
The dark side of immunotherapy: pancreatic cancer


Abstract
Since the journal Science deemed cancer immunotherapy as the “breakthrough of the year” in 2014, there has been an explosion of clinical trials involving immunotherapeutic approaches that, in the last decade - thanks also to the renaissance of the immunosurveillance theory (renamed the three Es theory) - have been continuously and successfully developed. In the latest update of the development of the immuno-oncology drug pipeline, published last November by Nature Review Drug Discovery, it was clearly reported that the immunoactive drugs under study almost doubled in just two years. Of the different classes of passive and active immunotherapies, “cell therapy” is the fastest growing. The aim of this review is to discuss the preclinical and clinical studies that have focused on different immuno-oncology approaches applied to pancreatic cancer, which we assign to the “dark side” of immunotherapy, in the sense that it represents one of the solid tumors showing less response to this type of therapeutic strategy.
Keywords
Introduction
Pancreatic ductal adenocarcinoma (PDAC) represents 90% of all pancreatic malignancies[1], with a 5-year survival rate of 10%[2]. According to the American Cancer Society, the estimated number of new pancreatic cancer cases in the USA during 2020 will be 57,600 and the anticipated deaths caused by pancreatic cancer will be 47,050[2]. PDAC is predicted to be the second leading cause of cancer-related death by 2030[3]. Similar to other epithelial malignancies, a progression model from precursor lesions to PDAC has been elaborated. These lesions are unified under the collective term of pancreatic intraepithelial neoplasia (PanIN)[4]. Activating mutations in the KRAS proto-oncogene have an initiating function and are present in over 90% of invasive PDAC cases. Consequently, genetic and epigenetic inactivation of suppressor genes act as promoting factors: these frequently involve p16 (90%), SMAD4 (55%) and BRCA2 (10%)[5]. Around 50%-70% of PDAC cases carry mutations in P53, which occur at later stages of PanIN, contributing to the malignant progression of PDAC rather than its initiation[5].
The median survival of PDAC patients is 4-6 months, partly because the disease usually only becomes clinically evident at a late stage, and due to its resistance to all forms of chemotherapy and radiotherapy because of its prominent desmoplastic reaction and immunosuppressive microenvironments[6]. Single agent gemcitabine, and chemotherapy combinations as well, such as FOLFIRINOX (irinotecan, oxaliplatin, 5-fluorouracil and leucovorin), prolong life expectancy only moderately[7]. Surgical resection remains the only curative option for PDAC patients but is restricted to earlier disease stages and is applicable to less than 20% of newly diagnosed patients. Considering the few benefits provided by the approved therapies and the variety of dysregulated signaling pathways in pancreatic cancer, targeted therapies have emerged as a potential way to strategically direct treatment to tumor cells and increase the patient’s survival. Immunotherapy is another important non-chemotherapeutic strategy, and it is a treatment designed to induce or enhance the immune system response against cancer. Accordingly, the goal of immunotherapy is to stop or slow down cancer cell growth and metastatic events through the enhanced recognition of cancer cells by the host immune system[7].
In agreement with the concept of the “cancer immunity cycle”[8], the wide spectrum of cancer immunotherapies can be associated with each different step. Accordingly, dendritic cells (DCs) must capture neoantigens released by necrotic and apoptotic cancer cells (step 1) and present them to T cells (step 2) to induce effector T cell priming and activation against cancer-specific antigens (step 3). Activated T cells then travel through blood vessels (step 4) to infiltrate the tumo area (step 5). Here, T cells recognize specific antigens presented in association with major histocompatibility complex (MHC) class I molecules on the cancer cell surface (step 6) and kill their targets (step 7)[8]. The first steps inducing priming, clonal expansion and activation of immune cells can be facilitated by cancer vaccines, which play an active role in directing the patient’s own immune system towards tumor-specific antigens. In addition, adoptive immunotherapy acts in a passive way, delivering ready-to-use lymphocytes able to infiltrate tumor sites, recognize cancer cells and destroy them. Finally, monoclonal antibodies (mAbs) and immune checkpoint inhibitors (ICIs) can be designed to target different steps throughout the whole cycle, acting as potent adjuvant agents[8][Figure 1].
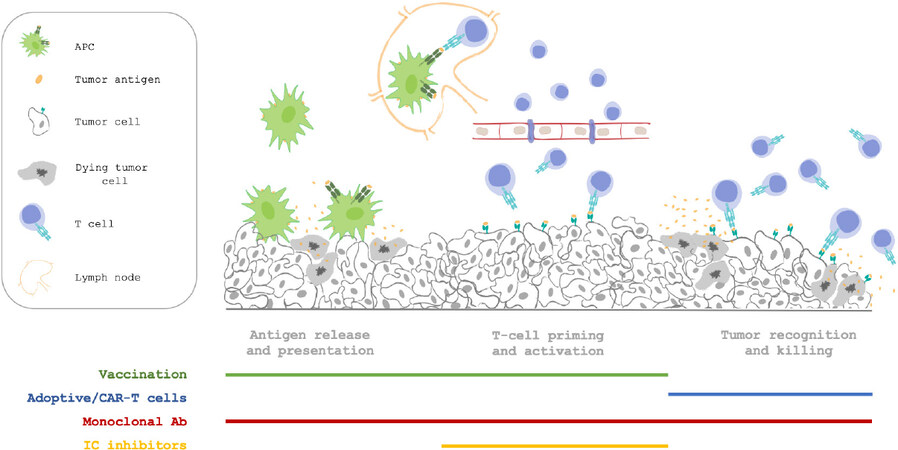
Figure 1. Immunotherapy intervention in the different steps of antitumor response. In this review, we will describe the current landscape of preclinical and clinical advances made in immunotherapy applied to pancreatic cancer. APC: antigen-presenting cell; CAR: chimeric antigen receptor
Cancer vaccines
Cancer vaccines have emerged to be very promising for treating different cancer types such as melanoma and lung, breast and renal cell carcinoma[9]. However, vaccine-based immunotherapy in PDAC has shown controversial results, thus requiring further studies[9]. Vaccines are designed to prime the patient’s own T cells against cancer-specific antigens by promoting effective anticancer immunity [Figure 1]. The most effective vaccines include different combinations to overcome the immunosuppressive microenvironment promoted by the tumor itself.
Vaccines deliver antigens either in the form of DNA, peptides or tumor cells that are captured by antigen-presenting cells (APCs). Once activated, loaded DCs migrate to the nearest draining lymph nodes and present antigens on MHC molecules that are recognized by T cells through their specific T cell receptors (TCRs). These crucial steps lead to the priming and activation of T cells. One specific form of vaccine is represented by antigen-pulsed DCs that directly migrate to the lymph nodes, thus avoiding the first steps of antigen expression and capture. Hence, activated antigen-specific T cells can recirculate and migrate into the tumor microenvironment (TME), where they can again recognize specific antigens expressed by tumor cells and thus destroy them. Consequently, dying cancer cells release new antigens that trigger adaptive immune responses and allow the expansion of other cancer-specific T cells. To be effective, it is crucial that the choice of antigen be restricted to the tumor or only minimally expressed by other tissues. Indeed, genes coding for tumor-associated antigens (TAAs) can also be found in normal cells but are aberrantly expressed in cancer cells or the encoded proteins have undergone abnormal posttranslational modifications during carcinogenesis.
Significant efforts have been made to identify new pancreatic cancer-associated antigens, preferably shared by the majority of patients, such as carcinoembryonic antigen (CEA)[10], mucin-1 (MUC-1)[10] and the product of mutated KRAS[10], to develop targeted vaccination strategies. More recently, novel TAAs have been associated with PDAC, including alpha-enolase (ENO1)[11-14], carcinoembryo antigen-related cell adhesion molecule 6 (CEACAM6)[15] and mucin-4 (MUC-4)[16], although they are not yet in clinical use.
GVAX (granulocyte-macrophage colony stimulating factor expressing vaccine)
One of the most promising vaccines in PDAC treatment is GVAX, a whole-cell vaccine consisting of allogeneic human pancreatic cancer cells engineered to express granulocyte-macrophage colony stimulating factor (GM-CSF)[17]. Several studies have highlighted that patients with advanced cancer have limited macrophage function, which could be restored by GM-CSF. Indeed, GM-CSF enhances DC activity and promotes antigen presentation to T cells[17]. GVAX was designed on the basis of preclinical data demonstrating that irradiated B16 melanoma cells expressing GM-CSF promoted the establishment of a long-lasting and specific antitumor response[17]. A phase I trial was conducted in surgically resected PDAC patients to assess its safety and effectiveness in inducing an antitumor immune response[18][Table 1]. At 8 weeks after pancreaticoduodenectomy, 14 patients received different doses of GVAX and a 6-month course of adjuvant radio- and chemotherapy. Afterwards, 6 out of 14 patients received additional vaccination. Since no dose-limiting toxicities were reported and the antitumor immunity increased in a dose-dependent manner, a follow-up phase II adjuvant study (NCT00084383) was conducted in 60 patients with resectable PDAC [Table 1]. GVAX was administered five times before 5-FU-based chemoradiotherapy and the combined treatment was administered monthly in disease-free patients for a total of four rounds[19]. Finally, a 5th and final dose was administered after 6 months. Data from this study showed that GVAX elicited the activation and expansion of tumor-specific CD8+ T cells, thus improving the patients’ OS. Interestingly, disease-free patients after the final GVAX treatment could elicit antigen spreading against a wide range of TAAs, suggesting that GVAX immunotherapy can be used both alone as an adjuvant therapy and/or in combination with conventional treatment[19].
Summary of clinical trials for the indicated immunotherapy approaches
| Immunotherapeutic treatment | ID number | Phase | No. of patients | Cohort composition | Strategy | Results |
|---|---|---|---|---|---|---|
| Whole cell-based vaccination | [18] | I | 14 | Surgically resected | GVAX + chemoradiotherapy | No toxicities; dose-dependent antitumor immunity increased |
| NCT00084383[19] | II | 60 | Surgically resected | GVAX + chemoradiotherapy | Expansion of CD8+ T cells; increased OS | |
| [20] | Pilot study | 50 | ADV | GVAX + cyclophosphamide | Minimal toxicities; increased T cell response; improved OS | |
| NCT00727441[21] | II | 87 | Surgically resectable | GVAX ± cyclophosphamide | TLS formation; increased T cell response; increased OS | |
| NCT00585845[23] | I | 17 | Treatment-refractory | CRS-207 | Dose-dependent antitumor immunity increased | |
| NCT01417000[24] | II | 93 | Pretreated metastatic | GVAX + cyclophosphamide ± CRS-207 | Minimal toxicities; increased T cell response; increased OS | |
| NCT02004262[25] | IIb | 213 | Pretreated metastatic | GVAX + cyclophosphamide + CRS-207; CRS-207 alone; CT | GVAX + CT + CRS-207 no improved survival over CT | |
| Peptide-based vaccination | [27] | I/II | 5 | ADV | Synthetic RAS-loaded APCs | 2/5 pts showed increased immune response and OS |
| CTN-95002/97004[28] | I/II | 48 | 10 resected/38 ADV | Synthetic RAS-loaded APCs + GM-CSF | Increased OS and immune response in 58% of pts | |
| CTN-95002/98010[29] | 10 yrs follow-up | 23 | Surgically resected | Synthetic RAS-loaded APCs + GM-CSF | Long-lasting vaccine-induced immune response in 85% of pts | |
| [31] | I/II | 48 | Unresectable | GV1001 + GM-CSF | Vaccination well tolerated; immune response in 75% of pts | |
| ISCRCTN-4382138[32] | III | 1062 | ADV/Metastatic | CT; GV1001 after or with CT | No differences between CT and combined treatment | |
| UMIN000000905[35] | I | 6 | Unresectable ADV | Survivin-2B80-88 + IFN-α | > 50% of pts with positive clinical and immunological responses | |
| UMIN000012146[36] | II | 83 | HLA-A24+ | Survivin-2B80-88 ± IFN-β | Combined treatment increased pts survival w/o toxicity | |
| Dendritic cell-based vaccination | [41] | I/II | 10 | Surgically resected | MUC1 peptide-loaded DCs | No toxicity; 3/10 pts alive after 4 yrs |
| 06DZ19009[42] | I | 7 | Metastatic | MUC1 peptide-loaded DCs | No toxicity; no clinical benefits | |
| UMIN000004855[44] | I | 10 | HLA-A2*2402 | WT1 peptide-loaded DCs | No toxicity; increased survival only in pts w/o liver metastasis | |
| UMIN000004063[45] | I | 10 | ADV | WT1 peptide-loaded DCs ± GEM | No toxicity; increased OS and PFS in 50% of pts | |
| NCT01410968[47] | I | 12 | Metastatic | hTERT/CEA/Survivin peptide loaded DCs + poly (IC:LC) | Fatigue and/or flu-like symptoms; 4/12 pts SD and 4/12 pts PD | |
| Adoptive cell transfer | NCT00965718[75] | II | 43 | Surgically resected | MUC1-CTLs + GEM | Reduction of liver and local recurrences |
| [77] | 20 | Treatment-refractory | CIK cells ± GEM | No differences observed between CT and combined therapy | ||
| [78] | 58 | Pretreated ADV | CIK cells ± S-1 | No differences observed between CT and combined therapy | ||
| CAR-T | NCT01897415[109] | I | 6 | Treatment-refractory | Anti-MSLN CAR-T | 2 pts with SD; MAV tumor lesions stable in 3 pts and decreased in 1 pt by 69.2% |
| Monoclonal antibody | NCT00711191[154] | I | 22 | CT-naïve ADV | CP-870,893 + GEM | 4 pts PR; 11 pts SD |
| Immune checkpoint inhibitors | NCT00112580[177] | II | 27 | ADV/metastatic | Ipilimumab (anti-CTLA-4) | 1/27 subject showed a significant delayed response |
| NCT00729664[180] | I | 14 | ADV | BMS-936559 (anti-PD-L1) | No response observed | |
| NCT01876511[181] | II | 8 | MMR-deficient | Pembrolizumab (anti PD-1) | 2 CR; 3 PR; 1 SD | |
| NCT00556023[187] | Ib | 34 | ADV | GEM ± Tremelimumab (anti-CTLA-4) | 6% PR; 21% SD; OS 7.4 months | |
| NCT01473940[188] | Ib | 21 | Unresectable ADV | GEM ± Ipilimumab (anti-CTLA-4) | Safe and tolerable regimen; 14% ORR; 33% SD; OS 6.9 months | |
| NCT023311251[189] | Ib/II | 17 | Pretreated/CT naive | Pembrolizumab (anti-PD-1) + GEM/nab-P | 25% PR; 67% SD; OS 15 months (CT naïve pts); no PD pts | |
| NCT02309177[190] | I | 42 | CT-naïve ADV | Nivolumab (anti-PD-1)/nab-P ± GEM | 2% CR; 16% PR; 46% SD; OS 9.9 months | |
| [197] | I | 30 | Pretreated ADV | Ipilimumab (anti-CTLA-4) ± GVAX | OS 5.7 months in pts with combined therapy, compared to 3.6 months in pts treated with ipilimumab alone |
To further assess the efficacy of the combination of another chemotherapeutic drug and GVAX, a study was conducted administering two allogeneic GM-CSF-secreting pancreatic cell lines (CG8020/CG2505), alone or with cyclophosphamide, in 50 patients with advanced PDAC[20][Table 1]. Again, data showed minimal treatment-related toxicity and higher specific T cell responses in patients who received the combined treatment compared to those who received vaccine or cyclophosphamide as monotherapy, resulting in improved overall survival (OS)[20].
A subsequent neoadjuvant and adjuvant clinical trial (NCT00727441) was conducted in 87 resectable PDAC patients to compare the efficacy of GVAX alone or in combination with low-dose cyclophosphamide to study how the TME is altered by immunotherapy[21][Table 1]. In most patients, combined treatment caused the formation of intratumoral tertiary lymphoid aggregates, as a regulatory structure of adaptive immunity. Moreover, combined treatment showed improved survival and an increased specific T cell response[21]. Regarding combined GVAX and cyclophosphamide treatment, an ongoing phase II clinical trial (NCT01088789) is now in the recruiting step and will be completed by 2023. The aim of this study is to assess the safety and viability of a long-term GVAX vaccination alone or in combination with cyclophosphamide in surgically resected pancreatic adenocarcinoma. This trial will evaluate the effect of GVAX vaccination administered every 6 months for a period of 10 years.
In a different approach, the bacterium Listeria monocytogenes, due to its ability to induce potent innate and adaptive immunity, was engineered to express the human mesothelin TAA (CRS-207) and used as a cancer vaccine[22]. A phase I study (NCT00585845) was conducted in 17 treatment-refractory PDAC patients, in which safety, toxicity and clinical activity were evaluated [Table 1]. Results showed that CRS-207 was safe and tolerable, and that the immune response increased in a dose-dependent manner[23]. These promising results prompted investigators to combine the CRS-207 vaccine with GVAX. A phase II clinical trial (NCT01417000) was conducted in 93 previously treated patients with metastatic PDAC[24][Table 1]. Half of patients received two doses of GVAX with low-dose cyclophosphamide (Cy/GVAX) followed by four doses of CRS-207 every 3 weeks, while the second half of patients received six doses of Cy/GVAX every 3 weeks. The results demonstrated that combined treatment significantly enhanced OS, inducing a mesothelin-specific T cell response, with minimal toxicity[24]. However, the ECLIPSE Study (NCT02004262), a phase IIB clinical trial conducted in 303 patients, in which Cy/GVAX plus CRS-207, CRS-207 alone, and single-agent chemotherapy were compared in previously treated patients with metastatic pancreatic adenocarcinoma, suggested that the combined treatment (even if well-tolerated) did not ameliorate survival compared to chemotherapy alone[25][Table 1].
Peptide-based cancer vaccines
In addition to cell lines, mutated peptides have also been used to immunize pancreatic cancer patients to enhance tumor-specific T cell responses. Peptide-based vaccines represent a promising tool for immunotherapy. Peptides are easily synthesized on the basis of the most immunogenic portions of TAAs or specifically identified mutations that are associated with each patient (personalized vaccine). The advantage of a peptide-based vaccine compared to the whole protein is to increase the number of peptide-MHC complexes available for T cell recognition and improve the immune response.
The most well-known driving mutation is on codon 12 of the KRAS gene, which has therefore been employed in a peptide-based cancer vaccine. Indeed, pioneer clinical studies assessed the safety and feasibility of synthetic RAS peptides loaded ex vivo on autologous antigen-presenting cells in five PDAC patients[26,27][Table 1]. Two clinical trials (CTN-95002 and CTN-97004) exclusively based on peptides, were conducted in 48 PDAC patients by combining synthetic mutant RAS peptide vaccination with GM-CSF[28][Table 1]. The results showed that specific T cell responses and increased survival were induced in almost 58% of evaluable patients. Furthermore, to evaluate potential long-term response, a follow-up study was conducted (CTN-95002 and CTN-98010)[29][Table 1]. A total of 23 patients were followed for more than 10 years and 85% of them showed a long-lasting vaccine-induced immune response, underlining the importance of combined treatments in patients carrying RAS mutations[29].
Regarding tumor-specific antigens, the telomerase reverse transcriptase (TERT) protein was shown to be overexpressed by the majority of pancreatic cancers[30]. Based on this TAA-like feature, the GV1001 vaccine, consisting of 16 amino acids derived from human TERT, was developed[31]. To assess vaccine safety, stability and toxicity, a phase I/II study was conducted in 48 unresectable PDAC patients receiving scalar doses of the vaccine in combination with GM-CSF. As well as being well-tolerated, the vaccine induced a specific immune response in 75% of patients[30][Table 1]. These promising results paved the way to design the first phase III clinical trial based on GV1001 administration, called TeloVac (ISRCTN-4382138)[32][Table 1]. A total of 1062 patients with locally advanced or metastatic PDAC were subdivided into three groups, namely patients receiving gemcitabine and capecitabine, those receiving gemcitabine + capecitabine followed by GV1001 vaccination (sequentially), and those receiving gemcitabine + capecitabine and GV1001 vaccination (concurrently). Unfortunately, OS was not significantly different between chemotherapy alone and the combined treatments. In addition, it was shown that gemcitabine and capecitabine combined with GV1001 vaccination did not reduce C-reactive protein, IL6 or GM-CSF levels[33]. Despite the discouraging results, the study of a large number of patients enrolled in the study resulted in the identification of 19 cytokines that were significantly reduced in patients receiving sequential chemo-immunotherapy but not in the group treated concurrently with GV1001 and chemotherapy. These cytokines could potentially be used as biomarkers in clinical follow-up[34].
Several studies have also highlighted survivin as one of the most promising candidates for tumor-specific immunotherapy in pancreatic cancer. Survivin is a member of the inhibitor of apoptosis protein family that prevents apoptotic cell death and its expression is high in numerous tumors, including PDAC. In particular, an HLA-A24-restricted antigenic peptide, survivin-2B80-88 (AYACNTSTL), was identified to develop a peptide-based vaccine. A phase I clinical trial (UMIN000000905) was conducted in unresectable advanced HLA-A24-positive PDAC patients, who received survivin-2B80-88 peptide in combination with interferon alpha (IFN-α)[35][Table 1]. Encouraging results emerged from the study, as more than 50% of patients had positive clinical and immune responses with stabilization of the disease. Despite the clinical results, some IFN-α-related side effects were detectable. Therefore, a phase II clinical trial (UMIN000012146) in which 83 HLA-A24-positive PDAC patients were vaccinated with survivin-2B peptide alone or in combination with interferon beta (IFN-β) was performed[36][Table 1]. Patients receiving the combined treatment had longer survival with no notable adverse effects, highlighting the reduced toxicity of IFN-β. Furthermore, immunohistochemical analyses revealed a strong infiltration of CD8+ T cells in lesions of vaccinated patients and a high rate of programmed cell death protein (PD-1), revealing the potential for a new combination of survivin-2B peptide-vaccine with immune checkpoint inhibitors[37].
Another emerging target for immunotherapy is gastrin, a gastric hormone able to bind to the CCK-B receptor, which is overexpressed in numerous types of gastrointestinal cancers, including pancreatic cancer. Recently, a gastrin-specific peptide-based vaccine, called polyclonal antibody stimulator, was assessed for its therapeutic potential in a transplantable PDAC mouse model. It was demonstrated to elicit both a humoral and cellular immune response, with the generation of specific target antibodies and T cell activation, respectively. Furthermore, the vaccine was effective alone, and also in synergy with a PD-1 antibody. In particular, combined therapy decreased tumor burden, fibrosis and immunosuppressive elements of the TME such as regulatory T lymphocytes (Tregs) and macrophages[38].
A more tailored approach based on personalized peptide vaccination strategies has also been developed for PDAC patients at the M.D. Anderson Cancer Center in collaboration with the National Cancer Institute and applied in a phase I clinical trial (NCT02600949). This approach relies on the availability of tumor samples and dedicated on-site technology. To date, however, there are no posted results that confirm that this this strategy is truly effective.
Dendritic cell-based vaccines
Pancreatic cancer is characterized by a limited number of functional DCs, which impairs the mounting of an effective antitumor response. DCs are professional antigen-presenting cells with a pivotal role in priming T cells. Thus, in vitro generated mature DCs can be used to develop a cancer vaccine, aimed at increasing antigen presentation, enhancing T cell priming and expanding antigen-specific T cells. DC loading can be achieved with different sources of tumor antigens such as tumor lysates, whole tumor cells, synthetic peptides and purified proteins or transfection with mRNA or cDNA[39].
One investigated target has been MUC-1, a PDAC-associated antigen that correlates with poor prognosis, metastasis and chemoresistance[10]. Since the MUC-1-targeted vaccine induced antitumor immunity in murine pancreatic cancer models[40], its potential in inducing specific antitumor immune responses was assessed in different clinical trials. A phase I/II clinical trial evaluated the therapeutic potential of DCs loaded with MUC-1 peptide in ten surgically resected PDAC patients. No toxicity was observed, and patients were followed for more than 4 years, after which three patients were still alive without recurrences[41][Table 1]. The same approach was used in another phase I clinical trial (06DZ19009), in which seven patients with metastatic disease received the MUC-1-targeted vaccine[42][Table 1]. A specific immune response was reported, and there was no toxicity, but there were unfortunately no clinical benefits. These studies indicate MUC-1 as a promising tumor antigen, which requires further investigation[42].
Another PDAC-associated antigen is the Wilms tumor gene (WT1), which is highly expressed in more than 75% of PDAC cases and has been reported to be highly immunogenic. For investigating the therapeutic potential of WT1, DCs were loaded with MHC class I- and/or II-restricted peptides. A retrospective analysis was conducted in 225 Japanese patients with pancreatic cancer refractory to standard treatment who received chemotherapy combined with MHC class I-restricted WT1 peptides. Interestingly, the median survival time was prolonged to 16.5 months, and WT1-targeted vaccination combined with standard chemotherapy was shown to be feasible and safe[43]. Another phase I pilot study (UMIN000004855) was conducted in ten PDAC patients who were treated with DCs loaded with MHC-I-restricted WT1 peptides combined with gemcitabine [Table 1]. The therapy was feasible, safe and well-tolerated; however, only patients without liver metastasis and high levels of inflammatory markers benefited from the treatment[44]. The phase I trial (UMIN000004063), instead used DCs loaded with both MHC class I- and MHC class II-restricted WT1 peptides combined with gemcitabine in advanced PDAC patients [Table 1]. The combined therapy was well tolerated. Half of the patients showed specific T cell responses, increased OS and progression-free survival. Interestingly, among the non-responsive patients with the combined treatment, increased levels of interleukin (IL)-6 correlated with poor prognosis[45,46].
In a pilot study (NCT01410968), Mehrotra et al.[47] evaluated the feasibility and safety of a DC-based triple vaccine: autologous HLA-A2-restricted DCs of 12 HLA-A2+ patients with unresectable or metastatic PDAC were pulsed for an hour with hTERT, CEA or survivin peptides [Table 1]. DCs were administered intradermally three times, followed immediately by intramuscular administration of a TLR3 agonist (polyinosinic:polycytolitic acid stabilized with polylysine and carboxymethylcellulose; poly IC:LC), as an adjuvant to boost T cell antitumor activity. Treatment was well tolerated, with some cases of fatigue and flu-like symptoms such as fever, night sweats and myalgia. A total of eight patients completed the study, half of them with stable disease and the other half with progressive disease. Compared to the control cohort treated with a second-line chemotherapy, which had a median survival of 4.9 months, the eight patients undergoing vaccination showed an increase in median survival by 2.8 months[47].
DNA vaccines
In the broad cancer vaccine landscape, DNA vaccination has emerged as a promising immunotherapeutic approach due to its stability and safety[48]. Two main findings have triggered the development of antitumor DNA vaccines, namely the ability of mammalian cells to express genes encoded by DNA plasmids and the in vivo humoral and cellular immune response obtained against the plasmid products[49,50]. An important benefit of DNA vaccines is that the antigen can be presented by both MHC class I and MHC class II, which can activate CD4+ and CD8+ T cells, and also possibly deliver several antigen genes in the same construct to obtain a wider specific immune response[51].
Our laboratory is focused on identifying new antigens to employ in DNA vaccination strategies for PDAC treatment[52]. In particular, we have identified the glycolytic enzyme ENO1 as a promising PDAC TAA by showing that antibodies against ENO1 are detected in more than 60% of PDAC patients[53]. Notably, this antibody response is absent or low in healthy donors and also in patients with non-pancreatic cancer and chronic pancreatitis[14]. ENO1 plays a critical role in cancer proliferation, metastasis and spreading, thus therapies targeting ENO1 may be effective in hindering tumor progression[54]. A preclinical study demonstrated that the ENO1 DNA vaccine elicited an integrated antitumor immune response in PDAC[55]. In particular, the ENO1-vaccinated PDAC mouse model showed a specific cellular response that significantly prolonged survival, as well as a higher amount of anti-ENO1 immunoglobulin (IgG) and a reduction of myeloid-derived suppressor cells (MDSCs) and Tregs. This correlated with a reduced amount of PDAC lesions in ENO1-vaccinated 8-month-old tumor-bearing mice[55].
An alternative DNA-vaccination method is being used in a phase I study to evaluate the safety and immunogenicity of an integrated vaccine comprising neoantigen plus mesothelin (MSLN) epitopes in PDAC patients following surgical resection and adjuvant chemotherapy (NCT03122106). The DNA vaccine combines prioritized neoantigens plus MSLN epitopes in a pING plasmid, which is administered with an electroporation device. Hopefully, neoantigen DNA vaccines will be safe and capable of generating neoantigen-specific CD4+ and CD8+ T cell responses, which will be evaluated by ELISPOT and multiparametric flow cytometry.
DNA-based approaches also include all those involving transfection of tumor cells or dendritic cells with plasmids encoding TAAs, cytokines, co-stimulatory molecules and neoantigens, which are discussed below.
Other alternative approaches
Neoantigens are tumor-specific antigens that are not expressed by normal cells, which result from de novo gene alterations that lead to potentially immunogenic peptides showing MHC molecules[56]. Neoantigen-specific immune responses do not follow central and peripheral tolerance laws and do not affect healthy tissues. Neoantigen-based immunotherapies are patient-related and, therefore, defined as tailored-made therapies. Mutated peptides are identified by whole exome sequencing and the most immunogenic epitopes matching MHC with peptide binding prediction algorithms, i.e., NetMHC[57]. The whole process relies on the availability of tumor samples, and therefore, this type of approach can be applied only to surgically resected patients. Nowadays, personalized neoantigen-based vaccines are being tested in phase I clinical trials with the purpose of assessing their safety and effectiveness in pancreatic cancer patients following surgical resection and adjuvant chemotherapy (NCT03122106; NCT03558945; NCT03956056), alone or combined with programmed cell death protein 1 ligand (PD-L1) inhibitors (NCT04161755). These studies are still in the recruiting phase.
The exosome-based vaccine is another innovative immunotherapy strategy. Exosomes are small, non-toxic, and non-immunogenic endosomal-derived vesicles secreted in the extracellular space that can carry lipids, proteins, RNA or DNA. Due to their phospholipid membrane and small size, their content is protected from degradation and able to penetrate natural barriers[58]. Exosomes can be obtained from the patient’s own cells, thus avoiding allograft reaction and can be engineered to carry specific tumor antigens to directly target cancer cells only. Taking these advantages into account, Xiao et al.[59] assessed the effectiveness of vaccination with tumor exosome-loaded dendritic cells in PDAC-bearing mice. Specifically, DCs loaded with PDAC-derived exosomes were administered intravenously in mice, either alone or in combination with gemcitabine, all-trans retinoic acid and sunitinib (tyrosine kinase inhibitor). Data showed that the vaccine effectively prolonged survival by inhibiting metastases and reducing the presence of MDCSs in the tumor infiltrate.
Another approach relies on virally-infected cell vaccines, replicating oncolytic viruses that are natural or genetically-modified viruses with a specific tropism for cancer cells, which generate a specific T cell activation. In this type of vaccination, cancer cells are pre-infected with tropic viruses and infused. In particular, Lu et al.[60] generated a virus-infected reprogrammed somatic cell-derived tumor cell vaccination (VIReST), consisting of murine fibroblasts transduced with lentiviruses to obtain induced pluripotent stem cells (iPSCs). These cells were then transduced to express mutated KRAS-G12D and p53-R172H, and then differentiated into pancreatic tumor cells. Cells obtained in this way were then infected with either oncolytic adenovirus (AdV) or vaccinia virus (VV). The authors showed how the order of viruses used to infect cells was fundamental for obtaining correct antitumor activity; mice spontaneously developing PDAC were first immunized with AdV-infected mitomycin C-treated cancer cells, followed by VV-infected cancer cells after 4 weeks in a prime-boost regimen. The importance of the order is due to the characteristics of the viruses; AdV is an extremely effective Toll-like receptor (TLR) activator, while the wide range of immunomodulatory proteins expressed by VV can boost T cell responses. VIReST was able to extend the lifespan of mice by 51%, halving the percentage of mice with detectable tumors at 6 weeks. Moreover, the authors detected an increase in CD8+ and CD4+ inside tumors, together with central memory populations. Importantly, no signs of autoimmunity were detected (colitis, ileitis and antinuclear antibodies). However, the protocol was not able to completely prevent tumor progression, indicating the need to combine this approach with other strategies[60].
Adoptive cell transfer
Another important trend in cancer immunotherapy focuses on T cell-mediated activity, namely adoptive autologous T cell transfer (ACT) and chimeric antigen receptor (CAR) cells. The fundamental purpose of autologous ACT is to generate a vigorous immune-mediated antitumor response via the infusion of ex vivo expanded or engineered leukocytes[61][Figure 1]. Tumor infiltrating lymphocytes (TILs) isolated from freshly resected tumors were the first source of T cells for ACT. However, more recently, peripheral blood mononuclear cells (PBMCs), tumor-primed draining lymph nodes, and iPSCs have also been used[62]. TIL generation techniques vary slightly in each institution, but small fragments are generally kept in culture medium supplemented with IL-2 and other cytokines and cells are harvested from supernatants over 2 to 3 weeks[63,64]. Generally, ACT is carried out with a mix of CD8+ and CD4+ T cells, depending on the patient’s T cell ratio and their expansion ex vivo, but certain protocols involve defined ratios, based on indications that this may lead to more favorable outcomes[65]. Even if CD8+ T cells are more powerful in exerting their antitumor effect through secreted cytokines and direct tumor cell killing, studies have demonstrated that CD4+ T cells play a pivotal role in these phenomena[66-68]. TILs have to be extensively cultured ex vivo and this can result in terminally differentiated cells that no longer exert their effect when re-infused[69]. The best responses to ACT therapy were in metastatic melanoma patients, in which substantial results were long-lived, and the majority of patients achieved a disease-free state for many years after treatment[61]. In other tumor types, even if promising preliminary results had been reported, the majority of clinical responses were transitory, ultimately leading to tumor relapse. This is probably due to the limited survival of the reinfused cells and their exhaustion[70]. To overcome these difficulties, immune checkpoint-blocking antibodies seem to enhance ACT activity, as reported below[71].
Concerning PDAC, one of the first targeted strategies applied to ACT exploited TP53 mutations present in cancer cells. This preclinical study proved that p53 peptide-specific cytotoxic T lymphocytes adoptively transferred in immunocompromised mice, were effective in suppressing PDAC growth in vivo and prolonged survival[72]. Other strategies focus on telomerase, a reverse transcriptase used to prevent senescence and maintain chromosomal stability, reported to be overexpressed in a significant percentage of PDAC (85%-90%) and considered a marker of poor prognosis[73]. For instance, adoptive telomerase-specific T cells obtained from peptide-immunized donor mice and stimulated with either IL-2, IL-15, APCs or allogeneic DCs, significantly slowed tumor growth and metastasis[74].
One of the first PDAC-associated targets investigated in clinical trials was MUC-1. Autologous PBMCs from resectable pancreatic cancer patients obtained either before surgery or during gemcitabine treatment, were cultured with MUC-1-expressing pancreatic cancer YPK-1 cells for 10 days and re-infused in the patients for a total of three rounds[75,76][Table 1]. It was shown that post-surgery gemcitabine-based regimens combined with adoptive transfer of MUC-1-specific cytotoxic T cells reduced the incidence of local recurrence and liver recurrence as well (33% compared to 60%)[75]. Overall, this type of therapy had no adverse events attributable to immunotherapy and could thus be a potential therapeutic strategy for pancreatic cancer.
Cytokine-induced killer (CIK) cells are another source of cells for ACT. CIK cells are generated from peripheral blood lymphocytes and exert an HLA-unrestricted antitumor activity. In a phase II trial (NCT00965718), advanced PDAC patients under gemcitabine regimens received adoptive transfer of CIK cells as a second-line therapy[77][Table 1]. Another study tried to combine S-1 (an oral fluorouracil anticancer drug comprising tegafur, gimeracil, and oteracil potassium) with or without CIK in advanced PDAC patients who previously received gemcitabine as a first-line therapy[78][Table 1]. Unfortunately, both studies showed a limited effect of CIK-based therapy in improving patient survival and disease control rate.
As ACT is more widely used, it is crucial that all centers be aware and become familiar with managing adverse events such as cytokine release syndrome, which may be primarily related to the preconditioning regimen, particularly to the administration of high doses of IL-2 after cell transfer[79-82]. Due to its relatively high cost, it is also important to stratify patients who may clinically benefit from ACT. In this regard, reports have shown how the tumor mutational burden and the number of neoantigens may be good predictive criteria for ACT response[83].
CAR-T and TCR-engineered T cells
As explained above, adoptive transfer of autologous TILs has some limitations such as the low presence of specific antitumor T cells and the time needed to isolate and expand these cells from patients. For this reason, engineered T cells strategies have been developed, namely CAR-T cells and TCR-gene transfer among them. CAR-T cells are lymphocytes genetically engineered to express a chimeric receptor made by a single-chain variable fragment (scFv) of an antibody linked to an intracellular signaling domain by a spacer region and a transmembrane domain. The TCR-gene transfer technique consists in isolating an antitumor TCR sequence and its transfection inside lymphocytes[84]. Lentiviruses[85] and DNA or mRNA transposon systems[86] are the most common methods to transduce genetic materials.
The ectodomain of CAR can recognize surface antigens in an MHC-independent way, covering approximately 1% of the proteome[87]. This limit can be overcome by using TCR-like antibody fragments that specifically recognize a haplotype-restricted peptide complexed with MHC-I molecules[88]. The spacer region derives from CD28, CD8a of Fc IgG molecules, which changes CAR flexibility and off-target activities. Transmembrane regions can derive from different proteins such as CD3, CD28, CD8a or CD4. Finally, the intracellular domain allows classifying CAR-T cells into three generations, namely first-generation CARs containing only the CD3z internal region, second-generation CARs containing CD28 or 4-1BB (CD137) intracellular tails, and third-generation CARs including both costimulatory domains. Inclusion of both costimulatory domains does not always produce better results than the second-generation CARs. Most clinical trials, in fact, use second-generation CARs. A fourth generation, also referred to as TRUCK T cells, are CAR-T cells genetically engineered to constitutively express cytokines or their receptors[89].
T cells can also be made insensitive to immunosuppressive signals. Different clinical trials (NCT02065362, NCT00368082, NCT03089203 and NCT01955460) are investigating the effects of engineered T cells expressing a truncated transforming growth factor beta (TGF-β) receptor 2 acting in a dominant negative manner in the tetramer receptor. New generations of CARs also contain suicide genes, co-expressed with the CAR construct to improve treatment safety. When off-target activities begin, genes can be induced by drugs or antibodies that trigger CAR cell apoptosis. The most commonly used are inducible caspase 9 and herpes simplex virus thymidine kinase[90].
Until now, the success of CAR-T cell therapies has been strictly related to hematological malignancies; Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel) are two CD19-targeting CAR treatments approved by the FDA[91,92] and EMA[93] to treat, respectively, pediatric relapsed or refractory acute lymphoblastic leukemia, and relapsed or refractory diffuse large B cell lymphoma. CAR-T cell therapies for solid malignancies are not yet as successful, with many ongoing preclinical studies and clinical trials. Solid tumors are more difficult to target than hematological ones due to differences in the tumor-specific antigen burden and the presence of multiple physical barriers, such as the aberrant tumor vasculature, the extracellular matrix composition and TME suppressive elements[94]. Strategies are in place to overcome all these obstacles to effector functions, such as engineering of CAR-T cells to constitutively express catalase[95] or hypoxia inducible factor 1 alpha (HIF1a)[96], combination of CAR-T cell therapy with checkpoint inhibitors or, preferably, CAR-T cells co-engineered to secrete antibodies such as anti-PD-L1[97] or anti-PD-1[98] antibodies. Moreover, evidence from leukemia[99] and melanoma patients[100] treated with CARs shows how lymphodepletion before infusion with CAR-T cells increases the expansion, persistence and activity of these engineered cells. The standard regimen includes cyclophosphamide and fludarabine alone or combined[101]. Regarding PDAC, different antigens have been explored as targets for CAR-T and TCR-gene transfer strategies, namely mesothelin, HER2, altered glycosylated protein MUC-1, differentiation antigens, CEA, prostate stem cell antigen and CD24.
MSLN
MSLN is a cell surface protein expressed in the pleura, pericardium and peritoneum; it is also minimally expressed on epithelial cells of the tonsils, trachea, ovaries and testes[102]. Discovered in 2014, the MSLN gene encodes a pre-pro-protein proteolytically processed by furin protease to release two fragments, namely megakaryocyte potentiating factor, a cytokine capable of stimulating megakaryocyte colony formation inside the bone marrow, and MSLN, a glycosylphosphatidylinositol (GPI)-anchored cell surface protein that is important for cell adhesion. A soluble form of MSLN, known as soluble MSLN-related protein has been found in the serum of patients with solid tumors[102]. MSLN overexpression has been discovered in different tumors, including pancreatic cancer, in which almost 85% of cells express this protein[103], and of course up to 90% of mesothelioma cells are positive for MSLN[104]. MSLN contributes to cell proliferation and resistance to apoptosis[105], and by activating matrix metalloproteases 9 (MMP-9)[104] and MMP-7[106], it can induce tumor cell migration and invasion. Immunotherapy strategies targeting MSLN are under investigation in various tumor subsets, such as mesothelioma and lung and breast cancer[107]. A 2009 preclinical study evaluated the use of engineered TCR against MSLN to treat PDAC[108]. The authors isolated T cell clones specific for different epitopes of MSLN in both MSLN-/- and WT mice; both mouse groups generated T cells against the MSLN fragment (406-414). T lymphocytes transfected with the highest affinity MSLN-/--TCR (TCR1045) were able to bind MHC on target cells, independently from CD8 or CD4, displaying a higher lytic activity against PDAC MHC-I+ tumor cells compared to the highest affinity TCR from WT mice. At 8 days after the infusion of TCR1045-expressing T cells, which preferentially infiltrated the pancreatic mass, the authors observed increased tumor cell apoptosis. This was, however, a transient antitumor response, as T cell number decreased 18-fold in PDAC between 8 and 28 days after transfusion and those remaining, progressively upregulated the inhibitory receptors PD1, Tim3, Lag3 and 2B4[108]. In a randomized, blinded, placebo-controlled experiment, the authors confirmed how serial T cell infusions were able to enhance mouse survival and reduce metastatic disease and malignant ascites[108]. Concerning the use of CAR-T against MSLN in human pancreatic cancer, a phase I clinical trial was completed (NCT01897415) in March 2017[109][Table 1]. In this study, autologous T cells were transfected with RNA to transiently express the anti-MSLN CAR construct. Cells were injected intravenously three times per week for 3 weeks. Following RECIST 1.1 criteria, the authors observed using PET stable disease in two patients and a stabilization of metabolically active volume and maximum standardized uptake value (SUVmax) in three patients. At 2 months after the final injections, the presence of IgG antibodies was evaluated against an array of 9000 proteins; all patients generated a response against immune-related proteins, such as BCMA, PD-1 and PD-L1, but also against oxidative stress-related proteins[109]. Three other clinical trials are currently ongoing, including an interventional clinical trial at Nanjing Medical University (NCT03638193) started in August 2018. PDAC patients will be treated with CAR-T-meso lymphocytes 3 days after lymphodepletion with cyclophosphamide, to assess adverse events as the primary outcome and general clinical response as the secondary outcome. Patients’ own T cells will be isolated and lentivirally transduced to express a second-generation 4-1BB CAR construct containing a partial murine single-chain variable fragment, which is a feature that could easily lead to antibody-mediated elimination[110]. For this reason, a single arm phase I clinical trial started by Wenzhou Medical University in April 2018 (NCT03497819) is evaluating the combination of this second-generation 4-1BB CART-meso therapy with or without CAR-T cells directed against the CD19 biomarker of B lymphocytes. The novelty of this study is the comparison between two modalities of administration, i.e., 3 days after cyclophosphamide treatment, CAR-T cells will be injected either intravenously or via the pancreatic artery. A third observational trial (NCT04203459) started in October 2019 at Harbin Medical University for evaluating how gut microbiota can modify the proliferation, migration and functions of MSLN CAR-T cells.
Her2 and CD24
Her2 is a member of the epidermal growth factor receptor (EGFR) family. Her2 can heterodimerize with other members of the family, mediating cell growth and proliferation by activation of the MAPK and PI3K pathways. Known to be hugely amplified in breast cancer, Her2 is also overexpressed in about 60% of pancreatic cancers, correlating with a higher metastatic disease and worse outcome in resected patients[111]. CD24 is a GPI-sialoglycoprotein anchored to the cell surface, and it is essential for the growth and differentiation of granulocytes and B cells. In the tumor context, CD24, together with CD44 and CD133, identifies cancer stem cells[112]. Both antigens have been targeted by a CAR-T cell approach. Maliar et al.[113] used a second-generation CD28 CAR-T against Her2 in a PDAC xenograft mouse model in which outcome was strictly related to Her2 expression on the tumor cell surface. In a second attempt, CD28 CAR-T cells targeting CD24 were injected after a preconditioning sub-lethal irradiation. CD24-specific CAR-T cells slowed xenograft growth and prolonged mouse survival. Treatments were also effective against high tumor burden and multiple metastases[113]. Unfortunately, a metastatic colon cancer patient treated with a third-generation CAR targeting Her2 died because of respiratory failure due to the recognition of Her2 in the lung epithelium[114]. The strong potency of CARs reduces the timeframe, after which antigens expressed on healthy tissues are targeted, increasing off-target toxicity. To extend this timeframe, Raj et al.[115] developed a switchable CAR-T. The “switch” consisted in combining an anti-Her2 Fab tagged with a peptide neoepitope (PNE) and T cells engineered with a second-generation 4-1BB CAR directed against the PNE tag. In this way, CARs can be “switched on” only when the Fab binds Her2, acting as a bridge between target and effector. In vitro experiments confirmed how switchable CAR-T cells were able to lyse both patient-derived tumor cells and cancer stem cells[115]. Moreover, the authors demonstrated how switchable CARs worked as well as conventional CARs against stage IV PDAC human cells orthotopically injected in mice[115]. Tumor cell growth was monitored with live imaging and after 17 days, when tumors were detectable, mice were enrolled for treatment. Treatment with inactive Fab was ineffective, but at day 10, mice administered with either switchable or conventional CAR-T cells were tumor- and metastasis-free[115]. Moreover, no signs of graft-versus-host disease (GvHD) were noticed in mice treated with the switchable form of CARs, while two mice treated with conventional CARs developed GvHD-like symptoms[115].
A phase I clinical trial referred to as VISTA (NCT03740256) is aimed at investigating a concurrent treatment - the intratumoral injection of oncolytic adenovirus (CAdVEC) modified to express genes coding for immune system stimulators, followed by injection of Her2 CAR-modified adenovirus cytotoxic T cells (Her2-AdVST). This approach will be tested on patients with Her2+ solid tumors, including pancreatic cancer as well as bladder, lung, breast, gastric and colorectal cancers. CAdVEC will be administered intratumorally with ultrasound guided percutaneous injection, while AdVST will be intravenously injected.
CEA
CEA, discovered by Gold and Freedman in 1965[116], is a cell surface protein with a GPI domain and is expressed from an early fetal period (9-14 weeks) throughout life. Detected at very low levels in healthy individuals, it is used as a tumor marker for colon, breast, liver, lung, stomach and pancreatic cancers, with 75% of the last expressing CEA. CEA can act as a homophilic or heterophilic adhesion tool in cancer cells[117]. CEA was previously exploited as a target in other immunotherapeutic strategies such as vaccines, while the CAR approach for this antigen began in 2012: Chmielewski et al.[118] used second-generation CD28 CAR-T cells containing the murine antibody SCA431scFv directed against an epitope of the membrane proximal CEA domain. This strategy avoided dose-limiting autoimmune colitis and pneumonia observed in a pilot study for metastatic colorectal cancer[119]. Panc02 pancreatic carcinoma cells were injected into the pancreas of immune-competent transgenic mice, expressing CEA in the gastrointestinal and pulmonary tracts[118]. Without any lymphodepletion preconditioning, 10 days after tumor transplantation, autologous T cells were inoculated via the tail vein in a single dose[118]. A specific antitumor activity was generated without any kind of autoimmune pathology[118] observed in the trial[119]. This was achieved due to the low-affinity CAR-T cells used in this preclinical model. CAR-T cells at cancer lesions were characterized by an exhausted phenotype (CD62L-, CD127- and CD279-) because of the low concentration of cytokines in the TME responsible for T cell activation, proliferation and degranulation[120]. To bypass these limitations, Chi et al.[121] combined the administration of an anti-CEA second-generation 4-1BB CAR with the intravenous injection of rhIL-12 to increase interferon gamma (IFN-γ) secretion and cytotoxicity of both natural killer (NK) and T cells. CAR-T cells co-cultured in vitro with CEA+ pancreatic cell lines, and treated with rhIL-12, showed a higher expression of cell surface proteins CD69 and CD25, a higher production of IL-2 and IFN-γ, greater cell proliferation and stronger cytotoxic activity than CAR-T cells not treated with the cytokine. The same CEA+ pancreatic cell lines were intravenously injected into mice, followed on day 7 by intravenous injection of anti-CEA CAR-T cells with or without rhIL-12 treatment. Adding the cytokine rhIL-12 enhanced the anti-metastatic activity of CAR-T cells, together with a higher CD8+/CD4+ circulating cell ratio and production of the serum cytokines: IFN-γ, IL-2 and tumor necrosis factor alpha (TNF-α) compared to mice not treated with the cytokine[121]. Similarly, Chmielewski and Abken[122] exploited second-generation CD28 anti-CEA CAR-T cells transduced with an inducible IL-18 cassette under the control of IL-2 promoter. In this way, engineered T cells were able to express IL-18 only after antigen recognition and dephosphorylation of nuclear factor of activated T cells, an inducer of IL-2 expression. IL-18 was found to be responsible for the effector T cell signature acting on Tbet and FoxO1[122]. Double-engineered CARs were able to prolong the survival of mice with long-term established orthotopic disease, together with a reduction of M2 macrophages and Tregs. Zhang et al.[123] also developed dual second-generation 4-1BB CAR-T cells targeting both MSLN and CEA. CD3-mediated activation was provided by the construct that recognized CEA, while 4-1BB mediated the co-stimulation signal by the construct that recognized MSLN. These dual CAR-T cells showed both in vitro and in vivo activities comparable to conventional CAR-T cells[123]. The off-target toxicities were strongly reduced, but dual CARs were only able to exert antitumor activity when both antigens were expressed on the tumor cell surface[107,123]. Two clinical trials are currently evaluating the effects of anti-CEA CAR: Roger Williams Medical Center started an open-label, single-arm phase 1b clinical trial (NCT03818165) in January 2019 for liver metastatic PDAC. This trial will involve infusion of second-generation anti-CEA CAR-T cells together with IL-2, and is expected to be completed in February 2022. Still concerning liver metastatic PDAC, Sorrento Therapeutics in California started an open-label phase 2b clinical trial (NCT04037241) in July 2019. Patients with progressive disease after first-line treatment with FOLFIRINOX or gemcitabine will receive second-generation anti-CEA CAR-T cells alone or combined with gemcitabine or nab-paclitaxel.
MUC-1
MUC-1 is a transmembrane protein composed of a tandem repeat (TR) containing an external domain, bound to the membrane through non-covalent interactions with the MUC-1-C domain, made of a short extracellular domain and transmembrane and cytoplasmic domains. In tumors, MUC-1 shows a different O-glycosylation of the serine and threonine residues in the TR domains[124]. The most common aberrant glycosylated form can bind the sialic acid-binding protein Siglec-9 expressed by monocytes and macrophages; after this interaction, tumor-associated macrophages (TAMs) increase expression of PD-L1[125]. The aberrant form of MUC-1 is expressed in more than 80% of PDAC cases, which makes MUC-1 a good target for cancer immunotherapy[126]. Second-generation 4-1BB CAR-T cells exploiting the domains of the 5E5 antibody, which specifically recognizes the aberrantly glycosylated MUC-1, were used to treat disseminated tumor xenografts of intraperitoneally injected Hs766T pancreatic cells. All mice treated with anti-MUC-1 CAR-T cells survived, while the survival of mice treated with untransduced CAR-T cells or unrelated CD19 CAR-T cells was 40% and 33%, respectively[125]. CD28 anti-MUC-1 CAR exploiting the variable fragments of another tumor-specific antibody (TAB004) was used in a preclinical study published in Cells in 2019[127]. A study using a xenograft model with the MiaPaCa2 cell line demonstrated how anti-MUC-1 CAR-T cells were able to reduce tumor weight and metastasis[127]. From the in vitro assay, five genes were found to be upregulated in two non-responding human cell lines (CFPAC and HPAFII), namely IDO-1, COX1 and COX2, ADAR1 and Gal-9. Exploiting small molecule inhibitors and antibodies against these genes, investigators were able to restore sensitivity to CAR-T cell treatment in vitro[127]. No clinical trials are currently testing anti-MUC-1 CAR-T cells in PDAC patients.
Prostate stem cell antigen
Prostate stem cell antigen (PSCA) is a glycoprotein composed of 123 amino acids anchored to the cell surface membrane by GPI. PSCA is undetectable in the healthy pancreas but overexpressed in PDAC, as well as in prostatic, urinary and bladder tumors, where its functions remain unclear. The difference in PSCA expression between normal and tumor tissues is more pronounced than with MSLN. Second-generation CD28 and third-generation CARs were developed by Abate-Daga et al.[128] and functionally compared to anti-MSLN CARs currently used in clinical trials. Anti-PSCA CARs induced larger amounts of IFN-γ than did anti-MSLN CARs when co-cultured with pancreatic cancer cell lines. Moreover, preclinical experiments demonstrated that second-generation CARs, but not third-generation CARs, were able to strongly reduce subcutaneous growth of HPAC pancreatic cells, with tumor rejection in two out of five mice[128]. To overcome the immunosuppression in the TME, Mohammed et al.[129] generated a first-generation of anti-PSCA CAR-T cells co-transduced with a chimeric switch receptor, in which the extracellular domain of IL-4R is linked to the immunostimulatory intracellular domain of IL-7R, to exploit the IL-4 inhibitory signal in the activation of the IL-7 pathway. The double transduction was able to increase the destruction of CAPAN-1 cells subcutaneously engrafted in NOD/SCID/IL-2Rgnull (NSG) mice[129]. Bellicum Pharmaceuticals in Houston started a phase I clinical trial (NCT02744287) in April 2016 to evaluate the efficacy and safety of BPX-601, an anti-PSCA CAR that also contains a rimiducid-inducible signaling domain able to enhance CAR-T cell proliferation and activation. The estimated primary completion date is set for February 2022.
Other alternative antigens
Some “unconventional” antigens were tested as targets for CAR-T cell therapies, including CD47, B7-H3 and integrin αvβ6. CD47 is a cell surface glycoprotein belonging to the immunoglobulin superfamily and represents a “don’t eat me signal”, able to inhibit phagocytosis by binding to signaling regulatory protein α (SIRP-α). CD47 is overexpressed in leukemia and lymphoma, and in ovarian cancer, small cell lung cancer, glioma, glioblastoma and pancreatic cancer as well, where it represents a negative prognostic factor[130]. A second-generation CD28 anti-CD47 CAR-based treatment was able to significantly decrease the volume and weight of tumors developed in mice subcutaneously injected with BxPC3 pancreatic cells[130]. B7-H3, instead, is a member of the B7 costimulatory or co-inhibitory family delivering an immunosuppressive signal. With a limited expression in normal tissue, B7-H3 is expressed by the tumor-associated vasculature and fibroblasts, and correlates with poor prognosis and outcome[131]. Two second-generation, CD28 or 4-1BB, anti-B7-H3 CARs were able to completely reject PANC1 orthotopic tumors, with all mice remaining tumor-free for more than 80 days[131]. Finally, αvβ6 is an integrin found overexpressed in pancreatic, head and neck, skin, lung, stomach, colon and breast cancers[132]. This integrin activates TGF-β, promoting the migration and activation of MMPs and the epithelial-to-mesenchymal transition. A second-generation CD28 anti-αvβ6 CAR was used to target BxPC3 and PANC0403 cell lines intraperitoneally inoculated into severe combined immunodeficient beige mice. These CAR-T cells were able to cause the tumor regression growth with regard to both cell lines[132].
These data support this tailored approach as a powerful tool for targeting solid tumors, including PDAC.
Monoclonal antibodies
Passive immunotherapies, mainly represented by monoclonal antibodies, are efficacious in many types of cancers, and the first used in clinical practice were EGFR- or VEGF-antibodies in colorectal cancer, and trastuzumab in Her-2-positive breast cancer. Her-2 is also widely expressed on PDAC cells, and trastuzumab therapies employ antibody-dependent cellular cytotoxicity (ADCC) mediated by NK cells for destruction of tumor cells. In 2018, Iwakiri’s group reported that combined therapy of trastuzumab with anti-CD137 (4-1BB) could improve NK-mediated ADCC of human PDAC cell lines in vitro[133]. This was related to an increased expression of CD137 on NK cells in response to trastuzumab, associated with the Fcg-RIIIA-VV/VF polymorphism[133]. Other targeted molecules have, so far, been unsuccessful in pancreatic cancer, such as cetuximab and bevacizumab[134].
Myeloid targeting approaches
A promising molecule combining adaptive and innate immunity is CD40, a member of the TNF receptor family[135]. CD40 is expressed on monocytes and DCs and also B cells, platelets and non-hematopoietic cells and tumor cells[136,137]. The ligand of CD40 (CD40L or CD154) is expressed following activation by CD4+ and CD8+ T cells, B cells, NK cells and macrophages[138]. The significance of CD40 in tumor immunology was highlighted by a series of pioneering articles that showed that administering an agonistic antibody induced protective T cell immunity in murine cancer models of T-cell lymphoma and renal carcinoma[139,140]. CD40 agonists have been exploited to ameliorate the three immune cell cycle steps: tumor killing, T-cell immunity induction and immunosurveillance activation[141][Figure 1]. The most widely used agonist is the fully human CD40-specific monoclonal antibody called CP-870,893. Even if CD40 targeting is effective in activating the immune system, its clinical activity is fairly limited. Therefore, it is clear that combination with other treatments is pivotal for effective translation of the CD40 agonist. Some preclinical modeling has aimed to combine CD40 antibodies with TLR agonists, showing how the combined therapy achieved 10- to 20-fold greater responses than the agonists alone by synergizing to stimulate CD8+ T cell responses, as demonstrated by increased lytic activity and IFN production[142]. Alternatively, CD40 antibodies have been combined with IL-2 immunotherapy as a strategy to induce tumor-specific T cell immunity in tumor models of renal cell adenocarcinoma or Lewis lung carcinoma. CD40 stimulation and IL-2 synergize to induce complete regression of metastatic tumors due to potentiation of T cell survival and CD40 expression of DCs, stimulating an immunomodulatory cascade[143].
To understand the mechanisms of action of the anti-CD40 antibody, many studies have used a genetically engineered mouse model of pancreatic cancer[5]. Using an agonist rat anti-mouse mAb as a prototype for CP-870,893, Beatty et al.[138] demonstrated a T cell-dependent and -independent mechanism induced by the agonist. In orthotopic PDAC models, anti-CD40 treatment was associated with an increased control of tumor growth. Notably, in a second set of experiments, mice spontaneously developing pancreatic tumors treated with the anti-CD40 mAb FGK45 and gemcitabine showed a major tumor regression compared to gemcitabine alone, supporting that observed in the previous model[144]. In addition, the same rate of tumor regression was obtained with anti-CD40 mAb alone as that produced by combination with gemcitabine. Previous studies have suggested that CD40-activated macrophages can inhibit tumor growth due to IFN-γ production, indicative of a classically-activated macrophage phenotype[144]. Administration of anti-CD40 in PDAC-bearing mice did not produce a significant change in the number of TAMs within the tumor, but a transitory change in macrophage activation (i.e., CD86 and MHC class II expression) was seen within 24-48 h of treatment[138]. Moreover, macrophages isolated from the pancreas of tumor-bearing animals treated in vivo with anti-CD40 mAb were capable of lysing tumor cells in vitro[138]. Furthermore, treatment of PDAC-bearing mice with clodronate abrogated anti-CD40-mediated tumor regression[138]. These findings reinforce the idea that CD40 immune therapy may be dependent on macrophages, and that the CD40 pathway can be harnessed to restore tumor immunosurveillance by targeting tumor-infiltrating macrophages. This particular study did not investigate the role of B cells and DCs, although both cells can be activated by the CD40 agonist or anti-CD40 mAb[145]. Indeed, Schultze et al.[146] discovered that fibroblasts expressing CD40L could be used in association with IL-4 to generate large numbers of human B cells that act as APCs to generate antitumor CD8+ T cells. More recently, CD40L-expressing fibroblasts have been replaced with soluble CD40L[147].
Regarding DCs, monocyte-derived DCs treated with CP-870,893 exhibited a mature phenotype, with upregulation of CD80, CD83, CD86 and HLA-DR, and increased Mip1α and IL-12 secretion, indicating that CP-870,893 binds and activates DCs in vitro[148]. In PDAC mouse models, classic DCs are a rare population in the TME expressing CD40[149]. Upon activation of CD40 on DCs, many other agonistic pathways such as GITR, OX40 and 41BB are activated. As DCs have an enhanced capacity to take-up antigens when immature[150], chemotherapy followed by CD40 activation but not vice versa, elicited effective T-cell dependent immunity in tumor-bearing mice compared with anti-CD40 alone[151,152]. In subcutaneously implanted PDAC tumors, gemcitabine followed by agonistic CD40 is effective in inducing IFN-γ and TNF-α secreting T cells, activated DCs, and limiting M2 macrophages and Tregs[153]. However, the addition of nab-paclitaxel to gemcitabine, which is more effective in patients than gemcitabine alone, synergizes to trigger tumor regression and improve survival in spontaneous PDAC mouse models[152]. Notably, chemotherapy plus anti-CD40 failed both in CD40-/- and Batf3-/- mice (which lacked cross-presenting DCs), suggesting a critical role of DCs[152]. Given the preclinical success, CD40-targeting agents have progressed to the clinical phases. In collaboration with Pfizer Corporation, a two-center trial at the University of Pennsylvania (PI, Peter O’Dwyer) and Indiana University (PI, Elena Chiorean) examined the combination of gemcitabine and CP-870,893 in untreated patients with advanced pancreatic adenocarcinoma (NCT00711191) [Table 1]. Though gemcitabine alone has, historically, produced a partial response of approximately 5% or less in the chosen settings, 4 patients out of 21 were reported to have a partial response and 11 patients had stable disease as the best response[154]. A follow-up clinical study of CP-870,893 with gemcitabine is now ongoing for patients with resectable PDA at the University of Pennsylvania (NCT01456585)[138,155].
Granulocyte-MDSC (Gr-MDSC) are another interesting target; these cells are abundant in the TME and their increase correlates with PDAC progression[156]. Stromnes et al.[156] depleted these populations using a mAb targeting Ly6G+ cells in a spontaneous PDAC mouse model. Treatment with this antibody achieved effective intratumoral Gr-MDSC depletion for over 2 weeks accompanied by an accumulation of CD8+ T cells. Although there were no observed differences in tumor growth, Gr-MDSC depletion decreased ECM deposition and enhanced caspase-3 cleavage in cancer cells.
Colony-stimulating factor-1 (CSF-1) acts as a monocyte attractant and is one of the main growth factors for monocyte-lineage cells. Moreover, CSF-1 induces polarization of macrophages into a tumor-promoting phenotype[157,158]. CSF-1 tyrosine kinase receptor (CSF-1R), has been the focus of attention by scientists and companies who have developed a number of small molecules and antagonists to block its action[159]. In PDAC preclinical mouse models, blocking the CSF-1/CSF-1R pathway was achieved through either CSF-1-neutralizing antibodies or CSF-1R inhibitors[160]. Abrogation of the CSF-1 axis was sufficient to redirect TAMs towards an M1 pro-inflammatory phenotype in metastatic lesions[160]. Zhu et al.[161] showed that treatment with an anti-CSF-1 mAb resulted in enhanced PD-1 and cytotoxic T-lymphocyte ntigen 4 (CTLA-4) expression in TILs. Therefore, they suggested combining inhibition of the CSF-1/CSF-1R axis with ICI therapy, particularly for single-agent resistant tumors. In preclinical PDAC models, this combined therapy led to regression of established orthotopic tumors[161]. Similarly, Mitchem et al.[162] showed how the CSF-1R inhibitor reduced the amount of CD11b+ Ly6G- Ly6Clow MHC-IIhigh F480+ macrophages that infiltrated into murine PDAC tumors. Moreover, combination treatment with the CSF-1R inhibitor and gemcitabine restored antitumor activity to TILs, demonstrated as increased levels of IFN-γ, IFN-β1, granzyme B, perforin, and IL-12 p35 and decreased expression of TGF-β and Arg-1 mRNA in tumors[162]. To evaluate the safety and clinical activity of a combined treatment, a recruiting early phase I trial (NCT03153410) is evaluating the combination of cyclophosphamide, pembrolizumab, GVAX and IMC-CS4 (LY3022855) (an anti-CSF-1R mAb) in patients with borderline resectable pancreatic cancer.
Immune checkpoints
In physiological conditions, the activation of T cells is counterbalanced by inducing inhibitory stimuli to maintain immune homeostasis and to limit tissue damage and autoimmune reactions. Signals and regulators involved in these kinds of inhibitory responses are known as immune checkpoints (IC). Among the molecules responsible for the negative regulatory processes, the most studied and characterized are CTLA-4 and PD-1. CTLA-4 is mainly expressed on the surface of Tregs, naïve T cells and effector T cells. CTLA-4 competes with the co-stimulatory receptor CD28 in binding CD80 (also known as B7-1) and CD86 (also known as B7-2) molecules, to modulate the strength of the activated T cell responses or to enhance Treg-mediated immunosuppression[163]. PD-1, expressed on the surface of T cells, B cells and NK cells, interacts with PD-1 ligand 1 (B7-H1) and ligand 2 (B7-DC), influencing both central and peripheral tolerance mechanisms[164-166]. Activating IC pathways combined with the recruitment of suppressive cell populations, is a typical immune-escape mechanism adopted by different types of cancers[167,168]. PDAC is considered an “immunologically cold” tumor because of the high infiltration of Tregs and MDSCs and the consequent impairment of CD4+ and CD8+ cell-mediated antitumor responses[169,170]. We have previously demonstrated that the tumor area is infiltrated by Tregs and Th17 cells that frustrate effector T cells much more than the normal surrounding mucosa[171]. However, the presence of CD4+ and CD8+ T cells in the tumor has been demonstrated to correlate with better prognosis[172]. A recent retrospective study of 453 PDAC samples investigated PD-L1 mRNA expression levels and revealed an upregulation in 19% of the samples[173]. Rahn and colleagues suggest that in pancreatic cancer, PD-L1 is mainly expressed on the surface of stromal and MDSC cells[174].
Taking these premises into account, the use of ICIs to disrupt Treg-mediated immunosuppression and to block the inhibitory pathways on effector T cells, seems to be potentially effective in PDAC treatment [Figure 1].
ICI monotherapy
The mechanism of action of ICIs is to stimulate immune-mediated recognition and elimination of the tumor. Ipilimumab is the first anti-CTLA-4 monoclonal antibody approved for treating metastatic melanoma patients[175]. Although the use of anti-CTLA-4 antibody in PDAC mouse models suggests potential benefits such as the increase of CD4+ cell recruitment[176], no effects were observed in tumor growth reduction or overall survival[149]. These results were also confirmed by a phase II trial (NCT00112580) in which the single agent ipilimumab was tested for the treatment of locally advanced or metastatic pancreatic adenocarcinoma [Table 1]. Only one out of 27 subjects showed a significantly delayed response, suggesting that ipilimumab monotherapy is ineffective for treating advanced PDAC[177]. Even anti-PD-1 mAbs (such as pembrolizumab and nivolumab) and anti-PD-L1 mAbs (such as ateolizumab, durvalumab and avelumab) have been approved by the FDA for treating different solid tumors[178]. In mice subcutaneously injected with PAN02 (PDAC cell line), anti-PD-L1 treatment significantly reduced tumor growth and increased the number of CD8+ cells producing IFN-γ, granzymes and perforins[179]. Unfortunately, opposite results were observed in a genetically engineered PDAC mouse model, in which anti-PD-L1 treatment alone was ineffective[149]. Moreover, as described previously for anti-CTLA-4, anti-PD-L1 monotherapy failed in the clinical approach. All 14 patients with advanced pancreatic cancer who were enrolled in a phase I clinical trial (NCT00729664) showed no responses to treatment with BMS-936559 (an anti-PD-L1 antibody)[180][Table 1]. Despite this, the anti-PD-1 antibody seemed to be effective in a small PDAC patient cohort with mismatch repair (MMR) deficiency. In a study conducted by Le et al.[181] (NCT01876511), eight PDAC patients were also enrolled for evaluating PD-1 blockade efficacy in patients with advanced MMR-deficiency in different kinds of cancers [Table 1]. Treatment with pembrolizumab was able to induce a complete response in two patients, a partial response in three patients and stabilization of the disease in one patient (the remaining two patients could not be evaluated)[181]. The high mutation rate observed in MMR-deficient tumors, due to the failure of the DNA repair mechanisms, increases their immunogenicity and makes them suitable for checkpoint inhibitor treatments. Unfortunately, this is the case for only 1% of PDAC patients[182].
One of the reasons for the failure of ICIs in treating pancreatic cancer may be the highly immuno-suppressive TME, which makes it comparable to an immune-privileged site[183]. To overcome these adverse conditions, ICIs were tested in combination with treatments that have been shown to affect steps of the cancer immunity cycle that are deficient in PDAC.
ICIs and chemotherapy
To date, the gold standard of care for PDAC treatment is based on the use of gemcitabine (with or without nab-paclitaxel) and FOLFIRINOX[184]. Chemotherapeutic agents not only have a direct effect on cancer cells (by impairing cell division or increasing the DNA-mutation rate and therefore their death), but can also boost the immune system response by inducing so-called “immunogenic cell death” (ICD). Briefly, upon chemotherapy, cancer cells undergo necrosis thereby releasing molecules that act as danger signals which, in turn, activate the innate immune system. Furthermore, it has been shown that some chemotherapeutic agents can increase MHC-I expression on the surface of the cancer cells and promote maturation of DCs. Overall, chemotherapy improves the immunogenicity and antigenicity of the tumor[185] by enhancing potential benefits from the coupling with ICIs. Nomi and colleagues demonstrated that combining gemcitabine with PD-L1 blockade in PDAC resulted in a complete antitumor response in a subcutaneous injection PDAC mouse model[179]. In another study, Winograd et al.[149] by using a combination of gemcitabine/nab-paclitaxel plus anti-PD-1 and anti-CD40 mAbs observed regression of the tumor mass and an improved median overall survival of tumor-bearing mice. Many clinical trials evaluate the potential of combining ICIs with chemotherapy in PDAC patients[186]. Gemcitabine combined with the anti-CTLA-4 antibody has been evaluated in two different phase Ib trials. Respectively, 34 and 16 patients were treated with gemcitabine with or without tremelimumab[187] and ipilimumab[188]. There was a partial response in 6% of patients, and 21% of patients achieved stable disease with an overall survival of 7.4 months in the tremelimumab trial (NCT00556023). In the ipilimumab trial (NCT01473940), 14% of patients achieved an objective response rate and 33% of patients achieved stable disease with an overall survival of 6.9 months[187,188][Table 1]. Blocking PD-1 through pembrolizumab, used in combination with gemcitabine/nab-paclitaxel, was evaluated in 17 patients with metastatic PDAC (5 out of 17 had received prior chemotherapy) in a phase Ib/II study (NCT02331251) [Table 1]. For the chemotherapy naïve patients, the overall survival was 15 months with a partial response and stable disease in 25% and 67% of patients, respectively. Interestingly, no patients had progressive disease[186,189]. A combination of nivolumab/nab-paclitaxel with or without gemcitabine was evaluated in 42 naïve treatment patients with advanced pancreatic cancer (NCT02309177) [Table 1]. The overall survival was 9.9 months and 2% of patients showed a complete response, 16% a partial response and 46% stable disease[190]. Many different trials with ipilimumab, avelumab, pembrolizumab, nivolumab and durvalumab are ongoing, either given alone or combined with chemotherapy and other biological drugs[186].
ICIs and radiotherapy
As previously described for chemotherapy, radiotherapy (RT) can also induce ICD of cancer cells and enhance the antitumor immune response[191]. In particular, it has been documented that RT can induce the so-called “abscopal effect”, by which treatment of a primary tumor can induce the immune-mediated shrinkage of secondary lesions[192]. This mechanism occurs for CD8+ T cells that migrate to secondary lesions after activation in the RT-treated primary tumor, where they recognize the same antigen by which they were primed[193]. Therefore, even if RT is not the gold standard treatment in PDAC, some studies have shown the benefits that derive from combining RT with ICIs[184]. Azad and colleagues demonstrated that RT and gemcitabine can induce overexpression of PD-L1 in syngeneic PDAC tumor allografts. Considering these findings, they decided to investigate RT in combination with anti-PD-L1 and/or gemcitabine. PD-L1 blockade in association with RT significantly delayed tumor growth, which was enhanced by the addition of gemcitabine[194]. Similarly, Victor et al.[195], demonstrated the efficacy of combined treatment with RT plus anti-PD-1 and anti-CTLA-4 in a subcutaneous PDAC model. Furthermore, more positive results were obtained in tumor-bearing mice treated with a combination of RT, anti-CD40 and anti-CTLA-4/PD-1[196]. To date, some clinical trials are ongoing to better evaluate different combinations of ICIs and RT in pancreatic cancer; for example, a combination of nivolumab, ipilimumab and RT are used in the trials NCT03104439 and NCT02866383; and tremelimumab and/or MEDI4736 (anti-PD-L1) in combination with RT are used in the trials NCT02311361 and NCT02639026.
ICIs and vaccination
As previously mentioned, one of the most used cancer vaccines in PDAC is GVAX. In a phase II study, its ability to induce cancer-specific CD8+ T cell expansion and formation of tertiary lymphoid organs was reported, suggesting the potential of GVAX to reverse PDAC from a “non-immunogenic” into an “immunogenic” state[19,21,197]. According to these findings, it was suggested that a combination based on vaccination and ICI could induce an effective anticancer response. In a phase I study, ipilimumab alone, or combined with GVAX was evaluated in 30 previously treated advanced PDAC patients [Table 1]. The median OS of patients receiving the combined treatment was 5.7 months compared to 3.6 months in patients receiving ipilimumab alone. In patients with an OS longer than 4.3 months, there was an increase in the peak of mesothelin-specific T cells, and an expansion of the T cell repertoire[198]. Soares and colleagues[199] observed that GVAX significantly upregulated PD-L1, and that combining GVAX with anti-PD-1 mAb improved mouse survival compared to monotherapy. This result was also supported by an increase in recruitment of tumor-specific effector CD8+ T cells producing IFN-γ[199]. Similarly to that observed in preclinical experiments, Lutz et al.[200] reported that 33 out of 39 resected tumor tissues of GVAX-vaccinated patients were characterized by the presence of intratumoral tertiary lymphoid aggregates, together with an upregulation of the PD-1/PD-L1 pathway. Hence, many clinical trials are evaluating the most efficacious combination therapies, for example, GVAX combined with nivolumab alone (NCT02243271, NCT02451982, NCT03161379, NCT03767582) or combined with either ipilimumab (NCT03190265) or pembrolizumab (NCT02648282)[201].
ICIs and adoptive T cell therapy
The lymphocytes used for adoptive T cell therapy (both autologous and allogenic T cells) face the same problem deriving from TME immunosuppression described above. Therefore, in this case as well, combined treatment with ICIs could increase ACT efficacy. ACT flop is often connected to enhanced expression of the T cell inhibitory marker, namely PD-1[202,203]. Ironically, recognition of tumor cells by transferred T cells leads to T cell activation and consequently upregulation of PD-1 on their surface; similarly, tumor cells enhance PD-L1 expression in response to T cells. This ultimately leads to abrogation of T cell activity and thus ACT failure[204]. Evidence that this anergic state could be reverted when the PD-1/PD-L1 axis is blocked has emerged from several phase III clinical trials in non-small cell lung cancer and melanoma[205,206]. On the basis of these reports, a similar approach has also been assessed in PDAC mouse models[207], testing an approach to transform the PD-1/PD-L1 immunosuppressive axis for stimulation of TILs through transfection of a fusion protein, consisting of the extracellular PD-1 domain fused with the intracellular T-cell-activating CD28 protein, in the transferred T cells. It was shown that murine CD4+ T cells expressing the PD-1-CD28 fusion protein can overcome PD-L1-induced T cell inhibition.
Furthermore, John and coworkers described a significant improvement in the growth inhibition of two different Her2+ tumors treated with anti-Her2 T cells combined with an anti-PD-1 mAb in tumor-bearing mice with subcutaneous injection of tumor cells. The therapeutic effects correlated with an increased function of anti-Her2 T cells and a significant decrease in tumor-infiltrating MDSCs[208]. Regarding pancreatic cancer, a phase II clinical trial is investigating the combination of ACT therapy and ICI in patients with metastatic PDAC. These patients will receive an infusion of autologous PBMC-derived T cells with IL-2 and pembrolizumab after chemotherapy (NCT01174121)[209]. In addition, novel studies are focused on autologous CAR-T cells engineered to recognize specific solid tumor antigens and to simultaneously express anti-PD-1 and anti-CTLA-4 antibodies in non-hematologic malignancies (NCT03182803, NCT03030001, NCT03179007). Moreover, a new strategy based on PD-1 gene depletion by using CRISPR/Cas9 technology is being developed to engineer CAR-T cells and make them insensitive to checkpoint inhibitory stimuli[210].
The unique pancreatic cancer microenvironment
The PDAC TME is characterized by cells exerting both anti-tumor and pro-tumor functions such as endothelial cells, fibroblasts, lymphocytes and myeloid-derived cells. These populations are able to influence therapeutic responses and to sustain tumor progression by compromising the antitumor activities of effector cells. Therefore, PDAC TME is considered one of the most relevant causes of immunotherapy failure in the treatment of this disease. TILs may encounter a suppressive environment involving inhibitory cytokines, hypoxia, as well as a dense fibrotic stroma and abnormal blood vessels that act as physical barriers preventing them from functioning properly. Other negative regulators, as already discussed, are the immune checkpoints expressed by tumor cells and myeloid-derived cells, such as PD-L1 and CD80/CD86, respectively, which interact with PD-1 and CTLA-4 expressed on the T cell surface. These interactions generate negative signals leading to immune suppression[157]. In particular, Candido et al.[211] confirmed that TAMs are the principal source of PD-L1 expression in the PDAC TME. TAMs can derive from the differentiation of monocytic-MDSCs (M-MDSCs), recruited by cancer-produced CCL-2 and CCL-5, and differented in macrophages inside the TME by stabilizing HIF1α in the tumor hypoxic condition. Furthermore, HIF1α binds to the PD-L1 promoter hypoxia-responsive element motifs, rendering those cells capable of suppressing T cell activation in an antigen-nonspecific manner[212]. In addition, macrophages can induce fibrosis, a key element of T cell exclusion from the TME[160]. TAMs with a M2-like polarization can directly and indirectly suppress CD8+ activity; by expressing TGF-β, they are able to repress levels of perforin and granzymes, and with the help of IL-10 they induce Tregs. These CD4+CD25+Foxp3+ T cells can derive from maturation in the thymus or by conversion of conventional T cells upon TCR stimulation together with TGF-β. Treg cells express CTLA-4 and predominantly consume IL-2 due to the high expression of CD25, depriving this cytokine of effector cells. Moreover, it has been demonstrated that Tregs are able to mediate an immunosuppressive mechanism through the co-expression of CD39 and CD73 molecules on their surface. In fact, CD39 converts ATP and ADP into cAMP that, in turn, is converted into adenosine by CD73. Adenosine binds to specific receptors on CD4 and CD8 T cells as well as NK cells, macrophages and DCs by inhibiting antitumor cell response[213,214]. In addition, Tregs can directly kill effector cells via perforin or granzyme release[215].
A similar pro-tumor microenvironment renders pancreatic cells capable of evading immune surveillance, especially because of the impaired tumor antigen presentation by DCs, which are not “classically activated”[216,217]. The immunosuppressive state, responsible for the failure of most of immunotherapeutic approaches, can be directly sustained also by tumor cells. Fogar et al.[218] showed how pancreatic cancer cells themselves release soluble mediators able to suppress the proliferation and migration of CD4+ T cells, expanding CD69+CD4+ T cells with an inhibitory role on conventional CD4+ in a contact-dependent way. Therefore, different from more immunogenic tumors such as renal cell carcinoma or melanoma, which attract tumor-infiltrating effector cells[219], pancreatic cancer shows poor immunogenicity, which results from both dysfunctional antitumor response and surmounting immunosuppression.
Deeper genetic characterization of solid tumors has led to the identification of specific subtypes of the same histotype tumor that benefit from different treatments. Thanks to an integrated genomic analysis of 456 cases of pancreatic ductal adenocarcinoma, 4 different tumor subtypes were identified: squamous, pancreatic progenitor, immunogenic and aberrantly differentiated endocrine exocrine. In particular, the immunogenic subtype is associated with a significant immune infiltrate, B cell signaling pathways, antigen presentation, CD4+ T cell, CD8+ T cell and Toll-like receptor signaling pathways. Of note, this subgroup displayed upregulated CTLA-4 and PD-1 suppressive pathways, suggesting a higher potential response to the inhibition of immune checkpoints compared to the other subtypes. However, stratification based on tumor immunological background might identify those patients who will benefit from immunotherapy strategies in general[220].
Indeed, immunotherapy responsiveness can be explained and predicted thanks to the combination of two independent elements: the “hot” or “cold” status, according to the range of inflammatory cell infiltration and IFN-γ signature, and the tumor mutation burden, which can lead to neoantigen expression. Melanoma and non-small cell lung cancer are hot tumors with a high tumor mutation burden, making them highly antigenic and immunogenic and thus potentially more sensitive to immunotherapy[221]. Unfortunately, PDAC, alongside a strong immunosuppression, has a low number of genomic mutations in protein coding regions. This small antigen mutational burden may be another reason for the difficulty of inducing an effective antitumor T cell response[201].
Conclusion
“Dark side” in the title can be considered a reference to the evil force in the famous Star Wars saga. Due to many of its intrinsic characteristics, including a well-established desmoplastic reaction and an immunosuppressive microenvironment, pancreatic cancer actually represents the dark side of immunotherapy. Compared to other solid tumors, indeed, many immunotherapy approaches drastically fail when applied to pancreatic cancer patients. However, thanks to intensive worldwide research focused on this deadly disease, in the last decade our knowledge has greatly increased, which has paralleled an increase in 5-year survival rate to 10% at least in the USA. The scientific community has developed sophisticated tools to study the biology of pancreatic cancer such as Avatars, genetically engineered mice, and organoids, in which novel therapeutic drugs can be tested. Genetically engineered mouse models represent the best choice for studying antitumor immune responses and novel immunotherapy approaches, as they mimic the complex relationship between the immune system and tumor cells. Of equal importance is the possibility to develop novel clinical studies and to enroll patients in clinical trials.
Therefore, our title better refers to the famous Pink Floyd album, “The Dark Side of the Moon”, which experimented with multiple sounds to create this masterpiece. The huge increase in funds focused on PDAC, together with advanced efforts by the scientific community, can provide hope for patients affected by this tumor.
Declarations
AcknowledgmentsWe thank Radhika Srinivasan for carefully reading and editing our manuscript.
Authors’ contributionsContributed in the collection and discussion of all preclinical and clinical studies and in writing the manuscript: Mucciolo G, Roux C, Scagliotti A, Brugiapaglia S, Novelli F, Cappello P
Revised the manuscript: Cappello P
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from Associazione Italiana Ricerca sul Cancro (5 x mille no. 12182 and IG no. 19931) to Novelli F; University of Turin-Progetti Ateneo 2014-Compagnia di San Paolo PANTHER (to Cappello P); the University of Turin-Progetti Ricerca Locale (to Novelli F and Cappello P); Fondazione Ursula e Giorgio Cytron, Associazione Nadia Valsecchi; Brugiapaglia S is a recipient of a fellowship granted by Fondazione Onlus Ricerca Molinette.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913-21.
4. Ren B, Liu X, Suriawinata AA. Pancreatic ductal adenocarcinoma and its precursor lesions. Am J Pathol 2019;189:9-21.
5. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437-50.
6. Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers (Basel) 2018;10:316.
7. Adamska A, Domenichini A, Falasca M. Pancreatic ductal adenocarcinoma: current and evolving therapies. IJMS 2017;18:1338.
10. Laheru D, Jaffee EM. Immunotherapy for pancreatic cancer - science driving clinical progress. Nat Rev Cancer 2005;5:459-67.
11. Tomaino B, Cappello P, Capello M, Fredolini C, Ponzetto A, et al. Autoantibody signature in human ductal pancreatic adenocarcinoma. J Proteome Res 2007;6:4025-31.
12. Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer 2009;125:639-48.
13. Cappello P, Principe M, Novelli F. Pancreatic cancer vaccine: a unique potential therapy. Gastrointestinal Cancer Targets Ther 2016;6:1-11.
14. Tomaino B, Cappello P, Capello M, Fredolini C, Sperduti I, et al. Circulating autoantibodies to phosphorylated α-enolase are a hallmark of pancreatic cancer. J Proteome Res 2011;10:105-12.
15. Pandey R, Zhou M, Islam S, Chen B, Barker NK, et al. Carcinoembryonic antigen cell adhesion molecule 6 (CEACAM6 ) in Pancreatic Ductal Adenocarcinoma (PDA): an integrative analysis of a novel therapeutic target. Sci Rep 2019;9:1-14.
16. Gautam SK, Kumar S, Dam V, Ghersi D, Jain M, et al. MUCIN-4 (MUC4) is a novel tumor antigen in pancreatic cancer immunotherapy. Semin Immunol 2020;47:101391.
17. Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA 1993;90:3539-43.
18. Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol 2001;19:145-56.
19. Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg 2011;253:328-35.
20. Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res 2008;14:1455-63.
21. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2:616-31.
22. Brockstedt DG, Giedlin MA, Leong ML, Bahjat KS, Gao Y, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci USA 2004;101:13832-7.
23. Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase i studies of safety and immune induction. Clin Cancer Res 2012;18:858-68.
24. Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, et al. Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 2015;33:1325-33.
25. Le DT, Picozzi VJ, Ko AH, Wainberg ZA, Kindler H, et al. Results from a phase IIb, randomized, multicenter study of GVAX pancreas and CRS-207 compared with chemotherapy in adults with previously treated metastatic pancreatic adenocarcinoma (ECLIPSE Study). Clin Cancer Res 2019;25:5493-502.
26. Gjertsen MK, Breivik J, Saeterdal I, Thorsby E, Gaudernack G, et al. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet 1995;346:1399-400.
27. Gjertsen MK, Bakka A, Breivik J, Saeterdal I, Gedde-Dahl T, et al. Ex vivo ras peptide vaccination in patients with advanced pancreatic cancer: results of a phase I/II study. Int J Cancer 1996;65:450-3.
28. Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer 2001;92:441-50.
29. Wedén S, Klemp M, Gladhaug IP, Møller M, Eriksen JA, et al. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer 2011;128:1120-8.
30. Bernhardt SL, Gjertsen MK, Trachsel S, Møller M, Eriksen JA, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 2006;95:1474-82.
31. Shaw VE, Naisbitt DJ, Costello E, Greenhalf W, Park BK, et al. Current status of GV1001 and other telomerase vaccination strategies in the treatment of cancer. Expert Rev Vaccines 2010;9:1007-16.
32. Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol 2014;15:829-40.
33. Middleton G, Greenhalf W, Costello E, Shaw V, Cox T, et al. Immunobiological effects of gemcitabine and capecitabine combination chemotherapy in advanced pancreatic ductal adenocarcinoma. Br J Cancer 2016;114:510-8.
34. Neoptolemos JP, Greenhalf W, Cox TF, Costello E, Shaw V, et al. Predictive cytokine biomarkers for survival in patients with advanced pancreatic cancer randomized to sequential chemoimmunotherapy comprising gemcitabine and capecitabine (GemCap) followed by the telomerase vaccine GV1001 compared to concurrent chemoimmunotherapy in the TeloVac phase III trial. JCO 2014;32:4121.
35. Kameshima H, Tsuruma T, Kutomi G, Shima H, Iwayama Y, et al. Immunotherapeutic benefit of α-interferon (IFN-α) in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci 2013;104:124-9.
36. Shima H, Tsurita G, Wada S, Hirohashi Y, Yasui H, et al. Randomized phase II trial of survivin 2B peptide vaccination for patients with HLA-A24-positive pancreatic adenocarcinoma. Cancer Sci 2019;110:2378-85.
37. Kubo T, Tsurita G, Hirohashi Y, Yasui H, Ota Y, et al. Immunohistological analysis of pancreatic carcinoma after vaccination with survivin 2B peptide: Analysis of an autopsy series. Cancer Sci 2019;110:2386-95.
38. Osborne N, Sundseth R, Burks J, Cao H, Liu X, et al. Gastrin vaccine improves response to immune checkpoint antibody in murine pancreatic cancer by altering the tumor microenvironment. Cancer Immunol Immunother 2019;68:1635-48.
39. Kajihara M, Takakura K, Kanai T, Ito Z, Matsumoto Y, et al. Advances in inducing adaptive immunity using cell-based cancer vaccines: Clinical applications in pancreatic cancer. World J Gastroenterol 2016;22:4446-58.
40. Mukherjee P, Ginardi AR, Madsen CS, Sterner CJ, Adriance MC, et al. Mice with spontaneous pancreatic cancer naturally develop MUC-1-specific CTLs that eradicate tumors when adoptively transferred. J Immunol 2000;165:3451-60.
41. Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther 2008;6:955-64.
42. Rong Y, Qin X, Jin D, Lou W, Wu L, et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exp Med 2012;12:173-80.
43. Kobayashi M, Shimodaira S, Nagai K, Ogasawara M, Takahashi H, et al. Prognostic factors related to add-on dendritic cell vaccines on patients with inoperable pancreatic cancer receiving chemotherapy: a multicenter analysis. Cancer Immunol Immunother 2014;63:797-806.
44. Mayanagi S, Kitago M, Sakurai T, Matsuda T, Fujita T, et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci 2015;106:397-406.
45. Koido S, Homma S, Okamoto M, Takakura K, Mori M, et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin Cancer Res 2014;20:4228-39.
46. Takakura K, Koido S, Kan S, Yoshida K, Mori M, et al. Prognostic markers for patient outcome following vaccination with multiple MHC class I/II-restricted WT1 peptide-pulsed dendritic cells plus chemotherapy for pancreatic cancer. Anticancer Res 2015;35:555-62.
47. Mehrotra S, Britten CD, Chin S, Garrett-Mayer E, Cloud CA, et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J Hematol Oncol 2017;10:82.
48. Hobernik D, Bros M. DNA vaccines - how far from clinical use? International J Mol Sci 2018;19:3605.
49. Raz E, Carson DA, Parker SE, Parr TB, Abai AM, et al. Intradermal gene immunization: the possible role of DNA uptake in the induction of cellular immunity to viruses. PNAS 1994;91:9519-23.
50. Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, et al. Direct gene transfer into mouse muscle in vivo. Science 1990;247:1465-8.
51. Wang Z, Troilo PJ, Wang X, Griffiths TG, Pacchione SJ, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther 2004;11:711-21.
52. Cappello P, Curcio C, Mandili G, Roux C, Bulfamante S, et al. Next generation immunotherapy for pancreatic cancer: DNA vaccination is seeking new combo partners. Cancers 2018;10:51.
53. Capello M, Ferri-Borgogno S, Cappello P, Novelli F. α-enolase: a promising therapeutic and diagnostic tumor target. FEBS J 2011;278:1064-74.
54. Cappello P, Principe M, Bulfamante S, Novelli F. Alpha-Enolase (ENO1), a potential target in novel immunotherapies. Front Biosci (Landmark Ed) 2017;22:944-59.
55. Cappello P, Rolla S, Chiarle R, Principe M, Cavallo F, et al. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology 2013;144:1098-106.
56. Pan RY, Chung WH, Chu MT, Chen SJ, Chen HC, et al. Recent development and clinical application of cancer vaccine: targeting neoantigens. J Immunol Res 2018;2018:1-9.
57. Chen F, Zou Z, Du J, Su S, Shao J, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest 2019;129:2056-70.
58. Batista I, Melo S. Exosomes and the future of immunotherapy in pancreatic cancer. IJMS 2019;20:567.
59. Xiao L, Erb U, Zhao K, Hackert T, Zöller M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology 2017;6:e1319044.
60. Lu S, Zhang Z, Du P, Chard LS, Yan W, et al. A virus-infected, reprogrammed somatic cell-derived tumor cell (VIReST) vaccination regime can prevent initiation and progression of pancreatic cancer. Clin Cancer Res 2020;26:465-76.
61. Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol 2019;41:49-58.
62. Visioni A, Skitzki J. Technical considerations for the generation of adoptively transferred T cells in cancer immunotherapy. Cancers (Basel) 2016;8:86.
63. Wickström S, Lövgren T. Expansion of tumor-infiltrating lymphocytes from melanoma tumors. Methods Mol Biol 2019;1913:105-18.
64. Meng Q, Liu Z, Rangelova E, Poiret T, Ambati A, et al. Expansion of tumor-reactive T cells from patients with pancreatic cancer. J Immunother 2016;39:81-9.
65. Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J 2014;20:141-4.
66. Wang LX, Shu S, Disis ML, Plautz GE. Adoptive transfer of tumor-primed, in vitro-activated, CD4+ T effector cells (TEs) combined with CD8+ TEs provides intratumoral TE proliferation and synergistic antitumor response. Blood 2007;109:4865-76.
67. Moeller M, Haynes NM, Kershaw MH, Jackson JT, Teng MWL, et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 2005;106:2995-3003.
68. Kennedy R, Celis E. T helper lymphocytes rescue CTL from activation-induced cell death. J Immunol 2006;177:2862-72.
69. Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev 2014;257:264-76.
70. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, et al. A human memory T-cell subset with stem cell-like properties. Nat Med 2011;17:1290-7.
71. Hall M, Liu H, Malafa M, Centeno B, Hodul PJ, et al. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer 2016;4:61.
72. McCarty TM, Liu X, Sun JY, Peralta EA, Diamond DJ, et al. Targeting p53 for adoptive T-cell immunotherapy. Cancer Res 1998;58:2601-5.
73. Zhou JH, Zhang HM, Chen Q, Han DD, Pei F, et al. Relationship between telomerase activity and its subunit expression and inhibitory effect of antisense hTR on pancreatic carcinoma. World J Gastroenterol 2003;9:1808-14.
74. Hassanin H, Serba S, Schmidt J, Märten A. Ex vivo expanded telomerase-specific T cells are effective in an orthotopic mouse model for pancreatic adenocarcinoma. Clin Exp Immunol 2009;158:125-32.
75. Matsui H, Hazama S, Sakamoto K, Shindo Y, Kanekiyo S, et al. Postoperative adjuvant therapy for resectable pancreatic cancer with gemcitabine and adoptive immunotherapy. Pancreas 2017;46:994-1002.
76. Kawaoka T, Oka M, Takashima M, Ueno T, Yamamoto K, et al. Adoptive immunotherapy for pancreatic cancer: cytotoxic T lymphocytes stimulated by the MUC1-expressing human pancreatic cancer cell line YPK-1. Oncol Rep 2008;20:155-63.
77. Chung MJ, Park JY, Bang S, Park SW, Song SY. Phase II clinical trial of ex vivo-expanded cytokine-induced killer cells therapy in advanced pancreatic cancer. Cancer Immunol Immunother 2014;63:939-46.
78. Wang M, Shi S, Qi J, Tang X, Tian J. S-1 plus CIK as second-line treatment for advanced pancreatic cancer. Med Oncol 2013;30:747.
79. D’Ippolito E, Schober K, Nauerth M, Busch DH. T cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer Immunol Immunother 2019;68:1701-12.
80. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 2013;19:4792-800.
81. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, et al. Brief communication: efficacy of adoptive cell transfer of tumor infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother 2012;35:615-20.
82. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T cell transfer immunotherapy. Clin Cancer Res 2011;17:4550-7.
83. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun 2017;8:1-11.
84. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol 2019;10:128.
85. Dufait I, Liechtenstein T, Lanna A, Bricogne C, Laranga R, et al. Retroviral and lentiviral vectors for the induction of immunological tolerance. Scientifica (Cairo) 2012;2012.
86. Kebriaei P, Izsvák Z, Narayanavari SA, Singh H, Ivics Z. Gene therapy with the sleeping beauty transposon system. Trends Genet 2017;33:852-70.
87. da Cunha JPC, Galante PAF, de Souza JE, de Souza RF, Carvalho PM, et al. Bioinformatics construction of the human cell surfaceome. Proc Natl Acad Sci U S A 2009;106:16752-7.
88. Cohen CJ, Denkberg G, Segal D, Reiter Y. Generation of recombinant immunotoxins for specific targeting of tumor-related peptides presented by MHC molecules. Methods Mol Biol 2003;207:269-82.
89. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014;257:83-90.
90. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol 2014;5:254.
91. FDA. Research C for BE and KYMRIAH (tisagenlecleucel). Available from: http://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel [Last accessed on 15 Apr 2020].
92. FDA. Research C for BE and YESCARTA (axicabtagene ciloleucel). Available from: http://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel [Last accessed on 15 Apr 2020].
93. European Medicines Agency. First two CAR-T cell medicines recommended for approval in the European Union. Available from: https://www.ema.europa.eu/en/news/first-two-car-t-cell-medicines-recommended-approval-european-union [Last accessed on 15 Apr 2020].
94. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012;21:309-22.
95. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, et al. Coexpressed catalase protects chimeric antigen receptor–redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol 2016;196:759-66.
96. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep 2017;7:39833.
97. Suarez ER, Chang DK, Sun J, Sui J, Freeman GJ, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016;7:34341-55.
98. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol 2018;36:847-56.
99. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011;118:4817-28.
100. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002;298:850-4.
101. DeSelm CJ, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol 2017;116:63-74.
102. Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res 2014;74:2907-12.
103. Shimizu A, Hirono S, Tani M, Kawai M, Okada KI, et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci 2012;103:739-46.
104. Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori J, et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res 2012;18:2478-89.
105. Bharadwaj U, Marin-Muller C, Li M, Chen C, Yao Q. Mesothelin confers pancreatic cancer cell resistance to TNF-α-induced apoptosis through Akt/PI3K/NF-κB activation and IL-6/Mcl-1 overexpression. Mol Cancer 2011;10:106.
106. Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep 2013;3:1870.
107. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 2016;6:133-46.
108. Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015;28:638-52.
109. Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 2018;155:29-32.
110. Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer 2018;4:418-28.
112. Ishiwata T, Matsuda Y, Yoshimura H, Sasaki N, Ishiwata S, et al. Pancreatic cancer stem cells: features and detection methods. Pathol Oncol Res 2018;24:797-805.
113. Maliar A, Servais C, Waks T, Chmielewski M, Lavy R, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology 2012;143:1375-84.e5.
114. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843-51.
115. Raj D, Yang MH, Rodgers D, Hampton EN, Begum J, et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019;68:1052-64.
116. Gold P, Freedman SO. Specific carcinoembryonic antigens of the human digestive system. J Exp Med 1965;122:467-81.
117. Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 1999;9:67-81.
118. Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, et al. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012;143:1095-107.e2.
119. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011;19:620-6.
120. Jewett A, Kos J, Fong Y, Ko MW, Safaei T, et al. NK cells shape pancreatic and oral tumor microenvironments; role in inhibition of tumor growth and metastasis. Semin Cancer Biol 2018;53:178-88.
121. Chi X, Yang P, Zhang E, Gu J, Xu H, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med 2019;8:4753-65.
122. Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep 2017;21:3205-19.
123. Zhang E, Yang P, Gu J, Wu H, Chi X, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol 2018;11:102.
124. Taylor-Papadimitriou J, Burchell JM, Graham R, Beatson R. Latest developments in MUC1 immunotherapy. Biochem Soc Trans 2018;46:659-68.
125. Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, et al. Engineered CAR T cells targeting the cancer-associated Tn-Glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 2016;44:1444-54.
126. Levi E, Klimstra DS, Adsay NV, Andea A, Basturk O. MUC1 and MUC2 in pancreatic neoplasia. J Clin Pathol 2004;57:456-62.
127. Yazdanifar M, Zhou R, Grover P, Williams C, Bose M, et al. Overcoming immunological resistance enhances the efficacy of a novel anti-tMUC1-CAR T cell treatment against pancreatic ductal adenocarcinoma. Cells 2019;8:1070.
128. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther 2014;25:1003-12.
129. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther 2017;25:249-58.
130. Golubovskaya V, Berahovich R, Zhou H, Xu S, Harto H, et al. CD47-CAR-T cells effectively kill target cancer cells and block pancreatic tumor growth. Cancers (Basel) 2017;9:139.
131. Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell 2019;35:221-37.e8.
132. Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RMG, et al. Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol Ther 2017;25:259-73.
133. Masu T, Atsukawa M, Nakatsuka K, Shimizu M, Miura D, et al. Anti-CD137 monoclonal antibody enhances trastuzumab-induced, natural killer cell-mediated cytotoxicity against pancreatic cancer cell lines with low human epidermal growth factor-like receptor 2 expression. PLoS One 2018;13:e0200664.
134. Springfeld C, Jäger D, Büchler MW, Strobel O, Hackert T, et al. Chemotherapy for pancreatic cancer. Presse Med 2019;48:e159-74.
135. Posner MR, Cavacini LA, Upton MP, Tillman KC, Gornstein ER, et al. Surface membrane-expressed CD40 is present on tumor cells from squamous cell cancer of the head and neck in vitro and in vivo and regulates cell growth in tumor cell lines. Clin Cancer Res 1999;5:2261-70.
136. Hess S, Engelmann H. A novel function of CD40: induction of cell death in transformed cells. J Exp Med 1996;183:159-67.
137. Weiss JM, Gregory Alvord W, Quiñones OA, Stauffer JK, Wiltrout RH. CD40 expression in renal cell carcinoma is associated with tumor apoptosis, CD8(+) T cell frequency and patient survival. Hum Immunol 2014;75:614-20.
138. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612-6.
139. French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med 1999;5:548-53.
140. Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med 1999;5:780-7.
141. Beatty GL, Li Y, Long KB. Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 2017;17:175-86.
142. Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, et al. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med 2004;199:775-84.
143. Murphy WJ, Welniak L, Back T, Hixon J, Subleski J, et al. Synergistic anti-tumor responses after administration of agonistic antibodies to CD40 and IL-2: coordination of dendritic and CD8+ cell responses. J Immunol 2003;170:2727-33.
144. Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, et al. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol 2005;174:6013-22.
145. Banchereau J, de Paoli P, Valle A, Garcia E, Rousset F. Long-term human B cell lines dependent on interleukin-4 and antibody to CD40. Science 1991;251:70-2.
146. Schultze JL, Michalak S, Seamon MJ, Dranoff G, Jung K, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest 1997;100:2757-65.
147. Mathieu M, Cotta-Grand N, Daudelin JF, Boulet S, Lapointe R, et al. CD40-activated B cells can efficiently prime antigen-specific naïve CD8+ T cells to generate effector but not memory T cells. Plos One 2012;7:e30139.
148. Hunter TB, Alsarraj M, Gladue RP, Bedian V, Antonia SJ. An agonist antibody specific for CD40 induces dendritic cell maturation and promotes autologous anti-tumour T-cell responses in an in vitro mixed autologous tumour cell/lymph node cell model. Scand J Immunol 2007;65:479-86.
149. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 2015;3:399-411.
150. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 2001;19:47-64.
151. Nowak AK, Robinson BWS, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003;63:4490-6.
152. Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep 2016;15:2719-32.
153. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology 2015;149:201-10.
154. Vonderheide RH, Bajor DL, Winograd R, Evans RA, Bayne LJ, et al. CD40 immunotherapy for pancreatic cancer. Cancer Immunol Immunother 2013;62:949-54.
155. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res 2013;19:6286-95.
156. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769-81.
157. Quaranta V, Schmid MC. Macrophage-mediated subversion of anti-tumour immunity. Cells 2019;8:747.
158. Lenzo JC, Turner AL, Cook AD, Vlahos R, Anderson GP, et al. Control of macrophage lineage populations by CSF-1 receptor and GM-CSF in homeostasis and inflammation. Immunol Cell Biol 2012;90:429-40.
159. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014;25:846-59.
160. Quaranta V, Rainer C, Nielsen SR, Raymant ML, Ahmed MS, et al. Macrophage-derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer. Cancer Res 2018;78:4253-69.
161. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057-69.
162. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73:1128-41.
163. Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, et al. CD86 and CD80 Differentially modulate the suppressive function of human regulatory T Cells. J Immunol 2004;172:2778-84.
164. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252-64.
165. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007;8:239-45.
166. Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015;15:457-72.
167. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008;8:467-77.
168. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24:541-50.
169. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518-27.
170. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013;108:914-23.
171. Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother 2013;62:1249-60.
172. Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004;28:e26-31.
173. Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget 2016;7:71198-210.
174. Rahn S, Krüger S, Mennrich R, Goebel L, Wesch D, et al. POLE Score: a comprehensive profiling of programmed death 1 ligand 1 expression in pancreatic ductal adenocarcinoma. Oncotarget 2019;10:1572-88.
175. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
176. Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother 2017;66:1609-17.
177. Royal RE, Levy C, Turner K, Mathur A, Hughes M, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33:828-33.
178. Looi CK, Chung FFL, Leong CO, Wong SF, Rosli R, et al. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res 2019;38:162.
179. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 2007;13:2151-7.
180. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65.
181. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
182. Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68-74.e2.
183. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 2015;21:687-92.
184. Kabacaoglu D, Ciecielski KJ, Ruess DA, Algül H. Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: current limitations and future options. Front Immunol 2018;9:1878.
185. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer 2018;118:312-24.
186. Henriksen A, Dyhl-Polk A, Chen I, Nielsen D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat Rev 2019;78:17-30.
187. Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol 2014;25:1750-5.
188. Kamath SD, Kalyan A, Kircher S, Nimeiri H, Fought AJ, et al. Ipilimumab and gemcitabine for advanced pancreatic cancer: a phase Ib study. Oncologist 2019; doi: 10.1634/theoncologist.2019-0473.
189. Weiss GJ, Blaydorn L, Beck J, Bornemann-Kolatzki K, Urnovitz H, et al. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Invest New Drugs 2018;36:96-102.
190. Wainberg ZA, Hochster HS, Kim EJH, George B, Kalyan A, et al. Phase I study of nivolumab (Nivo) + nab-paclitaxel (nab-P) + gemcitabine (Gem) in advanced pancreatic cancer (APC). JCO 2019;37:298.
191. Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol 2013;23:273-80.
192. Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer 2016;2:286-94.
193. Weichselbaum RR, Liang H, Deng L, Fu YX. Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol 2017;14:365-79.
194. Azad A, Yin Lim S, D’Costa Z, Jones K, Diana A, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med 2017;9:167-80.
195. Victor CTS, Rech AJ, Maity A, Rengan R, Pauken KE, et al. Radiation and dual checkpoint blockade activates non-redundant immune mechanisms in cancer. Nature 2015;520:373-7.
196. Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, et al. Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res 2018;78:4282-91.
197. Guo S, Contratto M, Miller G, Leichman L, Wu J. Immunotherapy in pancreatic cancer: unleash its potential through novel combinations. World J Clin Oncol 2017;8:230-40.
198. Le DT, Lutz E, Uram JN, Sugar EA, Onners B, et al. Evaluation of Ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother 2013;36:382-9.
199. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T cell infiltration into pancreatic tumors. J Immunother 2015;38:1-11.
200. Lutz ER, Kinkead H, Jaffee EM, Zheng L. Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology 2014:3e962401.
201. Hilmi M, Bartholin L, Neuzillet C. Immune therapies in pancreatic ductal adenocarcinoma: where are we now? World J Gastroenterol 2018;24:2137-51.
202. Griffith KD, Read EJ, Carrasquillo JA, Carter CS, Yang JC, et al. In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J Natl Cancer Inst 1989;81:1709-17.
203. Bourquin C, von der Borch P, Zoglmeier C, Anz D, Sandholzer N, et al. Efficient eradication of subcutaneous but not of autochthonous gastric tumors by adoptive T cell transfer in an SV40 T antigen mouse model. J Immunol 2010;185:2580-8.
204. Abate-Daga D, Hanada K, Davis JL, Yang JC, Rosenberg SA, et al. Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood 2013;122:1399-410.
205. Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol 2016;34:4102-9.
206. Iafolla MAJ, Juergens RA. Update on programmed death-1 and programmed death-ligand 1 inhibition in the treatment of advanced or metastatic non-small cell lung cancer. Front Oncol 2017;7:67.
207. Rataj F, Kraus FBT, Chaloupka M, Grassmann S, Heise C, et al. PD1-CD28 fusion protein enables CD4+ T cell help for adoptive T cell therapy in models of pancreatic cancer and non-hodgkin lymphoma. Front Immunol 2018;9:1955.
208. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 2013;19:5636-46.
209. Blair AB, Zheng L. Rational combinations of immunotherapy for pancreatic ductal adenocarcinoma. Chin Clin Oncol 2017;6:31.
210. Hu B, Zou Y, Zhang L, Tang J, Niedermann G, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Human Gene Therapy 2018;30:446-58.
211. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, et al. CSF1R+ macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep 2018;23:1448-60.
212. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 2016;37:208-20.
213. Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, et al. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene 2013;32:1743-51.
214. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007;204:1257-65.
215. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can treg cells be a new therapeutic target? Cancer Sci 2019;110:2080-9.
216. Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol 2007;148:127-35.
217. Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 2007;67:5479-88.
218. Fogar P, Basso D, Fadi E, Greco E, Pantano G, et al. Pancreatic cancer alters human CD4+ T lymphocyte function: a piece in the immune evasion puzzle. Pancreas 2011;40:1131-7.
219. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer 2011;11:805-12.
220. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47-52.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mucciolo G, Roux C, Scagliotti A, Brugiapaglia S, Novelli F, Cappello P. The dark side of immunotherapy: pancreatic cancer. Cancer Drug Resist 2020;3:491-520. http://dx.doi.org/10.20517/cdr.2020.13
AMA Style
Mucciolo G, Roux C, Scagliotti A, Brugiapaglia S, Novelli F, Cappello P. The dark side of immunotherapy: pancreatic cancer. Cancer Drug Resistance. 2020; 3(3): 491-520. http://dx.doi.org/10.20517/cdr.2020.13
Chicago/Turabian Style
Mucciolo, Gianluca, Cecilia Roux, Alessandro Scagliotti, Silvia Brugiapaglia, Francesco Novelli, Paola Cappello. 2020. "The dark side of immunotherapy: pancreatic cancer" Cancer Drug Resistance. 3, no.3: 491-520. http://dx.doi.org/10.20517/cdr.2020.13
ACS Style
Mucciolo, G.; Roux C.; Scagliotti A.; Brugiapaglia S.; Novelli F.; Cappello P. The dark side of immunotherapy: pancreatic cancer. Cancer Drug Resist. 2020, 3, 491-520. http://dx.doi.org/10.20517/cdr.2020.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 56 clicks
Cite This Article 56 clicks

 Like This Article 33
likes
Like This Article 33
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.