alterations. Epigenetic events such as methylation and acetylation affect genetic expression leading to the silencing, overexpression, or amplification of oncogenes or tumor suppressor genes, resulting in the development of cancer drug resistance | [54] |
Researchers have suggested an alternative to reduce the possibility of developing acquired anticancer drug resistance. A patient’s biopsy sample of cancerous tissue can be screened to identify genetic anomalies that could lead to cancer treatment resistance[55]. This can contribute to determining the best suitable treatment, lowering the chances to acquire resistance after general chemotherapy sessions, and prevent the failure or risks of subsequent more toxic treatments. In addition to the standard pathological analysis, several clinicians have included these genetic screenings as part of the diagnostics to guide the selection of drug combinations on different types of cancers[55]. This approach could lead to a personalized therapeutic alternative based on the patient’s genetic pattern.
Investigations have been focusing on alternative drug delivery systems (DDS) designed to overcome cancer drug resistance. Efficiency in delivery and target specificity are the characteristics in consideration for drug delivery vehicle designs. DDS could increase bioavailability, diminish side effects, and improve therapeutic indexes when compared to current clinical drugs used for treatments[27,56,57]. Consequently, DDS could also help overcome acquired resistance induced by chemotherapy or radiotherapy[58].
In this review, we summarize the most significant genes that contribute to drug resistance till date, discuss anticancer drug inefficacy, and present DDS as an alternative to overcome this clinical challenge.
Cancer drug resistance related genes
Cancer cells can grow, develop, and survive in defiance of anticancer treatment due to intrinsic or acquired causes. Genes are key players to resistance to many common cytotoxic anticancer drugs. There is strong evidence pointing that most of these resistance-related genes are involved in DNA repair and apoptosis pathways[59]. In this regard, the most well-known and significant genes that contribute to anticancer drug resistance, based on our understanding, are outlined in this review. We present in the list of genes below their general information, the cancer types affected by drug resistance, how these genes are regulated in general, and recent research studies that incorporate drug delivery system techniques to combat cancer drug resistance.
B-cell lymphoma-2 family proteins
Evasion of apoptosis supports the development of cancer, and it is an important resistance mechanism for cancer cells against chemotherapy. Apoptosis is characterized by two established pathways: an extrinsic pathway mediated by death receptors at the cell membrane and an intrinsic pathway mediated by the mitochondria. Gene products that influence apoptosis include B-cell lymphoma-2 (Bcl-2) family proteins. This large multigene family encodes proteins that are capable of inhibiting apoptosis (BCL-2, BCL-XL, BCL-W, BFL-1, BRAG-1, MCL-1, and A1) or promoting it (BAX, BAD, BAK, BCL-XS, BID, BIK, BIM, and HRK). In mammalian cells, Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1, are the Bcl-2 proteins that block the apoptosis promoting proteins Bak and Bax, inhibiting their action by interacting with them[60-62]. The cellular outcome of undergoing intrinsic apoptosis or surviving depends on the balance and interaction between the pro- and anti-apoptotic proteins inside the cell. These Bcl-2 proteins show four homologous domains in their sequence (BH1, BH2, BH3, and BH4) and are called BCL-2 homology motifs[63,64], except the BH3-only proteins; Bim, Bid, and Bad. Genetic alterations associated with cancer and tumor growth often affect programmed cell death regulation in a way that favors cell proliferation[65]. These genetic changes are either inherited or acquired during the cell cycle. These include substitutions, insertions, or deletions of small or large fragments of DNA, genomic amplification, and rearrangements[66,67]. For example, chromosomal translocation t (14;18) activates the BCL-2 gene in most non-Hodgkin’s lymphomas[68,69]; nucleotide substitution and a frameshift mutation[70] inactivates the BAX gene in some colon, hematological, and stomach malignancies[71-73]; retrovirus gene insertion activates BCL-XL gene in murine leukemia[74]. These BCL-2 family gene alterations result in overexpression of either apoptosis-suppressing or apoptosis-inducing proteins of the Bcl-2 family. Similarly, the Bcl-2 protein is overexpressed in numerous breast and prostate cancers[75-77]. Other studies have shown that mutations found on the coding sequence of the BCL-2 gene in patients at the time of diagnosis were associated with a shortened time to its transformation into an aggressive lymphoma, and subsequently, earlier death due to the lymphoma[78].
Additionally, it was found that the Bcl-2 protein induces cell migration and invasion in a breast cancer cell line and also promotes metastasis to the lungs in a mouse model[79]. BCL-2, MCL-1, and BCL-XL are also overexpressed in several non-small cell lung carcinomas (NSCLC)[80-82]. Amplification of the apoptotic inhibitors Bcl-2-like genes, MCL-1 and BCL-XL, and deletion of apoptotic-promoter genes BOK and PUMA are presented in the somatic copy number variations in over 3000 cancer specimens across 26 human cancer types[83].
Besides malignancy, the imbalance ratio between apoptosis-suppressing and apoptosis-inducing proteins of the Bcl-2 family often makes cancer cells more resistant to a number of cell death inducers, including chemotherapeutic drugs, by impeding drug-induced damage from successfully translating into cell death[84]. Multidrug resistance (MDR) is reported to be associated with the overexpression of specific proteins such as P-glycoprotein and the anti-apoptotic genes of the BCL-2 family, where the former plays the role of expelling the drug out of the cells while the latter induces their proliferation[85]. Bcl-2 affects cancer drug resistance by inhibiting the apoptotic effect on cancer cells by dimerizing Bax and Bad, pro-apoptotic members of the Bcl-2 family. Also, overexpression of Bcl-2 can prevent chemotherapy treatment by blocking paclitaxel-induced apoptosis, and the translocation of nuclear factor of activated T lymphocytes. Additionally, BCL-2 antagonizes apoptosis induced by drugs through the inhibition of calcineurin protein activation, thereby preventing the activation of T cells from the immune system[86].
In breast cancer cells, overexpression of Bcl-2 has been correlated to the formation of polyploid cells, which confer MDR properties to cancer cells[51]. In colorectal cancer, it has been shown that the cytokine interleukin 17 (IL-17) plays an important role in promoting the development of resistance to cisplatin by inhibiting the expression of several pro-apoptotic proteins, including those from the Bcl-2 family such as Bax[87]. In breast cancer, IL-17 has also been shown to promote paclitaxel resistance through activation of the ERK1/2 pathway[88]. Defects in splicing events lead to resistance against selected therapy agents. Studies provide evidence that BIM alternative splicing products play a key role in drug resistance. In one study, inhibition of the three major protein products (BimEL, BimS, and BimL) resulted in different levels of resistance to glucocorticoid treatment in acute lymphoblastic leukemia cells[74]. Using paired-end DNA sequencing, Ng et al.[89] (2012) discovered an intronic deletion polymorphism in BIM that was sufficient to confer intrinsic resistance to the tyrosine kinase inhibitors, imatinib and gefitinib in chronic myeloid leukemia (CML) and epidermal growth factor receptor-mutated non-small-cell lung cancer (EGFR NSCLC). In summary, these studies point at targeting the anti-apoptotic members of the Bcl-2 family as a strategy to prevent MDR.
Chromodomain-helicase-DNA-binding protein 4
Chromodomain-helicase-DNA-binding protein 4 (CHD4) is the main component of the nucleosome-remodeling and histone-deacetylation (NuRD) complex. The NuRD complex’s primary function is to regulate gene expression and promote DNA repair. This complex is expressed throughout all tissues and is composed of multiple subunits, including ATP-dependent chromatin remodeling helicases CHD3/CHD4. The NuRD complex contributes to several cellular processes such as stem cell differentiation, cell cycle regulation, genome integrity maintenance, DNA damage repair, and development of the immune system[90]. NuRD subunits contribute to oncogenesis and cancer progression through DNA-damage repair, impacting the tumor’s microenvironment[91].
The CHD4 gene plays a critical role in epigenetic transcriptional repression[91]. This gene has been associated with oncogenic effects such as promoting cancer cell stemness, renewal and altering cell-cycle[92], and poor prognosis of advanced-stage cancer[91]. In collaboration with the histone deacetylases (HDACs), which allow the histones to wrap the DNA more tightly, and DNA methyltransferases, which mostly repress genes, CHD4 contributes to silencing as well as reducing and blocking the transcription of tumor suppressor genes. One of the main reasons for tumor recurrence is the resistance to DNA damage, and genes such as CHD4 enable this repair in cancer cells. CHD4 promotes DNA repair from insults such as oxidative damage in cancer cells[93,94].
Drug resistance is promoted in cancers associated with breast cancer genes (BRCA), which are sensitive to DNA-damaging agents, once the CHD4 expression decreases. Furthermore, CHD4 expression reduction affects cancer cell’s autophagy process as well as the ERBB2 gene, which is an epithermal growth factor member, resulting in a drug resistance effect[93]. Expression of CHD4 can increase stem-cell characteristics in cancer cells, stimulating anticancer drug resistance to DNA-damaging drugs[44]. CHD4 can regulate cancer cell behavior through post-transcriptional modifications. CHD4 is associated with transcriptional repression of genes involved in the repair of double-strand break DNA-damage. It has been considered a potential biomarker present in biopsies of patients (with significant upregulation) in cancers such as liver, renal, osteosarcoma, breast, and ovarian[93].
Wang et al.[93] (2019) showed that CHD4-increased expression was associated with advanced tumor invasion during metastasis and increased vascularity, promoting a more aggressive cancer phenotype. This group also reported that increased expression of the CHD4 gene was proportional to cancer treatment resistance by suppressing the expression of the cell cycle inhibitor and anti-proliferative effector, p21, which works together with the DNA-repair gene BRCA to cause an overall decrease in the sensitivity of cells to anticancer treatment. Furthermore, a decrease in the gene expression of CHD4 promotes radiotherapy sensitivity of head and neck cell carcinoma. CHD4 cooperates with DNA methyltransferases (DNMTs) in the silencing of many tumor suppressor genes; therefore, its decreased expression inhibits cell proliferation and sensitizes cells to radiotherapy[93,94]. In ERBB2+ breast cancer cells, which are resistant to Trastuzumab, a monoclonal antibody anticancer treatment, the depletion of CHD4 was shown to induce the cell’s sensitivity to this antibody by reducing ERBB2 signaling, affecting the autophagy process, and decreasing cell proliferation[93,95].
The CHD4 gene has a crucial role in colorectal cancer, and it is important to consider the activity of this gene to establish a treatment for colorectal cancer patients[93]. Overexpression of CHD4 led to pronounced radiotherapy-resistance by maintaining DNA hypermethylation transcription silencing on colorectal cancer patients[96]. In addition, CHD4 knockdown increased the chemosensitivity of breast cancer cells towards cisplatin[94] and increased the sensitivity of hepatocellular carcinoma cells towards epirubicin, an antitumor antibiotic[44].
The DNA-repair promoting gene, CHD4, is responsible for the transcriptional activity of the anti-proliferative gene, cyclin-dependent kinase inhibitor 1 (CDKN1A or p21); therefore, these genes have opposed functions regarding cell survival. CHD4 deficiency debilitates cell survival by not-suppressing and increasing p21 levels[94]. Inhibition of CHD4 results in the restoration of p21 expression and recovery of breast cancer cell sensitivity to cisplatin and poly ADP ribose polymerase (PARP) inhibitors[93].
Unfortunately, knockdown of CHD4 subunits can negatively affect the chromatin-remodeling ability of the NuRD complex, promoting cell proliferation, migration, and invasion, which represses apoptosis pathways and allows cancer cells to resist drugs that lead to DNA-damage[91]. Therefore, if CHD4 inhibitors are therapeutically tested, a targeted drug delivery system must be developed to direct this drug into the tumor to decrease the chances of affecting healthy cells or other unwanted secondary effects. Many efforts have been made in the development of therapeutic strategies against cancer that are likely to develop resistance. The combination of radiotherapy, together with an inhibitor of the NuRD complex subunit CHD4, should be a viable alternative to treat colorectal and liver cancer[44,91,93].
p53
TP53 was the first tumor suppressor gene identified in 1979. Since then, this gene has been extensively studied. p53 works mainly as a transcription factor, and its most important function is to induce or suppress the transcription of effector genes that will inhibit cancer cell proliferation, promote apoptosis, and impede tumor development[97]. DNA integrity is maintained by p53 through activation of the transcription of genes inducing cell cycle arrest as a DNA damage response[40]. Once DNA damage is detected in the cell, p53 favors the elimination of the affected cell, inducing activity of pro-apoptotic genes such as FAS (Fas Cell Surface Death Receptor) and BAX (from the BCL-2 family), and downregulating anti-apoptotic genes such as BCL-2[40]. The activation of p53 occurs in response to cellular stress and can induce cell cycle arrest to ensure genomic integrity[97-99]. Once p53 is activated, several effectors and p53-responsive genes such as CDKN1A, GADD45α, p21, MDM2, and RIT42, among others, work to inactivate cyclin-dependent kinases on the cell cycle[99]. Cancer drug resistance is influenced by loss-of-function p53 gene mutations, affecting mainly its transcriptional activity. The function of p53 is lost through the modifications that the gene undergoes (i.e., single point mutations and some hotspot mutations), leading to sensitivity loss to cytotoxic agents[100].
In contrast, we also found studies that show gain-of-function p53 mutations by inducing new interactions with other transcription factors that further promote chemoresistance[101]. Mutations of the p53 gene will obstruct cancer treatment. Hypoxia promotes upregulation of p53 in cancer cells blocking the cell cycle, and this event leads to the downstream activation of the p21 gene, decreasing the cytotoxic effect of anticancer drugs like cisplatin[102]. An in-vitro study reported that when p53 is mutated on cancer cells, anticancer drug 5-fluorouracil sensitivity is reduced[103]. The most common cancer types affected by the mutation of p53 are ovarian serous carcinoma, lung cancer, pancreatic cancer, head and neck squamous cell carcinoma, and breast carcinoma[104].
Studies have found small molecules that can restore the conformation and function of mutated p53 to prevent drug resistance. These include derivatives of the thiosemicarbazone family, PRIMA-1 and MIRA-1[100]. Also, treatment with blockers of p53-inhibitory proteins such as MDM2 could help restore p53’s function in the cases where there is an under-expression of p53 or an overexpression of MDM2[100]. To recover p53’s decreased function that leads to drug resistance, interventions with nanomedicine to deliver small molecules or MDM2 inhibitors, plus the specific treatment against the tumor, could help advance the battle against cancer drug resistance.
Cyclin-dependent kinase inhibitor 1
Cyclin-dependent kinase interacting protein 1, also known as p21, is encoded by the CDKN1A gene. p21 is capable of controlling cyclin complexes, including Cycling dependent kinase 2 (CDK2), a catalytic subunit that can restrict cell cycle and DNA replication. In healthy cells, p21 prevents proliferation, while in several cancer cells, this function is dysregulated. Among p21 functions, its role in maintaining genomic stability, DNA-damage repair, apoptosis, and tumor-suppressing functions is worth mentioning[105]. In cancer cells, p21 functions as a tumor suppressor and an anti-apoptotic protein, and its relationship with the tumor suppressor protein p53 have been under study due to its potential contribution to cancer therapy. Studies report various roles for p21, depending on its subcellular localization. P21 can be considered as an oncogenic protein inside the cytoplasm, while it can operate as a tumor suppressor inside the nucleus[106]. As part of an anti-apoptotic protein, p21 can promote cancer tumor evolution and growth by diminishing DNA damage accumulation[107]. A study incorporated human leukemia cells treated with SP600125, an anti-inflammatory and anticancer drug that inhibits c-Jun N-terminal kinase, to generate an increase in p21 expression as well as p21 phosphorylation, thereby preventing its binding with proliferating cell nuclear antigen, a DNA polymerase cofactor, while also inactivating caspase-3 and consequently apoptosis[95]. The anti-apoptotic role of p21 is inhibiting the ability of pro-apoptotic proteins to affect apoptosis. Differentially, p21 in the nucleus has a tumor suppressor role due to the regulations to the cell cycle on CDK/cyclin complexes suppression. In a study, p21 and p53 were introduced through a nanoparticle injection, and cells were introduced into a breast cancer mouse model resulting in a reduction in cell proliferation and tumor growth[108]. Once the DNA is damaged, an increase in p53 levels leads to the activation of p21 transcription. Subsequently, p21 can either inhibit cell cycle binding CDK/cyclin complexes or block DNA replication via its interaction with DNA polymerase cofactors[109][Figure 1].
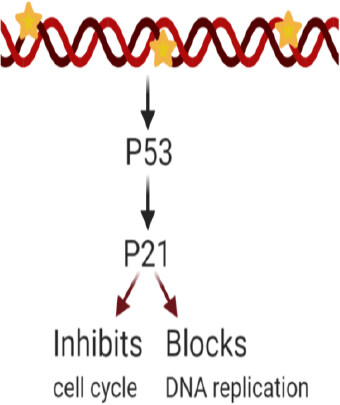
Figure 1. p21 overexpression effects after DNA damage. Excess p21 induces cell cycle inhibition or blockade of DNA replication
The p21 and p53 relationship has been under investigation to consider treatment for cancer cell drug resistance. p21 mediated p53-dependent apoptotic pathways, and p53-independent pathways[110,111] have been recently studied. These pathways lead to transcription induction of p21 and DNA-damage in cancer cells[112]. Reduced levels of p21 are associated with tumorigenesis on several cancers such as squamous cell carcinoma of the lung, colorectal, ovarian, cervical, and head and neck[113]. P21 is an important downstream target of p53 and is rarely mutated. This means that the resistance induced by p21 could be a consequence of different factors: deficiency of the p21 gene[114], or high expression of cytoplasmic p21; and as a result, the p21 binding to procaspase-3 blocking caspase cascade and apoptosis[115,116]. However, p21 overexpression is also correlated to the aggressiveness and invasiveness of different cancers [Figure 1][109]. Other studies have reported that p21 collaborates with anticancer, DNA-damaging agents to promote cell cytotoxicity. DNA-damaging agents can be combined with the anti-apoptotic p21 function as a possible target for anticancer treatment[105]. Considering the controversy around p21’s various responses, more research is needed to further understand its mechanism of action on specific cancer types. Research is being conducted to systemically study the regulation of p21’s expression upstream and downstream at different levels (transcriptionally, post-transcriptionally, and post-translationally) and contribute with therapeutic approaches against cancer and drug resistance treatments[117].
Multidrug resistance gene or P-glycoprotein-1
The multidrug resistance gene or P-glycoprotein-1 (MDR1) gene is responsible for the expression of P-glycoprotein (P-gp), a transmembrane glycoprotein that mediates ATP-dependent efflux with permeability properties to expulse cytotoxic drugs into the extracellular space[118]. Multidrug resistance protein 1 is a member of the ATP-Binding Cassette (ABC) transporter protein family. ABC transporters are transmembrane proteins that move compounds into or out of the cell. These transporter proteins are composed of a pair of transmembrane domains and two nucleotide-binding domains. They are essential in the elimination of toxins from the human body. MDR1 is normally expressed in healthy tissue (usually on the liver, kidney, colon, pancreas, uterus, placenta, testis, and brain), although its overexpression has been associated with cancerous cells[119,120].
Before treatment, it is important to evaluate the presence of MDR1 mutations in cancer patients to predict the tumor’s sensitivity to therapy. Patients with mutated MDR1 can be intrinsically resistant to drugs or could develop resistance over time[121]. It is worth mentioning that when P-gp is pharmacologically inhibited, thyroid hormones can promote its transcription and its function in the body[122]. MDR1/P-gp1 causes resistance to chemotherapeutic agents in different ways; for example, by direct interaction, in the case of paclitaxel and doxorubicin, and by indirect interaction, in the case of cisplatin, carboplatin, and oxaliplatin[123]. The overexpression of MDR1 contributes to drug resistance, particularly when genetic polymorphism variations are present. MDR1 G1199A variation exhibits a serine-to-asparagine transition in amino acid 400 in a Pgp cytoplasmic domain, producing an alteration on the efflux and transepithelial transport as well as drug sensitivity to chemotherapeutic agents[124].
Consequently, overexpression of P-gp decreases intracellular anticancer drug accumulation, which helps prevent the generation of MDR[121]. An increase in the expression of MDR1 by vitamin C has also been associated with the inhibition of the anti-tumor action of doxorubicin in ovarian and prostate cancer cells[17]. Recently, a study defined the resistance mechanism of paclitaxel and olaparib (inhibitor of PARP1) in resistant ovarian cancer cells that was reversible with the MDR1 inhibitors, verapamil and elacridar. They found that paclitaxel-resistant cells were cross-resistant to Olaparib, Rucaparib (PARP inhibitors), and doxorubicin, but not to the PARP inhibitors, Veliparib or AZD2461[123].
MDR1 gene expression can also be regulated through small interfering RNA (siRNA), which are lower in toxicity to healthy cells, and show higher specificity to the cells containing the mutated gene. This targeted siRNA therapy downregulates the MDR1 gene transcription, leading to a decreased amount of P-gp transporter proteins and a reduction of anticancer drug expelled from the cell[119]. Other strategies to decrease the efflux of anticancer drugs through P-gp consist of developing compounds that either compete with anticancer drugs for transport or act as direct inhibitors of P-gp. Up to date, no P-gp blockers are being used in the clinic, possibly due to the toxic effects of such inhibition. Several alternative approaches could include nanotechnology to specifically target the cancer cells and deliver P-gp inhibitors, molecules that reduce the expression of P-gp in cancer cells, or anticancer drugs.
Glioma pathogenesis-related protein 1
The glioma pathogenesis-related protein 1 (GLIPR1) is a member of the cysteine-rich secretory proteins (CRISPS), consisting of the following members: antigen 5 (Ag5), and pathogenesis-related 1 protein (Pr-1) CAP superfamily containing three core members, GLIPR1, GLIPR1-like 1 (GLIPR1L1), and GLIPR1-like 2 (GLIPR1L2)[125]. GLIPR1, a p53 target gene cluster found on human chromosome 12q21, is located in the endoplasmic reticulum (ER) membrane, and it is involved in the ER secretory pathway[126,127]. GLIPR1 is reported to contain an amino-terminal peptide sequence and a transmembrane domain that indicates its secretion or its location on the surface of the cell membrane[128].
Downregulation of GLIPR1 in prostate cancer and other malignant cell lines has been observed, largely in part to the methylation of the human GLIPR1 promoter[129]. Initially identified as a tumor-suppressor gene with apoptosis-inducing activities in prostate cancer, GLIPR1 pleiotropic effects have been reported to be highly expressed and upregulated, and it acts as an oncogene specifically in glioblastomas and gliomas, thus promoting cell proliferation[127,130,131]. The underlying mechanism of upregulated GLIPR1 cell growth stimulation has been studied in human lung adenocarcinoma A549 cells and correlates with inducing anti-apoptotic Bcl-2 protein expression[130]. In glioma cells, GLIPR1 overexpression reduced c-Jun N-terminal kinase (JNK) phosphorylation and induced Bcl-2 expression, thus increasing cell survival and glioma cells’ protective effect against apoptotic stimuli such as Fas ligation, chemotherapy, and radiation treatment[132]. Conversely, the mechanism contributing to GLIPR1-induced apoptosis is dependent on Bcl-2 downregulation and phosphorylation at Thr56 and Ser70, which support p53-induced apoptosis; and on the increase in ROS, signaling by apoptosis signal-regulated kinase 1 (ASK1), mitogen-activated protein-extracellular signal-regulated kinase kinase (MEK), and the consequent activation of JNK. Thereby, GLIPR1 acts through the ROS-ASK1-MEK4/7-JNK signaling pathway[133,134]. Moreover, GLIPR1-mediated apoptosis through the Bcl-2 family proteins and caspases may occur through caspase-dependent and caspase-independent pathways[134].
Originally identified as a tumor-suppressor gene with apoptosis-inducing activities in prostate cancer, GLIPR1 has been reported to be upregulated in glioblastomas, enhancing cell proliferation[127]. The mechanism contributing to GLIPR1-induced apoptosis is associated with an increase in ROS and consequent activation of the c-Jun N-terminal kinase (JNK) pathway[133].
Downregulation of c-Myc protein and CK1a-mediated targeted destruction of c-Myc and b-catenin in prostate cancer cell lines contributes to apoptosis induction by GLIPR1. Also, serine/threonine-protein kinase AURKA and Xenopus kinase-like TPX2 protein signaling pathway suppression by GLIPR1 interaction with heat shock cognate protein 70 (Hsc70) also contribute to apoptosis induction[135]. TPX2 has been associated with metastasis and the prognosis of bladder cancer. New findings have identified GLIPR1 as part of a regulatory circuit composed of TPX2 and p53, which modulates cell proliferation, migration, invasion, and tumorigenicity of bladder cancer cells.
The GLIPR1 gene has been identified in different forms of human cancers, including prostate, lung, ovarian, Wilms’ tumor, acute myeloid leukemia, and in the most aggressive types, brain cancer, glioblastoma multiforme/astrocytoma, and within glioma cell lines[133,136]. Overexpression of GLIPR1 induces apoptosis in prostate and lung cancer cells. In contrast, GLIPR1 overexpression in glioma and osteosarcoma cells leads to an increase in the proliferation, survival, invasion, migration, and anchorage-independent growth[130,131,137]. In a study by Dong et al.[137] (2015), overexpression of GLIPR1 induced the differentiation of osteosarcoma cancer-initiating cells and upregulated miR-16, thus blocking anti-apoptotic BCL-2 genes. GLIPR1 promotes an increase in Bcl-2 expression to subsequently decrease the apoptosis of A549/DDP lung cancer cells. The upregulation of GLIPR1 increases and affects drug resistance by promoting cell proliferation. Otherwise, if GLIPR1 is silenced in A549/DDP cells, caspase-3 dependent apoptosis is induced by mitochondrial signaling pathways through the decreased expression of the Bcl-2 protein[130].
Downregulation of GLIPR1 and gene knockdown experiments in various leukemia cell lines treated with the small drug SB225002 (N-(2-hydroxy-4-nitrophenyl)-N’-(2-bromophenyl)urea) resulted in elevated production of ROS, a decrease in cell proliferation linked to an increased level of apoptosis due to GLIPR1 silencing, and amplified drug resistance[138]. In another study, the siRNA-mediated knockdown of GLIPR1 expression induced a reduction in the number of melanomas, glioma cell invasion and proliferation[136]. In human lung adenocarcinoma A549 cells, upregulation of GLIPR1 stimulated cell proliferation by inducing the increased expression of Bcl-2, thus increasing resistance to the chemotherapeutic drug cisplatin[130]. To increase the apoptotic effects of docetaxel in prostate cancer cells and overcome resistance, synergistic treatment with recombinant GLIPR1 (GLIPR1-DTM) inhibited tumor growth, consequently enhancing the chemotherapy effect[139]. In summary, a decrease in GLIPR1 expression is another recommended strategy to diminish resistance to anticancer drugs such as cisplatin and docetaxel.
Human epidermal growth factor receptor-2
The ERBB family comprises four receptor tyrosine kinase members named EGFR, ERBB2 (HER2), ERBB3 (HER3), and ERBB4 (HER4), located on the cell surface. These four members share structure similarities, such as an extracellular binding domain, a transmembrane lipophilic segment, and an intracellular tyrosine kinase domain[140,141]. Of the ERBB family, HER2 is a proto-oncogene located on the long arm of chromosome 17, whose activation relies upon homodimerization when expressed at high levels and by hetero-dimerization with EGFR or kinase-inactive HER3][141-145]. After ligand binding, intracellular cell signaling pathways result in the inhibition of apoptosis, promoting proliferation, and tumorigenesis[146]. The molecular mechanisms of HER2-mediated tumorigenesis encompass various models, including the overexpression of HER2, which induces an increase in the HER2-containing dimers, maximizing, and sustaining signaling activity[147]. Among the dimer complexes formed, HER2/HER3 is the most critical activator of the PI3K/Akt signaling pathway (crucial for cell survival)[148]. Transcript variants of HER2 manifest higher dimerization, increased ligand-independent signaling activity, and a significant presence in HER2 amplified tumors[147,149]. Activation of src kinases, second messengers of HER2, exhibit increased Src protein levels and protein kinase activity in many human tumor tissues when combined with EGFR, to yield a synergistic tumorigenic effect[147]. HER2 involvement in G1/S cell cycle checkpoint control is regulated by cyclin D1 and its cyclin-dependent kinases (CDK), which play a critical proliferative role in cell cycle progression, and the CDK inhibitor p27 as a cell-cycle regulator through the induction of G1 arrest, halting cell growth[150,151]. HER2 tumorigenic signaling also appears to be potentiated by a stable interaction via one of two EGF-like domains with the transmembrane mucin glycoprotein Muc4, known to frequently display an altered expression in many cancer types, thus promoting tumor cell proliferation and metastasis[152,153]. Gene amplification of HER2 is known to occur in a variety of tumor types and in approximately 25% of human breast cancers, where it manifests as an early event[141,144]. HER2 gene amplification is the primary mechanism prior to protein overexpression of HER2, consequently activating the HER2 signaling network leading to uncontrolled cell proliferation and poor prognosis[154-156]. HER2 overexpression and activity drive a tumorigenic signaling cascade in breast cancer when homodimerization and HER2/HER3 heterodimerization events arise[157].
HER2 overexpression has been associated with resistance to chemotherapeutic agents[158]. This has been observed in malignancies other than breast cancer such as gastric, ovarian, colon, lung, cervical, pancreatic, and esophageal cancers; presenting, in general, a more aggressive disease, a lower survival rate, and a higher recurrence risk[141,159]. In the case of HER2 knockdown, reduced proliferation and apoptosis induction was observed in vitro on breast cancer tumors that overexpressed HER2; also, tumor regression after HER2 silencing with shRNA has been observed using in vivo a mouse xenograft model[147,160]. The potential of HER2 as a target for cancer therapeutic strategies mostly involves the use of various antibody-based agents and tyrosine kinase inhibitors (TKIs), either as single agents or in combination with other therapies.
The upregulation of HER2 in metastatic breast cancer to the uterus, in combination with tamoxifen therapy, stimulates aggressive growth and invasiveness of tumors, as HER2 overexpression is associated with relative resistance to tamoxifen, and increased sensitivity to anthracycline chemotherapy, usually 5-fluorouracil and doxorubicin[161-164]. In another study, treatment with gemcitabine (GEM) enhanced HER2 expression on low HER2 expression breast cancer cell lines, while paclitaxel treatment induced a low and moderate HER2 upregulation. Related studies in HER2-positive breast cancer cells demonstrated that overexpression of HER2 induced paclitaxel chemotherapy resistance[165]. The therapeutic outcome of the monoclonal anti-HER2 antibody-drug, Trastuzumab, is known to downregulate HER2 signaling PI3K/Akt and MAPK pathways and exhibit primary resistance in HER2-positive tumors as a monotherapy[166]. Some breast cancers even contain an abnormal form of HER2, lacking the extracellular domain needed for Trastuzumab binding, thereby causing resistance to the drug[141]. To overcome resistance, conjugation of Trastuzumab with the cytotoxic agent emtansine (T-DM1) requires elevated HER2 expression levels. Thus, pretreatment with GEM was used to increase HER2 upregulation, and T-DM1 binding to HER2 on breast cancer cell surface was used as a strategy to induce antiproliferative effects[167-170].
In malignant pleural mesothelioma (MPM) cancer cells, the TKIs, lapatinib and afatinib, prevented cell proliferation, upregulating and downregulating HER2 expression, respectively. Furthermore, lapatinib enhanced the monoclonal anti-EGFR antibody drug cetuximab and Trastuzumab binding with MPM cancer cells. As a result of heightened cetuximab- and Trastuzumab treatment, antibody cellular cytotoxicity (ADCC) in MPM cell lines was observed. Likewise, lapatinib enhances Trastuzumab-mediated ADCC in HER2-positive breast cancer and esophageal and gastric cancer cell lines[148,168]. Cisplatin is the standard treatment for gastric cancer; however, high expression of HER2 is associated with resistance to cisplatin-based chemotherapy[171]. An improvement to HER2 downregulation, as well as an increased tumor cell binding and blockade of ligand-dependent and independent- tumor growth, was accomplished with the use of the antibody ZW25[172]. In summary, several strategies can be used to target HER2’s cancer drug resistance effects, from antibodies such as ZW25 to disulfide bond disrupting agents such as RBF3 or a combination of drugs that allow HER2-overexpressing cells to regain their sensitivity to tamoxifen or cisplatin.
N-myc downstream-regulated gene
Cancer metastasis is the process in which cancer cells from an organ disseminate to another through circulation[173]. The N-myc downstream-regulated gene (NDRG) family has been identified as one of several metastasis suppressors involved in cancer cell invasion. The NDRG family of proteins contains four members: NDRG 1-4. The family functions are not well known, but they are associated with tumor suppression, cell proliferation, and stress response[174]. NDRG1 has shown to be an iron-regulated growth suppressor and metastasis inhibitor, exhibiting anti-oncogenic activity, decreased cell proliferation, migration, invasion, and angiogenesis[175]. NDRG1 is mainly located in the cytoplasm and translocates to the nucleus after DNA damage, hypoxia, and cell differentiation signals[176]. This protein is a downstream target of p53, and it is involved in cancer cell resistance to hypoxia and retinoic acid (anticancer activity and chemo-preventive properties)[177]. Nevertheless, it has not been established if NDRG1’s expression is inversely related to the survival of cancer cells[178]. In one study, NDRG1 demonstrated its capacity to suppress metastasis progress without altering tumor progression in an in vivo prostate cancer model[179]. NDRG1 has a pleiotropic behavior, considerable similar effects were observed on colon and pancreatic cancer[180,181].
NDRG1 can associate with other genes and proteins such as KAI1 and ATF3. NDRG1 expression is elevated in non-small cell lung carcinoma and contributes to cancer growth while having a variety of functions. NDRG1’s overexpression reduces anticancer drug-induced cytotoxicity in lung cancer by downregulating the stress-inducible gene ATF3. The ATF3 protein, located in the cytoplasm and nucleus, promotes apoptosis and inhibits cisplatin-induced cytotoxicity in lung cancer A549 cells[182]. Thus, by inhibiting ATF3’s cisplatin-induced cytotoxicity, NDRG1 can also regulate anticancer drug sensitivity to cisplatin. On the other hand, suppression of NDRG1-mediated metastasis occurs upon loss of KAI1 expression in vitro and in vivo, demonstrating that KAI1 is a functional downstream target of the NDRG1 pathway on prostate cancer[179]. These results suggest that inhibition or suppression of KAI1 could also be a target to decrease NDRG1’s mediated anticancer drug resistance.
Hypoxia-inducible factors
Hypoxia induces chemoresistance by two major factors: (1) low drug concentration in hypoxic cells and (2) impaired cell proliferation of hypoxic cells by starvation[183]. When hypoxia is induced through carcinogenic pathways, the cellular response is mediated by hypoxia-inducible factors (HIF-1α, -2α -3α, and -β). Hypoxia-inducible factors (HIFs) are transcription factors that form heterodimers. The α -subunit implies degradation and sensitivity to oxygen, the β-subunit means oxygen independence and the -3α serves as a suppressor or negative regulator for HIF-1α and HIF-2α (tumor promoters due to cellular response to low oxygen). HIFsare involved in different cancer stages - HIF-2α is responsible for chronic and prolonged phases of metastasis and anticancer drug resistance that occur in later stages of cancer; whereas HIF-1α is involved in the early stages of cancer that later can switch to HIF-2α through the upregulation of signaling proteins. HIF-1α and HIF-2α function can overlap during tumor development[184,185].
HIF-2α is a transcription factor localized in the cell nucleus, and it is expressed under hypoxic stimulation. HIF-2α activation controls the intracellular hypoxic response around the body due to its expression in endothelial, parenchyma, and interstitial cells in multiple organs. HIF-2α can modulate the expression of cytochrome c oxidase isoforms to enhance the electron transport chain. Because HIF-2α is expressed in multiple organs, it affects many different types of cancer. The cancer types affected as a consequence of low oxygen availability in cellular and organismal levels are breast, colon, ovarian, pancreatic, prostate, renal, and hepatocellular cancers. The solid tumor cancer types, where HIF-2α is frequently detected, include: head and neck, renal, bladder, glial, breast, ovarian, prostate, and the digestive system[186].
HIF-2α’s most important role is to control vascular morphogenesis, integrity, and assembly; and mediate p53’s suppression to maintain the human embryonic stem cells. HIF-2α downregulates P53 activity under hypoxic conditions and regulates cell proliferation, angiogenesis, metabolism, metastasis, and resistance to chemotherapy as a part of tumorigenesis[186]. Overexpression of HIF-2α enhances the expression of the endothelial kinase receptor, Tie2. Tie2 helps to develop the embryonic vasculature, which persists in adulthood, and it increases cytokine protein levels and mRNA in endothelial cells, promoting angiogenesis and tumor growth[187-189]. HIF-2α overexpression inhibits xenobiotic sensing nuclear receptors and their gene expression, affecting the expression of MDR1 and Cytochrome P450 3A4 (which oxidizes small foreign organic molecules expression). Furthermore, HIF-2α overexpression reduces the pharmacological effects of paclitaxel, mitomycin C, imatinib, and sorafenib on gastric cancer cells[186]. Currently, the evaluation of PT2385, a HIF-2α inhibitor, in combination with nivolumab targeted therapy to programmed death receptor-1 (PD-1) in patients with advanced clear cell renal cell carcinoma previously treated with one VEGFR targeted therapy is in clinical trial Phase I (NCT02293980). The combination of both drugs has demonstrated promising anti-tumor activity in ccRCC patients[190]. EZN-2208 is a transcriptional inhibitor of HIF-1α, which in combination with All-trans retinoic acid-arsenic trioxide (ATRA-ATO) was highly effective in treating patients with acute promyelocytic leukemia who develop resistance to ATO[138].
In general, the downregulation of HIFs in tumors overexpressing this protein can be another strategy to prevent tumor drug resistance. A decrease in HIF-2α activity can be acheived by drug delivery strategies that introduce small molecule inhibitors of HIF-2α, interference RNA, or by inhibiting its downstream effectors.
Breast cancer gene
There are two breast cancer genes, BRCA1 and BRCA2, and each one has different tumor suppressor characteristics. Their main function is to indirectly maintain the genomic integrity collaborating with recombination repair proteins[191]. Estrogen receptor signaling is the guardian of genome stability, together with the BRCA genes and proteins that control and repair DNA damage[192]. Both (BRCA1 and BRCA2) form complexes with Rad51, a recombination protein that controls the S/G2 phase in the cell cycle process. The BRCA proteins also form complexes with each other to collaborate in the tumor suppression process[191]. The BRCA1 performs several tasks, including DNA replication, cell cycle control, apoptosis, regulation of transcription, and chromatin unfolding[193]. Concurrently, BRCA2 activity is mainly focused on DNA repair by Rad51-mediated homologous recombination. When BRCA1 or BRCA2 genes are mutated, cancer cell lines diminish the DNA double-strand break repair ability through the process of homologous recombination (HR), promoting tumorigenesis due to genome instability[191,194]. When patients exhibit a BRCA mutation, they usually reveal p53 mutations as well. As previously discussed, p53 gene mutations prevent further p21 expression, favoring BRCA-mutated cells to avoid apoptosis, and perpetuate the development of cancer tumors[195]. Mutations in BRCA2 increase the risk of developing breast, prostate, pancreas, gall-bladder/bile duct, stomach and malignant melanoma[191]. Meanwhile, BRCA1 mutations increase the incidence of ovarian cancer and breast tumors[196].
BRCA1 gene upregulation is caused by estrogen-induced cell proliferation and differentiation, supporting the effect of DNA stabilization. Upregulation of estrogen receptor expression is inhibited when BRCA genes are mutated, repressing the estrogen receptor’s function. Simultaneously, BRCA gene mutations upregulate defective estrogen signaling that leads to tumorigenesis[192]. CHD4 acts as a tumor suppressor gene in female cancers (i.e., ovarian cancer), promoting DNA repair similar to the BRCA gene, reducing proliferation, and increasing sensitivity to DNA damaging agents. CHD4 modulates therapeutic responses to DNA-damaging agents in BRCA mutant cancer cells. A previous study from Guillemette et al.[197] (2015) revealed that mRNA expression levels from CHD4 contribute to the prediction of BRCA mutation cancers. When BRCA-associated cancer exhibited CHD4 depletion, a DNA-damaging agent (e.g., cisplatin) resistance was observed.
Meanwhile, the downregulation of p53 transcriptional activity is related to the overexpression of BRCA2[198]. In another study, BRCA2 inactivation decreased cell cycle progression and DNA replication and lowered cell proliferation compared to BRCA. BRCA2 knockdown is related to an innate immune response upregulation, promoting cell survival[199]. The anticancer drugs commonly used to treat breast cancer are taxanes and platinum agents. Taxane drugs include paclitaxel and docetaxel for BRCA1 gene mutations or hormone-negative cancers. The positive-hormone cancers are less sensitive to taxanes. Thus, platinum agent anticancer drugs such as cisplatin and doxorubicin are included as an alternative to triple-negative breast cancer (lack of estrogen receptors, progesterone receptors, and ERBB2 receptors)[195].
Occludin
Tight junctions (TJs) are structural proteins that control transportation across the cell membrane. These proteins regulate cellular permeability while maintaining cell polarity, restricting the diffusion of molecules through the membrane. Tight junctions also control cellular functions, including cellular responses to environmental stimuli, intracellular gene expression, cell differentiation, and proliferation. TJs are composed of membrane proteins that can interact with adjacent cells, functioning as a barrier[200,201]. An integral component of TJs that provides structure and function is the protein occludin, encoded by the occludin (OCLN) gene[202,203]. Occludin oxidizes NADH[204], which is essential for TJ morphology, stability, barrier function, and localization of the plasma membrane on endothelial cells[200]. Occludin contains a transmembrane domain with four membrane-spanning regions and other protein domains such as a C-terminus coiled-coil domain to interact with other proteins[200,203]. OCLN’s protein expression can influence the development of several cancer types, including ovarian cancer[200], lung adenocarcinoma[203,205], and breast cancer metastasis[206]. Zhang et al.[200] (2018) reported that OCLN overexpression increased transepithelial resistance, which indicates stronger TJs, while downregulation of OCLN resulted in a decreased cell to cell adhesion phenotype (a common characteristic of tumors). Another study reported that OCLN overexpression stimulates malignant growth of lung cancer cells, thereby promoting proliferation and blocking apoptosis[203]. On the other side, eliminating the OCLN gene has been shown to promote tumorigenic factors and reduce susceptibility to apoptosis in squamous cell carcinoma[201]. OCLN expression increases on A549 lung cancer cells promote their resistance to cisplatin, doxorubicin, and gemcitabine. As an anticancer drug resistance mechanism, there is an increased expression of OCLN in the TJs of lung cancer cells. The overexpression of OCLN induces drug resistance by inhibiting the flux of doxorubicin, thus lowering drug concentration within the cell. OCLN may not be related to cancer drug resistance acquisition directly, but it limits the chemosensitivity of anticancer drugs to lung cancer cells[205].
In the A549 lung cancer cell line, OCLN knockdown was not related directly to their resistance to anticancer drugs, yet it suppressed their chemosensitivity on a multicellular spheroid assay. OCLN overexpression on A549 cells decreased doxorubicin permeability due to their effect on signaling pathways, lowering the drug’s accumulation and cytotoxicity, leading to anticancer drug resistance. Interestingly, spheroid cancer cells with an increased OCLN expression developed cisplatin resistance, showing the importance of this gene in MDR[205].
Drug delivery systems using nanoparticles to improve the effectiveness of chemotherapeutic drugs in resistant tumors
Researchers have adopted several strategies to incorporate carriers to deliver a drug or a combination of drugs intracellularly. The development of nanoparticles has become an outstanding application of nanotechnology into medicine, where a nano-sized carrier efficiently delivers its payload of anticancer drug moieties. Using this type of therapy, researchers and clinicians take advantage of the irregular vasculature of the tumor to selectively deliver the drug and diminish the drug’s toxic side effects[207,208]. Another important advantage supporting the use of nanoparticles as a drug delivery system in cancer therapy is that it overcomes drug resistance by deactivating or avoiding various drug efflux pumps[209,210]. This could be accomplished by designing a selective (targeted) uptake of an endogenously endocytosed compound and promoting an intracellular accumulation of the drug, driven by the delivery system.
A remarkable characteristic of most of the nanoparticles for drug delivery systems includes a spherical shape and a large surface area-to-volume ratio. This property allows the nanocarriers to be absorbed through the cell’s membrane while carrying an anticancer agent. Also, most nanocarriers’ surface provide an alternative to add modifications, thus improving the nanoparticle’s targetability. Chemotherapeutic nanocarriers have two major categories for both active-targeted and passive-targeted delivery systems: 1) inorganic nanocarriers (metal core) and 2) organic nanocarriers (polymers, lipids, or liposomes)[57]. Currently, all the clinically approved nanocarriers are passive-targeted delivery systems[208]. However, the clinical approval of these DDS for cancer therapy was not based on their effect against anticancer drug resistance, but instead on their potential to specifically target tumors based on their irregular vasculature. Based on this, we focus our next sections on studies of organic and inorganic nanocarriers, which showed significant results against resistance.
Organic nanocarriers
Nanoparticles containing an organic core are biocompatible, solid, and often biodegradable. Organic nanocarriers are accessible for synthesis and viable for surface modifications. These characteristics increase the efficiency and biodistribution of the delivery system[211]. Based on our knowledge, currently, all the FDA-approved nanoparticle-based drugs are in the category of organic nanocarriers, i.e., protein-based polymers and liposomes; also, various nanoparticle-based drugs are in clinical trials[208]. The following section will discuss organic nanocarriers and their applications in cancer resistance, which is triggered by the genes discussed.
Polymers
In polymer nanoparticles, anticancer agents can be encapsulated through conjugation, or polymer attachments can be added to promote their release after a stimulus-response[57]. Risnayanti and his collaborators incorporated polylactic-co-glycolic acid (PLGA) and carboxylic acid-based particles to encapsulate both MDR1 and BCL2 siRNA[79]. Their design tackled drug efflux and cell death defense pathways. This dual MDR1 and BCL2 siRNA-loaded PLGA nanoparticle system was a viable strategy to overcome the chemoresistance on ovarian cancer cells (paclitaxel-resistant cell line SKOV3-TR and cisplatin-resistant cell line A2780-CP20) by enhancing cellular drug sensitivity[85]. Wang Z et al.[212] (2017) developed PLGA nanoparticles to encapsulate the anticancer drug Disulfiram to protect it from degradation due to its short half-life in the bloodstream. The nanoparticles were combined with copper, to inhibit liver cancer stem cells. In addition, the nanoparticles were combined with 5-fluorouracil, thus resulting in a synergistic cytotoxicity and anti-metastasis effect on a mouse model of liver cancer. Another research group’s delivery system used PLGA as a water-soluble carrier. Chang et al.[213] (2013) designed nanoparticles to encapsulate curcumin, a low water-soluble compound with anti-tumor, anti-metastasis, and anti-angiogenesis properties. The curcumin nanoparticles were used to treat cisplatin-resistant human oral cancer cells. As a result, curcumin nanoparticles induced apoptosis of the resistant cancer cells and showed low cytotoxicity to normal human oral epidermal cells. Moreover, these curcumin nanoparticles caused DNA fragmentation, upregulation of caspase-3/9, cytochrome c, and Apaf-1, while increasing ROS levels, which are known to induce apoptosis. In addition, Bcl-2 was downregulated, and the protein and mRNA expression levels of MDR1 were suppressed.
In other studies, Xiao et al.[214] (2015) designed a double functionalized PLGA nanoparticle delivery system that included chitosan (to enhance endocytic uptake) and two drugs - Pluronic (an MDR1 inhibitor) and Camptothecin (a topoisomerase 1 inhibitor) - that were encapsulated into the nanoparticle to treat colon cancer, both in vitro and in vivo. The outcomes for this nanocarrier design were advantageous due to the downregulation of MDR1 expression and the inhibition of P-gp, which induces apoptosis and reduces systemic toxicity. Another study registered hyaluronic acid-modified Paclitaxel nanoparticles to encapsulate and deliver MDR1 siRNA inside ovarian cancer cells. This formulation inhibited tumor growth and induced apoptosis by decreasing P-gp and MDR1 expression. These particular nanoparticles take advantage of the cluster of differentiation 44 (CD44), targeting the hyaluronic acid receptor[121]. This active targeting strategy is an additional feature to increase drug accumulation into cells, promoting nanoparticle endocytosis instead of drug internalization by influx pumps. Finally, synthetic and natural polymers can be used to encapsulate and deliver drugs to overcome MDR.
Liposomes
Liposomes are drug delivery nanocarriers that exhibit biodegradable and biocompatible properties, with the ability to encapsulate water-soluble agents, e.g., DNA and RNA, in their aqueous inner core and insoluble agents into their bilayer membrane[57]. Those specific characteristics make the liposome a versatile therapeutic nanocarrier. In this way, a research group worked on a self-assembling nanocomplex to carry the p53 gene and targeted glioblastoma multiforme. They reported a cationic liposome composed of 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) and di-oleoyl phosphatidylethanolamine (DOPE) as the carrier encapsulating an oligonucleotide. This nano-delivery platform successfully crosses the blood-brain barrier to target glioblastoma multiforme (GBM) and cancer stem cell lines: U87, T98G, and LN-18. The nanoparticles carried p53 and Temozolomide. This treatment revealed an increase in anti-tumor efficiency, increased sensitivity of cancer stem cells and tumor cells to the drug, activation of apoptosis, and decrease in cancer drug resistance in human cancer[215]. In another recent study, researchers developed curcumin loaded into a cationic liposome-polyethylene-glycol-polyethyleneimine complex (LPPC), together with the drug Herceptin (Trastuzumab) non-covalently intercalated on the surface of the carrier. Curcumin-LPPC-Herceptin and doxorubicin-LPPC-Herceptin complexes dramatically enhanced the cytotoxic effects of LPPC-encapsulated curcumin on HER2-positive cells, with a potent therapeutic effect on SKBR3 (HER2 positive) as compared to Hs578T (HER2 negative) breast cancer cells[216].
Micelles
Micelles as spherical drug nanocarriers are self-assembly systems of water-soluble components in an aqueous solution that results in a hydrophobic core and a hydrophilic shell. The hydrophilic shell can stabilize the hydrophobic core while keeping a non-water-soluble drug inside. The resulting nanoparticle is an excellent candidate to carry non-water-soluble drugs that can be incorporated in the polymeric micelle through physical, chemical, or electrostatic interactions[211]. In a study, researchers prepared micelles of mPEG modified with transferrin and containing R547 as a drug delivery system. R547 is an ATP-competitive CDK inhibitor that specifically induces cell-cycle arrest and apoptosis. However, R547 is poorly soluble in an aqueous solution at physiological pH conditions, making it a candidate molecule to be integrated into a drug delivery system. These transferrin-modified micelles showed cytotoxicity against ovarian carcinoma cells, A2780, and inhibited tumor growth on A2780 tumor-bearing mice compared to non-drug and non-modified micelles[217]. Another research group showed the synergistic effect of using the combination of verapamil (P-gp inhibitor) and paclitaxel targeted-delivery into breast carcinoma cell lines, using a folate-conjugated deoxycholic acid micelle to overcome MDR. The cells used were MCF-7 and MCF-7/ADR (multi-drug-resistant variants). Synergistic effects of the folate receptor, whichmediates internalization, and the drugs diminished MDR. Side effects and toxicities to healthy tissues or organs were reduced[50]. In another study, a preparation of Herceptin conjugated to micellar nanoparticles consisting of d-a-tocopherol polyethylene glycol succinate (TPGS) was evaluated for the concomitant targeted delivery of docetaxel drug and the Polo-like kinase 1 siRNA to MCF7 and SKBR3 breast cancer cells. The synergistic effects of the co-delivery of drugs into the cells with different HER2 expression levels resulted in a sustained and controlled delivery of docetaxel, thereby increasing its therapeutic effect[218]. Finally, theranostic iron oxide-coated nanoparticles combined with cisplatin and with a tumor imaging infrared-dye- labeled HER2 antibody were presented in an interesting study. The study used HER2-positive chemo-resistant ovarian cancer cells (SKOV3) in female athymic nude mice. The results of this in vivo study showed inhibition of primary tumor growth and metastasis, and the downregulation of HER2 in an ovarian cancer xenograft model[219].
Solid lipid nanoparticles
Solid lipid nanoparticles (SLN) are nanocarriers parallel to liposomes and lipid emulsions. SLN can incorporate drugs and perform targeted and controlled drug delivery[220]. Eskiler et al.[221] (2018) developed solid lipid nanoparticles to treat resistant triple-negative breast cancer that is due to BRCA1 mutation. These nanoparticles are composed of Poly (ADP-ribose) polymerase (PARP) inhibitors to induce DNA damage and overcome HR-mediated resistance in HCC1937 and HCC1937R cell lines while delivering the anticancer drug Talazoparib (BMN673). PARP is a family of proteins associated with the regulation of many cellular processes such as genomic stability, DNA repair, and apoptosis[222]. The results from this study indicated DNA double-stranded breakage, G2/M cell cycle arrest, and PARP cleavage[221]. Differently, Choi et al.[223] (2008) reported a cationic SLN design to deliver a non-viral vector-mediated p53 gene into H1299 lung cancer. SLN enters the cell to deliver the p53 gene through cell membrane permeabilization. After treatment, the in vitro and in vivo results showed an increase and restoration of p53 function and apoptosis, and decreased cancer cell growth. These nanoparticles reported higher efficiency of p53 gene delivery than wild type p53 mRNA and protein expression levels in lung cancer cells. The nanoparticles also promoted lung cancer cell restoration of apoptotic pathways and reversed deficiencies in both in vitro and in vivo tumor models[223].
Inorganic nanocarriers
Inorganic nanocarriers have shown various advantages over organic nanocarriers, including, for example, high stability on most organic solvents, large surface area, superior drug loading capacity, enhanced bioavailability, low toxicity, and controlled drug release[57]. Based on our knowledge, to date, there are several inorganic DDS under clinical trials for cancer therapy, i.e., gold nanoparticles, but none of them have been approved yet[208].
A study incorporated an inorganic material carrier (i.e., nanodiamond) to effectively deliver the anticancer drug Epirubicin to the hepatic cancer stem cell line LT2-MYC (murine hepatoblastoma)[224]. These nanodiamonds reduced toxicity primarily through passive targeting so as to increase tumor-specific drug accumulation. This nanodiamond-Epirubicin complex exhibited high stability and adsorption, and promoted a significant uptake and retention of the drug in tumor cells. Also, these nanodiamonds prevented the efflux of Epirubicin by ABC transporters, enhancing drug retention that led to overcoming resistance triggered by the CHD4 gene[224]. In contrast, Zhang et al.[225] (2020) developed PEGylated tetrasulfide organosilica shell nanoparticles, exploring the co-delivery of cisplatin and Acriflavine drugs to suppress HIF functions and inhibit metastasis. This delivery system was able to synergistically co-deliver the drugs into A549 adenocarcinoma lung cancer cells in vitro and in vivo. The results revealed the versatility of this system to combat anticancer drug resistance[225,226]. These nanocarrier designs demonstrate the effective incorporation of inorganic materials as a viable method to overcome anticancer drug resistance.
Transitional metals
Another type of inorganic nanocarriers include transitional metals. Most nanomaterials are metallic compounds; because of their inherent properties as nano-sized particles, they facilitate transportation through biologicals barriers. In this regard, nickel (Ni) is considered as a highly abundant metallic material candidate to be developed for health-based applications. Ingestion, inhalation, and skin absorption are the best routes for nickel to enter the human body, making the lung and kidney its primary targets[227]. Researchers have studied Ni-containing nanoparticles. Bioavailability and toxicological properties of metallic Ni nanoparticles and NiO nanoparticles have been examined on lung cancer cells. NiO nanoparticles promoted nuclear translocation of HIF-1α, consequently leading to the upregulation of NDRG1 in H460 human lung epithelial cells. However, these metallic nanoparticles showed moderate toxicity. Both Ni nanoparticles generated the activation of apoptosis through caspase and poly (ADP-ribose) polymerase[228]. On the other hand, a study using zinc oxide nanoparticles as treatment reported the upregulation of NDRG1 expression, and other cell growth and differentiation proteins essential in specific pathways to the ovarian granulosa cells of hens[229].
Some studies have been incorporating metallic-based nanomedicine to treat cancer resistance caused as a response to hypoxia. Silver nanoparticles were incubated with the MCF7 (breast cancer cells) and HeLa (ovarian cancer cells) in hypoxic conditions. These silver nanoparticles promoted the inhibition of HIF-1 reporter and vascular endothelial growth factor (VEGF) to disrupt angiogenesis. In addition, silver nanoparticles disrupted the cellular function of HIF signaling pathway[230]. Satapathy et al.[231] (2013) reported the implementation of silver-based nanoparticles as an alternative to treat HCT116 human colon cancer cells. These nanoparticles contributed to growth inhibition and increased cytotoxicity. This research group reported an increase in BAX/BCL-XL ratio, and p53, p21, caspase 3, 8, and 9 activations, leading to apoptosis; as well as a decrease in AKT and NF-κB levels[231]. The NF-κB and AKT levels were determined due to their importance in cell proliferation, promoting survival in resistant cancer. Silver-based nanoparticles can also be considered as an anticancer strategy to treat p53-dependent cancer cell development.
In addition, the study of titanium dioxide nanoparticles to determine their effects on the blood-brain barrier has increased. Disdier et al.[232] (2015) study revealed that titanium (Ti) is internalized in the liver, lungs, and spleen, persisting for up to a year. However, in brain epithelial cells, Ti circulated for a short period and had no effect on blood-brain barrier integrity, although brain inflammation was reported. Interestingly, the presence of Ti in the liver increased TJ protein concentration, including OCLN, and promoted the modulation of P-gp mRNA expression[232]. Although the principal objective of this study was to analyze the effect of nanoparticles on the blood-brain barrier, their results showed the influence of Ti on the regulation of OCLN. The incorporation of Ti nanoparticles to influence cancer resistance by the modulation of OCLN protein has excellent potential for future studies.
In parallel to all the results presented in this section, the incorporation of nanomedicine to increase intracellular drug concentration, changing the cell uptake route of the drug (e.g., endocytosis instead of passive diffusion), and targeting cancer resistance-related genes show great potential as the next steps to improve cancer therapy. Due to this, there are many nano-sized DDS under different stages of translational research for cancer therapy.
Tumors are very heterogeneous in their cell population. In this way, the physiological barriers, e.g., microenvironment, that must overcome these DDS are more complex than challenges seen in the use of in-vitro models. In addition, it is still unknown whether the inherent physicochemical impact of the DDS nanoparticles inside the human body will completely change the fate and therapeutic effect of the drug. Thus, the uptake of drugs delivered by nanocarriers is not significantly higher than that of the free chemotherapeutic drug in patients. Due to this, researchers are still working on the development of more robust DDS to overcome these limitations.
Table 4 summarizes the different nanocarriers that have been studied to treat cancer resistance triggered by genes.
Drug delivery system nanoparticles# and their effect on cancer resistance
| DDS carrier | Nanoparticles modification | Encapsulated drug or toxic agent | Cells or tumor treated | Genes affected | Effect over resistance | Ref. |
|---|---|---|---|---|---|---|
| PLGA* | Dual RNAi delivery system (MDR1 and BCL2 siRNA) | Paclitaxel* and cisplatin* | ovarian cancer cells: SKOV3-TR and A2780-CP20 | MDR1 and BCL2 | Stimuli inhibition of drug efflux and cell defense pathways (enhanced drug sensitivity) | [85] |
| PLGA* | PLGA-encapsulated Disulfiram | Disulfiram* | Hepatocellular carcinoma (Huh7, PLC/PRF/5) | CHD4 | Extended the half-life of Disulfiram | [212] |
| PLGA* | Pluronic and chitosan surface- functionalized PLGA nanoparticles | Camptothecin* | Colon-26 cells (Colon cancer cells) | MDR1 | Downregulate the expression of MDR1 expression and enhanced tumor uptake. Induced tumor cell apoptosis, reduced systemic toxicity, and inhibited P-gp. | [214] |
| PLGA* | PLGA-curcumin nanoparticles | Curcumin* | CAL27-cisplatin-resistant human oral cancer cells | MDR1
Bcl-2 | Suppress the protein and mRNA expression levels of MDR1. Downregulate the protein levels of Bcl-2. Intrinsic apoptotic pathway through regulating the function of MDR1 and the production of ROS | [213] |
| PEG* and PEI | hyaluronic acid (HA) based nanoparticle | MDR1 siRNA with paclitaxel* | SKOV-3TR and OVCAR8TR
Ovarian cancer cells | MDR1 | Down-regulation of MDR1 and P-gp expression. Inhibitory effect on the tumor growth. Decreased P-gp expression and increased apoptosis in MDR ovarian cancer mice model | [121] |
| ModifiePEG-PE micelles | Tf-conjugated polymeric micelles | R547 (a potent and selective ATP-competitive CDK inhibitor) | A2780 ovarian carcinoma cells | P21 | In vitro and in vivo studies in ovarian cancer confirmed cytotoxicity and tumor growth inhibition. | [217] |
| Deoxycholic acid micelles | Folate-conjugated | Verapamil*, a P-gp inhibitor, and Paclitaxel* | MCF-7 and MCF-7/ADR (multi-drug-resistant variant), human breast carcinoma cell lines | MDR and P-gp | Verapamil-mediated overcome MDR solid tumors by targeting the delivery of micellar Paclitaxel into tumor cells. | [50] |
| Cationic liposome DOTA/DOPE* | systematic nanodelivery platform encapsulating human p53 or oligonucleotide | Temozolomide* and p53 therapy | Human GBM cell lines U87, T98G, and LN-18 | p53 | DDS crosses the blood-brain barrier and efficiently targets cancer stem cells and tumor cells, activating apoptosis. | [215] |
| Cationic liposome-PEG-PEI complex | Herceptin was non-covalently associated onto the surface of the nanocarrier | Curcumin* and doxorubicin* | SKBR3 (HER2-positive) and Hs578T (HER2-negative) breast cancer cells | HER2 | Cytotoxicity improved. Anti-proliferative effect increased. | [216] |
| Micells TPGS* and siRNA | Herceptin-conjugated micelles | Docetaxel* and polo-like kinase 1 siRNA | MCF7 and SK-BR-3 cell lines Breast cancer cell | HER2 | Co-delivery of drugs was sustained and controlled | [218] |
| amphiphilic polymer nanoparticle | coated magnetic iron oxide | Cisplatin* and near-infrared dye labeled HER2 antibody | SKOV3 ovarian cancer cell line. In vivo models female athymic nude mice | HER2 | Inhibited the growth of the primary tumor, peritoneal, and lung metastasis in ovarian cancer. Shrinkage of tumor and primary tumors that had low levels of HER2. | [219] |
| Nanodiamond | Epirubicin* nanodiamond complex | Epirubicin* | LT2-MYC cell line from murine hepatoblastoma tumor model | CHD4 | Nanodiamond-drug complex with epirubicin exhibited high stability and adsorption, promoting uptake and retention on tumor cells | [224] |
| Nickel oxide | Nickel-containing nanoparticles | H460 human large cell lung cancer | NDRG1
and HIF-1a | Activate a toxicity pathway characteristic of carcinogenic Ni compounds | [228] | |
| Zinc oxide | ZnO nanoparticles | Jinghong-1 laying hen’s ovarian granulosa cells | NDRG1 | Upregulated the expression of NDRG1
and regulate proteins | [229] | |
| Silver nanoparticles | Ag nanoparticles | MCF7 (breast cancer) and HeLa (cervical cancer) cells | HIF-1 | HIF-1a signaling pathway disrupted and vascular endothelial growth factor to inhibit angiogenesis. | [230] | |
| Silica matrix | microporous organosilica shell-coated cisplatin nanoparticle* | Cisplatin* and acriflavine | A549 lung cancer cells | HIF-1 | Synergistic co-delivery of drugs. Inhibit metastasis and enhancing cisplatin efficiency | [226] |
| Solid lipid nanoparticles | PARP inhibitor to induce toxicity | Talazoparib* (BMN 673) | HCC1937 and HCC 1937R Triple-negative breast cancer | BRCA1 | DNA double-stranded breakage, G2/M cell cycle arrest and PARP (protein regulator of genomic stability) cleavage | [222] |
| Titanium dioxide
nanoparticles | TiO2* nanoparticles | Titanium | Brain epithelial cells (brain microvasculature endothelial cells) and Male Fisher F344 rats | OCLN | Occludin protein is regulated while crossing blood-brain barrier with not affected integrity. Upregulation of tight junction proteins, modulation of P-gp mRNA expression | [232] |
Conclusion
Resistance to therapy continues to be the most significant medical challenge in cancer today. The current multimodal approach of cancer treatment is not enough to cure many tumor types and to decrease relapse. Since there are many underlying mechanisms of resistance, it is vital to understand the biological determinants. Identifying the biological drivers of drug resistance will result in new therapeutic strategies, which can focus on targeting the internal tumor characteristics that develop malignancies. The introduction of targeted and specific drug delivery systems such as nanocarriers is a big step towards the correct path in drug design. Non-toxic and targeted new cancer treatment alternatives will help overcome anticancer drug therapy resistance, thus providing hope to patients who are victims of this devastating disease.
Declarations
AcknowledgmentsThis publication was made possible by the support of the University of Puerto Rico Rio Piedras Campus, Universidad Central del Caribe, and San Juan Bautista School of Medicine (SJBSM). The authors thank Dr. Estela Estapé (Director of the SJBSM Research Center) for her outstanding dedication and support in the writing process of this review.
Authors’ contributionsdesigned the study, selected the topics, and revised the article: Torres-Martinez Z, Ferrer-Acosta Y, Suarez-Arroyo IJ, Delgado Y
contributed to drafting, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work: Torres-Martinez Z, Delgado Y, Ferrer-Acosta Y, Suarez-Arroyo IJ, Joaquín-Ovalle FM, Delinois LJ, Griebenow K
coordinated and conceived the review article: Delgado Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was financially supported by the National Institutes of Health, RISE program, grant (no 5R25GM061151-18) to Torres-Martinez Z. Institutional Development Award (IDeA) from the NIGMS/NIH under grant (number P20 GM103475-17) to Suarez-Arroyo IJ. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019;2:141-60.
2. Liu FS. Mechanisms of chemotherapeutic drug resistance in cancer therapy--a quick review. Taiwan J Obstet Gynecol 2009;48:239-44.
3. Haider T, Tiwari R, Vyas SP, Soni V. Molecular determinants as therapeutic targets in cancer chemotherapy: An update. Pharmacol Ther 2019;200:85-109.
4. Mills CC, Kolb EA, Sampson VB. Development of Chemotherapy with Cell-Cycle Inhibitors for Adult and Pediatric Cancer Therapy. Cancer Res 2018;78:320-5.
5. Gelbert LM, Cai S, Lin X, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014;32:825-37.
6. Bukowski K, Kciuk M, Kontek R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int J Mol Sci 2020;21:3233.
7. Chorawala M, Oza P, Shah G. Mechanisms of anticancer drugs resistance: an overview. Int J Pharm Sci Drug Res 2012;4:1-9.
8. Zhou J, Kang Y, Chen L, et al. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front Pharmacol 2020;11:343.
9. Singh RK, Kumar S, Prasad DN, Bhardwaj TR. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur J Med Chem 2018;151:401-33.
10. Myers AL, Kawedia JD, Champlin RE, et al. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: a comprehensive review. Expert Opin Drug Metab Toxicol 2017;13:901-23.
12. Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front Pharmacol 2018;9:1697.
13. Madaan K, Kaushik D, Verma T. Hydroxyurea: a key player in cancer chemotherapy. Expert Rev Anticancer Ther 2012;12:19-29.
14. Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: a review. Talanta 2011;85:2265-89.
15. Henriques AC, Ribeiro D, Pedrosa J, Sarmento B, Silva PMA, Bousbaa H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett 2019;440-441:64-81.
16. Sun Y, Saha S, Wang W, Saha LK, Huang SN, Pommier Y. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair (Amst) 2020;89:102837.
17. Varela-Lopez A, Battino M, Navarro-Hortal MD, et al. An update on the mechanisms related to cell death and toxicity of doxorubicin and the protective role of nutrients. Food Chem Toxicol 2019;134:110834.
18. Saleem T, Kasi A. Daunorubicin. StatPearls [Internet]: StatPearls Publishing; 2020.
19. Wahdan-Alaswad R, Liu B, Thor AD. Targeted lapatinib anti-HER2/ErbB2 therapy resistance in breast cancer: Opportunities to overcome a difficult problem. Cancer Drug Resist 2020;3:1-20.
20. Rodon Ahnert J, Gray N, Mok T, Gainor J. What it takes to improve a first-generation inhibitor to a second-or third-generation small molecule. American Society of Clinical Oncology Educational Book 2019;39:196-205.
21. Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer 2018;17:36.
22. Martin-Broto J, Moura DS. New drugs in gastrointestinal stromal tumors. Curr Opin Oncol 2020;32:314-20.
23. Han W, Li L, Qiu S, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 2007;6:1641-9.
24. Lau MT, Ghazanfar S, Parkin A, et al. Systematic functional identification of cancer multi-drug resistance genes. Genome Biol 2020;21:27.
25. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 2017;7:339-48.
26. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6:1769-92.
27. Tang Y, Soroush F, Tong Z, Kiani MF, Wang B. Targeted multidrug delivery system to overcome chemoresistance in breast cancer. Int J Nanomedicine 2017;12:671-81.
28. Min Y, Mao CQ, Chen S, Ma G, Wang J, Liu Y. Combating the drug resistance of cisplatin using a platinum prodrug based delivery system. Angew Chem Int Ed Engl 2012;51:6742-7.
30. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 2014;740:364-78.
31. Chen SH, Chang JY. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int J Mol Sci 2019;20:4136.
32. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012;12:587-98.
34. Smith L, Watson MB, O’Kane SL, Drew PJ, Lind MJ, Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol Cancer Ther 2006;5:2115-20.
35. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 2013;65:157-70.
36. Prados J, Melguizo C, Ortiz R, et al. Doxorubicin-loaded nanoparticles: new advances in breast cancer therapy. Anticancer Agents Med Chem 2012;12:1058-70.
37. Taymaz-Nikerel H, Karabekmez ME, Eraslan S, Kirdar B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci Rep 2018;8:13672.
38. Huizing MT, Misser VH, Pieters RC, ten Bokkel Huinink WW, Veenhof CH, Vermorken JB, et al. Taxanes: a new class of antitumor agents. Cancer Invest 1995;13:381-404.
39. Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci U S A 1997;94:9679-83.
40. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003;3:330-8.
41. Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008;13:1551-69.
42. Wang W, Cassidy J, O’Brien V, Ryan KM, Collie-Duguid E. Mechanistic and predictive profiling of 5-Fluorouracil resistance in human cancer cells. Cancer Res 2004;64:8167-76.
43. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci 2018;25:20.
44. Nio K, Yamashita T, Okada H, Kondo M, Hayashi T, Hara Y, et al. Defeating EpCAM(+) liver cancer stem cells by targeting chromatin remodeling enzyme CHD4 in human hepatocellular carcinoma. J Hepatol 2015;63:1164-72.
45. Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat 2012;15:39-49.
46. Jo Y, Choi N, Kim K, Koo HJ, Choi J, Kim HN. Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-Cancer Drug Development. Theranostics 2018;8:5259-75.
47. Lippert TH, Ruoff HJ, Volm M. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittelforschung 2008;58:261-4.
48. Ye Q, Liu K, Shen Q, Li Q, Hao J, Han F, et al. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front Oncol 2019;9:487.
49. Wang J, Seebacher N, Shi H, Kan Q, Duan Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017;8:84559-71.
50. Wang F, Zhang D, Zhang Q, et al. Synergistic effect of folate-mediated targeting and verapamil-mediated P-gp inhibition with paclitaxel -polymer micelles to overcome multi-drug resistance. Biomaterials 2011;32:9444-56.
51. Yuan B, Hao J, Zhang Q, Wang Y, Zhu Y. Role of Bcl-2 on drug resistance in breast cancer polyploidy-induced spindle poisons. Oncol Lett 2020;19:1701-10.
52. Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol 2015;35 Suppl:S78-S103.
53. Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol 2019;12:134.
54. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer 2014;14:747-53.
55. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346:1480-6.
56. Kalaydina RV, Bajwa K, Qorri B, Decarlo A, Szewczuk MR. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int J Nanomedicine 2018;13:4727-45.
57. Senapati S, Mahanta AK, Kumar S, Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Target Ther 2018;3:7.
58. Li C, Wang J, Wang Y, Gao H, Wei G, Huang Y, et al. Recent progress in drug delivery. Acta Pharm Sin B 2019;9:1145-62.
59. Lonning PE, Knappskog S. Mapping genetic alterations causing chemoresistance in cancer: identifying the roads by tracking the drivers. Oncogene 2013;32:5315-30.
60. Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008;135:1074-84.
61. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006;9:351-65.
62. Moore VDG, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. The Journal of clinical investigation 2007;117:112-21.
63. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008;9:47-59.
64. Delgado Y, Torres A, Milián M. Apoptosis’ activation associated to BH3 only domain and BCL-2 homology domain proteins: new way to design anti-cancer drugs. J Cancer Prev Curr Res 2019;10:54-9.
65. Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Rev 1995;14:3-15.
66. Chakravarthi BV, Nepal S, Varambally S. Genomic and Epigenomic Alterations in Cancer. Am J Pathol 2016;186:1724-35.
68. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985;228:1440-3.
69. Duan H, Heckman CA, Boxer LM. The immunoglobulin heavy-chain gene 3’ enhancers deregulate bcl-2 promoter usage in t(14;18) lymphoma cells. Oncogene 2007;26:2635-41.
70. Thomas J, Leverrier Y, Marvel J. Bcl-X is the major pleiotropic anti-apoptotic gene activated by retroviral insertion mutagenesis in an IL-3 dependent bone marrow derived cell line. Oncogene 1998;16:1399-408.
71. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997;275:967-9.
72. Meijerink JP, Smetsers TF, Sloetjes AW, Linders EH, Mensink EJ. Bax mutations in cell lines derived from hematological malignancies. Leukemia 1995;9:1828-32.
73. Yamamoto H, Sawai H, Perucho M. Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997;57:4420-6.
74. Abrams MT, Robertson NM, Yoon K, Wickstrom E. Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. J Biol Chem 2004;279:55809-17.
75. Callagy GM, Pharoah PD, Pinder SE, Hsu FD, Nielsen TO, Ragaz J, et al. Bcl-2 is a prognostic marker in breast cancer independently of the Nottingham Prognostic Index. Clin Cancer Res 2006;12:2468-75.
76. Bhargava V, Kell DL, van de Rijn M, Warnke RA. Bcl-2 immunoreactivity in breast carcinoma correlates with hormone receptor positivity. Am J Pathol 1994;145:535-40.
78. Correia C, Schneider PA, Dai H, Dogan A, Maurer MJ, Church AK, et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood 2015;125:658-67.
79. Du C, Zhang X, Yao M, Lv K, Wang J, Chen L, et al. Bcl-2 promotes metastasis through the epithelial-to-mesenchymal transition in the BCap37 medullary breast cancer cell line. Oncol Lett 2018;15:8991-898.
80. Hata AN, Engelman JA, Faber AC. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov 2015;5:475-87.
81. Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther 2005;4:267-76.
82. Ikegaki N, Katsumata M, Minna J, Tsujimoto Y. Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res 1994;54:6-8.
83. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899-905.
85. Risnayanti C, Jang YS, Lee J, Ahn HJ. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Sci Rep 2018;8:7498.
86. Srivastava RK, Sasaki CY, Hardwick JM, Longo DL. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J Exp Med 1999;190:253-65.
87. Sui G, Qiu Y, Yu H, Kong Q, Zhen B. Interleukin-17 promotes the development of cisplatin resistance in colorectal cancer. Oncol Lett 2019;17:944-50.
88. Laprevotte E, Cochaud S, du Manoir S, et al. The IL-17B-IL-17 receptor B pathway promotes resistance to paclitaxel in breast tumors through activation of the ERK1/2 pathway. Oncotarget 2017;8:113360-72.
89. Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 2012;18:521-8.
90. O’Shaughnessy A, Hendrich B. CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochem Soc Trans 2013;41:777-82.
91. Shao S, Cao H, Wang Z, et al. CHD4/NuRD complex regulates complement gene expression and correlates with CD8 T cell infiltration in human hepatocellular carcinoma. Clin Epigenetics 2020;12:31.
92. Xia L, Huang W, Bellani M, et al. CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer Cell 2017;31:653-668.e7.
93. Wang HC, Chou CL, Yang CC, et al. Over-Expression of CHD4 Is an Independent Biomarker of Poor Prognosis in Patients with Rectal Cancers Receiving Concurrent Chemoradiotherapy. Int J Mol Sci 2019;20:4087.
94. Hou MF, Luo CW, Chang TM, et al. The NuRD complex-mediated p21 suppression facilitates chemoresistance in BRCA-proficient breast cancer. Exp Cell Res 2017;359:458-65.
95. D’Alesio C, Bellese G, Gagliani MC, et al. The chromodomain helicase CHD4 regulates ERBB2 signaling pathway and autophagy in ERBB2+ breast cancer cells. Biol Open 2019;8:bio038323.
96. Lin CY, Lin CY, Chang IW, Sheu MJ, Li CF, Lee SW, et al. Low thrombospondin 2 expression is predictive of low tumor regression after neoadjuvant chemoradiotherapy in rectal cancer. Am J Transl Res 2015;7:2423-32.
98. Han ES, Muller FL, Pérez VI, et al. The in vivo gene expression signature of oxidative stress. Physiol Genomics 2008;34:112-26.
99. Kurdistani SK, Arizti P, Reimer CL, Sugrue MM, Aaronson SA, Lee SW. Inhibition of tumor cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res 1998;58:4439-44.
100. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017;8:8921-46.
101. Zhou X, Hao Q, Lu H. Mutant p53 in cancer therapy-the barrier or the path. J Mol Cell Biol 2019;11:293-305.
102. Wang Z, Deng Z, Zhu G. Emerging platinum(iv) prodrugs to combat cisplatin resistance: from isolated cancer cells to tumor microenvironment. Dalton Trans 2019;48:2536-44.
103. Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999;104:263-9.
104. Blandino G, Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res 2018;37:30.
105. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009;9:400-14.
107. Viale A, De Franco F, Orleth A, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009;457:51-6.
108. Yeganeh M, Gui Y, Kandhi R, et al. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene 2016;35:4200-11.
109. Piccolo MT, Crispi S. The dual role played by p21 may influence the apoptotic or anti-apoptotic fate in cancer. J Can Res Updates 2012;1:189-202.
110. Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D. Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res 1994;54:3391-5.
111. Gartenhaus RB, Wang P, Hoffman M, Janson D, Rai KR. The induction of p53 and WAF1/CIP1 in chronic lymphocytic leukemia cells treated with 2-chlorodeoxyadenosine. J Mol Med (Berl) 1996;74:143-7.
112. Macleod KF, Sherry N, Hannon G, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 1995;9:935-44.
113. Huang Y, Wang W, Chen Y, et al. The opposite prognostic significance of nuclear and cytoplasmic p21 expression in resectable gastric cancer patients. J Gastroenterol 2014;49:1441-52.
114. Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med 2008;10:e19.
115. Koster R, di Pietro A, Timmer-Bosscha H, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest 2010;120:3594-605.
116. Martinez-Rivera M, Siddik ZH. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem Pharmacol 2012;83:1049-62.
117. Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010;22:1003-12.
118. Chen C, Chin JE, Ueda K, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986;47:381-9.
119. Montazami N, Aghapour M, Farajnia S, Baradaran B. New insights into the mechanisms of multidrug resistance in cancers. Cell Mol Biol (Noisy-le-grand) 2015;61:70-80.
120. Alfarouk KO, Stock CM, Taylor S, et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int 2015;15:71.
121. Yang X, Iyer AK, Singh A, et al. MDR1 siRNA loaded hyaluronic acid-based CD44 targeted nanoparticle systems circumvent paclitaxel resistance in ovarian cancer. Sci Rep 2015;5:8509.
122. Nishio N, Katsura T, Inui K. Thyroid hormone regulates the expression and function of P-glycoprotein in Caco-2 cells. Pharm Res 2008;25:1037-42.
123. Vaidyanathan A, Sawers L, Gannon AL, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 2016;115:431-41.
124. Yang Z, Wu D, Bui T, Ho RJ. A novel human multidrug resistance gene MDR1 variant G571A (G191R) modulates cancer drug resistance and efflux transport. J Pharmacol Exp Ther 2008;327:474-81.
125. Asojo OA, Koski RA, Bonafé N. Structural studies of human glioma pathogenesis-related protein 1. Acta Crystallogr D Biol Crystallogr 2011;67:847-55.
126. Thompson TC. Exploiting the complex biology of prostate cancer. Cancer Biol Ther 2006;5:1573-6.
127. Capalbo G, Mueller-Kuller T, Koschmieder S, et al. Endoplasmic reticulum protein GliPR1 regulates G protein signaling and the cell cycle and is overexpressed in AML. Oncol Rep 2013;30:2254-62.
128. Thompson TC. Glioma pathogenesis-related protein 1: tumor-suppressor activities and therapeutic potential. Yonsei Med J 2010;51:479-83.
129. Li L, Ren C, Yang G, et al. GLIPR1 suppresses prostate cancer development through targeted oncoprotein destruction. Cancer Res 2011;71:7694-704.
130. Gong X, Liu J, Zhang D, et al. GLIPR1 modulates the response of cisplatin-resistant human lung cancer cells to cisplatin. PLoS One 2017;12:e0182410.
131. Sheng X, Bowen N, Wang Z. GLI pathogenesis-related 1 functions as a tumor-suppressor in lung cancer. Mol Cancer 2016;15:25.
132. Rosenzweig T, Ziv-Av A, Xiang C, et al. Related to testes-specific, vespid, and pathogenesis protein-1 (RTVP-1) is overexpressed in gliomas and regulates the growth, survival, and invasion of glioma cells. Cancer Res 2006;66:4139-48.
133. Gibbs GM, Roelants K, O’Bryan MK. The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr Rev 2008;29:865-97.
134. Li L, Abdel Fattah E, Cao G, et al. Glioma pathogenesis-related protein 1 exerts tumor suppressor activities through proapoptotic reactive oxygen species-c-Jun-NH2 kinase signaling. Cancer Res 2008;68:434-43.
135. Li L, Yang G, Ren C, et al. Glioma pathogenesis-related protein 1 induces prostate cancer cell death through Hsc70-mediated suppression of AURKA and TPX2. Mol Oncol 2013;7:484-96.
136. Awasthi A, Woolley AG, Lecomte FJ, et al. Variable Expression of GLIPR1 Correlates with Invasive Potential in Melanoma Cells. Front Oncol 2013;3:225.
137. Dong J, Bi B, Zhang L, Gao K. GLIPR1 inhibits the proliferation and induces the differentiation of cancer-initiating cells by regulating miR-16 in osteosarcoma. Oncol Rep 2016;36:1585-91.
138. de Vasconcellos JF, Laranjeira AB, Leal PC, et al. SB225002 Induces Cell Death and Cell Cycle Arrest in Acute Lymphoblastic Leukemia Cells through the Activation of GLIPR1. PLoS One 2015;10:e0134783.
139. Karanika S, Karantanos T, Kurosaka S, et al. GLIPR1-ΔTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol Cancer 2015;14:122.
140. Sousa V, Santo JE, Silva M, et al. EGFR/erB-1, HER2/erB-2, CK7, LP34, Ki67 and P53 expression in preneoplastic lesions of bronchial epithelium: an immunohistochemical and genetic study. Virchows Arch 2011;458:571.
141. Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int 2014;2014:852748.
142. Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med 2011;135:55-62.
143. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989;244:707-12.
144. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog 2012;17:1-16.
145. Hsu JL, Hung M-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev 2016;35:575-88.
146. Carpenter RL, Lo HW. Regulation of Apoptosis by HER2 in Breast Cancer. J Carcinog Mutagen 2013;2013:003.
147. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007;26:6469-87.
148. Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009;9:463-75.
149. Castagnoli L, Ladomery M, Tagliabue E, Pupa SM. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers 2019;11:902.
150. Filipits M, Dafni U, Gnant M, et al. Association of p27 and cyclin D1 expression and benefit from adjuvant trastuzumab treatment in HER2-positive early breast cancer: a TransHERA study. Clin Cancer Res 2018;24:3079-86.
151. Yang H-Y, Shao R, Hung M-C, Lee M-H. p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene 2001;20:3695-702.
152. Ponnusamy MP, Singh AP, Jain M, Chakraborty S, Moniaux N, Batra SK. MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br J Cancer 2008;99:520-6.
153. Xia P, Choi AH, Deng Z, et al. Cell membrane-anchored MUC4 promotes tumorigenicity in epithelial carcinomas. Oncotarget 2017;8:14147-57.
154. Wolff AC, Hammond MEH, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 2007;131:18-43.
155. Wolf-Yadlin A, Kumar N, Zhang Y, et al. Effects of HER2 overexpression on cell signaling networks governing proliferation and migration. Mol Syst Biol 2006;2:54.
156. Le XF, Pruefer F, Bast RC Jr. HER2-targeting antibodies modulate the cyclin-dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell cycle 2005;4:87-95.
157. Yan M, Schwaederle M, Arguello D, Millis SZ, Gatalica Z, Kurzrock R. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev 2015;34:157-64.
158. English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther 2013;17:85-99.
160. Ludyga N, Anastasov N, Rosemann M, et al. Effects of simultaneous knockdown of HER2 and PTK6 on malignancy and tumor progression in human breast cancer cells. Mol Cancer Res 2013;11:381-92.
161. Moey MYY, Hassan OA, Papageorgiou CN, Schnepp SL, Hoff JT. The potential role of HER2 upregulation in metastatic breast cancer to the uterus: a case report. Clin Case Rep 2016;4:928-34.
162. Gabos Z, Sinha R, Hanson J, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol 2006;24:5658-63.
163. Ding W, Li Z, Wang C, Dai J, Ruan G, Tu C. Anthracycline versus nonanthracycline adjuvant therapy for early breast cancer: A systematic review and meta-analysis. Medicine 2018;97:e12908.
164. Cooke T, Reeves J, Lanigan A, Stanton P. HER2 as a prognostic and predictive marker for breast cancer. Ann Oncol 2001;12 Suppl 1:S23-8.
165. Haghnavaz N, Asghari F, Elieh Ali Komi D, Shanehbandi D, Baradaran B, Kazemi T. HER2 positivity may confer resistance to therapy with paclitaxel in breast cancer cell lines. Artif Cells Nanomed Biotechnol 2018;46:518-23.
166. Ferreira RB, Law ME, Jahn SC, et al. Novel agents that downregulate EGFR, HER2, and HER3 in parallel. Oncotarget 2015;6:10445-59.
167. Kan S, Koido S, Okamoto M, et al. Gemcitabine treatment enhances HER2 expression in low HER2-expressing breast cancer cells and enhances the antitumor effects of trastuzumab emtansine. Oncol Rep 2015;34:504-10.
168. Okita R, Shimizu K, NojIma Y, et al. Lapatinib enhances trastuzumab-mediated antibody-dependent cellular cytotoxicity via upregulation of HER2 in malignant mesothelioma cells. Oncol Rep 2015;34:2864-70.
169. Bhatnagar S, Tushir-Singh J. Targeting HER2 beyond breast cancer. Mol Cell Oncol 2019;6:1571984.
170. Jiang N, Lin JJ, Wang J, et al. Novel treatment strategies for patients with HER2-positive breast cancer who do not benefit from current targeted therapy drugs. Exp Ther Med 2018;16:2183-92.
171. Huang D, Duan H, Huang H, et al. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep 2016;6:20502.
172. Pernas S, Tolaney SM. HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol 2019;11:1758835919833519.
173. Berger JC, Vander Griend DJ, Robinson VL, Hickson JA, Rinker-Schaeffer CW. Metastasis suppressor genes: from gene identification to protein function and regulation. Cancer Biol Ther 2005;4:805-12.
174. Melotte V, Qu X, Ongenaert M, et al. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 2010;24:4153-66.
175. Kovacevic Z, Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis 2006;27:2355-66.
176. Shi XH, Larkin JC, Chen B, Sadovsky Y. The expression and localization of N-myc downstream-regulated gene 1 in human trophoblasts. PLoS One 2013;8:e75473.
177. Jung EU, Yoon JH, Lee YJ, et al. Hypoxia and retinoic acid-inducible NDRG1 expression is responsible for doxorubicin and retinoic acid resistance in hepatocellular carcinoma cells. Cancer Lett 2010;298:9-15.
178. Weiler M, Blaes J, Pusch S, et al. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc Natl Acad Sci U S A 2014;111:409-14.
179. Liu W, Iiizumi-Gairani M, Okuda H, et al. KAI1 gene is engaged in NDRG1 gene-mediated metastasis suppression through the ATF3-NFkappaB complex in human prostate cancer. J Biol Chem 2011;286:18949-59.
180. Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM, Pardee AB. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res 2000;60:749-55.
181. Maruyama Y, Ono M, Kawahara A, et al. Tumor growth suppression in pancreatic cancer by a putative metastasis suppressor gene Cap43/NDRG1/Drg-1 through modulation of angiogenesis. Cancer Res 2006;66:6233-42.
182. Du A, Jiang Y, Fan C. NDRG1 Downregulates ATF3 and Inhibits Cisplatin-Induced Cytotoxicity in Lung Cancer A549 Cells. Int J Med Sci 2018;15:1502-7.
183. Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov 2019;14:667-82.
184. Murugesan T, Rajajeyabalachandran G, Kumar S, Nagaraju S, Jegatheesan SK. Targeting HIF-2α as therapy for advanced cancers. Drug Discov Today 2018;23:1444-51.
185. Jarman EJ, Ward C, Turnbull AK, et al. HER2 regulates HIF-2α and drives an increased hypoxic response in breast cancer. Breast Cancer Res 2019;21:10.
186. Zhao J, Du F, Shen G, Zheng F, Xu B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis 2015;6:e1600.
187. Willam C, Koehne P, Jürgensen JS, et al. Tie2 receptor expression is stimulated by hypoxia and proinflammatory cytokines in human endothelial cells. Circ Res 2000;87:370-7.
188. Tang N, Wang L, Esko J, et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004;6:485-95.
189. Florczyk U, Czauderna S, Stachurska A, et al. Opposite effects of HIF-1α and HIF-2α on the regulation of IL-8 expression in endothelial cells. Free Radic Biol Med 2011;51:1882-92.
190. Rini BI, Appleman LJ, Figlin RA, et al. Results from a phase I expansion cohort of the first-in-class oral HIF-2α inhibitor PT2385 in combination with nivolumab in patients with previously treated advanced RCC. JCO 2019;37:558-558.
191. Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res 2000;2:324-30.
192. Suba Z. DNA stabilization by the upregulation of estrogen signaling in BRCA gene mutation carriers. Drug Des Devel Ther 2015;9:2663-75.
193. Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 2002;21:8981-93.
194. Murphy CG, Moynahan ME. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J 2010;16:39-47.
195. Godet I, Gilkes DM. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr Cancer Sci Ther 2017;4.
196. Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998;62:676-89.
197. Guillemette S, Serra RW, Peng M, et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev 2015;29:489-94.
198. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 2004;95:866-71.
199. Reisländer T, Lombardi EP, Groelly FJ, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun 2019;10:3143.
200. Zhang L, Feng T, Spicer LJ. The role of tight junction proteins in ovarian follicular development and ovarian cancer. Reproduction 2018;155:R183-98.
201. Rachow S, Zorn-Kruppa M, Ohnemus U, et al. Occludin is involved in adhesion, apoptosis, differentiation and Ca2+-homeostasis of human keratinocytes: implications for tumorigenesis. PLoS One 2013;8:e55116.
202. O’Driscoll MC, Daly SB, Urquhart JE, et al. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet 2010;87:354-64.
203. Wang M, Liu Y, Qian X, Wei N, Tang Y, Yang J. Downregulation of occludin affects the proliferation, apoptosis and metastatic properties of human lung carcinoma. Oncol Rep 2018;40:454-62.
204. Castro V, Bertrand L, Luethen M, et al. Occludin controls HIV transcription in brain pericytes via regulation of SIRT-1 activation. FASEB J 2016;30:1234-46.
205. Eguchi H, Akizuki R, Maruhashi R, et al. Increase in resistance to anticancer drugs involves occludin in spheroid culture model of lung adenocarcinoma A549 cells. Sci Rep 2018;8:15157.
206. Martin TA, Mansel RE, Jiang WG. Loss of occludin leads to the progression of human breast cancer. Int J Mol Med 2010;26:723-34.
207. Soares S, Sousa J, Pais A, Vitorino C. Nanomedicine: Principles, Properties, and Regulatory Issues. Front Chem 2018;6:360.
208. Morales-Cruz M, Delgado Y, Castillo B, et al. Smart Targeting To Improve Cancer Therapeutics. Drug Des Devel Ther 2019;13:3753-72.
209. Bar-Zeev M, Livney YD, Assaraf YG. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist Updat 2017;31:15-30.
210. Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist Updat 2011;14:150-63.
211. Park JH, Lee S, Kim J-H, Park K, Kim K, Kwon IC. Polymeric nanomedicine for cancer therapy. Progress in Polymer Science 2008;33:113-37.
212. Wang Z, Tan J, McConville C, et al. Poly lactic-co-glycolic acid controlled delivery of disulfiram to target liver cancer stem-like cells. Nanomedicine 2017;13:641-57.
213. Chang PY, Peng SF, Lee CY, et al. Curcumin-loaded nanoparticles induce apoptotic cell death through regulation of the function of MDR1 and reactive oxygen species in cisplatin-resistant CAR human oral cancer cells. Int J Oncol 2013;43:1141-50.
214. Xiao B, Zhang M, Viennois E, et al. Inhibition of MDR1 gene expression and enhancing cellular uptake for effective colon cancer treatment using dual-surface-functionalized nanoparticles. Biomaterials 2015;48:147-60.
215. Kim SS, Rait A, Kim E, et al. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014;8:5494-514.
216. Lin YL, Tsai NM, Chen CH, et al. Specific drug delivery efficiently induced human breast tumor regression using a lipoplex by non-covalent association with anti-tumor antibodies. J Nanobiotechnology 2019;17:25.
217. Sawant RR, Jhaveri AM, Koshkaryev A, Zhu L, Qureshi F, Torchilin VP. Targeted transferrin-modified polymeric micelles: enhanced efficacy in vitro and in vivo in ovarian carcinoma. Mol Pharm 2014;11:375-81.
218. Zhao J, Mi Y, Feng SS. Targeted co-delivery of docetaxel and siPlk1 by herceptin-conjugated vitamin E TPGS based immunomicelles. Biomaterials 2013;34:3411-21.
219. Satpathy M, Wang L, Zielinski RJ, et al. Targeted drug delivery and image-guided therapy of heterogeneous ovarian cancer using her2-targeted theranostic nanoparticles. Theranostics 2019;9:778-95.
220. Mishra V, Bansal KK, Verma A, et al. Solid Lipid Nanoparticles: Emerging Colloidal Nano Drug Delivery Systems. Pharmaceutics 2018;10:191.
221. Eskiler GG, Cecener G, Egeli U, Tunca B. Synthetically Lethal BMN 673 (Talazoparib) Loaded Solid Lipid Nanoparticles for BRCA1 Mutant Triple Negative Breast Cancer. Pharm Res 2018;35:218.
222. Herceg Z, Wang Z. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2001;477:97-110.
223. Choi SH, Jin SE, Lee MK, et al. Novel cationic solid lipid nanoparticles enhanced p53 gene transfer to lung cancer cells. Eur J Pharm Biopharm 2008;68:545-54.
224. Wang X, Low XC, Hou W, et al. Epirubicin-adsorbed nanodiamonds kill chemoresistant hepatic cancer stem cells. ACS Nano 2014;8:12151-66.
225. Zhang X, He C, Liu X, et al. One-pot synthesis of a microporous organosilica-coated cisplatin nanoplatform for HIF-1-targeted combination cancer therapy. Theranostics 2020;10:2918-29.
226. Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A 2009;106:17910-5.
227. Magaye R, Zhao J, Bowman L, Ding M. Genotoxicity and carcinogenicity of cobalt-, nickel- and copper-based nanoparticles. Exp Ther Med 2012;4:551-61.
228. Pietruska JR, Liu X, Smith A, et al. Bioavailability, intracellular mobilization of nickel, and HIF-1α activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol Sci 2011;124:138-48.
229. Zhao Y, Li L, Zhang PF, et al. Differential Regulation of Gene and Protein Expression by Zinc Oxide Nanoparticles in Hen’s Ovarian Granulosa Cells: Specific Roles of Nanoparticles. PLoS One 2015;10:e0140499.
230. Yang T, Yao Q, Cao F, Liu Q, Liu B, Wang XH. Silver nanoparticles inhibit the function of hypoxia-inducible factor-1 and target genes: insight into the cytotoxicity and antiangiogenesis. Int J Nanomedicine 2016;11:6679-92.
231. Satapathy SR, Mohapatra P, Preet R, et al. Silver-based nanoparticles induce apoptosis in human colon cancer cells mediated through p53. Nanomedicine (Lond) 2013;8:1307-22.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Torres-Martinez Z, Delgado Y, Ferrer-Acosta Y, Suarez-Arroyo IJ, Joaquín-Ovalle FM, Delinois LJ, Griebenow K. Key genes and drug delivery systems to improve the efficiency of chemotherapy. Cancer Drug Resist 2021;4:163-91. http://dx.doi.org/10.20517/cdr.2020.64
AMA Style
Torres-Martinez Z, Delgado Y, Ferrer-Acosta Y, Suarez-Arroyo IJ, Joaquín-Ovalle FM, Delinois LJ, Griebenow K. Key genes and drug delivery systems to improve the efficiency of chemotherapy. Cancer Drug Resistance. 2021; 4(1): 163-91. http://dx.doi.org/10.20517/cdr.2020.64
Chicago/Turabian Style
Torres-Martinez, Zally, Yamixa Delgado, Yancy Ferrer-Acosta, Ivette J Suarez-Arroyo, Freisa M. Joaquín-Ovalle, Louis J. Delinois, Kai Griebenow. 2021. "Key genes and drug delivery systems to improve the efficiency of chemotherapy" Cancer Drug Resistance. 4, no.1: 163-91. http://dx.doi.org/10.20517/cdr.2020.64
ACS Style
Torres-Martinez, Z.; Delgado Y.; Ferrer-Acosta Y.; Suarez-Arroyo IJ.; Joaquín-Ovalle FM.; Delinois LJ.; Griebenow K. Key genes and drug delivery systems to improve the efficiency of chemotherapy. Cancer Drug Resist. 2021, 4, 163-91. http://dx.doi.org/10.20517/cdr.2020.64
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 30
likes
Like This Article 30
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.