
Manipulation of the immune system by non-small cell lung cancer and possible therapeutic interference

Abstract
Pulmonary carcinomas have developed mechanisms by which they escape the attack of immune cells. Immune checkpoint molecules programmed death 1 - programmed death ligand 1 (PD1-PDL1) and the cytotoxic T-lymphocyte antigen 4 system have gained attention. The expression of PDL1 by tumor cells causes immune tolerance, and further influences the microenvironment via orchestration by cytokines. Therapy with PDL1 antibodies could restore the cytotoxicity of T-lymphocytes towards tumor cells. Many patients will respond to this treatment. However, resistance mechanisms will counteract this therapy. New investigations have identified additional immune checkpoint inhibitors such as lymphocyte activation gene 3 and T cell immunoglobulin and mucin-domain containing-3. Tumor cells also induce tolerance by manipulating cells of the innate immune system. Macrophages are polarized to tumor-friendly M2, neutrophils into N2 types, and dendritic cells and myeloid suppressor cells are switched to assist tumor cells. Regulatory T cells enter the tumor microenvironment and signal tolerance to cytotoxic cells, inhibiting the influx of NK cells. Soluble mediators either released by tumor cells or cells of the tumor stroma induce immune tolerance, examples including tryptophan and indolamine dioxygenases, arginine and adenosine. Treatment options to counteract these molecules are currently being tested. The tumor stroma has been classified as immune-inflamed, immune-excluded, and immune-desert types. The latter might be switched to an inflamed type by induction of tertiary lymph follicles. Dendritic cells and macrophages normally phagocytose tumor antigens, but inhibitors of phagocytosis can block this. Interference with these molecules is another option for re-establishing the cytotoxic action of the immune system against tumor cells. In this review we will discuss these aspects with a special emphasis on non-small cell lung cancer.
Keywords
Introduction
Immunotherapy was first applied to melanomas, which is one of the malignancies with the highest mutational burden. But shortly after it was reported that non-small cell carcinoma (NSCLC) can be targeted by antibodies for immune checkpoint inhibitors. A major breakthrough in establishing this type of therapy was the discovery of immunomodulation of T cells response through immune checkpoints inducing immune evasion of cancer cells. Cytotoxic T-lymphocyte antigen 4 (CTLA-4) was not only found in dendritic cells and regulatory T cells (Tregs) in lymph nodes, but also circulating in the blood, inducing apoptosis and cell death of cytotoxic T-lymphocytes[1]. Programmed cell death 1 (PD1) and its ligand PDL1 was found on tumor cells as well as on lymphocytes inducing immune tolerance[2]. Based on these findings, antibodies for PDL1 and PD1 were tested for their ability to interfere with this mechanism of immune tolerance against tumor antigens. Important efforts are under way and have focused on this new treatment modality[2-10]. At present, several pharmaceutical companies have developed checkpoint inhibitors (humanized monoclonal antibodies) for the treatment of NSCLC patients and several phase II and III studies have been performed. The FDA and EMA have recently approved three of these drugs for treatment of NSCLC. Whereas some drugs are selectively approved for squamous cell carcinomas (SCC), others may be used in all NSCLC[9,11-14].
Immunotherapy of NSCLC
Like other solid tumors, pulmonary carcinomas have developed several mechanisms by which they escape the attack of cytotoxic immune cells. Some of these mechanisms have recently gained special attention: the programmed death 1 - programmed death ligand 1 and 2 (PDL1/2-PD1) and the CTLA4 system. Tumor cells as well as lymphocytes can express ligands for PD1[4]. Through this process, they interact with surface molecules on CD8+ T cells causing apoptosis, and furthermore influence the microenvironment via orchestration by cytokines, which all together cause immune tolerance. Therapy using antibodies against PDL1 have shown significant improvement experimentally as well as in clinical studies to restore the cytotoxic attack of T-lymphocytes towards tumor cells in several solid malignancies[3,6,10]. Parallel to targeting PDL1 by anti-PDL1 antibodies, antibodies against the receptor PD1 have also been created and used experimentally with success[2,4,5,7,8]. In some clinical trials, specific immunohistochemistry tests for the expression of PD1 and PDL1 were used to select those patients, who might best respond to the antibody-based therapy. A strong staining in at least 5% of tumor cells and/or lymphocytes - or 50% in another trial[15]- was regarded as a positive result and predictive for outcome [Figure 1]. The clinical data of these trials showed that majority of patients are detected by this simple stain. This however, created problems: In the clinical studies each company used a different antibody for PDL1 immunohistochemistry, and even more different staining platforms were used. This created problems for the pathology laboratories: purchasing all antibodies and also the staining platforms is impossible or would significantly increase the price for the tests. Finally, in a multi-institutional study, all antibodies and platforms were investigated, and three antibodies showed a similar staining performance[16]. But soon after, other anomalies emerged: patients who do not respond to treatment although being positive for PD1/PDL1 exist, as well as patients who do respond despite being negative or low positive for these immunohistochemical tests[17-19].
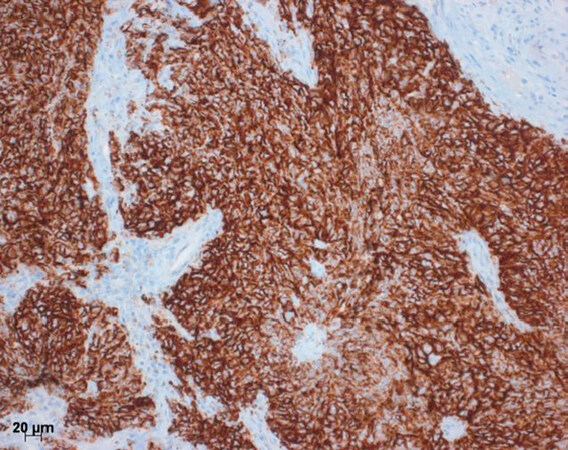
Figure 1. Immunohistochemistry for PDL1. Here a squamous cell carcinoma to almost 100% positive for PDL1. This would be an ideal candidate for the PDL1 antibody therapy. Bar 20 µm. PDL1: programmed death ligand 1
New research has addressed this issue. Even if the reaction of cytotoxic lymphocytes (Tcyt) against tumor cells is restored, that does not mean the Tcyt will recognize tumor cells, because the tumor cells do not express many neoantigens[20]. In addition, some neoantigens might be low immunogenic, i.e., the epitopes are recognized, but elicit only a mild reaction with low affinity antibodies, or antibodies, which do not induce a combined cytotoxic reaction together with T cells[21-23]. The evaluation of mutational burden (MTB) was therefore proposed as an additional test to select those patients who will benefit from immunotherapy[24,25]. However, MTB alone was not sufficient to identify patients profiting by immunotherapy: a combination of PDL1 expression on tumor cells and a high MTB better characterized these patients[24,26,27]. Even these combined tests do not fully cover all patients and resistance to this treatment is possible. Processing of neoantigens for presentation to Tcyt also requires the cooperation of molecules from the major histocompatibility complex (MHC). Tumor antigens can be non- or low-immunogenic, for example, aberrant expressed/overexpressed proteins from trophoblast, cancer-testis proteins (an example is the MAGE cluster; some of these have been used for vaccination). These antigens usually bind to MHC-I molecules and most often only elicit a transient immune reaction[20,28,29]. Recently, a report was published showing that the MHC-I complex might be digested by an autophagy mechanism, therefore inhibiting the procession of an antigen[30]. Mutated neoantigens are presently those eliciting a good and lasting immune reaction; examples are proteins derived from mutated TP53 gene in cigarette smokers.
To recognize a tumor neoantigen as non-self and immunogenic, MTB was established as a method to analyze the number of mutations in a given tumor[27,31]. Different types of next generation sequencing platforms can be used: whole exome sequencing, whole genome, or sequencing of a cancer gene panel. Cutoffs have been established to separate high- from low-mutational burden [Figure 2]. One problem in using MTB is the necessity to have normal control tissue from the patient.
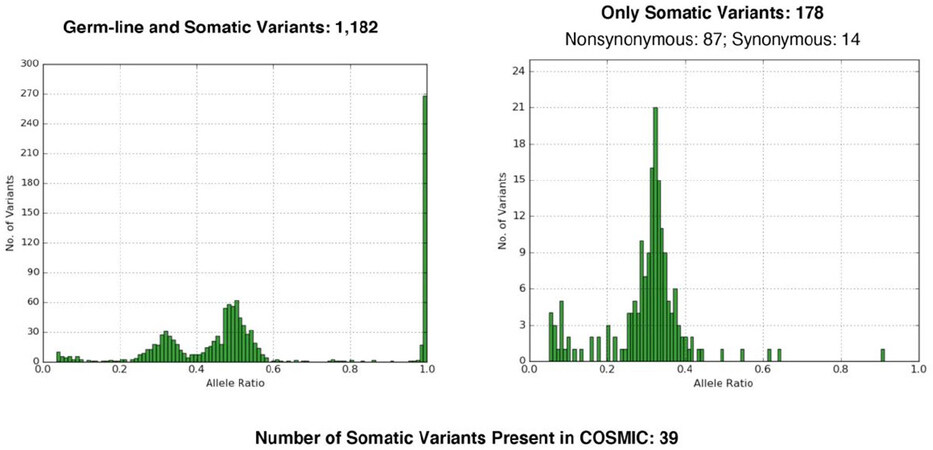
Figure 2. Tumor mutational burden. On the left side germline and somatic mutations are shown for the normal control tissue, to the right high numbers of mutations are seen in the tumor. Such a finding might help to decide for an immunotherapy
New results were reported from the analysis of T-cell receptors on Tcyt: The recognition of tumor neoantigens could be evaluated, and this method showed a good correlation with response to immunotherapy[20,26,32-34]. In another approach, tumor infiltrating lymphocytes were screened. If there was a high number of tumor-infiltrating CD8+ lymphocytes, in most cases a good response to immunotherapy was seen [Figure 3]. Even more sensitive was the analysis of these lymphocytes: the best response to therapy and increased survival was seen in those cases where the lymphocytes predominantly expressed CD8+ together with TCF7+20. Polymorphisms of the HLA genes might also influence the immune reaction against cancer neoantigens. The presence of HLAB44 corresponded favorable with immunotherapy resulting in prolonged survival[35,36]. On the contrary, patients with some germline mutations on HLA-I loci have shown a reduced response to immunotherapy[37].
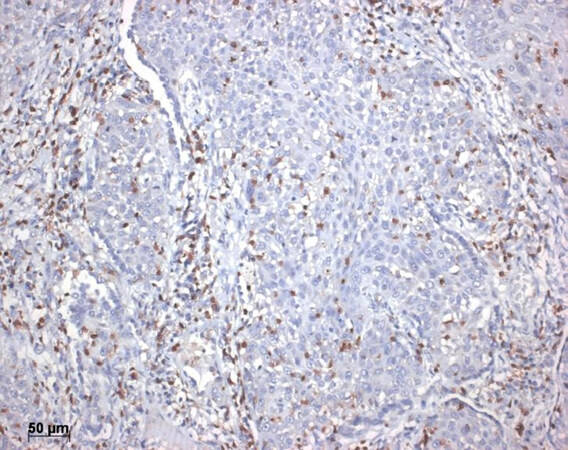
Figure 3. CD8+ lymphocytes infiltrating this solid adenocarcinoma. Bar 50 µm
Before we proceed into new protocols of how to restore the immune reaction against cancer cells, we need to recapitulate the principals of an immune reaction This will not be an extensive review on the subject but a brief and simplified presentation, helping to understand the following paragraphs.
Tumor cells are either destroyed by cells of the innate or specific immune system, or die due to hypoxia. These debris need to be removed by phagocytosis; macrophages are the main population, which will take up the debris and process the neoantigens from tumor cells. Macrophages subsequently present these processed antigen fragments to dendritic cells or directly to lymphocytes/NK cells. In conjunction with antigens, costimulatory and MHC molecules have to be present to stimulate an immune reaction. Immune check point molecules will next decide if there is tolerance for these antigens (self-antigen), or if this antigen-bearing cells should be eliminated. Different cells of the immune system will cooperate during an immune reaction, including: cytotoxic T cells either CD8+ or CD4+, CD20+ B cells, NK cells, but also N1 neutrophils, and M1 macrophages. Along all these steps, interventions are possible, which we will cover in the following paragraphs.
Another approach was to analyze the microenvironment (TME) of the tumor in more detail. Three types were recognized so far: the immune inflamed, the immune excluded, and the immune desert tumor [Figure 4][38]. In the inflamed type there are CD4+ and CD8+ T cells within the tumor stroma and between tumor cells. These lymphocytes and tumor cells usually express PD1/PDL1 (besides other regulatory molecules; see below). In the excluded type there are different immune cells in the surrounding stroma but not within the tumor. In this case most often the WNT-β catenin pathway and TGF is activated[39,40]. Lymphocytes are absent or scarce in the desert type. These patients do not respond to immunotherapy[20,38]. In this type, molecules within the circulation or expressed on endothelia inhibit the extravasation of immune cells; there is ongoing research to explore these mechanisms and find possibilities to interfere with the blockade[41-44]. We will come back to this later on.
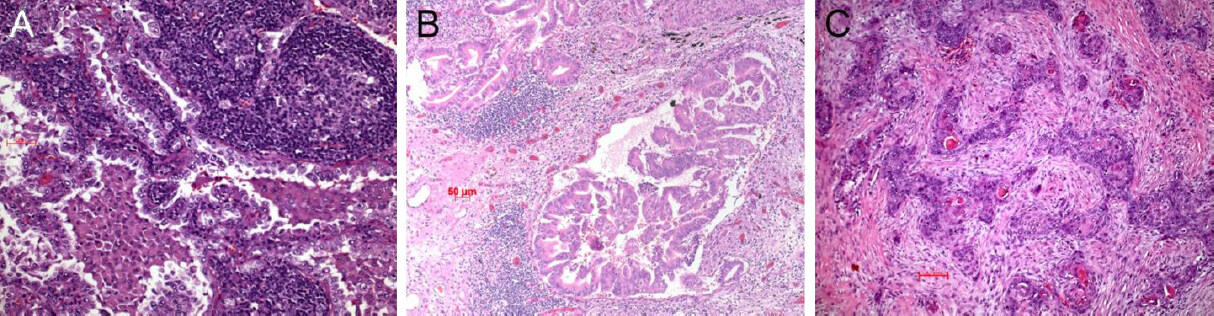
Figure 4. Immune inflamed adenocarcinoma. Note the intimate association of lymphocytes with the carcinoma cells (A); immune excluded type, an acinar adenocarcinoma. The lymphocytes are seen respecting the tumor boundaries (B); immune desert type of carcinoma, here a squamous cell carcinoma (C). Bars 50 and 100 µm
Although PD1-PDL1 has been proven as targetable molecules within the immune regulatory system, it is well known from non-neoplastic diseases, that there are many more ways the immune system can be shifted towards antigen tolerance or even lymphocyte exhaustion. Several of these regulatory mechanisms are also targetable. Subsequent development of resistance towards the PD1-PDL1 therapy might be due to upregulation of one of these immunomodulators. In addition, patients might have developed tolerance against their tumor cells not only by expression of PD1-PDL1, but also by other tolerance mechanisms - this could occur in patients who do not respond to anti-PDL1 therapy despite having PD1-PDL1 upregulated. Finally, in some cases tumor cells escape the immune cell attack by shedding or losing some of their neoantigens[45].
Other checkpoint inhibitors/regulators
The search for other molecules functioning as immune check-point controls has identified several new molecules, such as lymphocyte activation gene 3 (LAG3), T cell immunoglobulin and mucin-domain containing-3 (TIM3), tumor necrosis factor OX40 (CD134). While OX40 is a stimulating molecule, LAG3 and TIM3 are inhibitory. Treatment protocols have been developed (analogs and inhibitors) and are currently used in clinical studies usually in combination with PDL1 inhibition. Similar to PD1/PDL1 axis, LAG3 and TIM3 can also induce immunotolerance. TIM3 is suspected to even signal super-exhaustion to Tcyt[46].
So far there are preliminary data on these molecules and OX40 does not seem to be very effective. There are conflicting reports on the combination of PDL1 and OX40 therapies: firstly, the expression of PDL1, OX40 and OX40L varies considerably among NSCLC tested, and secondly low expression of OX40 was associated with longer overall survival and better prognosis. Other studies also reported on OX40, GITR and 4-1BB, other receptors implicated in immune homeostasis. Although tumor regression was seen in experimental setting, this was associated with severe autoimmune events (so called cytokine storm) due to loss of Treg homeostasis[47-50]. Other regulatory molecules including LAG3 and TIM3 were studied in combination with PDL1 blockade. The most advanced involved LAG3, which results in the exhaustion of T cells. Several in-vivo approaches demonstrated a highly significant clinical benefit under dual blockade of PDL1 and LAG3, whereas the efficacy was very low in cases of single agent targeting[51]. Human tumor tissues showed co-expression of LAG3 and PD-1 in infiltrated lymphocytes. The ongoing clinical studies mainly used dual blockage of LAG3/PD-1, which demonstrated promising survival benefits and long duration of response rates. Combining immunotherapy of anti-LAG-3 and anti-PD-1 has shown exciting efficacy in fighting PD-1 resistance[52,53]. Clinical studies on the combination of PDL1 and T-cell Immunoglobulin- and Mucin-domain-containing molecule 3 (TIM3) are ongoing, however, the significance has not been proven so far. TIM3 is expressed on lymphocytes and seems to be a marker for super-exhaustion of these cells. If the combination therapy might overcome the resistance will be seen[46,54]. Primarily not much attention was given to cells of the innate immune system and cells cooperating with lymphocytes.
Systems known to be able to induce immune tolerance towards foreign antigens
One of the most important cell types in this regard are antigen-presenting cells from the dendritic cell lineage. There are several populations, such as conventional dendritic cells, which promote immune attack by processing foreign antigens and presenting them to CD8+ T-lymphocytes. Others such as plasmocytoid and granulocytic dendritic cells cause immune tolerance by either cooperating with regulatory T cells or by inducing an inflammatory environment, which promotes tumor cell invasion, spread, and finally metastasis[55,56]. The function of Langerhans cells, another dendritic cell type, is entirely unknown, but most probably act against tumor antigens. We will come back to other aspects of dendritic cell function.
Plasmocytoid dendritic cells [Figure 5A] can also interact with monocytoid cells by promoting the differentiation of macrophages into the tumor-promoting M2 lineage[57]. Macrophages play a major role early on in carcinogenesis. Macrophages can induce angiogenesis, prepare and modulate stroma proteins in favor of invading tumor cells, thus promoting tumor growth, invasion, and metastasis[58,59]. On the contrary, M1 macrophages might inhibit tumor progression not only in the early phase but also during metastasis formation[60,61].
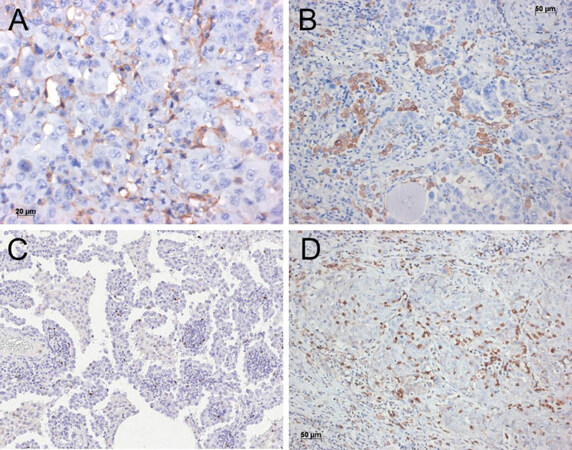
Figure 5. Plasmocytoid dendritic cells closely intermingled with the carcinoma cells (A); macrophages of M2 type are intimately associated with the adenocarcinoma cells. Immunohistochemistry for CD206 (B); increased numbers of regulatory T cells within the tumor stroma of this adenocarcinoma. Immunohistochemistry for FOXP3 (C); myeloid derived suppressor cells closely associated with cells of this squamous cell carcinoma. Immunohistochemistry for CD11b (D). Bars 20 and 50 µm, and magnification 200×
There are additional pathways by which tumor cells can influence the differentiation of M0-macrophages into M2 lineage. Whereas activation of Notch signaling increased M1 macrophages, blocking of Notch induced an M2 response. Wang et al.[60] showed that RBP-J-mediated Notch signaling regulates the M1 versus M2 polarization through SOCS3. Notch is inactivated by mutation in several lung carcinomas, however, the link with tumor-associated macrophages has not been studied so far. Tumor cells might use SOCS3 signaling to induce M2 macrophage polarization [Figure 5B]. Studies are on the way to influencing the differentiation of macrophages, as it is known that M2 macrophage polarization can be reverted[62-64].
Regulatory T cells (Treg) can accumulate at the tumor site and block CD8+ T-lymphocytes and NK cells [Figure 5C] in their anti-tumor action as well as inhibit influx of the cells into the tumor[65,66].
Myeloid-derived suppressor cells have been shown in pulmonary and squamous cell adenocarcinomas [Figure 5D]. These cells not only inhibit Tcyt, but also assist tumor cell metabolism under hypoxic condition[67-70].
Neutrophils have recently gained recognition as a part of the innate immune system. Neutrophils can often be seen in squamous cell carcinomas, probably attracted by keratinized tumor cells and cell-free keratin, which can activate the complement cascade and in turn induce influx of neutrophils [Figure 6]. Moreover, there are two types of neutrophils: N1 and N2[71-73]. The N2 type seems to favor and promote lung cancer progression and escape of immune cell attack. These tumor-associated neutrophils interact with CD47, inhibiting phagocytosis by macrophages and increase inflammation. In patients refractory to PD1/PDL1 therapy, a myeloid-rich group of lung cancer cases was seen: in these cases, neutrophils outnumbered CD8+ and CD4+ lymphocytes and a neutrophil antagonizing strategy might help to overcome resistance to immunotherapy.
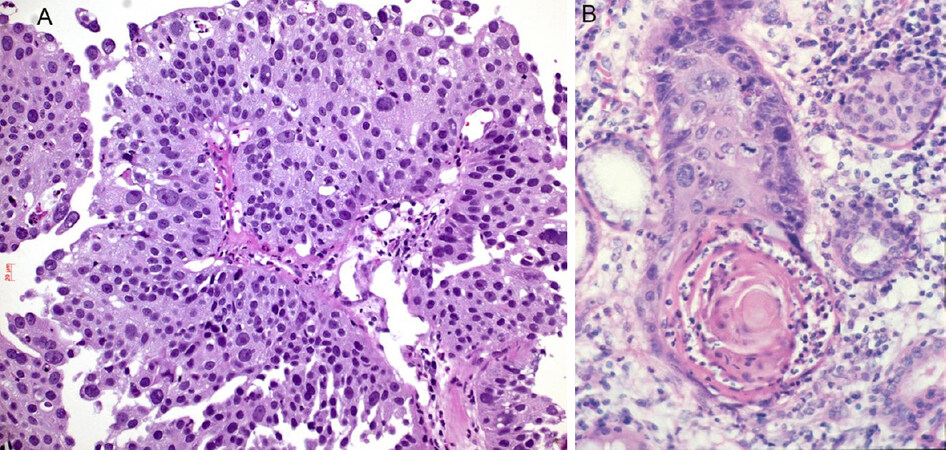
Figure 6. Infiltration of neutrophils in squamous cell carcinomas: neutrophils within the carcinoma (A), neutrophils concentrated in the keratin pearl (B). Magnification 200×
Autophagy is another mechanism associated with immune cell modulation, although this mechanism is presently not fully understood. Autophagy is seen in many lung carcinoma types. Increase of autophagy in cancer cells liberates nutrients, decreases the formation of reactive oxygen species, and aids in the clearance of misfolded proteins. This provides a survival advantage for cancer cells in the TME. However, immune cells also infiltrate the tumor environment and encounter hypoxia, resulting in hypoxia-induced autophagy. Due to the fact that autophagy is crucial for immune cell proliferation as well as for antigen presentation and T cell-mediated killing of tumor cells, anticancer treatment strategies based on autophagy modulation will need to consider the impact of autophagy on the immune system[74]. In melanomas, it has been shown that hypoxia leads to instability of gap-junctional CX43 and impairs melanoma cell killing by NK cells. Inhibition of autophagy by pharmacological approaches might restore NK cell mediated lysis of hypoxic melanoma cells[75].
Soluble mediators
News with a potential to interfere with immunotolerance signals from the tumor and the tumor-associated macrophages have been identified in metabolic studies. The amino acids tryptophan (trp) and arginine (arg), if present in sufficient concentration within the tumor MEV? can elicit immune tolerance. Trp is catabolized by tryptophan-2,3-dioxygenase or by indolamine-2,3-dioxygenase (IDO) to kynurenic and picolinic acid. Arg is metabolized predominantly by macrophages in two different ways: M1 macrophages metabolize Arg into nitric oxide and citrulline via nitric oxide synthase, whereas M2 macrophages will metabolize Arg into ornithine and urea via arginase. These latter metabolites will inhibit T cells and NK function[76]. A therapy using an arginase inhibitor showed promising results by inhibiting tumor growth, especially when combined with standard chemotherapy[77]. It was also shown that IDO also affects dendritic cells. Experimentally silencing IDO can create a robust anti-tumor immunity with activation of anti-tumor acting dendritic cells cooperating with cytotoxic T cells[78]. This could lead to the discovery of a new line of NSCLC immunotherapy.
Another factor involved in resistance mechanism of immunotherapy is adenosine. Adenosine is created from ATP via ADP-AMP by the ectonucleotidases CD73 and CD38. Either tumor cells or macrophages can express these enzymes. Metabolites as well as arginine itself can induce immunotolerance in Tcyt[79-85]. As these amino acids and their enzymes are part of the immune machinery in inflammation, these are most likely involved in immune reactions of the inflamed and the excluded types and interference with these molecules can be an additional strategy to overcome immune tolerance to cancer cells. However, these amino acids are also necessary for T cell proliferation, which should not be blocked. Accumulation of adenosine is seen especially in hypoxic areas. Adenosine blocks NK cells by different mechanisms, such as cytolytic activity, expression of cytotoxic granules, and interferon g release[86]. Extracellular adenosine suppresses pro-inflammatory activities upon binding with adenosine receptors on the surface of various immune cells. In addition, signaling through adenosine receptors upregulates a number of anti-inflammatory molecules and immunoregulatory cells, leading to the establishment of a long-lasting immunosuppressive environment[87]. Treatment options are available to block CD73 but not for CD38 [Figure 7A]. Some treatment protocols use a combination of anti-PD1/PDL1. A CD73 inhibitor MEDI-9,447 influences myeloid and lymphocyte infiltrations in the TME: an increase of CD8+ lymphocytes and activated macrophages was noted. A combination with PD1 antibodies was also tested. Another study analyzed the concentrations of ATP and ADP in BAL fluid and found a significant increase in cancer patients, and in patients with metastatic disease. This study again showed a rationale for the blockade of the CD73-CD38-arginine system. Finally, a study also concluded that blocking the adenosine receptor A2AR together with anti-PD1 was effective in the treatment of metastatic and residual disease[88-91].
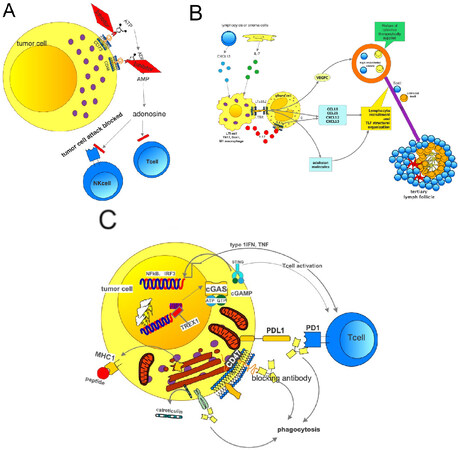
Figure 7. Mechanism of adenosine generation and its effects blocking T and NK cells. The generation of adenosine can be inhibited by either blocking the CD73 or CD38 exonucleases. Trials have been started (A); how tertiary lymph follicles can be formed. On the left side the natural mechanisms are shown. On top right the possible therapeutic intervention is shown (B); how to block the “no-eat-me” signal: a therapy using a blocking antibody for CD47 in combination with antiPDL1 will result in antigen processing by a macrophage, presentation of the antigen to cytotoxic T cells; as the PDL1 on expressing tumor cell is also blocked by an antiPDL1 antibody, the T lymphocyte can act and finally kill the tumor cell (C). PDL1: programmed death ligand 1
Adenosine receptors have another function which was recently studied. NK cells need an antigen stimulation together with costimulatory signals such as OX40 or GITR. Adenosine receptors A2AR and A2BR both decrease proliferation of NK cells. Blockade of CD73 increases NK cell function. NK cells can also be stimulated by Fms-like tyrosine kinase3-ligand, GM-CSF, IL2, and IL15[92]. Many pulmonary adenocarcinomas express A2A receptors. If these receptors are inhibited, this results in impaired growth of tumor cells and carcinoma associated myofibroblasts. If a carcinoma is positively tested for high expression of A2A receptors, an inhibitory therapy might inhibit tumor cell growth[93]. Another effect of adenosine is in the interaction of tumor cells and platelets: Tumor cells interacting with platelets induce a release of adenosine, and this induces an opening of the endothelial barrier, subsequently allowing transendothelial migration of tumor cells. The primary mediator of this effect is the endothelial P2Y2 receptor, which is activated by ATP[94]. Another ATP-gated receptor P2X(7) is expressed in multiple malignant tumors. Prolonged ATP-mediated activation of P2X(7) leads to formation of pores, increases membrane permeability and eventually causes cell death. This is a paradoxon: The TME contains high levels of extracellular ATP, which can activate the P2X(7) and trigger cell death. However, P2X(7) expression is associated with enhanced cancer cell survival, proliferation and metastatic potential. The explanation might be within a non-pore functional P2X(7)[95]. ATP and activation of P2 receptors are also linked to amplification of TGF-β1 induced migration of lung cancer cells[96,97]. Exposure to high ATP concentration induces P2X(7) expression and is essential for tumor cell survival, as ATP is rapidly internalized by micropinocytosis and thus supply energy to the tumor cells. This opens the possibility of another line of therapy[98,99].
Tertiary lymph follicles
A new option was recently discussed for carcinomas of the immune-desert type. Tertiary lymph follicles (TLF) are formed in some carcinomas. In these TLFs, there is a T cell zone with CD3 expressing lymphocytes and dendritic cells (DC) expressing LAMP3, a follicular zone with CD20+ lymphocytes and mature DCs. This can be simulated by CXCL13 and IL17, which will result in the recruitment of lymphoid tissue. Bcells and M1 macrophages can substitute this effect. An interaction with stroma cells by lymphotoxin-α1β2 and binding to lymphotoxin receptor leads to the secretion of VEGFC by the stroma cells. This in turn induces the formation of high endothelial venules, which cells secrete VCAM1 or MADCAM1 [Figure 7B]. As a result, lymphocytes are recruited to this area. A form of therapy might be beneficial using a mixture of cytokines (analogs) to induce high endothelial venule formation and finally influx of cytotoxic lymphocytes[100,101].
Another approach is the sequential combination of chemotherapy or radiotherapy, which can create TLFs, followed by the application of PDL1 therapy combined with either LAG3 or TIM3[100,102].
Prostaglandins
The inhibition of the prostaglandin system has always been a topic of discussion. Cyclooxygenase 2 (COX2) and PGE2 inhibit maturation of DC and activation of NK and T cells, and promotes differentiation of macrophages towards the M2 type. Inhibition of COX2 had shown some benefit in cancer therapy[103].
Design of cytotoxic lymphocytes and killer cells
A new way to target the immune system is the Car-technology (chimeric antigen receptor, Car). A fusion molecule is produced from the neoantigen with a fragment of the T cell receptor. This fusion molecule is expanded in patients’ lymphocytes. This was effectively applied in Bcell lymphomas. However, severe cytotoxicity (cytokine release syndrome) was seen when this therapy was applied in solid tumors[104,105]. A progress was made by combining Car T cell therapy with the gene editing system Crispr/Cas[106]. This field has expanded: instead of modified lymphocytes, the focus has shifted to natural killer (NK) cells and macrophages. These cells could also be modified to detect neoantigens from cancer cells and attack them. This therapy has much less toxicity, less cytokine release and less neurotoxicity[107-110]. NK cells could be harvested from pluripotent stem cells; in addition, macrophages could be polarized to M1 type.
Phagocytosis and the immune system
A very recent focus was drawn to phagocytosis. Phagocytosis is essential for macrophages and antigen presenting cells. A neoantigen needs to be phagocytosed to be processed. Don’t-eat-me signals were detected, which support cancer development and progression. This don’t-eat-me signal involves CD24 binding to sialic acid binding IG-like lectin 10 (Siglec10). This signal blocks innate immune cells, especially macrophages. A blockade of the CD24-Siglec10 binding decreased tumor growth[111]. Thus, another option for immunotherapy of cancer has been explored. Interestingly this is not the one-and-only phagocytosis checkpoint: Feng et al.[112] described in their review another phagocytosis checkpoint, namely the axis CD47-SIRP, another inhibitor of phagocytosis [Figure 7C]. On the other end, calreticulin promotes phagocytosis by macrophages. If the CD47-SIRPa axis is disrupted by a CD47 antibody, phagocytosis was increased and tumor progression inhibited. Under this treatment, an increase of M1 macrophages was observed. This treatment approach is also now tested in combination with PDL1.
Expression of CD47 has recently been reported in EGFR-mutated pulmonary adenocarcinoma cell lines. A treatment with an EGFR inhibitor combined with an CD47-blocking antibody induced downregulation of CD47 and increased phagocytosis by monocyte-derived dendritic cells (probably interdigitating dendritic cells)[113]. However, there are some limitations in this study: the results were based on cell cultures, focused especially on EGFR-mutated adenocarcinomas, and the dendritic cells were derived from blood monocytes, which do not reflect the dendritic cell population seen within NSCLC tissues[67,114]. Treatment trials have been installed, and first results seem promising. One study used intra-tumoral vaccination with autologous dendritic cells[115] and resulted in an increase of anti-tumor response by T cell infiltration. An ongoing trial is focusing on patients with surgical resected stage II and III NSCLC, who were also postoperatively treated by chemotherapy; pulsed dendritic cells are infused into patients to install an anti-tumor immune reaction[116]. Another study focused on the ICOS/ICOSL system, which is expressed on cells of the innate immune system, especially on dendritic cells. Blockade of the ICOS-system might improve T cell mediated cytotoxicity against lung cancer[117].
Bronchoalveolar lavage (BAL) is used for the evaluation of inflammatory diseases of the lung by washing out the lung lobes affected by an inflammatory/immune disease. Typing of lymphocytes allows for evaluation of disease activity, impact of therapy, and in few diseases also the diagnosis of the disease[118-122]. In cases of lung tumors, BAL is most often used to collect tumor cells from peripheral tumors not otherwise accessible. However, this tool might also be used to analyze the percentage of immune cells in lung lobes bearing lung carcinomas or molecules associated with immune reactions.
Response to immunotherapy cannot be assessed with any of the present-day markers. Neither PDL1, nor tumor mutational burden by itself can predict response with certainty. Expression of different modulators of immune responses such as CD73, CD38, CD24, CD47 and many more can be evaluated on tumor cells, immune cells, or as soluble markers in BAL.
Another approach recently tested by many investigators is to harvest circulating tumor cells together with platelets, lymphocytes, macrophages, and neutrophils and to study their interaction (for example by cDNA or proteomic analysis). In addition, organ cultures cultivated together with their stroma/microenvironment opens such an insight into the immune mechanisms acting in a given carcinoma[123,124].
HLA types associated with response/resistance (HLA diversity) should be included into the tests, as well as the analysis of the TMA with infiltrating T-lymphocytes (Treg, T-CD8), neoantigen diversity, clonal/subclonal neoantigen diversity, etc.[20,26]. The landscape of tumor immunology is almost daily expanding, and we are only at the beginning of understanding the multitude of mechanisms which play a role in the tumor immune system interplay.
Conclusion
We have discussed the PD1/PDL1 checkpoint inhibition and the mechanisms of resistance. We focused on cells of the specific and innate immune system and their role in immune tolerance, as well as possibilities to introduce new therapy options. We finally focused on the new field of soluble mediators involved in regulating the tumor-immune system interaction, and new therapy avenues. At the end, some thoughts on methods for better evaluating this tumor immune system interaction were discussed.
Declarations
Author’s contributionsDesigned and wrote the article: Popper HH
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Peggs KS, Quezada SA. PD-1 blockade: promoting endogenous anti-tumor immunity. Expert Rev Anticancer Ther 2012;12:1279-82.
3. Velcheti V, Rimm DL, Schalper KA. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1). J Thorac Oncol 2013;8:803-5.
4. Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res 2013;19:1021-34.
5. Hamid O, Carvajal RD. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin Biol Ther 2013;13:847-61.
6. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65.
7. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54.
8. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013;3:1355-63.
9. Davies M. New modalities of cancer treatment for NSCLC: focus on immunotherapy. Cancer Manag Res 2014;6:63-75.
10. Yang CY, Lin MW, Chang YL, Wu CT, Yang PC. Programmed cell death-ligand 1 expression in surgically resected stage I pulmonary adenocarcinoma and its correlation with driver mutations and clinical outcomes. Eur J Cancer 2014;50:1361-9.
11. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med 2015;21:24-33.
12. Pan ZK, Ye F, Wu X, An HX, Wu JX. Clinicopathological and prognostic significance of programmed cell death ligand1 (PD-L1) expression in patients with non-small cell lung cancer: a meta-analysis. J Thorac Dis 2015;7:462-70.
13. Karachaliou N, Cao MG, Teixidó C, Viteri S, Morales-Espinosa D, et al. Understanding the function and dysfunction of the immune system in lung cancer: the role of immune checkpoints. Cancer Biol Med 2015;12:79-86.
14. Teixidó C, Karachaliou N, González-Cao M, Morales-Espinosa D, Rosell R. Assays for predicting and monitoring responses to lung cancer immunotherapy. Cancer Biol Med 2015;12:87-95.
15. Ochoa CE, Mirabolfathinejad SG, Ruiz VA, Evans SE, Gagea M, et al. Interleukin 6, but not T helper 2 cytokines, promotes lung carcinogenesis. Cancer Prev Res (Phila) 2011;4:51-64.
16. Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol 2017;12:208-22.
17. Lu J, Lee-Gabel L, Nadeau MC, Ferencz TM, Soefje SA. Clinical evaluation of compounds targeting PD-1/PD-L1 pathway for cancer immunotherapy. J Oncol Pharm Pract 2015;21:451-67.
18. O’Byrne K. Stimulating immune responses to fight cancer: basic biology and mechanisms. Asia Pac J Clin Oncol 2015;11:9-15.
19. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, et al. Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015;373:123-35.
20. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133-50.
21. Pujol JL, De Pas T, Rittmeyer A, Vallières E, Kubisa B, et al. Safety and immunogenicity of the PRAME cancer immunotherapeutic in patients with resected non-small cell lung cancer: a phase I dose escalation study. J Thorac Oncol 2016;11:2208-17.
22. Tveita AA, Schjesvold F, Haabeth OA, Fauskanger M, Bogen B. Tumors escape CD4+ T-cell-mediated immunosurveillance by impairing the ability of infiltrating macrophages to indirectly present tumor antigens. Cancer Res 2015;75:3268-78.
23. Martin K, Schreiner J, Zippelius A. Modulation of APC function and anti-tumor immunity by anti-cancer drugs. Front Immunol 2015;6:501.
24. Scheel AH, Ansén S, Schultheis AM, Scheffler M, Fischer RN, et al. PD-L1 expression in non-small cell lung cancer: correlations with genetic alterations. Oncoimmunology 2016;5:e1131379.
25. Deeb KK, Hohman CM, Risch NF, Metzger DJ, Starostik P. Routine clinical mutation profiling of non-small cell lung cancer using next-generation sequencing. Arch Pathol Lab Med 2015;139:913-21.
26. Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med 2019;25:767-75.
27. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8.
28. Saito K, Nakayama E, Valmori D, Kato T. Immune responses to the cancer testis antigen XAGE-1b in non small cell lung cancer caucasian patients. PLoS One 2016;11:e0150623.
29. Ohue Y, Kurose K, Nozawa R, Isobe M, Nishio Y, et al. Survival of lung adenocarcinoma patients predicted from expression of PD-L1, Galectin-9, and XAGE1 (GAGED2a) on tumor cells and tumor-infiltrating T cells. Cancer Immunol Res 2016;4:1049-60.
30. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020;581:100-5.
31. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, et al. Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell 2017;171:934-49.e16.
32. Sarkizova S, Hacohen N. How T cells spot tumour cells. Nature 2017;551:444-6.
33. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551:512-6.
34. Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017;551:517-20.
35. Pereira C, Gimenez-xavier P, Pros E, Pajares MJ, Moro M, et al. Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin Cancer Res 2017;23:3203-13.
36. Bilsborough J, Panichelli C, Duffour MT, Warnier G, Lurquin C, et al. A MAGE-3 peptide presented by HLA-B44 is also recognized by cytolytic T lymphocytes on HLA-B18. Tissue Antigens 2002;60:16-24.
37. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018;359:582-7.
38. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017;541:321-30.
39. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res 2015;3:1082-95.
40. Domagala-Kulawik J. The role of the immune system in non-small cell lung carcinoma and potential for therapeutic intervention. Transl Lung Cancer Res 2015;4:177-90.
41. Ager A, Watson HA, Wehenkel SC, Mohammed RN. Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem Soc Trans 2016;44:377-85.
42. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 2015;6:7458.
43. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res 2014;74:7168-74.
44. Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, et al. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 2010;29:1093-102.
45. Aithal A, Rauth S, Kshirsagar P, Shah A, Lakshmanan I, et al. MUC16 as a novel target for cancer therapy. Expert Opin Ther Targets 2018;22:675-86.
46. Gonzalez-Gugel E, Saxena M, Bhardwaj N. Modulation of innate immunity in the tumor microenvironment. Cancer Immunol Immunother 2016;65:1261-8.
47. He Y, Zhang X, Jia K, Dziadziuszko R, Zhao S, et al. OX40 and OX40L protein expression of tumor infiltrating lymphocytes in non-small cell lung cancer and its role in clinical outcome and relationships with other immune biomarkers. Transl Lung Cancer Res 2019;8:352-66.
48. Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with Anti-OX40. Clin Cancer Res 2017;23:6165-77.
49. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, et al. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer 2016;52:50-66.
50. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 2016;50:312-32.
51. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): the next immune checkpoint receptor. Semin Immunol 2019;42:101305.
52. Puhr HC, Ilhan-Mutlu A. New emerging targets in cancer immunotherapy: the role of LAG3. ESMO Open 2019;4:e000482.
53. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev 2017;276:80-96.
54. Antonia SJ, Vansteenkiste JF, Moon E. Immunotherapy: beyond anti-PD-1 and anti-PD-L1 therapies. Am Soc Clin Oncol Educ Book 2016;35:e450-8.
56. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, et al. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol 2009;182:6207-16.
57. Liu Y, Chen K, Wang C, Gong W, Yoshimura T, et al. Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res 2013;73:550-60.
58. Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med 2011;17:579-87.
59. Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci 2006;97:439-47.
60. Wang YC, He F, Feng F, Liu XW, Dong GY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res 2010;70:4840-9.
61. Wu H, Shang LQ, Chen RL, Yang SM, Wang SL, et al. Significance of Trask protein interactions in brain metastatic cohorts of lung cancers. Tumour Biol 2015;36:4181-7.
62. Huang WC, Chan ML, Chen MJ, Tsai TH, Chen YJ. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget 2016;7:39363-75.
63. Zanin-Zhorov A, Flynn R, Waksal SD, Blazar BR. Isoform-specific targeting of ROCK proteins in immune cells. Small GTPases 2016;7:173-7.
64. Olsson A, Nakhlé J, Sundstedt A, Plas P, Bauchet AL, et al. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J Immunother Cancer 2015;3:53.
65. Wang Y, Sparwasser T, Figlin R, Kim HL. Foxp3+ T cells inhibit antitumor immune memory modulated by mTOR inhibition. Cancer Res 2014;74:2217-28.
66. Kim PS, Jochems C, Grenga I, Donahue RN, Tsang KY, et al. Pan-Bcl-2 inhibitor, GX15-070 (obatoclax), decreases human T regulatory lymphocytes while preserving effector T lymphocytes: a rationale for its use in combination immunotherapy. J Immunol 2014;192:2622-33.
67. Brcic L, Stanzer S, Krenbek D, Gruber-Moesenbacher U, Absenger G, et al. Immune cell landscape in therapy-naïve squamous cell and adenocarcinomas of the lung. Virchows Arch 2018;472:589-98.
68. Adah D, Hussain M, Qin L, Qin L, Zhang J, et al. Implications of MDSCs-targeting in lung cancer chemo-immunotherapeutics. Pharmacol Res 2016;110:25-34.
69. McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting ph regulation, angiogenesis and immune dysfunction. Front Cell Dev Biol 2016;4:27.
70. Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, et al. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res 2015;75:996-1008.
71. Orozco-Morales M, Soca-Chafre G, Barrios-Bernal P, Hernández-Pedro N, Arrieta O. Interplay between cellular and molecular inflammatory mediators in lung cancer. Mediators Inflamm 2016;2016:3494608.
72. Kargl J, Busch SE, Yang GH, Kim KH, Hanke ML, et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat Commun 2017;8:14381.
73. Kargl J, Zhu X, Zhang H, Yang GHY, Friesen TJ, et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 2019;4:130850.
74. Schlie K, Spowart JE, Hughson LR, Townsend KN, Lum JJ. When cells suffocate: autophagy in cancer and immune cells under low oxygen. Int J Cell Biol 2011;2011:470597.
75. Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S. The selective degradation of synaptic Connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J Biol Chem 2015;290:23670-9.
76. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol 2014;5:532.
77. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 2017;5:101.
78. Zhang Y, Fu J, Shi Y, Peng S, Cai Y, et al. A new cancer immunotherapy via simultaneous DC-mobilization and DC-targeted IDO gene silencing using an immune-stimulatory nanosystem. Int J Cancer 2018;143:2039-52.
79. Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer 2019;19:162-75.
80. Mellor AL, Lemos H, Huang L. Indoleamine 2,3-Dioxygenase and tolerance: where are we now? Front Immunol 2017;8:1360.
81. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res 2016;76:2076-81.
82. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity 2017;46:233-44.
83. Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy 2017;9:83-97.
84. Feng PH, Lee KY, Chang YL, Chan YF, Kuo LW, et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med 2012;186:1025-36.
85. Raber P, Ochoa AC, Rodríguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest 2012;41:614-34.
86. Wang J, Matosevic S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J Mol Med (Berl) 2018;96:903-13.
87. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 2016;7:109.
88. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer 2017;17:709-24.
89. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016;5:e1208875.
90. Schmid S, Kübler M, Korcan Ayata C, Lazar Z, Haager B, et al. Altered purinergic signaling in the tumor associated immunologic microenvironment in metastasized non-small-cell lung cancer. Lung Cancer 2015;90:516-21.
91. Mittal D, Young A, Stannard K, Yong M, Teng MW, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res 2014;74:3652-8.
92. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, et al. Harnessing innate immunity in cancer therapy. Nature 2019;574:45-56.
93. Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, et al. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol Ther 2013;14:860-8.
94. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013;24:130-7.
95. Gilbert SM, Oliphant CJ, Hassan S, Peille AL, Bronsert P, et al. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019;38:194-208.
96. Takai E, Tsukimoto M, Harada H, Kojima S. Autocrine signaling via release of ATP and activation of P2X7 receptor influences motile activity of human lung cancer cells. Purinergic Signal 2014;10:487-97.
97. Takai E, Tsukimoto M, Harada H, Sawada K, Moriyama Y, et al. Autocrine regulation of TGF-β1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J Cell Sci 2012;125:5051-60.
98. Qian Y, Wang X, Liu Y, Li Y, Colvin RA, et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett 2014;351:242-51.
99. Qian Y, Wang X, Li Y, Cao Y, Chen X. Extracellular ATP a new player in cancer metabolism: NSCLC cells internalize ATP in vitro and in vivo using multiple endocytic mechanisms. Mol Cancer Res 2016;14:1087-96.
100. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 2014;189:832-44.
101. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 2019;19:307-25.
102. Sautès-Fridman C, Cherfils-Vicini J, Damotte D, Fisson S, Fridman WH, et al. Tumor microenvironment is multifaceted. Cancer Metastasis Rev 2011;30:13-25.
103. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015;4:e954829.
104. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med 2017;68:139-52.
105. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov 2018;8:1219-26.
106. Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol 2018;79:876-82.
107. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018;23:181-92.e5.
108. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol 2018;9:283.
109. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther 2017;25:1769-81.
110. Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol 2019;41:59-68.
111. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019;572:392-6.
112. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 2019;19:568-86.
113. Nigro A, Ricciardi L, Salvato I, Sabbatino F, Vitale M, et al. Enhanced expression of CD47 is associated with off-target resistance to tyrosine kinase inhibitor gefitinib in NSCLC. Front Immunol 2019;10:3135.
114. Stankovic B, Bjørhovde HAK, Skarshaug R, Aamodt H, Frafjord A, et al. Immune cell composition in human non-small cell lung cancer. Front Immunol 2018;9:3101.
115. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8+ T-cell infiltration. Clin Cancer Res 2017;23:4556-68.
116. Saka H, Kitagawa C, Ichinose Y, Takenoyama M, Ibata H, et al. A randomized phase II study to assess the effect of adjuvant immunotherapy using α-GalCer-pulsed dendritic cells in the patients with completely resected stage II-IIIA non-small cell lung cancer: study protocol for a randomized controlled trial. Trials 2017;18:429.
117. Marinelli O, Nabissi M, Morelli MB, Torquati L, Amantini C, et al. ICOS-L as a potential therapeutic target for cancer immunotherapy. Curr Protein Pept Sci 2018;19:1107-13.
118. Popper H, Pongratz M. Value and indications for bronchoalveolar lavage combined with transbronchial lung biopsy. Wien Klin Wochenschr 1987;99:848-55.
119. Robinson PC, Watters LC, King TE, Mason RJ. Idiopathic pulmonary fibrosis. Abnormalities in bronchoalveolar lavage fluid phospholipids. Am Rev Respir Dis 1988;137:585-91.
120. Oshima M, Maeda A, Ishioka S, Hiyama K, Yamakido M. Expression of C-C chemokines in bronchoalveolar lavage cells from patients with granulomatous lung diseases. Lung 1999;177:229-40.
121. Magi B, Bini L, Perari MG, Fossi A, Sanchez JC, et al. Bronchoalveolar lavage fluid protein composition in patients with sarcoidosis and idiopathic pulmonary fibrosis: a two-dimensional electrophoretic study. Electrophoresis 2002;23:3434-44.
122. Bozinovski S, Jones JE, Vlahos R, Hamilton JA, Anderson GP. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J Biol Chem 2002;277:42808-14.
123. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am J Respir Crit Care Med 2018;198:777-87.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Popper HH. Manipulation of the immune system by non-small cell lung cancer and possible therapeutic interference. Cancer Drug Resist 2020;3:710-25. http://dx.doi.org/10.20517/cdr.2020.40
AMA Style
Popper HH. Manipulation of the immune system by non-small cell lung cancer and possible therapeutic interference. Cancer Drug Resistance. 2020; 3(4): 710-25. http://dx.doi.org/10.20517/cdr.2020.40
Chicago/Turabian Style
Popper, Helmut H.. 2020. "Manipulation of the immune system by non-small cell lung cancer and possible therapeutic interference" Cancer Drug Resistance. 3, no.4: 710-25. http://dx.doi.org/10.20517/cdr.2020.40
ACS Style
Popper, HH. Manipulation of the immune system by non-small cell lung cancer and possible therapeutic interference. Cancer Drug Resist. 2020, 3, 710-25. http://dx.doi.org/10.20517/cdr.2020.40
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 1
likes
Like This Article 1
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.