
Intracrine androgen biosynthesis and drug resistance

Abstract
Castration-resistant prostate cancer is the lethal form of prostate cancer and most commonly remains dependent on androgen receptor (AR) signaling. Current therapies use AR signaling inhibitors (ARSI) exemplified by abiraterone acetate, a P450c17 inhibitor, and enzalutamide, a potent AR antagonist. However, drug resistance to these agents occurs within 12-18 months and they only prolong overall survival by 3-4 months. Multiple mechanisms can contribute to ARSI drug resistance. These mechanisms can include but are not limited to germline mutations in the AR, post-transcriptional alterations in AR structure, and adaptive expression of genes involved in the intracrine biosynthesis and metabolism of androgens within the tumor. This review focuses on intracrine androgen biosynthesis, how this can contribute to ARSI drug resistance, and therapeutic strategies that can be used to surmount these resistance mechanisms.
Keywords
Introduction
Prostate cancer is a leading cause of cancer in the U.S. male population resulting in 160,000 new cases per year and 34,000 deaths annually[1]. The seminal discoveries of Charles Huggins showed that surgical castration followed by adrenalectomy with replacement glucocorticoids led to remission and that the disease could be treated with androgen deprivation treatment (ADT)[2,3]. It is noteworthy that these studies implied that there could be two sources of androgens for the tumor to continue to grow, the testis and the adrenal glands. These observations led to the introduction of surgical castration and later leuprolide and other LH-RH agonists to cause a chemical castration as the mainstay of first-line ADT[4-6]. R-Biaclutamide (Casodex), an androgen receptor antagonist, is often added to this regimen[7]. Furthermore, R-biaclutamide can now be replaced with more potent AR antagonists such as enzalutamide[8]. Following, ADT, there is a period of remission, however, the cancer invariably returns to give rise to castration-resistant prostate cancer (CRPC)[9,10].
CRPC is the lethal form of prostate cancer and is often detected by a rising serum prostatic-specific antigen (PSA). PSA is an androgen-dependent gene and this implies that despite castrate levels of circulating androgens, the disease remains dependent on androgen receptor (AR) signaling[11]. Castrate levels of androgens does not indicate an absence of androgens but only the presence of very low androgen levels in the circulation. Dependence on AR signaling can arise due to changes in the AR[12,13]. These changes include AR gene amplification[14] so that it can respond to trace ligand, AR mutation so that the receptor becomes ligand promiscuous[15-17], the appearance of splice variants that have lost their ligand binding domain and thus are constitutively active in the absence of ligand[18], and phosphorylation to activate the receptor in the absence of ligand[19,20]. In addition, the tumor adapts to make its own androgens in response to castration. Traditional ADT does not address the adrenal source of androgens even though the work of Huggins identified that this was an important source. The ability of tumors to make their own steroid hormones is referred to as intracrine synthesis, and Labrie coined the phrase “intracrinology” to describe this process[21,22]. It is now recognized that enzymes and transporters involved in the regulation of ligands for steroid receptors determine the pre-receptor concentration of ligand for hormone action[23,24].
Changes in AR and intracrine androgen biosynthesis are both adaptive responses to ADT. To attenuate these adaptive responses, androgen receptor signaling inhibitors (ARSI) were introduced and belong to two classes of agents. One class of agents are the potent AR antagonists enzalutamide and its second-generation analogs apalutamide and darolutamide[25-28]; and the other class of agents were the P450c17 (17α-hydroxylase/17,20-lyase) inhibitors, represented by abiraterone acetate[11,29,30] and second-generation analogs galeterone[31,32] and orterenol. Both classes of ARSIs improve progression-free survival but drug resistance emerges so the increase in overall survival may only be 3-4 months compared to standard ADT. ARSI drug resistance can involve increases in intracrine androgen synthesis mediated by transporters and enzymes, which determine the amount of ligand for the AR, and these mechanisms are the subject of this review.
Intracrine androgen biosynthesis
In a castrate environment, the major source of precursors of potent androgens in the tumor are those derived from the adrenal glands. The adrenal androgens of interest are the C19 steroids dehydroepiandrosterone (DHEA), DHEA-SO4, Δ4-androstene-3,17-dione (Δ4-AD), and the 11-oxygenated androgens (11β-hydroxy-Δ4-AD and 11-oxo-Δ4-AD)[33]. Of these, DHEA-SO4 is the dominant serum steroid, and is present in a huge excess compared to all other circulating steroids[21,34].
In addition to the contribution from adrenal steroids, intracrine steroid hormone biosynthesis could start from cholesterol using the side-chain cleavage enzyme (P450Scc) to produce pregnenolone. Tumor P540c17 would then convert pregnenolone to DHEA. While changes in transcripts for these enzymes have been observed in prostate cancer cell lines and in CRPC tumor biospecimens[23], there is no compelling evidence to support the conversion of C27 steroids (cholestanes) into C19 steroids (androgens) by flux measurements[35-37]. Moreover, the large amount of DHEA-SO4 that exists in the systemic circulation with and without P450c17 inhibition makes it seem unlikely that the tumor needs to adapt to castration or ADT to make its own DHEA[34].
For DHEA-SO4 to be utilized by prostate tumors, two events have to take place. First, the anionic steroid needs to be transported by an organic anion transporter protein of the SLCO gene family and, second, steroid sulfatase (STS) needs to remove the sulfate group to generate free DHEA [Figure 1]. DHEA is then converted to Δ4-AD by the bifunctional enzyme HSD3B1, which has both 3β-hydroxysteroid dehydrogenase activity and ketosteroid isomerase activity. Δ4-AD represents a junction point; it can be converted by the canonical pathway to testosterone (T) by type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) and then reduced by steroid 5α-reductase (SRD5A1 and SRD5A2) to 5α-dihydrotestosterone (DHT) as demonstrated in clinical specimens[38,39]. Alternatively, Δ4-AD can be converted by steroid 5α-reductase (SRD5A1 and SRD5A2) to yield 5α-androstane-3,17-dione, which is then reduced by AKR1C3 to yield DHT. The latter pathway bypasses T altogether and has been demonstrated in prostate cancer cell lines and xenografts as being the preferred pathway[40] and was confirmed to occur in hormone refractory cancer[41]. However, in CRPC patients and soft tissue metastasis the ratio of T:DHT favors formation of T which can be explained by a down regulation of SRD5A2 and up-regulation of AKR1C3[42]. DHEA is also reduced to 5-androstene-3β,17β-diol by AKR1C3, and then converted to T by HSD3B1. Finally, there is a backdoor pathway in which 5α-androstane-3,17-dione is reduced to androsterone by AKR1C2, and then further reduced by AKR1C3 to 5α-androstane-3α,17β-diol[43]. The diol is then oxidized by HSD17B6 to DHT[44,45].
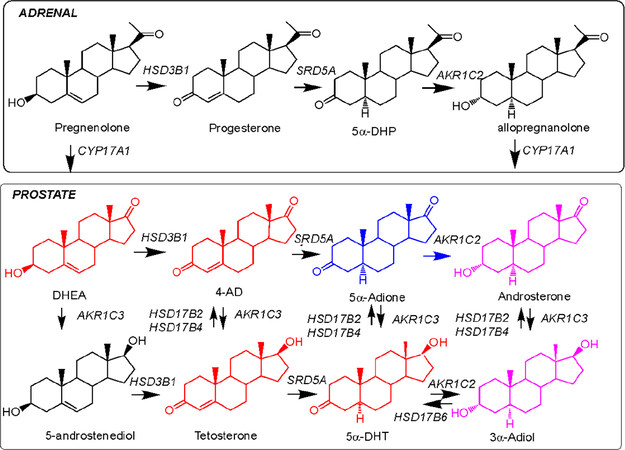
Figure 1. Intracrine androgen biosynthesis. Top panel shows reactions that occur in the adrenal reticularis. Bottom panel shows the conversion of adrenal steroids into the potent androgens testosterone and 5α-DHT in human prostate. The classical or canonical pathway is shown in red; the 5α-adione pathway is shown in blue; the backdoor pathway from allopregnanlone is shown in purple; the alternative pathway from 5-androstenediol is also shown. All enzymes are listed in italics by their gene names as follows: AKR1C1, 3α(20α)-hydroxysteroid dehydrogenase; AKR1C2, type 3 3α-hydroysteroid dehydrogenase; AKR1C3, type 5 17β-hydroxysteroid dehydrogenase; CYP17A1, 17α-hydroxylase17/20 lyase; HSD3B1, type 1 3β-hydroxysteroid dehydrogenase; HSD17B2 and 17HSD17B4, type 2 and type 4 17β-hydroxysteroid dehydrogenase; HSD17B6, type 6 17β-hydroxysteroid dehydrogenase and retinol dehydrogenase; and SRD5A1/2, type 1 and type 2 steroid 5α-reductase
The canonical pathway described above can also convert 11-keto-Δ4-AD to produce 11-keto-T and 11-keto-DHT. In addition, the bypass pathway could convert 11-keto-Δ4-AD to 11-keto-5α-androstane-3, 17-dione on route to 11-keto-DHT and thereby bypass 11-Keto-T altogether[46-48][Figure 2]. Transfection studies in HEK-293 cells and studies with recombinant AKR1C3 indicate that 11-keto-Δ4-AD and 11-keto-5α-androstane-3,17-dione are the preferred substrates for AKR1C3 over Δ4-AD and 5α-androstane-3,17-dione[49,50]. Both 11-keto-T and 11-keto-DHT are equipotent to T and DHT in AR driven luciferase reporter gene assays[46]. These pathways underscore the importance of AKR1C3 in all pathways to form potent ligands for the AR.
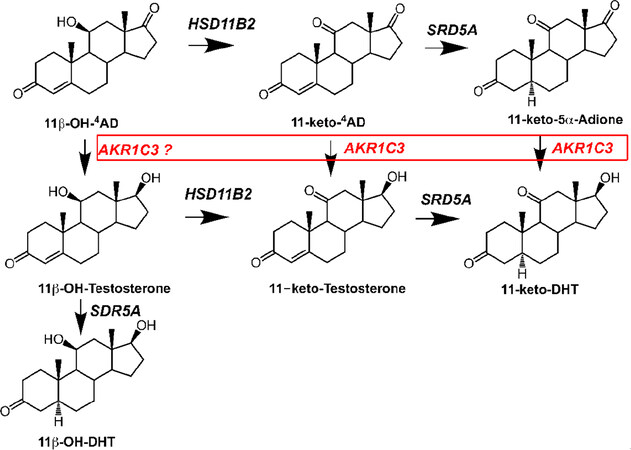
Figure 2. Conversion of 11-oxygenated androgens to 11-keto-Testosterone and 11-keto-DHT. The pathway from adrenal 11β-hydroxy-4AD and 11-keto-4AD to 11-keto-testosterone and 11-Keto-DHT, respectively is shown. The enzymes implicated in this conversion in the prostate are shown. All enzymes are listed in italics by their gene names. AKR1C3, type 5 17β-hydroxysteroid dehydrogenase; HSD11B2, type 2 11β-hydroxysteroid dehydrogenase; and SRD5A1/2, type 1 and type 2 steroid 5α-reductase
By contrast, the related AKR1C enzymes, AKR1C1 and AKR1C2 play important roles in the inactivation of DHT[51]. AKR1C1 works predominately as a 3-ketosteroid reductase on DHT leading to the formation of 5α-androstane-3β,17β-diol, a pro-apoptotic ligand for estrogen receptor β[52]. AKR1C2 works predominately as a 3-ketosteroid reductase on DHT leading to the formation of the inactive androgen 5α-androstane-3α,17β-diol[53]. Similar reactions are possible with 11-keto-DHT[50]. As described above, HSD17B6 is involved in the back conversion of 5α-androstane-3α,17β-diol to DHT, and compelling evidence exists that by working in opposite directions, AKR1C2 (3-ketosteroid reductase) and HSD17B6 (3a-hydroxysteroid oxidase) work as the molecular switch that determines DHT ligand access to the AR in the normal and diseased prostate[53]. HSD17B2 and HSD17B4 by working as 17β-hydroxysteroid oxidases convert T and DHT to their inactive counterparts, e.g., Δ4-AD and 5α-androstane-3,17-dione, respectively, and are implicated in the inactivation of these hormones[54,55]. The 5α-androstanediols once formed by AKR1C1 and AKR1C2, can then be glucuronidated by UGT family members UGT2B15 and UGT2B17[56,57]. Both UGT2B15 and UGT2B17 expression have been shown to be increased in CRPC.
Androgen biosynthesis and metabolism and drug resistance
Intracrine androgen biosynthesis has an important role in resistance to ARSI. Abiraterone acetate and other P450c17 inhibitors leave behind a significant reservoir of DHEA-SO4 in the serum following leuprolide treatment[58,59], while enzalutamide will have no effect on adrenal androgens following chemical or surgical castration. For example, the amount of DHEA-SO4 that remains even after combined leuprolide and abiraterone treatment is 4,000 times higher than the castrate amounts of T achieved. Resistance to ARSI can be enhanced when there is either upregulation or activating germline mutations in SLCO transporters or androgen biosynthesis genes or down-regulation or inactivating germline mutations in androgen metabolizing genes that result in elevated intra-tumoral T and DHT [Figure 3]. Thus, the serum levels of DHEA-SO4 that exist in patients on ARSI coupled with increased intracrine formation of T and DHT combined with increased AR expression create a “perfect-storm” for drug resistance.
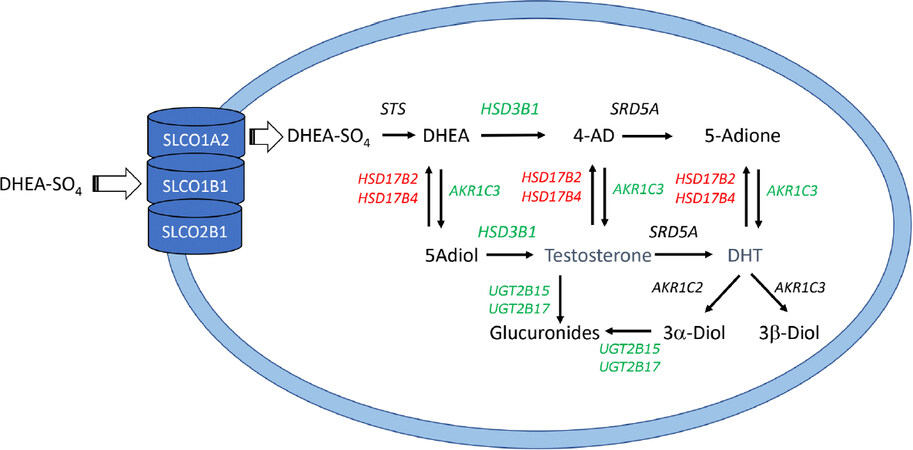
Figure 3. Mechanisms of drug resistance to androgen receptor signaling inhibitors (ARSI). Following uptake of DHEA-SO4 into the prostate by SLCO transporters a number of enzymes with either increased expression or germline mutations that result in increased activity/stability may contribute to ARSI drug therapy resistance (shown in green); similarly, a number of enzymes with either decreased activity or germline mutations that decrease activity/stability may contribute to drug resistance (shown in red). AKR1C3 may be an exception in that it is highly upregulated by androgen deprivation, and nsSNPs in conserved positions may reduce its activity. All enzymes are listed in italics by their gene names as described in the legends to Figures 1 and 2
Genes required for androgen biosynthesis
SLCO genes
Transcripts of SLCO1A2, SLCO1B1 and SLCO1B2 are increased 4-fold, 2.5-fold, and 1.74-fold respectively in LNCaP and 22Rv1 prostate cancer cells grown under conditions of androgen depletion to mimic ADT. Increased SLCO expression led to increased cellular uptake of [3H]-DHEA-SO4 and increased cellular proliferation stimulated by DHEA-SO4. The cellular proliferation in response to DHEA-SO4 was attenuated by SLCO1A2 knockdown, indicating the requirement of these transporters to facilitate intra-tumoral androgen biosynthesis[60]. SLCO1B1- and SLCO2B1-expressing prostate cancer xenografts showed a 3.9-fold and 1.9-fold increase in tumor accumulation of DHEA-SO4, respectively[61]. SLCO genes are also overexpressed in CRPC metastases. When 1,309 Caucasian prostate cancer patients were genotyped, nsSNPs in SLCO were associated with a poor progression-free survival[24]. For example, three SNPs in SLCO2B1 rs1242149, [935G>A; Arg312Gln], rs1789693 (intronic), and rs1077858 (intronic) were associated with decreased time to progression on ADT (P < 0.05). When the SLCO2B1 SNP variant rs1242149 (935G>A; Arg312Gln) was transfected into LNCaP cells, transfected cells exhibited higher maximal DHEA-SO4 accumulation when compared with the wild-type allele[62]. It is predicted that variants in the SLCO genes may serve as predictors of DHEA-SO4 uptake and ARSI resistance (see Table 1 for a summary of the common nsSNPs in genes involved in androgen biosynthesis and metabolism in the human prostate).
Common nsSNPs in genes involved in intracrine androgen biosynthesis and metabolism
| Gene | Accession No. | Nucleotide change | Minor allelic frequency | Amino-acid change | Change in function |
|---|---|---|---|---|---|
| SLCO2B1 | rs12422149 | 935G>A | 0.2099 | Arg312Gln | |
| HSD3B1 | rs1047303 | 1245 A>C | 0.2450 | Asn367Thr | Increase protein stability |
| AKR1C1 | rs149693250 | 116 A>G | 0.0046 | Lys39Arg | |
| rs540512862 | 440 C>T | 0.0022 | Thr147Ile | ||
| rs139588200 | 509 G>A | 0.0028 | Arg170His | ||
| rs142200840 | 840 C>A | 0.0076 | Asn280Lys | ||
| rs201500205 | 891 Ter | 0.0016 | Tyr323ns | ||
| AKR1C2 | rs149358453 | 68 C>T | 0.0002 | The23Ile | |
| rs2854482 | 137 T>A | 0.0649 | Phe46Tyr | ||
| rs147648222 | 209 C>T | 0.0034 | Ala70Val | ||
| rs142672563 | 211 G>T | 0.0026 | Asp71His | ||
| rs145967531 | 364 G>A | 0.0026 | Val122Ile | ||
| rs139521690 | 355 C>A | 0.0000 | Pro119Thr | ||
| rs114734 | 515 T>A | 0.0002 | Leu172Gln | ||
| rs2518043 | 553 A>G | ND | Lys185Glu | ||
| rs782195517 | 772 C>T | 0.064 | Arg258Cys | ||
| rs539972269 | 837 G>C | 0.001 | Gln279His | ||
| AKR1C3 | rs12529 | 15 C>G,T | 0.4203 | His5Gln | Decrease protein activity |
| rs1155177 | 230 A>G | 0.0367 | Glu77Gly | Decrease protein activity | |
| rs14888604 | 253 C>T | 0.0000 | Leu85Phe | ||
| 253 C>G | Lue85Val | ||||
| rs12387 | 312 G>C | 0.1518 | Lys104Asp | Decrease protein activity | |
| rs34186955 | 538 C>T | 0.0086 | Pro180Ser | ||
| rs61730879 | 548 A>G | 0.0026 | Lys183Arg | ||
| rs199934766 | 595 C>T | 0.0004 | Arg199Trp | ||
| rs139146411 | 596 G>A | 0.0004 | Arg199Gln | ||
| rs62621365 | 772 C>T | 0.0325 | Arg258Cys | Decrease protein activity | |
| rs1804059 | 879 G>T | Met293Ile | |||
| SRD5A2 | rs763681675 | 7 G>C | ND | Val3Ile | |
| rs766177308 | 42 C>A | ND | Ser14Arg | ||
| G>A | Ala49Thr | Increase protein stability | |||
| rs1465766670 | 187G>A | ND | Val63Met | ||
| rs753942411 | 354C>G | ND | Phe118Leu | ||
| rs759561106 | 433C>T | ND | Arg145Trp | Reduced affinity for NADPH | |
| rs756405261 | 513G>T | ND | Arg171Ser | Reduced affinity for NADPH | |
| G>A | Gly183Asp | ||||
| rs774564339 | 565G>A | ND | Val189Ala(Leu) | ||
| G>A | Gly191Glu | ||||
| T>C | Leu221Pro | ||||
| T>C | Leu226Pro | Increase protein stability | |||
| rs142602296 | 686 T>C | ND | Phe229Ser | ||
| rs145712014 | 734 C>A | 0.0006 | Ser245Tyr | Reduced affinity for NADPH | |
| C>T | Ala248Val | ||||
| UGT2B15 | rs1902923 | G>T | 0.32 | Asp85Tyr | |
| rs4148269 | 1568 A>C | 0.450 | Lys523Thr |
STS gene
There is only one steroid sulfatase gene (STS) which is required for the deconjugation of DHEA-SO4 to from free DHEA for downstream androgen biosynthesis. Knockdown of STS in castrate-resistant prostate cancer cell lines demonstrates the dependency of T and DHT synthesis on active STS. However, inhibition of STS for the treatment of CRPC is not a good strategy, since STS deficiency leads to ichthyosis which has been observed naturally in X-linked ichthyosis[63].
HSD3B1 gene
The transcriptional regulation of HSD3B1 has been examined in multiple metastatic prostate cancer cell models. HSD3B1 is induced by androgens in VCaP, CWR22Rv1, LNCaP, and LAPC4 models over 72 h but is then attenuated after 120 h. Thus, the enzyme does not appear to be regulated by androgen deprivation[64].
A common 1245 A→C missense- single nucleotide polymorphism in HSD3B1 (rs1047303) results in the amino acid change Asn367Thr. This mutation does not change the kinetic properties of the enzyme but leads to a more stable protein that is resistant to degradation suggesting that the variant will lead to increased intra-tumoral androgen biosynthesis and ARSI drug resistance[65]. Although it is not possible to determine how this mutation affects protein stability without a crystal structure for the enzyme, clinical studies show that the adrenal permissive mutation 1245 A→C is associated with inferior outcomes to ADT[66,67].
The ARSI abiraterone has a 3β-hydroxyl- Δ5-ene in its structure and can be metabolized to Δ4-abiratreone by HSD3B1, which is then converted to 5α-abiraterone by SRD5A1/SRD5A2. Each of these metabolites have their own activity. Δ4-Abiraterone has anti-tumor activity since it inhibits P450c17, HSD3B1, and is an antagonist of the AR[68], leading to the concept that abiraterone acetate may be a pro-drug and that Δ4-abiraterone is the active metabolite[69]. By contrast, 5α-abiraterone will activate the AR, potentially driving cancer progression. Thus, the mutant HSD3B1 allele can contribute to ARSI drug resistance by this mechanism.
AKR1C3 gene
AKR1C3 is one of the most highly expressed steroidogenic genes in CRPC; this has been seen by RT-PCR and Affymetrix microarray[70]. AKR1C3 is upregulated by androgen deprivation in prostate cancer cells[71], xenografts[71], and patient tumor samples[72-74]. Castration also induces up-regulation of AKR1C3 in an orthotopic VCaP human prostate cancer xenograft and leads to tumor growth[74]. Ten or more separate studies have replicated the finding that AKR1C3 is overexpressed in CRPC[38,39,70-73,75,76]. One such study showed AKR1C3 overexpression in primary lesion re-biopsies at the time of metastatic disease[77]. Consecutive prostate cancer specimens revealed increased AKR1C3 expression during progression to CRPC[78]. It is estimated that approximately 30% of CRPC patients will overexpress this enzyme. AKR1C3 has since been proposed as a biomarker for the CRPC. Experiments aimed at determining the mechanism of AKR1C3 over-expression have implicated the involvement of the TMPRSS2-ERG fusion protein in its regulation[75]. The TMPRSS2-ERG fusion protein arises in late-stage disease based on Gleason grade, where ERG acts as a transcription factor to increase AKR1C3 expression and remove the repressive effect of the AR on the AKR1C3 promoter. In this mechanism, ERG binds to the AKR1C3 promoter to induce gene transcription to increase T or DHT production which in turn increases TMPRSS2-ERG expression to induce AKR1C3 expression in a “feed-forward” manner[75].
Treatment of prostate cancer cells with abiraterone or enzalutamide all increase AKR1C3 expression consistent with androgen-dependent repression of the AKR1C3 gene[79,80]. C4-2B prostate cancer cells when grown in the presence of either abiraterone or enzalutamide over-express AKR1C3 and are resistant to growth inhibition by these drugs. The drug-resistant cell lines are, however, sensitive to growth inhibition in colony formation assays in soft agar and in xenografts to the AKR1C3 competitive inhibitor indomethacin[79,80]. The efficacy of indomethacin to improve progression-free survival and overall survival is being tested in a clinical trial in patients that progress on enzalutamide (NCT02935205 in clinical trials.gov), and in patients that progress on abiraterone acetate (NCT0254990 in clinical trials.gov). Similarly, AKR1C3 inhibitors could be used to surmount resistance to apalutamide and darolutamide[81], discussed later in this paper.
There are 14 AKR1C3 non-synonymous single nucleotide polymorphisms (nsSNPs) with varying global frequencies. The top-most frequently occurring variant (His5Gln) occurs in over 50% of the global population with an occurrence of 43% in African Americans. Lys104Asp, Glu77Gly, Arg258Cys have minor allelic frequencies of 15%, 3.7%, and 3.3%, respectively. All of these mutations were examined for their ability to conduct 17-ketosteroid reduction of the aromatase inhibitor exemestane, and each gave a significant reduction in catalytic efficiency that was 17-170 fold lower than the wild-type enzyme[82]. The remainder of the nsSNPs have a minor allelic frequency (MAF) of > 1%. It is unknown if these nsSNPs are enriched in prostate cancer patients. These nsSNPs can be mapped to the crystal structure of AKR1C3 and do not reside in the cofactor binding site, steroid binding cavity, or at the active site. AKR1C3 has a a/b8-barrel structure which is evolutionarily conserved across phyla in the AKR gene superfamily. In AKR1C3 there are seven nsSNPs that are in amino acids that are evolutionarily conserved in the structure (Leu85Phe; Pro180Ser; Lys183Arg; Arg199Trp; Arg199Gln; Arg258Cys; Met293Ile). These amino acids are conserved since they likely maintain protein folding and therefore mutations in these amino acids likely affect protein stability. It is predicted that carriers of these allelic variants may be more responsive to ARSI therapy.
HSD17B6 gene
The HSD17B6 gene has a number of aliases, e.g., RL-HSD, and actually functions as a 3α-hydroxysteroid dehydrogenase and epimerase rather than a 17β-HSD. It was identified as the major oxidoreductase responsible for the back conversion of 5α-andostane-3α,17β-diol to DHT in prostate cancer[44,45]. Few SNPs have been reported in this gene to date.
SRD5A genes
Abnormal SRD5A2 deficiency [46, XY SRD5A2 an autosomal recessive disorder of sex development (DSD)] has been well documented and associated with pseudo-hermaphroditism, lack of male pattern baldness, and an atrophied prostate gland[83]. There are more than 114 different mutations in the SRDA2 gene (including missense/nonsense mutations, deletions, insertions, and indels). However, less than 20 have been expressed to determine the consequences of these mutations on enzyme function. The protein is predicted to have two putative functional domains, an NADPH cofactor binding domain and a steroid binding domain. Ser14Arg, Arg145Trp, Arg171Ser, Phe229Ser, Ser245Tyr result in either complete loss of enzyme activity or seriously diminished enzyme activity. Arg145Trp, Arg171Ser, and Ser245Tyr are predicted to have diminished affinity for NADPH[84]. Many SNPs have been associated with prostate cancer and lead to the following amino acid changes: Val3Ile, Ala49Thr, Val63Met, Phe118Leu, Gly183Asp, Val189Ala, Gly191Glu, Leu221Pro, Leu226Pro, and Ala248Val, and have been kinetically characterized. Of these, Ala49Thre and Leu226Pro had significantly higher utilization ratios (Vmax/Km)[85]. The loss-of-function mutations in prostate cancer patients are likely to make the tumor more dependent on T rather than DHT, and may, therefore, have a neutral effect in response to ARSI therapy, and their occurrence is unlikely to be a major mechanism of drug resistance.
A third steroid 5α-reductase gene (SRD5A3) has also been identified. However, there has been controversy as to whether this reductase is really involved in the conversion of T to DHT. There is strong evidence that this gene instead encodes for a polyprenol reductase required for the synthesis of dolichol, and that mutations in this gene lead to a congenital glycosylation disorder[86].
Genes required for androgen metabolism
AKR1C1 and AKR1C2 genes
The AKR1C1 and AKR1C2 genes are highly induced by the Nrf2-Keap 1 pathway due to the presence of an antioxidant response element in their promoters[87]. However, the effect of oxidative stress and electrophilic induction of these genes on androgen metabolism has yet to be described. Increased expression of AKR1C1 and AKR1C2 in prostate cancer would lead to the elimination of DHT and may favor the unexpected high ratio of T:DHT in metastatic prostate cancer specimens. There are a several nsSNPs in AKR1C1: Lys39Arg, Thr147Ieu, Arg170His, Asn280Lys, and Thre323ns, with MAF less than 0.0076. The effects of these mutations on enzyme function have not been determined.
There are a number of nsSNPs in AKR1C2 (Phe46Tyr, Ala70Val, Asp71His, Val122Ile, Pro119Thr, Leu172Gln, Lys185Glu, Gln279His, and Arg258Cys). Three of these (Pro119Thr, Lys185Glu, and Arg258Cys) are in evolutionarily conserved residues, whereas Arg258Cys has a MAF of 0.064. In addition, Phe46Tyr has a MAF of 0.0649, but the MAF of the remainder are > 0.0034. The effect of some of these allelic variants on the in vitro metabolism of DHT has been examined. Unfortunately, the authors examined the effect of these variants following expression in Sf9 insect cell lysates and used a catalytic inactive mutant Tyr55Phe as a control. Under these conditions, a significant background turnover of DHT was noted in the presence of the catalytically inactive Tyr55Phe mutant making it difficult to interpret these data[88]. The authors concluded that Phe46Tyr (0.0649 MAF) and Leu172Gln (not in NCBI) reduced the apparent Vmax and that Leu172Gln, Lys185Glu and Arg258Cys all reduced the apparent Km. However, their effect on the utilization ratio Vmax/Km for reduction of DHT by these variants was modest and varied by only 2- to 3-fold.
HSD17B2 gene
HSD17B2 expression is reduced in prostate cancer patients, and consistently a HSD17B2 gene deletion was found in both primary and metastatic prostate cancer. In xenograft models over-expression of HSD17B2 suppressed androgen-induced cell proliferation and xenograft growth, consistent with its enzyme activity to inactivate 17β-hydroxyandrogens. Mechanisms responsible for this reduced expression included DNA methylation and mRNA alternative splicing. Two new catalytic-deficient shorter isoforms generated by alternative splicing were found to bind to the full-length enzyme promoting its degradation and are involved in the functional silencing of HSD17B2[89]. It is unknown if this alternative splicing mechanism occurs more frequently in ARSI drug resistance.
HSD17B4 gene
HSD17B4 expression can increase in CRPC and predicts poor prognosis which appears counterintuitive based on its role to inactive androgens. However, there are five alternatively spliced isoforms, and only isoform 2 inactivates T and DHT, and it is this isoform that is suppressed in CRPC. Genetic knockdown of HSD17B4 isoform 2 increases T and DHT to stimulate the AR and CRPC development in xenograft models[55]. It is unknown if reduced HSD17B4 expression contributes to ARSI drug resistance.
UGT2B15 and UGT2B17 genes
UGT2B15 and UGT2B17 are actively involved in the elimination of hydroxyandrogens as glucuronides from prostate cancer cells[56]. These genes are characterized by common polymorphisms and copy number variants that affect enzymatic activity and expression. Whole-gene deletions in UGT2B15 and UGT2B17 are observed in 27% and 13.5% of Caucasians and over 50% have a deletion of one of these genes. The nsSNP in UGT2B15 (Asp85Tyr, rs1902923 G.T) increases Vmax by several fold and has a MAF of 0.32. Both these changes have been associated with increased prostate cancer risk. The expression of UGT2B17 is inversely correlated with activation of the full-length AR receptor consistent with maintaining a homeostasis of AR signaling in normal prostate and castration sensitive prostate cancer. However, castration resistance leads to the expression of the constitutively active AR-V7 splice variant which upregulates UGT2B17 expression[90-92]. Although this seems counterintuitive, increased expression of UGT2B17 induced by AR-SV could be part of a coordinated response to make the tumor remain dependent on AR signaling in the absence of ligand[91]. Additional UGT2B15 polymorphisms (namely rs4148269, rs3100 (3’-UTR) rs9994887 (upstream), rs13112099 (upstream), rs7686914 (upstream), and rs7696472 (upstream) have been associated with an increased risk of PCa in a multiethnic study[93].
Non-classical properties of steroidogenic enzymes
Several of the steroidogenic enzymes have other properties that contribute to their role in ARSI drug resistance. One such enzyme is AKR1C3, that can act as a coactivator of the AR[94]. AR coactivators are AR-interacting proteins that amplify AR-dependent gene transcription in the presence of ligand[95]. AKR1C3 is a selective coactivator of AR, having no effect on the transcription mediated by other nuclear receptors. Furthermore, AKR1C3 acts as coactivator on AR with an activity comparable to SRC-2[94]. The co-activator domain of AKR1C3 maps to amino acid residues 171-237 which is distinct from the active-site. This region contains a putative p160 coactivator peptide of LxxIL buried within α-helix 5, were the consensus sequence to bind to the ligand binding domain of AR is LxxLL[96]. Although this motif can bind to the AF-2 region of the ligand binding domain of the AR, this interaction is out competed by a N/C interdomain interaction involving the FxxLF motif in the N-terminus of AR[96,97], leaving the identity of the coactivator peptide domain of AKR1C3 uncertain. The LxxLL motif present in AKR1C3 is also conserved within the AKR1C1 and AKR1C2 sequence and these proteins do not have coactivator properties. Comparison of the 171-237 region in all three enzymes shows a difference in amino acid residues in these enzymes. In AKR1C3, the sequence corresponds to 222QRDKRW227 whereas in AKR1C1 and AKR1C2 the sequence corresponds to 222HREEPW227. Mapping this region to the AKR1C3 crystal structure shows that this peptide is in a disordered loop. It is likely that this loop interacts with the NTD pf the AR [Figure 4].
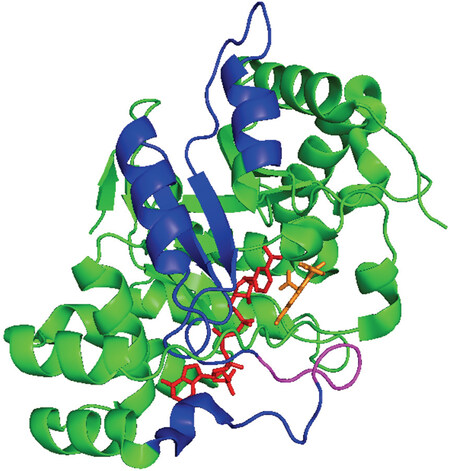
Figure 4. Putative Coactivator Domain of AKR1C3. Ribbon diagram of the AKR1C3•NADP+•Indomethacin complex (taken from PDB ID: 3UGB) showing the putative coactivator domain in the enzyme. Protein fold (green); NADP+ stick representation (red); indomethacin stick representation (orange); blue corresponds to amino acids 171-237; the disordered loop that may bind to the N-terminus of the AR (magneta). Cartoon prepared in PyMol
Confocal microscopy and proximity ligation assays have validated the interaction between AKR1C3 and the AR, and the co-translocation of both proteins to the nucleus. While many different coactivators exist for the AR, AKR1C3 has a unique property of being up-regulated by androgen deprivation in CRPC. AKR1C3 stabilizes AR-V7, a major AR-SV observed in clinical specimens[98]. In addition, AKR1C3 binds and stabilizes the ubiquitin ligase Siah-2, inhibiting its degradation, thereby enhancing Siah-2-dependent down-regulation of the AR corepressor NCoR in prostate cancer cells[99].
Another function of AKR1C3 is its ability to be the dominant prostaglandin F2α synthase in humans. This enzyme function leads to the formation of PGF2α epimers that can activate the FP1 receptor[100]. Activation of the receptor leads to activation of the MAPK pathway and phosphorylation of PPARγ, leading to its inactivation. Inactivation of PPARγ is considered a pro-proliferative signal since it prevents cellular differentiation. The involvement of this pathway in CRPC remains to be determined.
Inhibitors of androgen biosynthetic enzymes for CRPC
Drug resistance to ARSI could be attenuated by inhibitors of HSD3B1, AKR1C3, HSD17B6, and SRD5A2/A1. However, there are no inhibitors that have the desired selectivity for HSD3B1 over HSD3B2, and inhibition of these enzymes would block all steroid hormone biosynthesis. Both finasteride (SRD5A1 inhibitor) and dutasteride (dual SRD5A1/SRD5A2 inhibitor) have been shown in chemoprevention trials to give rise to a reduction in tumor incidence but also cause an increase in high-grade disease as determined by Gleason grade[101,102]. There has been much debate as to whether this was due to increased patient monitoring and frequency of needle biopsy, despite the increase in high-grade disease being small. Nonetheless, the FDA has issued black-box warnings for their use in prostate cancer therapy. Inhibitors of HSD17B6 appear to be in their infancy. Thus, the focus of our discussion will be on inhibitor development for AKR1C3.
AKR1C3 inhibitors
A large number of small-molecule AKR1C3 inhibitors have been developed for the potential treatment of CRPC as a monotherapy and to surmount ARSI drug resistance. Effective compounds need to show potency and selectivity for the target. Compounds must not inhibit AKR1C1 or AKR1C2 which would inactivate DHT. This can be challenging since the AKR1C enzymes exhibit 86% sequence identity and have similar crystal structures[103].
AKR1C3 inhibitors that have been developed are both non-steroidal and steroidal in nature. The non-steroidal inhibitors include re-purposed NSAID analogs that no longer inhibit Cox-1 and Cox-2, based on N-phenylanthranilic acid, indomethacin and R-naproxen analogs[104-106], and their bioisoteres[107], as well as 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids[108], caffeic acid phenethyl esters[109], berberine analogs[110], 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones[111], 2,3-diarylpropenoic acids[112], and ASP9521 (developed by Astellas)[113,114], GTx-560 (developed by GTx-therapeutics)[94]; the natural product analogs based on baccharin[115] and amaryllidaceae alkaloids[116]; and finally the steroidal based analogs developed by Bayer, e.g., BAY1128688, that are proprietary. Crystal structures of AKR1C3•NADP+•Inhibitor complexes exist for many of these agents, and there is a good structural basis for their mode of inhibition. Carboxylic acid analogs or those that contain an organic anion form a counter ion with the catalytic tetrad in which Tyr55 has TyrOH2+ character. Depending on the appendages to the anion, side chains can occupy three different pockets: the steroid binding cavity, sub-pocket (SP)1 (lined by Ser118, Phe306, Leu308, and Tyr319), SP2 (lined by Leu129, Phe306, and Phe311), and SP3 (lined by Tyr24, Ser221, Ser217, Gln222, Phe306)[117]. A recent patent review on many of these agents was published by our group[118]. Rather than review all these agents a few highlight points will be made.
First, indomethacin has made it into clinical trial for patients who progress on enzalutamide. Due to the gastrointestinal side effects associated with chronic use of this NSAID, indomethacin analogs, that are more potent and selective for AKR1C3 than the parent compound and that do not inhibit the COX isozymes, offer promise to replace indomethacin[106].
Second, ASP9521 is an AKR1C3 inhibitor that went into a phase 1/2 clinical trial and was found to be well tolerated but without efficacy[113,114]. However, there were several reasons why this trial failed. Of the small number of patients enrolled 6/13 failed to complete their drug regimen. Patients were excluded from the trial if they had been on abiraterone or R-biaclutamide before the commencement of the trial but these would be the very treatments that would upregulate AKR1C3. Moreover, there was no determination of whether the patient tumors were AKR1C3 positive, even though there is an IHC grade monoclonal Ab available[119].
Third, the baccharin anlogs (Kv37) are the only agents that have been shown to have synergistic anti-proliferative effects with enzalutamide in vitro, where the combination index shows up to a 200-fold synergistic effect[115]. However, this does not mean that other monofunctional AKR1C3 inhibitors will not behave the same way. They have not yet been screened in drug combinations.
Fourth, steroidal analogs, e.g., BAY1128688, run the risk of also inhibiting other steroid-transforming enzymes or steroid receptors. It is noteworthy that BAY1128688 went into a phase 2 clinical trial for endometriosis that was halted due to hepatic toxicity[120]. However, it is not clear whether the compound was counter screened against other AKRs, including AKR1C4 and AKR1D1 that are essential for bile acid biosynthesis[103].
Fifth, the N-phenylaminobenzoates based on N-phenylanthranilic acids gave rise to the N-naphthylaminobenzoate (BMT4-158) that is a “first-in-class” bifunctional AKR1C3 inhibitor and AR antagonist[121]. In a recent head to head comparison of this agent with ASP9521 and GTx-560, BMT-4-158 out-performed these agents since it was the only compound that acted as a competitive inhibitor in AR binding assays and displaced R1881 in AR competitive binding assays[122].
Sixth, GTx-560 is a “first-in-class” bifunctional AKR1C3 inhibitor that competitively inhibits the enzyme function of AKR1C3 but also blocks its coactivator function on the AR[94]. The agent had anti-tumor activity in xenograft models of prostate cancer, and it is unclear why this compound was not taken into clinical trials. This compound and its analogs offer promise to block two mechanisms of ARSI resistance, intra-tumoral androgen biosynthesis, and AR coactivation.
It is apparent that a large number of competitive inhibitors of AKR1C3 exist that have the desired potency and selectivity, however, if used as monofunctional agents, it is likely that other mechanisms of drug resistance will emerge, e.g., the appearance of AR splice variants (AR-SVs). Bifunctional inhibitors BMT4-158 and GTx-560 offer the promise of blocking intra-tumoral androgen biosynthesis and AR transactivation.
Alternative mechanisms of drug resistance
Upregulation of AKR1C3 in response to ADT and its contribution to drug resistance to ARSI is only one mechanism of resistance to these agents. Multiple mechanisms may contribute to the drug resistance phenotype, they may not be mutually exclusive, they may also synergize with each other, and different mechanisms may contribute differently based on the patient. One mechanism involves mutations in other steroidogenic genes such as HSD3B1, where the stability mutant enabled increased formation of Δ4-AD and 5α-DHT[65]. When combined with upregulation of AKR1C3 these changes could lead to increases in steroidogenesis that is greater than that achieved by either change alone. A second mechanism involves the appearance of AR-SVs as the disease emerges[18,123]. AR-SVs have lost their ligand binding domain and some of these AR-SVs are constitutively active in the absence of ligand. AR-V7 is stabilized by AKR1C3 so that overexpression of the latter will increase the steady state concentration of AR-V7[98]. AR-V7 is one of the most prominent AR-SVs, leading to attempts to develop clinical assays to detect this variant in biospecimens to determine whether ARSI drug therapy should be discontinued[124-126]. A third mechanism involves phosphorylation of the AR-MED1 transcriptional complex mediated by cyclin-dependent kinase 7 (CDK7)[127]. Phosphorylation on AR Ser81 is sufficient to activate the AR in the absence of ligand[128] and may contribute to resistance to enzalutamide, darolutamide, and apalutamide. It is unknown whether AKR1C3 could act as a coactivator of phosphorylated AR. A fourth mechanism involves the hijacking of the glucocorticoid receptor (GR) to substitute for the AR[129]. It is noteworthy that both the GR and AR bind to the same hormone response elements in the promoter of regulated genes[130].
Conclusion
It is apparent that intracrine androgen biosynthesis is an important component of resistance to ADT and ARSI inhibitors, and is clinically actionable. Knowledge of the nsSNPs that alter the activity or stability of the enzymes involved can determine who will benefit and who will not benefit from ARSI inhibitors. The enzyme that appears to be most important in mediating drug resistance is AKR1C3, and this is endorsed by the large number of small-molecule inhibitors that have been developed for this target both by academic laboratories and industry. Studies have moved beyond target validation and time will tell whether some of these compounds can transit preclinical development and make their way into well designed clinical trials for ARSI drug resistance. It is also apparent that AKR1C3 inhibitors do not necessarily have to be used as a monotherapy, but may improve progression-free survival in combination with other ARSIs.
Declarations
Author’s contributionsWrote and edited portions of the manuscript: Penning TM, Asangani IA, Sprenger C, Plymate S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the National Institute of Environmental Health Science (P30ES013508 ) (to Penning TM); by DoD Idea Development grant from the National Cancer Institute (W81XWH-17-1-0404 and R01CA249210) (Asangani IA); and by Department of Defense Prostate Cancer Research Program (W81XWH-17-1-0484, W81XWH-17-2-0323, W81XWH-20-1-0146), Lopker Foundation, Institute for Prostate Cancer Research, and Veterans Affairs Research Program (to Sprenger C and Plymate S).
Conflicts of interestPenning TM is Founder Penzymes, LLC, he receives sponsored research funding from Forendo and serves on the Expert Panel for Research Institute for Fragrance Materials. Plymate S is president of ProsTech, Inc. All other authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Huggins CB, Hodges CV. Studies on prostatic cancer 1. Effect of castration, estrogen and androgen injection on serum phosphatases in metatstatic carcinoma of the prostate. Cancer Res 1941;1:293-397.
3. Huggins CB. Two principles in endocrine therapy of cancers: Hormone deprival and hormone interference. Cancer Res 1965;25:1163-7.
4. Santen RJ, Demers L, Max DT, et al. Long term effects of administration of a gonadotropin-releasing hormone superagonist analog in men with prostatic carcinoma. J Clin Endocrinol Metab 1984;58:397-400.
6. Sharifi R, Soloway M. Clinical study of leuprolide depot formulation in the treatment of advanced prostate cancer. The Leuprolide Study Group. J Urol 1990;143:68-71.
7. Schellhammer PF, Sharifi R, Block NL, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate carcinoma. Analysis of time to progression. CASODEX Combination Study Group. Cancer 1996;78:2164-9.
8. Shah H, Vaishampayan U. Therapy of advanced prostate cancer: targeting the androgen receptor axis in earlier lines of treatment. Target Oncol 2018;13:679-89.
9. Yuan X, Cai C, Chen S, et al. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014;33:2815-25.
10. Cannata DH, Kirschenbaum A, Levine AC. Androgen deprivation therapy as primary treatment for prostate cancer. J Clin Endcorinol Metab 2012;97:360-5.
11. Attard G, Reid AHM, Yap TA, et al. Phase 1 clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 2008;28:4563-71.
12. Knudsen K, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res 2009;15:4792-8.
13. Knudsen K, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 2010;21:315-24.
14. Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995;9:401-6.
15. Buchanan G, Greenberg NM, Scher HI, et al. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res 2001;7:1273-81.
16. Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 1995;332:1393-8.
17. Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res 1999;59:2511-5.
18. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer 2011;18:R183-96.
19. Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A 2006;103:15969-74.
20. Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem 1992;267:968-74.
21. Labrie F, Belanger A, Simard J, et al. DHEA and peripheral androgen and estrogen formation: intracrinology. Ann N Y Acad Sci 1995;774:16-28.
22. Labrie F, Belanger A, Luu-The V, et al. DHEA and the intracrine formation of androgens and estrogens in peripheral target tissues: its role during aging. Steroids 1998;63:322-8.
23. Cai C, Chen S, Ng P, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011;71:6503-13.
24. Wright JL, Kwon EM, Ostrander EA, et al. Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev 2011;20:619-27.
25. Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012;72:1494-503.
26. Fizazi K, Shore N, Tammela TL, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med 2019;380:1235-46.
27. Fizazi K, Smith MR, Tombal B. Clinical development of darolutamide: a novel androgen receptor antagonist for the treatment of prostate cancer. Clin Genitourin Cancer 2018;16:332-40.
28. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787-90.
29. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995-2005.
30. Fizazi K, Scher HI, Molina A, et al; COU-AA-301 Investigators. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2012;13:983-92.
31. Kwegyir-Afful AK, Ramaligam S, Ramamurthy VP, et al. Galeterone and the next generation galeterone analogs, VNPP414 and VNPP433-3beta exert potent therapeutic effects in castration-/drug-resistant prostate cancer preclinical models in vitro and in vivo. Cancers (Basel) 2019;11:1637.
32. Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem 2015;58:2077-87.
33. Rege J, Turcu AF, Kasa-Vubu JZ, et al. 11-Ketotestosterone is the dominant circulating bioactive androgen during normal and premature adrenarche. J Clin Endocrinol Metab 2018;103:4589-98.
34. Penning TM. Dehydroepiandrosterone (DHEA)-SO4 depot and castration-resistant prostate cancer. Vitam Horm 2018;108:309-31.
35. Locke JA, Nelson CC, Adomat HH, et al. Steroidogenesis inhibitors alter but do not eliminate androgen synthesis mechanisms during progression to castration-resistance in LNCaP prostate xenografts. J Steroid Biochem Mol Biol 2009;115:126-36.
36. Locke JA, Guns E, Lubik AA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 2008;68:6407-15.
37. Kumagai J, Hofland J, Erkens-Schulze S, et al. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate 2013;73:1636-50.
38. Fankhauser M, Tan Y, Macintyre G, et al. Canonical androstenedione reduction is the predominant source of signaling androgens in hormone-refractory prostate cancer. Clin Cancer Res 2014;20:5547-57.
39. Mitsiades N, Sung CC, Schultz N, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res 2012;72:6142-52.
40. Chang KH, Li R, Papari-Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2011;108:13728-33.
41. Sharifi N, Auchus RJ. Androstenedione is the preferred androgen source in hormone refractory prostate cancer--letter. Clin Cancer Res 2014;20:4971.
42. Montgomery RB, Mostaghel E, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447-54.
44. Bauman DR, Steckelbroeck S, Williams MV, et al. Identification of the major oxidative 3a-hydroxysteroid dehydrogenase in human prostate that converts 5a-andostane-3a,17b-diol to 5a-dihydrotestosterone. A potential therapeutic target for androgen dependent disease. Mol Endocrinol 2006;20:444-58.
45. Mohler JL, Titus MA, Bai S, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res 2011;71:1486-96.
46. Pretorius E, Africander DJ, Vlok M, et al. 11-Ketotestosterone and 11-Ketodihydrotestosterone in castration resistant prostate cancer: potent androgens which can no longer be ignored. PLoS One 2016;11:e0159867.
47. Pretorius E, Arlt W, Storbeck KH. A new dawn for androgens: novel lessons from 11-oxygenated C19 steroids. Mol Cell Endocrinol 2017;441:76-85.
48. Schiffer L, Arlt W, Storbeck KH. Intracrine androgen biosynthesis, metabolism and action revisited. Mol Cell Endocrinol 2018;465:4-26.
49. Barnard M, Quanson J L, Mostaghel E, et al. 11-Oxygenated androgen precursors are the preferred substrates for aldo-keto reductase 1C3 (AKR1C3): implications for castration resistant prostate cancer. J Steroid Biochem Mol Biol 2018;183:192-201.
50. Endo S, Morikawa Y, Kudo Y, et al. Human dehydrogenase/reductase SDR family member 11 (DHRS11) and aldo-keto reductase 1C isoforms in comparison: substrate and reaction specificity in the reduction of 11-keto-C19-steroids. J Steroid Biochem Mol Biol 2020;199:105586.
51. Penning TM, Jin Y, Rizner TL, Bauman DR. Pre-receptor regulation of the androgen receptor. Mol Cell Endocrinol 2008;281:1-8.
52. Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3a-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3b-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. J Biol Chem 2003;279:10784-95.
53. Penning TM, Bauman DR, Jin Y, Rizner TL. Identification of the molecular switch that regulates access of 5a-DHT to the androgen receptor. Mol Cell Endocrinol 2007;265-266:77-82.
54. Gao X, Dai C, Huang S, et al. Functional silencing of HSD17B2 in prostate cancer promotes disease progression. Clin Cancer Res 2019;25:1291-301.
55. Ko H, Berk M, Chung YM, et al. Loss of an androgen-inactivating and isoform-specific HSD17B4 splice form enables emergence of castration-resistant prostate cancer. Cell Rep 2018;22:809-19.
56. Chouinard S, Barbier O, Bélanger A. UDP-glucuronosyltransferase 2B15 (UGT2B15) and UGT2B17 enzymes are major determinants of the androgen response in prostate cancer LNCaP cells. J Biol Chem 2007;282:33466-74.
57. Pâquet S, Fazil L, Grosse L, et al. Differential expression of the androgen-conjugating UGT2B15 and UGT2B17 enzymes in prostate tumor cells during cancer progression. J Clin Endcrinol Metab 2012;97:E428-32.
58. Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact 2015;234:332-8.
59. Taplin ME, Montgomery B, Logothetis CJ, et al. Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. J Clin Oncol 2014;32:3705-15.
60. Arakawa H, Nakanishi T, Yanagihara C, et al. Enhanced expression of organic anion transporting polypeptides (OATPs) in androgen receptor-positive prostate cancer cells: possible role of OATP1A2 in adaptive cell growth under androgen-depleted conditions. Biochem Pharmacol 2012;84:1070-7.
61. Green SM, Kaipainen A, Bullock K, et al. Role of OATP transporters in steroid uptake by prostate cancer cells in vivo. Prostate Cancer Prostatic Dis 2017;84:20-7.
62. Yang M, Xie W, Mostaghel E, et al. SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol 2012;29:2565-73.
64. Hettel D, Zhang A, Alyamani M, Berk M, Sharifi N. AR signaling in prostate cancer regulates a feed-forward mechanism of androgen synthesis by way of HSD3B1 upregulation. Endocrinology 2018;159:2884-90.
65. Chang KH, Li R, Kuri B, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 2013;154:1074-84.
66. Hearn JWD, AbuAli G, Reichard CA, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet Oncol 2016;17:1435-44.
67. Hearn JWD, Xie W, Nakabayashi M, et al. Association of HSD3B1 genotype with response to androgen-deprivation therapy for biochemical recurrence after radiotherapy for localized prostate cancer. JAMA Oncol 2018;4:558-62.
68. Li R, Evaul K, Sharma KK, et al. Abiraterone inhibits 3β-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res 2012;18:3571-9.
69. Li Z, Bishop AC, Alyamani M, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 2015;523:347-51.
70. Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006;66:2815-25.
71. Hofland J, van Weerden WM, Dits NFJ, et al. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res 2010;70:1256-64.
72. Hamid AR, Pfeiffer MJ, Verhaegh GW, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med 2012;18:1449-55.
73. Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med 2011;17:657-64.
74. Knuuttila M, Yatkin E, Kallio J, et al. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am J Pathol 2014;184:2163-73.
75. Powell K, Semaan L, Conley-LaComb MK, et al. ERG/AKR1C3/AR constitutes a feed-forward loop for AR signaling in prostate Cancer Cells. Clin Cancer Res 2015;21:2569-79.
76. Tian Y, Zhao L, Zhang H, et al. AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagnostic Pathol 2014;9:42-8.
77. Zhao J, Zhang M, Liu J, et al. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. Prostate 2019;79:1553-62.
78. Miyazaki Y, Teramoto Y, Shibuya S, et al. Consecutive prostate cancer specimens revealed increased Aldo-Keto reductase family 1 member C3 expression with progression to castration-resistant prostate cancer. J Clin Med 2019;8:E601.
79. Liu C, Lou W, Zhu Y, et al. Intracrine androgens and AKR1C3 activation confer resistance to Enzalutamide in prostate cancer. Cancer Res 2015;75:1413-22.
80. Liu C, Armstrong CM, Lou W, et al. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther 2017;16:35-44.
81. Morsy A, Trippier PC. Reversal of apalutamide and darolutamide aldo-keto reductase 1C3-mediated resistance by a small molecule inhibitor. ACS Chem Biol 2020;15:646-50.
82. Platt A, Xia Z, Liu Y, Chen G, Lazarus P. Impact of nonsynomynous single nucleotide polymorphsims on in vitro metabolism of exemestane by hepatic cytosolic reductases. Genomics 2016;26:370-80.
83. Russell DW, Wilson JD. Steroid 5apha-reductase: two genes/two enzymes. Annu Rev Biochem 1994;63:25-61.
84. Ramos L, Vichis F, Chávez B, Mares L. Mutational analysis of SRD5A2: from gene to functional kinetics in individuals with steroid 5α-reductase 2 deficiency. J Steroid Biochem Mol Biol 2020;200:105691.
85. Makridakis N, Akalu A, Reichardt JK. Identification and characterization of somatic steroid 5alpha-reductase (SRD5A2) mutations in human prostate cancer tissue. Oncogene 2004;23:7399-405.
86. Cantagrel V, Lefeber DJ, Ng BG, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 2010;142:203-17.
87. Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a Human Aldo-Keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH Activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res 1999;59:607-14.
88. Takahashi RH, Grigliatti TA, Reid RE, Riggs KW. The effectc of allelic variation in aldo-keto-reductase 1C2 on the in vitro metabolism of dihydrotestosterone. J Pharmaco Exp Ther 2009;329:1032-9.
89. Zhu Z, Chung YM, Sergeeva O, et al. Loss of dihydrotestosterone-inactivation activity promotes prostate cancer castration resistance detectable by functional imaging. J Biol Chem 2018;293:17829-37.
90. Gauthier-Landry L, Bélanger A, Barbier O. Multiple roles for UDP-glucuronosyltransferase (UGT)2B15 and UGT2B17 enzymes in androgen metabolism and prostate cancer evolution. J Steroid Biochem Mol Biol 2015;145:187-92.
91. Liu LL, Xie N, Sun S, et al. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014;33:3140-50.
92. Zhang A, Zhang J, Plymate S, Mostaghel EA. Classical and non-classical roles for pre-receptor control of DHT metabolism in prostate cancer progression. Horm Cancer 2016;7:104-13.
93. Vidal AC, Tucker C, Schildkraut JM, et al. Novel associations of UDP-glucuronosyltransferase 2B gene variants with prostate cancer risk in a multiethnic study. BMC Cancer 2013;13:556.
94. Yepuru M, Wu Z, Kyulkarni A, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res 2013;19:5613-25.
95. Culig Z, Comuzzi B, Steiner H, Bartsch G, Hobisch A. Expression and function of androgen receptor coactivators in prostate cancer. J Steroid Biochem Mol Biol 2004;92:265-71.
96. He B, Bowen NT, Minges JT, Wilson EM. Androgen-induced NH2- and COOH-terminal interaction inhibits p160 coactivator recruitment by activation function 2. J Biol Chem 2001;276:422293-301.
97. He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J Biol Chem 2002;277:25631-9.
98. Liu C, Yang JC, Armstrong CM, et al. AKR1C3 promotes AR-V7 protein stabilization and confers resistance to AR-targeted therapies in advanced prostate cancer. Mol Cancer Ther 2019;18:1875-96.
99. Fan L, Peng G, Hussain A, et al. The steroidogenic enzyme AKR1C3 regulates stability of the ubiquitin ligase Siah2 in prostate cancer cells. J Biol Chem 2015;290:20865-79.
100. Penning T. AKR1C3 (Type 5 17β-hydroxysteroid Dehydrogenase/Prostaglandin F Synthase): roles in malignancy and endocrine disorders. Mol Cell Endcorinol 2019;489:82-91.
101. Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. Engl J Med 2003;349:215-24.
102. Andriole GL, Bostwick DG, Brawley OW, et al; REDUCE Study Group. Effect of dutasteride on the risk of prostate cancer. N Engl J Med 2010;362:1192-202.
103. Penning TM, Wangtrakuldee P, Auchus RJ. Structural and functional biology of aldo-keto reductase steroid-transofrming enzymes. Endocrine Rev 2019;40:447-75.
104. Adeniji AO, Uddin MJ, Zang T, et al. Discovery of (R)-2-(6-Methoxynaphthalen-2-yl)butanoic acid as a potent and selective aldo-keto reductase 1C3 inhibitor. J Med Chem 2016;59:7431-44.
105. Adeniji AO, Twenter BM, Byrns MC, et al. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem 2012;55:2311-23.
106. Liedtke AJ, Adeniji AO, Chen M, et al. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17b-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem 2013;56:2429-46.
107. Lolli ML, Carnovale I, Pippione AC, et al. Bioisosteres of indomethacin as inhibitors of aldo-keto reductase 1C3. ACS Med Chem Lett 2019;10:437-43.
108. Jamieson SM, Brooke D, Heinrich D, et al. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids: highly potent and selective inhibitors of the type 5 17-β-hydroxysteroid dehydrogenase AKR1C3. J Med Chem 2012;55:7746-58.
109. Li C, Zhao Y, Zheng X, et al. In vitro CAPE inhibitory activity towards human AKR1C3 and the molecular basis. Chen Biol Inter 2016;253:60-5.
110. Tian Y, Zhao L, Wang Y, et al. Berberine inhibits androgen synthesis by interaction with aldo-keto reductase 1C3 in 22Rv1 prostate cancer cells. Asian J Androl 2016;18:607-12.
111. Heinrich DM, Flanagan J, Jamieson SM, et al. Synthesis and structure-activity relationships for 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones as novel non-carboxylate inhibitors of the aldo-keto reductase enzyme AKR1C3. Eur J Med Chem 2013;62:738-44.
112. Gazvoda M, Beranic N, Turk S, et al. 2,3-diarylpropenoic acids as selective non-steroidal inhibitors of type-5 17β-hydroxysteroid dehydrogenase (AKR1C3). Eur J Med Chem 2013;62:89-97.
113. Kikuchi A, Furutani T, Azami H, et al. In vitro and in vivo characterization of ASP9521: a novel selective, orally bioavailable inhibitor of 17b-hydroxysteroid dehydrogenase type 5 (17b-HSD5; AKR1C3). Invest New Drugs 2014;32:860-70.
114. Loriot Y, Fizazi K, Jones RJ, Brand Van den J, Molife RL, et al. Safety, tolerability and anti- tumor activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs 2014;32:995-1004.
115. Verma K, Gupta N, Zang T, Wangtrakluldee P, Srivastava SK, et al. AKR1C3 Inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. Mol Cancer Ther 2018;17:1833-45.
116. Hulcová D, Breiterová K, Zemanová L, et al. AKR1C3 inhibitory potency of naturally-occurring amaryllidaceae alkaloids of different structural types. Nat Prod Commun 2017;12:245-6.
117. Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol 2011;125:95-104.
118. Penning TM. Aldo-Keto Reductase (AKR) 1C3 inhibitors: a patent review. Expert Opin Ther Pat 2017;27:1329-40.
119. Lin HK, Steckelbroeck S, Fung KM, Jones AN, Penning TM. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3a-hydroxysteroid dehydrogenase/type 5 17b-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids 2004;69:795-801.
120. Rizner TL, Penning TM. Aldo-keto reductase 1C3-assessment as a new target for the treatment of endometriosis. Pharmacol Res 2020;152:104446.
121. Chen M, Adeniji AO, Twenter BM, et al. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett 2012;22:3492-7.
122. Wangtrakuldee P, Adeniji AO, Zang T, et al. A 3-(4-nitronaphthen-1-yl) amino-benzoate analog as a bifunctional AKR1C3 inhibitor and AR antagonist: Head to head comparison with other advanced AKR1C3 targeted therapeutics. J Steroid Biochem & Mol Biol 2019;192:105283.
123. Schweizer MT, Plymate SR. Targeting constitutively active androgen receptor splice variants in castration resistant prostate cancer. Expert Opin Ther Targets 2016;20:903-6.
124. Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028-38.
125. Antonarakis ES, Lu C, Luber B, et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol 2015;1:582-19.
126. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol 2017;35:2149-56.
127. Rasool RU, Natesan R, Deng Q, et al. A CDK7 inhibition suppresses castration-resistant prostate cancer through MED1 inactivation. Cancer Discov 2019;9:1538-55.
128. Russo JW, Liu X, Ye H, Calagua C, Chen S. Phosphorylation of androgen receptor serine 81 is associated with its reactivation in castration-resistant prostate cancer. Cancer Lett 2018;438:97-104.
129. Narayanan S, Srinivas S, Feldman D. Androgen-glucocorticoid interactions in the era of novel prostate cancer therapy. Nat Rev Urol 2016;13:47-60.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Penning TM, Asangani IA, Sprenger C, Plymate S. Intracrine androgen biosynthesis and drug resistance. Cancer Drug Resist 2020;3:912-29. http://dx.doi.org/10.20517/cdr.2020.60
AMA Style
Penning TM, Asangani IA, Sprenger C, Plymate S. Intracrine androgen biosynthesis and drug resistance. Cancer Drug Resistance. 2020; 3(4): 912-29. http://dx.doi.org/10.20517/cdr.2020.60
Chicago/Turabian Style
Penning, Trevor M., Irfan A. Asangani, Cynthia Sprenger, Stephen Plymate. 2020. "Intracrine androgen biosynthesis and drug resistance" Cancer Drug Resistance. 3, no.4: 912-29. http://dx.doi.org/10.20517/cdr.2020.60
ACS Style
Penning, TM.; Asangani IA.; Sprenger C.; Plymate S. Intracrine androgen biosynthesis and drug resistance. Cancer Drug Resist. 2020, 3, 912-29. http://dx.doi.org/10.20517/cdr.2020.60
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 20
likes
Like This Article 20
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.