
Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy

 ,
, Abstract
Development of drug resistance represents the major cause of cancer therapy failure, determines disease progression and results in poor prognosis for cancer patients. Different mechanisms are responsible for drug resistance. Intrinsic genetic modifications of cancer cells induce the alteration of expression of gene controlling specific pathways that regulate drug resistance: drug transport and metabolism; alteration of drug targets; DNA damage repair; and deregulation of apoptosis, autophagy, and pro-survival signaling. On the other hand, a complex signaling network among the entire cell component characterizes tumor microenvironment and regulates the pathways involved in the development of drug resistance. Gut microbiota represents a new player in the regulation of a patient’s response to cancer therapies, including chemotherapy and immunotherapy. In particular, commensal bacteria can regulate the efficacy of immune checkpoint inhibitor therapy by modulating the activation of immune responses to cancer. Commensal bacteria can also regulate the efficacy of chemotherapeutic drugs, such as oxaliplatin, gemcitabine, and cyclophosphamide. Recently, it has been shown that such bacteria can produce extracellular vesicles (EVs) that can mediate intercellular communication with human host cells. Indeed, bacterial EVs carry RNA molecules with gene expression regulatory ability that can be delivered to recipient cells of the host and potentially regulate the expression of genes involved in controlling the resistance to cancer therapy. On the other hand, host cells can also deliver human EVs to commensal bacteria and similarly, regulate gene expression. EV-mediated intercellular communication between commensal bacteria and host cells may thus represent a novel research area into potential mechanisms regulating the efficacy of cancer therapy.
Keywords
Introduction
The development of resistance to cancer therapies represents a major challenge in the treatment of cancer patients and the main cause of poor prognosis. Despite initial treatment response, cancer cells can eventually develop resistance, which can result from the acquisition of intrinsic characteristics of cancer cells, and the action of extrinsic factors. Such intrinsic cancer cell characteristics include genetic alterations (mutations, amplifications, deletions, translocations) and epigenetic modifications (methylation, acetylation) that can determine the aberrant expression of genes controlling drug metabolism (inactivation, efflux, target modification), cancer cell survival (inhibition of cell death, pro-survival signaling), or the acquisition of cancer stem cell phenotypes[1-9]. Extrinsic factors include the activation of signaling pathways in the tumor microenvironment (TME) that drive the acquisition of drug resistance in cancer cells. For instance tumor-associated immune cells, fibroblasts, and cancer cells can all secrete soluble signaling factors (e.g., cytokines and growth factors) that can activate cancer cell signaling pathways in autocrine or paracrine fashion to induce drug resistance (mTOR, NF-κB, AKT, STAT3)[10-12]. Furthermore, the combination of intrinsic and extrinsic factors plays a role in the regulation of tumor cell response to immunotherapy[13-17]. More recently, a new and important player is emerging in the regulation of the development of drug resistance: the gut microbiota.
Studies investigating the role of gut microbiota in regulating treatment response to chemotherapy and immunotherapy have been gaining interest. In particular, growing evidence highlights the importance of the interactions between commensal bacteria with both cancer and immune cells in modulating the efficacy of cancer treatment[18-21][Table 1]. The gut microbiota consists of a multispecies microbial community composed of bacteria, yeast, fungi protozoa, archea, and viruses that establishes symbiosis with the host organism. Our intestinal tract can host up to 100 trillion microbes with the number of expressed genes estimated to outnumber that of the host human’s genes by at least two orders of magnitude[22,23]. Recently, the bacterial to human cell ratio has been revised with an updated estimate of ≈ 1:1[24]. These studies provide evidence that gut microbiota may influence the physiology of the human host. The microbiota is an important source of metabolites (e.g., vitamins, organic acids, lipids, polyphenols, amino acids, and short chain fatty acids) that exert important functions on the regulation of intestinal epithelial and immune cell metabolism[25,26]. These metabolites are involved in the regulation of several biological functions, including modulation of chemotherapy and immunotherapy efficacy. Besides metabolites, bacterial components [lipopolysaccharides (LPS), bacterial cell wall constituents, DNA, and RNA] can also activate innate immune cells[27,28] and modulate the efficacy of cancer treatment (immunotherapy and chemotherapy).
Roles of bacteria in regulating the response to cancer therapies
| Bacterial species | Cancer type | Therapy | Mechanism | Ref. |
|---|---|---|---|---|
| Streptococci | Different types | Tumor cell killing | Unknown | [59] |
| Mycobacterium bovis | Human bladder | Reduction of tumor progression | Unknown | [60] |
| Lactobacillus casei | Human bladder | Decrease of recurrence | Unknown | [61] |
| B. thetaiotaomicron and B. fragilis | Mouse sarcoma, melanoma, colon | Response to anti-CTLA-4 | Th1 induction, DC maturation (IL-12 production) | [63] |
| Bifidobacterium | Mouse melanoma | Response to anti-PD-L1 | DC activation, stimulation of CD8+ T cell effector functions | [65] |
| A. muciniphila | Human NSCLC, RCC, UC | Response to anti-PD-1/PD-L1 | DC activation, increased CD4+ TCM tumor infiltration | [66] |
| Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium | Human melanoma | Response to anti-PD-1 | Cancer immune response | [68] |
| Faecalibacterium, Ruminococcaceae | Human melanoma | Response to anti-PD-1 | Increased CD8+ T cell tumor infiltration | [69] |
| Bacteroides thetaiotaomicron, Escherichia coli, Anaerotruncus colihominis | Human melanoma | Response to anti-PD-1 | Increased levels of Treg cells and MDSCs | [69] |
| B. caccae, B. thetaiotaomicron, B. vulgatus, B. massiliensis, P. distasonis, E. coli | Normal mouse colon | No therapy | Increase the proportion of Foxp3+ Treg cells among the CD4+ T cells | [70] |
| Commensal microbiota | Mouse lymphoma | Oxaliplatin | ROS-mediated cytotoxicity | [62] |
| Lactobacillus johnsonii, Lactobacillus murinus, Enterococcus hirae | Mouse melanoma, sarcoma | Cyclophosphamide | Induction of Th1, Th17-IFN-γ+, CD3+ T cell tumor infiltration | [93] |
| E. hirae, B. intestinihominis | Mouse sarcoma | Cyclophosphamide | Induction of Th1, CD8+ T cell tumor infiltration | [95] |
| Gammaproteobacteria | Mouse colon cancer | Gemcitabine | Intratumor gemcitabine deamination | [96] |
Extracellular vesicles (EVs) represent an additional level of complexity in the regulation of cancer treatment response. EVs are natural nanoparticles delimited by cellular membranous components that carry lipids, carbohydrates, signaling molecules, metabolites, proteins, DNA, RNA, and mediate cell-to-cell communication by delivering their cargo to recipient cells[29]. They are produced and secreted by virtually all cell types in all life kingdoms, including eukaryotic cells[30], prokaryotic cells[31], fungi[32], and archaea[33]. Growing evidence suggests that EVs may represent a universal mechanism of cell-to-cell communication in the same (intrakingdom) or different kingdoms (interkingdom) and regulate gene expression in recipient cells[34]. To illustrate, bacterial EVs have the ability to mediate horizontal transfer of nucleic acid molecules (intrakingdom communication) and confer antibiotic resistance to recipient cells[35-39]. Recently, the interkingdom interactions between bacteria and their human host have aroused particular interest, as bacterial EVs and their RNA cargos have the potential to regulate specific genes in the human host cells[34]. EVs produced by the commensal bacteria can cross the intestinal epithelium, where they can regulate the underlying immune cells. Furthermore, bacterial EVs carrying nucleic acids (DNA and RNA) can be secreted into the circulation[40-43], detected in human bodily fluids[44,45], and possibly be delivered to various tissues [Figure 1] after crossing intestinal epithelium[46]. Therefore, bacteria can potentially regulate the biological functions of host cells through the delivery of their EVs[34,46-49]. Indeed, bacterial EVs can be taken up by eukaryotic host cells and modulate the gene expression of recipient cells[34,45,50-53]. How can this be achieved? Bacterial EVs are enriched with short RNAs (sRNAs) ranging around 50-250 nucleotides[41]. These sRNAs have regulatory functions similar to miRNAs in eukaryotic cells, as they act by base-pairing with target mRNAs. However, different from eukaryotic miRNAs, sRNAs can either positively or negatively regulate the stability and translation of the target mRNA[54]. Recently, sRNAs comparable in size to miRNAs (15-28 nucleotides) named miRNA-sized sRNAs (msRNAs), have been identified in bacteria and may have a similar regulatory function to that of eukaryotic miRNAs[55-57]. Therefore, bacterial EVs have the potential ability to regulate specific genes in recipient cells. An intriguing speculation might consider that bacterial EVs could perhaps, similarly modulate the response to cancer therapy of human cells in the TME[58].
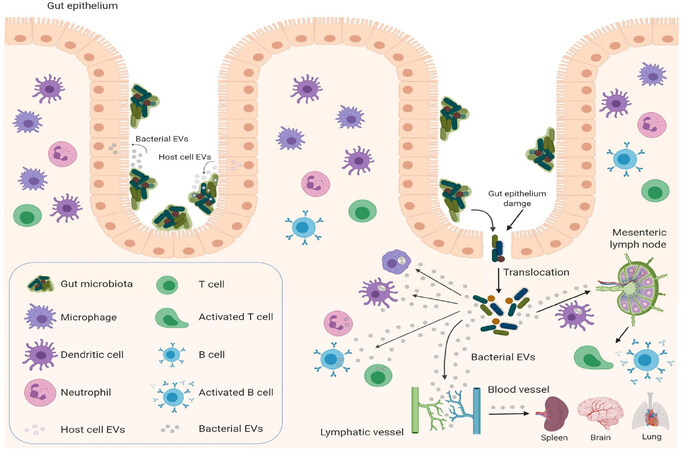
Figure 1. Gut microbiota and host cells: EV-mediated crosstalk. Bacterial EVs can deliver functional and active RNA molecules to host cells, such as gut epithelial cells, immune cells, and potentially cancer cells, and regulate their biological functions by affecting gene expression in recipient cells. Microbe-associated molecular patterns carried by bacterial EVs can activate the immune system locally (mesenteric lymph nodes) or systemically by reaching the circulatory system (spleen, peripheral lymph nodes) to modulate cancer immune responses. Host cells, such as intestinal epithelial cell can affect the gut microbiota by secreting EV-encapsulated miRNAs, which can regulate intestinal bacterial functions and composition by targeting microbial gene expression and potentially modulate the responses to cancer treatment. (Figure is created with BioRender.com). EVs: extracellular vesicles
Herein, we review the role of gut bacteria in the regulation of cancer treatment response to chemotherapy and immunotherapy, and the different mechanisms of inter-cellular communication between bacterial and host cells and their potential effect on treatment resistance.
Gut microbiota regulates effector functions and responses of immune cell to checkpoint inhibitors
The importance of microbes in cancer treatment was first discovered back in 1890, when heat-inactivated Streptococci were injected intratumorally[59]. Several decades later, it was found that intravesical administration of attenuated Mycobacterium bovis (strain Bacille Calmette-Guérin, BCG) was able to reduce tumor progression in muscle invasive bladder cancer[60]. More recently, it has been discovered that oral administration of Lactobacillus casei was able to decrease the recurrence of superficial bladder cancer[61]. These evidences highlight the potential role of bacteria in regulating cancer therapy.
Evidence of the functional role of gut microbiota in regulating the efficacy of immunotherapy was first provided by Iida et al.[62]. Antibiotic treatment (vancomycin, imipenem, neomycin) that disrupted the microbiota or germ-free condition before tumor inoculation impaired the efficacy of CpG-oligodeoxynucleotides and anti-IL-10R treatment in retarding tumor growth and prolonging survival. Gene expression analysis of tumors from mice pre-treated with antibiotics revealed downregulation of genes associated with inflammation, phagocytosis, antigen presentation, and adaptive immune responses, and upregulation of genes related to tissue development, cancer, and metabolism. In particular, antibiotic treatment reduced the infiltration of myeloid-derived cells (monocytes, macrophages, and dendritic cells) and their ability to produce pro-inflammatory cytokines (TNF-α, IL-1α, IL-1β, IL-12, and CXCL10), but not the expression of anti-inflammatory cytokines (IL-10, IL-1RA). The activation of tumor-associated myeloid-derived cells through LPS-mediated activation of TLR4 by commensal microbiota was necessary to prime these cells for the TLR9-dependent response to GpG-oligodeoxynucleotide therapy and achieve optimal response to cancer therapy.
Following this discovery, several studies have shown the key role of gut microbiota in regulating the efficacy of immune checkpoint inhibitor (ICI) therapy for cancer treatment. Vétizou et al.[63] found that gut microbiota composition could affect the response to anticancer immunotherapy by CTLA-4 blockade. In an animal model using three different cancer types (sarcoma, melanoma, and colon cancer), the administration of anti-CTLA-4 was able to control tumor growth in mice housed in specific pathogen-free conditions and in the presence of specific Bacteroides, but failed to inhibit tumor growth in germ-free mice and mice previously treated with antibiotics. These findings suggest that gut microbiota could potentially regulate the anticancer effect of anti-CTLA-4 therapy. Specific Bacteroides species (B. thetaiotaomicron and B. fragilis) were able to restore the anticancer response to anti-CTLA-4 treatment when used to recolonize germ-free mice and mice treated with antibiotics. The presence of B. fragilis induced T helper 1 (Th1) responses and promoted dendritic cell (DC) maturation (IL-12 production) in tumor-draining lymph nodes and within the tumor itself, respectively. Interestingly, treatment with anti-CTLA-4 was also able to induce an immune response against these Bacteroides species, which raised the hypothesis of potential molecular mimicry between tumor antigens and specific commensal bacterial species. Similar results were obtained when feces from melanoma patients treated with ipilimumab were transplanted into germ-free mice, which resulted in the induction of a marked response to anti-CTLA-4 and decreased tumor growth.
Heterogeneous and transitory responses to immune checkpoint inhibitors are often observed in patients with melanoma[64]. Sivan et al.[65] hypothesized that intestinal microbiota can be a possible modulator of the immune response in melanoma patients. In an animal model, tumor growth of subcutaneously injected B16.SIY cells was different in genetically similar C57BL/6 mice that harbored different commensal microbes. The difference in B16.SIY cell growth rate was due to different spontaneous antitumor immunity characterized by enhanced tumor-specific T cells responses and intratumoral CD8+ T cells infiltration. The authors found that Bifidobacterium application was associated with the antitumor effect, as it was able to activate DCs, possibly through soluble signaling factors disseminating systemically, which in turn elicited the antitumor effector functions of CD8+ T cells. Furthermore, oral administration of Bifidobacterium resulted to be effective in reducing tumor growth to the same extent as anti-PD-L1 treatment, and the combination of these two treatments almost completely inhibited tumor growth.
More recently, other studies focused on the role of the microbiota in regulating the efficacy and resistance of ICI therapy for cancer treatment. Routy et al.[66] found that primary resistance to ICI therapy can be associated with altered gut microbiota composition, as antibiotic treatment inhibited the efficacy of ICI therapy in patients with advanced cancer. Patients with advanced non-small cell lung carcinoma, renal cell carcinoma, and urothelial carcinoma treated with a combination of broad-spectrum antibiotics (including β-lactamase-inhibitors, fluoro-quinolones, or macrolides) 2 months before, or one month after the first administration of anti-PD1/PD-L1 therapy, had a poorer clinical prognosis compared to patients who did not receive antibiotics. These antibiotics are generally prescribed for common indications including dental, urinary, and pulmonary infections, and can transiently alter the composition of gut microbiota. A metagenomic analysis of patients’ stools revealed a relative abundance of Akkermansia muciniphila was associated with improved clinical responses to ICI. As one of the most abundant bacteria in the ileum microbiota[67], A. muciniphila can induce DCs to secrete IL-12 and increase recruitment of CD4+ T central memory cells expressing small intestine-associated chemokine receptor CCR9 and Th1-associated chemokine receptor CXCR3 to mice tumors.
Another work by Matson et al.[68] showed that the composition of microbiota could contribute towards identifying cancer patients who may benefit from anti-PD-1-based immunotherapy. A metagenomic analysis of baseline stool samples from metastatic melanoma patients before immunotherapy revealed an association between the composition of commensal microbes and clinical responses. Specifically, Bifidobacteriumlongum, Collinsellaaerofaciens, and Enterococcus faecium were more abundant in patients who responded to anti-PD-1 immunotherapy.
A similar study by Gopalakrishnan et al.[69] compared the gut microbiome between responder and non-responder metastatic melanoma patients treated with anti-PD-1 immunotherapy. Significant differences in both the diversity and composition of the microbiome were found between the two groups of patients, with a relative abundance of bacteria of Clostridiales order and Ruminococcaceae family in the responder group. Furthermore, a higher density of CD8+ T cell infiltration was observed in tumor tissues of responder patients and this was correlated with the abundance of the Faecalibacterium genus (Ruminococcaceae family). Analysis of the systemic immune response showed a higher frequency of effector CD4+ and CD8+ T cells in circulation and a preserved ability of cytokine production in patients with a higher abundance of Ruminococcaceae in gut microbiota. On the other hand, non-responder patients had enrichment in Bacteroides thetaiotaomicron, Escherichia coli, and Anaerotruncus colihominis. Furthermore, non-responder patients with a higher abundance of Bacteroidales had increased levels of regulatory T (Treg) cells and myeloid-derived suppressor cells in the systemic circulation, with reduced cytokine production ability.
A systematic analysis identified human-associated microbial species that have the specific ability to modulate Treg cell responses. Faith et. al[70] showed that, in in vivo study using mice monocolonized by specific bacterial strains, four members from the Bacteroides genus (B. caccae, B. thetaiotaomicron, B. vulgatus, and B. massiliensis), one from the Parabacteroides genus (P. distasonis), and E. coli from the Escherichia genus were able to significantly increase the proportion of Foxp3+ Treg cells among CD4+ T cells.
In summary, these results provide evidence to support the crucial role of the gut microbiota in regulating the effector functions of immune cells and thus, control of the efficacy of ICI therapy. The presence of specific microbial species is also essential for an effective response to, or failure of treatment.
Soluble signaling factors from gut microbiota regulate immune cells
Toll-like receptor activation
We have discussed above that soluble signaling factors released from commensal microbiota may regulate the activation of immune cells in the TME or tumor-draining lymph nodes, and thus the efficacy of immune therapy. These soluble signaling factors include microbe-associated molecular patterns (MAMPs) or pathogen-associated molecular patterns (PAMPs), which are conserved, specific molecules derived from microbes. They can bind to pattern recognition receptors (PRRs) such as Toll-like receptors, cytosolic NOD-like receptors, C-type lectin receptors, and RIG-I-like receptors. PRRs are expressed by immune cells of the innate (macrophages and DCs) and adaptive (T and B cells) immune system[28,71,72]. Therefore, microbiota-derived MAMPs or PAMPs, such as LPS[73], peptidoglycan[74], or flagellin[75], can enter the circulation through the mucosal barrier to modulate immune responses to cancer. Nucleic acids, such as unmethylated CpG released by bacteria, can bind to TLR9 and control differential T cell responses by regulating Th1 and Th2 polarization[76], the frequency of CD4+Foxp3+ Tregs, and IL-17-IFN-γ-producing effector T (Teff) cells[77]. Bacteroidetes are one of the major phyla of the commensal microbiota and they can control inflammation by regulating the differentiation of Tregs. Capsular polysaccharide A released by Bacteroides fragilis (B. fragilis) has immunoregulatory properties that mediate the conversion of CD4+ T cells into Foxp3+ Tregs through TLR2-mediated signaling. These cells have increased suppressive capacity through higher production of the anti-inflammatory cytokine IL-10[78].
In summary, soluble signaling factors released from specific bacterial species of the gut, which include structural components of bacteria, such as MAMPs, can induce different immune responses with opposing effects on the efficacy of cancer therapy.
Metabolite receptor activation
Other soluble signaling factors from gut microbiota can regulate cancer immune responses. Growing evidence highlight the importance of bacterial metabolites in the profound regulation of immune cells[79] and accordingly, the efficacy of cancer immunotherapy. Immune cells express receptors specific for bacterial metabolites: purinergic receptors (P2X7) detect adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide; GPR43 and GPR41 detect short-chain fatty acids (SCFAs); membrane bile acid receptor (M-BAR/TGR5) and farnesoid X receptor detect bile acid and xenobiotic metabolites; and aryl hydrocarbon receptor precursor and pregnane X receptor detect tryptophan, indole, bile acid and toxicant metabolites[80]. Gut bacteria produce SCFAs, such as butyrate, acetate, and propionate, through anaerobic fermentation of carbohydrates. SCFA receptors are expressed on macrophage, dendritic cells, and neutrhophils, which can regulate T and B cell-mediated responses. SCFAs also play an important role in the generation and modulation of Tregs[81]. Butyrate, produced during starch fermentation, induces extrathymic differentiation of Tregs and propionate potentiates the generation of Tregs in the periphery[82]. SCFAs can also regulate T cell differentiation into T helper 17 (Th17), Th1, and IL-10-producing T cells by their histone deacetylase inhibitor activity[83]. Indeed, butyrate can induce differentiation of Tregs both in vitro and in vivo by enhancing the acetylation of histone H3 in the promoter region and intragenic enhancer elements of the FoxP3 locus termed conserved noncoding sequence[84].
Roughly, one-third of patients treated with anti-CTLA-4 develop immune-mediated colitis induced by mucosal immune dysregulation[85]. Bacteroides can induce the expansion of Tregs[78], which play an important role in regulating the development of colitis[86]. Increased representation of members of the Bacteroidetes phylum (Bacteroidaceae, Rikenellaceae and Barnesiellaceae families) inhibit the development of colitis induced by anti-CTLA-4 immunotherapy and this is associated with the biosynthesis of B vitamins[87].
In summary, metabolites produced by bacteria have important effects on the regulation of immune cell functions and accordingly, the efficacy of cancer therapy.
Gut microbiota regulates the response to chemotherapy
Besides regulating the response to immunotherapy, gut microbiota can also modulate the efficacy of chemotherapy. As discussed above, antibiotic treatment can reduce the efficacy of CpG-oligodeoxynucleotides and anti-IL-10R treatment in retarding tumor growth and prolonging survival of mice[62]. Besides regulating the response to immunotherapy, the authors have shown that the absence of commensal bacteria, either by treatment with an antibiotic cocktail or rearing mice in germ-free conditions, resulted in an impaired response to treatment with oxaliplatin, a cytotoxic cancer chemotherapy that forms platinum DNA adducts and intrastrand cross-links[88,89]. The absence of gut microbiota was also associated with the reduction of the expression of Cybb gene, which encodes for NADPH oxidase 2 (Nox2) that in turn determined a decreased generation of reactive oxygen species (ROS) by tumor-associated myeloid cells (neutrophils and macrophages) and ROS-mediated oxaliplatin genotoxicity. The underlying mechanism involved could be associated with microbial products sensed by myeloid cells in a TLR4-independent fashion.
Cyclophosphamide (CTX) is an alkylating chemotherapy drug that interferes with DNA replication by forming intrastrand and interstrand DNA crosslinks[90]. Besides controlling tumor cell apoptosis[91,92], the therapeutic efficacy of CTX is also due to its ability to induce immunogenic cancer cell death, leading to stimulation of antitumor immune responses. Viaud et al.[93] showed that CTX treatment of mice bearing subcutaneous cancers (B16F10 melanoma cells and MCA205 sarcoma cells) induced increased permeability of the intestinal mucosa with consequent translocation of distinct commensal bacteria (gram-positive Lactobacillus johnsonii, Lactobacillus murinus and Enterococcus hirae) into mesenteric lymph nodes and the spleen. Gram-positive bacteria could induce polarization of CD4+ T cells towards IFN-γ-producing Th1 and “pathogenic” Th17 (pTh17, expressing both IFN-γ and IL-17) cells, which are important effector cells in controlling cancer growth[94]. Importantly, both broad-spectrum (ATB) and gram-positive specific antibiotics (e.g., vancomycin) considerably reduced the anticancer efficacy of CTX against MCA205 sarcoma and P815 mastocytoma. Similar results were obtained using a transgenic lung cancer mouse model, where a reduction of tumor-infiltrating CD3+ T and Th1 cells was seen.
A study by Daillère et al.[95] revealed that two different commensal bacteria located in different parts of the intestine could ameliorate the efficacy of CTX. In tumor-bearing mice treated with CTX, the gram-negative Barnesiella intestinihominis, residing in the colon, induced systemic CD4+ Th1 cells and type 1 CD8+ T (Tc1) cells and increased the proportion of intratumoral IFN-γ-producing γδT cells. In contrast, the gram-positive E. hirae, which resides in the small intestine, induced systemic pTh17 cells and increased the intratumoral CD8+ cytotoxic T lymphocytes (CTL): Tregs ratio. These effects were subsequently translated into effective anticancer responses to CTX. Furthermore, the presence of memory CD4+ CD45RO+ Th1 cells against E. hirae and B. intestinihominis was predictive of a longer progression-free survival in lung cancer patients after platinum-based chemotherapy and, in the treatment of ovarian cancer patients resistant to platinum-based chemotherapy with CTX.
More recently, the intratumor presence of bacteria was found to be responsible for chemotherapeutic drug resistance. Geller et al.[96] found that the presence of Mycoplasma hyorhinis (M. hyorhinis) in human dermal fibroblasts determined resistance to gemcitabine both in vitro and in vivo and this was due to the metabolization of gemcitabine into its deaminated inactive metabolite 2’,2’-difluorodeoxyuridine. However, Mycoplasma sp. was not the only bacteria capable of inducing resistance to gemcitabine. Indeed, the author revealed that bacteria belonging to the Gammaproteobacteria class expressed the long form bacterial enzyme cytidine deaminase (CDDL), which mediates gemcitabine deamination and its inactivation. In an animal model of colon cancer, the intratumor presence of CDDL-expressing bacteria conferred resistance to gemcitabine and the elimination of bacteria by ciprofloxacin treatment restored the response to the chemotherapeutic drug. Furthermore, analysis of a cohort of 113 pancreatic ductal adenocarcinoma (PDAC) tissues revealed the presence of Gammaproteobacteria (most of them belonging to Enterobacteriaceae and Pseudomonadaceae families) in 86 patients’ samples (76%), possibly due to retrograde migration from the duodenum to the pancreas. Bacteria from fresh PDAC could also render both colon cancer cell lines RKO and HCT116 fully resistant to gemcitabine, in an in vitro co-culture model.
In summary, gut microbiota can regulate the response to chemotherapy either by affecting the effector mechanism of the drug (e.g., ROS production), modulating the induction of its immunogenic properties, or metabolizing it into an inactive form.
Gut microbiota regulates the expression of host cells miRNAs that modulate the response to cancer therapy
Our discussion thus far has suggested that gut microbiota can regulate the response to cancer therapy by modulating the functions of immune cells against cancer cells, or affecting the efficacy of chemotherapeutics drugs. Growing evidence reveals that gut bacteria play a role in the modulation of gene expression in host cells by regulating their miRNA expression[97,98]. Dalmasso et al.[99] showed that one miRNA in the ileum (miR-298) and eight miRNAs in the colon (upregulated: miR-128, miR-200c, miR-342-5p; downregulated: miR-465c-5p, miR-466d-3p, miR-466d-5p, miR-665 and miR-683) were differentially expressed in the microbiota of colonized mice compared with germ-free mice. Importantly, the microbiota-mediated downregulation of miR-665 induced the upregulation of its target gene, the ATP-binding cassette transporter Abcc3, both at mRNA and protein levels in the colon. Notably, the overexpression of Abcc3 in colon cancer can contribute to the acquisition of drug resistance[100].
Commensal bacteria can also regulate miRNA expression in dendritic cells and accordingly their functions. For instance, the engagement of TLR1/2, TLR4, TLR5, TLR9 and NOD2 induces the downregulation of miR-10a through the MyD88-dependent pathway[101]. MiR-10a also targets the IL-12/IL-23p40 gene, which is a key regulator of the induction of Th1 cell-mediated immune responses and increasd cytotoxic activity of CD8+ T and natural killer cells. These immune cell-mediated functions are crucial for effective anti-cancer immune responses.
Recently, an over-representation of Fusobacteriumnucleatum (F. nucleatum) was found in the tissue of colorectal cancer (CRC) patients compared with matched normal tissue[102]. Interestingly, Yu et al.[103] found a higher abundance of F. nucleatum in the tissue of CRC patients with recurrent disease compared with non-recurrent patients after chemotherapy. Furthermore, high levels of F. nucleatum were associated with shorter recurrence-free survival. To investigate the potential mechanisms behind F. nucleatum-induced chemoresistance, the authors compared miRNA expression profiles between CRC tissues with a high amount of F. nucleatum from recurrent patients and CRC tissues with a low amount of F. nucleatum from non-recurrent patients. Among the 68 miRNAs that were differentially expressed, two specific miRNAs showed the most significant downregulation in CRC tissues with a high amount of F. nucleatum: miR-4802 and miR-18a*. These two miRNAs target ULK1 and ATG7, both of which are mediators of autophagy. The F. nucleatum-mediated downregulation of miR-4802 and miR-18a* was associated with reduced apoptosis induced by oxaliplatin and 5-FU through the activation of autophagy both in vitro and in vivo. The acquisition of chemoresistance mediated by F. nucleatum was dependent on the activation of TLR4/MYD88 signaling.
In summary, these studies provide insight into the role of soluble signaling factors released by gut microbiota on the modulation of drug resistance through the regulation of miRNA expression in host cells.
EVs mediated cross talk communication between bacteria and host cells
Due to the high number of molecules with regulatory ability, EVs play an important role in cell-to-cell communication and the regulation of biological function in both normal and cancer cells[29]. Bacteria are able to generate EVs carrying molecules with signaling properties. The presence of nucleic acids inside bacterial EVs and the ability to mediate intercellular genetic transfer was first detected in 1989[104]. The mechanism of biogenesis of EVs by bacteria remains poorly understood and less well-known compared to that of eukaryotic cells. Bacterial EVs can be generated by both gram-positive[105-107] and gram-negative bacteria[108]. Most of our understanding of the biogenesis of bacterial EVs originate from studies on gram-negative bacteria[108], which generate EVs from the outer membrane and accordingly, are named outer membrane vesicles (OMVs). These may range from 20 to 250 nm in diameter and contain components derived from the outer membrane (OM), inner membrane, and periplasmic space, which includes proteins, lipoproteins, phospholipids and LPS. OMVs are generated by blebbing outwards from the OM, during which process they include soluble components inside and adherent material, on the external surface[109]. EVs produced by gram-positive bacteria are structurally similar to OMVs, with sizes ranging from 10 to 400 nm[107]. They carry bacterial components including nucleic acid, proteins, lipids, enzymes and toxins. Compared with the well-elucidated mechanism of generating OMVs, the secretion of gram-positive EVs is still disputed, with an evidence-supported hypothesis that describes the enzyme-mediated degradation of the bacterial wall to facilitate EV release[105,106]. The loading of cargo inside gram-positive EVs and OMVs may represent an active metabolic process[110], as marked differences in RNA content were measured between bacterial EVs and their producer cells[43,111,112].
Bacterial EVs are enriched in sRNAs and msRNA[41,51,52] that have regulatory functions similar to miRNA in eukaryotic cells. Therefore, EV-mediated delivery of these RNA molecules can potentially regulate specific genes in recipient human cells[54]. A recent study reported that OMVs from Pseudomonas aeruginosa can mediate the delivery of a specific sRNAs (sRNA52320) to human bronchial epithelial cells and target two mitogen-activated protein (MAP) kinases (MAP3K7 and MAP2K4) to regulate the LPS-stimulated mitogen-activated protein kinase (MAPK) signaling pathway[51]. Another study showed that the periodontal pathogens Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, and Treponema denticola express msRNAs that can be delivered to fibroblast cells through OMVs. Furthermore, transfection of Jurkat cells with synthetic msRNAs reduced the expression of IL-5, IL-13 and IL-15[52]. These studies confirm that bacterial EVs have the ability to deliver short RNA molecules with regulatory functions to human cells and modulate gene expression. This microbial biological function may support the hypothesis that bacterial EVs might potentially modulate the expression of genes regulating resistance to chemotherapy and immunotherapy.
Interestingly, host cells can also participate in inter-kingdom communication through the delivery of miRNAs encapsulated within human cell-derived EVs to bacteria [Figure 1]. Host intestinal epithelial cells secrete EV-encapsulated miRNAs that can be detected in the feces of mouse (the most abundant: miR-1224, miR-2146, miR-2134, miR-483, miR-710, miR-2141, miR-720, miR-155 and miR-34c) and human (the most abundant: miR-1246, miR-601, miR-630, miR-2116-5p, miR-320e, miR-1224-5p, miR-155-5p and miR-194-5p). EV-encapsulated miRNAs can then be taken up by F. nucleatus and E. coli and alter their expression of genes controlling their growth, with the result of maintaining the normal physiological balance of gut microbiota. In particular, hsa-miR-515-5p promotes the growth of F. nucleatus while hsa-miR-1226-5p promotes the growth of E. coli[113].
Collectively, these studies provide supporting evidence that a potential two-way EV-mediated communication may exist between bacteria and human host cells, resulting in reciprocal regulation of gene expression and accordingly, biological cell function. An example of such reciprocal regulation between gut bacteria and host cells with an effect on the development of drug resistance is illustrated by F. nucleatus. High amounts of F. nucleatus in the gut of CRC patients induce the activation of autophagy in CRC cells through the TLR4/MYD88-mediated downregulation of miR-18a* and miR-4802, which in turn promotes chemoresistance of CRC cells to chemotherapeutic drugs (oxaliplatin and 5-FU)[103]. On the other hand, intestinal epithelial cells can control the growth of F. nucleatus through the delivery of miRNAs, such as hsa-miR-515-5p[113]. In CRC patients, this equilibrium can be potentially compromised by the altered expression of miR-515-5p, that might affect F. nucleatus proliferation and consequent response to chemotherapeutic drugs, and thus prognosis[114].
Conclusions
The development of drug resistance represents the most important cause of cancer treatment failure and is responsible for the poor prognosis of cancer patients. Growing evidence demonstrates that tumors are characterized by a high degree of molecular heterogeneity[115] and cancer treatment can promote therapy-induced selection for a previously minor subset of resistant cells[116]. Different mechanisms are responsible for drug resistance. Some arise from intrinsic modification of cancer cells causing the alteration of genes that control specific pathways involved in treatment resistance such as drug transport and metabolism, alteration of drug targets, DNA damage repair, deregulation of apoptosis, autophagy, and prosurvival signaling[117]. Other mechanisms are included in the complex network of signaling the TME[118].
Recently, the gut microbiota has received increasing attention for its role in regulating the efficacy of both immunotherapy and chemotherapy. Indeed, since the first evidence suggestive of the role of microbes in cancer treatment in 1890[59], recent work has started to show that gut microbiota can have a remarkable effect on cancer therapies. After the advent of ICI therapy and promising evidence of its curative potential when used in combination with chemotherapy[119], it has been shown that specific species of the gut microbiota can regulate the efficacy of ICI responses. Specific Bacteroides species (B. thetaiotaomicron and B. fragilis) are essential for an effective response to anti-CTLA-4[63]. The presence of Bifidobacterium in combination with anti-PD-L1 treatment can result in almost complete inhibition of melanoma tumor growth[65]. The presence of A. muciniphila, one of the most abundant bacteria in the ileum microbiota, is associated with clinical response to anti-PD-1/PD-L1 therapy[66]. Similarly, patients who respond to anti-PD-1 therapy have high levels of Bifidobacteriumlongum, Collinsellaaerofaciens, and Enterococcus faecium[68]. Patients with melanoma who respond to anti-PD-1 therapy have a relative abundance of bacteria of the Ruminococcaceae family, whereas patients who do not respond are enriched in Bacteroides thetaiotaomicron, Escherichia coli, and Anaerotruncus colihominis[69]. As reported in the studies above, it is important to note that the use of antibiotics induces dysbiosis or elimination of gut microbiota and accordingly, impairs the efficacy of ICI therapy. Cancer patients are already at higher risk of bacterial infection and sepsis during treatment. Indeed, chemotherapeutic drugs[120] and ICI therapy[121,122] often cause intestinal mucositis and treatment-induced intestinal barrier dysfunction due to injury of the mucosal barrier with consequent translocation of bacteria across the gut into the systemic environment [Figure 1]. Furthermore, neutropenia is often associated with cancer treatment[120,123]. These two complications require prophylactic antibiotic therapy to prevent sepsis during chemotherapy[124,125]. Because the alteration of the physiological equilibrium of gut microbiota induced by prophylactic antibiotic therapy can affect the efficacy of chemotherapy and immunotherapy, a careful approach should be considered with regard to prophylactic antibiotics for cancer patients. Furthermore, intestinal barrier dysfunction is associated with increased levels of systemic LPS-positive bacterial EVs that induce the secretion of pro-inflammatory mediators (IL-6, IL-8, MCP-1 and MIP-1α) by peripheral blood mononuclear cells[126] with consequent increased risk of sepsis or toxic shock syndrome[127]. Therefore, the use of selective antibiotic therapy to target specific harmful bacterial species and spare those associated with the effectiveness of cancer therapy along with a concomitant administration of probiotics, prebiotics to rebalance the gut microbiota should be taken in consideration[128].
It is noteworthy to observe that specific bacterial species are associated with effective responses to cancer therapy. Thus, it could be hypothesized that besides common MAMPs or PAMPs that are conserved molecules derived from bacteria, other specific bacteria-derived components may be involved in regulating the efficacy of cancer treatment. That bacteria-specific protein antigens are able to elicit and sustain a cancer immune response could be a possible explanation. Additionally, bacteria-specific nucleic acid sequences that are able to activate TLR3-8-9-mediated immune responses could also be involved.
An additional level of complexity is added by bacterial EVs, which can carry bacterial components and have the ability to regulate gene expression in host recipient cells. Potentially, shuttling of sRNAs through bacterial EVs to host cells could represent an additional mechanism to either inhibit or enhance the response to chemotherapy and immunotherapy. Thus, EV-mediated delivery of bacterial sRNAs to cancer cells has the potential to represent a new field of research for the study of response to cancer therapies. Furthermore, due to the two-ways nature of EV-mediated inter-kingdom communication, either normal or host cancer cells may potentially affect the microbiota composition and in turn, regulate the efficacy of cancer therapies.
In conclusion, the gut microbiota represents a new player in the regulation of cancer therapy efficacy. EV-mediated inter-kingdom communication could also represent a new research field to better understand the relationship between commensal bacteria and the human host to improve the efficacy and reduce toxicity of cancer therapies.
Declarations
Authors’ contributionsMade substantial contributions to the content, composition, and discussion: Anfossi S
Made substantial contributions to editing and discussion: Calin GA
Availability of data and materialsNot applicable.
Financial support and sponsorshipDr. Calin is the Felix L. Endowed Professor in Basic Science. Work in Dr. Calin’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943-01 through the NIH Common Fund, Office of Strategic Coordination (OSC) the NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, an NIGMS 1R01GM122775-01 grant, a U54 grant #CA096297/ CA096300 - UPR/MDACC Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Ladies Leukemia League grant, a Chronic Lymphocytic Leukemia Moonshot Flagship project, a Sister Institution Network Fund (SINF) 2017 grant, and the Estate of C. G. Johnson Jr.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6:1769-92.
3. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull 2017;7:339-48.
4. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15:81-94.
5. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010;11:220-8.
6. Chatterjee N, Bivona TG. Polytherapy and targeted cancer drug resistance. Trends Cancer 2019;5:170-82.
7. Pathania S, Bhatia R, Baldi A, Singh R, Rawal RK. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed Pharmacother 2018;105:53-65.
8. Calin GA, Trapasso F, Shimizu M, Dumitru CD, Yendamuri S, et al. Familial cancer associated with a polymorphism in ARLTS1. N Engl J Med 2005;352:1667-76.
10. Qu Y, Dou B, Tan H, Feng Y, Wang N, et al. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol Cancer 2019;18:69.
11. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013;13:759-71.
12. Fiedler E, Hemann MT. Aiding and abetting: how the tumor microenvironment protects cancer from chemotherapy. Ann Rev Cancer Biol 2019;3:409-28.
13. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707-23.
14. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99.
15. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8.
16. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819-29.
17. Saleh R, Elkord E. Acquired resistance to cancer immunotherapy: role of tumor-mediated immunosuppression. Semin Cancer Biol 2019. S1044-579X(19)30171-3
18. Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome 2018;6:92.
19. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol 2017;14:356-65.
20. Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108.
21. Li H, He J, Jia W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol 2016;12:31-40.
22. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006;124:837-48.
23. Lepage P, Leclerc MC, Joossens M, Mondot S, Blottière HM, et al. A metagenomic insight into our gut’s microbiome. Gut 2013;62:146-58.
24. Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016;164:337-40.
25. Vernocchi P, Del Chierico F, Putignani L. Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front Microbiol 2016;7:1144.
26. Levy M, Thaiss CA, Elinav E. Metabolites: messengers between the microbiota and the immune system. Genes Dev 2016;30:1589-97.
27. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol 2001;1:135-45.
29. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018;19:213-28.
30. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014;30:255-89.
31. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun 2012;80:1948-57.
32. Peres da Silva R, Puccia R, Rodrigues ML, Oliveira DL, Joffe LS, et al. Extracellular vesicle-mediated export of fungal RNA. Sci Rep 2015;5:7763.
33. Choi DH, Kwon YM, Chiura HX, Yang EC, Bae SS, et al. Extracellular Vesicles of the Hyperthermophilic archaeon “thermococcus onnurineus” NA1T. Appl Environ Microbiol 2015;81:4591-9.
34. Tsatsaronis JA, Franch-Arroyo S, Resch U, Charpentier E. Extracellular vesicle RNA: a universal mediator of microbial communication? Trends Microbiol 2018;26:401-10.
35. Lee J, Lee EY, Kim SH, Kim DK, Park KS, et al. Staphylococcus aureus extracellular vesicles carry biologically active β-lactamase. Antimicrob Agents Chemother 2013;57:2589-95.
36. Ciofu O, Beveridge TJ, Kadurugamuwa J, Walther-Rasmussen J, Høiby N. Chromosomal beta-lactamase is packaged into membrane vesicles and secreted from Pseudomonas aeruginosa. J Antimicrob Chemother 2000;45:9-13.
37. Rumbo C, Fernández-Moreira E, Merino M, Poza M, Mendez JA, et al. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob Agents Chemother 2011;55:3084-90.
38. Medvedeva ES, Baranova NB, Mouzykantov AA, Grigorieva TY, Davydova MN, et al. Adaptation of mycoplasmas to antimicrobial agents: Acholeplasma laidlawii extracellular vesicles mediate the export of ciprofloxacin and a mutant gene related to the antibiotic target. Sci World J 2014;2014:150615.
39. Yaron S, Kolling GL, Simon L, Matthews KR. Vesicle-mediated transfer of virulence genes from Escherichia coli O157:H7 to other enteric bacteria. Appl Environ Microbiol 2000;66:4414-20.
40. Bitto NJ, Chapman R, Pidot S, Costin A, Lo C, et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci Rep 2017;7:7072.
41. Lambertz U, Oviedo Ovando ME, Vasconcelos EJ, Unrau PJ, Myler PJ, et al. Small RNAs derived from tRNAs and rRNAs are highly enriched in exosomes from both old and new world Leishmania providing evidence for conserved exosomal RNA Packaging. BMC Genomics 2015;16:151.
42. Blenkiron C, Simonov D, Muthukaruppan A, Tsai P, Dauros P, et al. Uropathogenic escherichia coli releases extracellular vesicles that are associated with RNA. PLoS One 2016;11:e0160440.
43. Sjöström AE, Sandblad L, Uhlin BE, Wai SN. Membrane vesicle-mediated release of bacterial RNA. Sci Rep 2015;5:15329.
44. Beatty M, Guduric-Fuchs J, Brown E, Bridgett S, Chakravarthy U, et al. Small RNAs from plants, bacteria and fungi within the order Hypocreales are ubiquitous in human plasma. BMC Genomics 2014;15:933.
45. Wang K, Li H, Yuan Y, Etheridge A, Zhou Y, et al. The complex exogenous RNA spectra in human plasma: an interface with human gut biota? PLoS One 2012;7:e51009.
46. Stentz R, Carvalho AL, Jones EJ, Carding SR. Fantastic voyage: the journey of intestinal microbiota-derived microvesicles through the body. Biochem Soc Trans 2018;46:1021-7.
47. Choi JW, Um JH, Cho JH, Lee HJ. Tiny RNAs and their voyage via extracellular vesicles: Secretion of bacterial small RNA and eukaryotic microRNA. Exp Biol Med (Maywood) 2017;242:1475-81.
48. Ho MH, Chen CH, Goodwin JS, Wang BY, Xie H. Functional Advantages of Porphyromonas gingivalis Vesicles. PLoS One 2015;10:e0123448.
49. Kunsmann L, Rüter C, Bauwens A, Greune L, Glüder M, et al. Virulence from vesicles: Novel mechanisms of host cell injury by Escherichia coli O104:H4 outbreak strain. Sci Rep 2015;5:13252.
50. Thay B, Damm A, Kufer TA, Wai SN, Oscarsson J. Aggregatibacter actinomycetemcomitans outer membrane vesicles are internalized in human host cells and trigger NOD1- and NOD2-dependent NF-kappaB activation. Infect Immun 2014;82:4034-46.
51. Koeppen K, Hampton TH, Jarek M, Scharfe M, Gerber SA, et al. A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog 2016;12:e1005672.
52. Choi JW, Kim SC, Hong SH, Lee HJ. Secretable small RNAs via outer membrane vesicles in periodontal pathogens. J Dent Res 2017;96:458-66.
53. Kim MR, Hong SW, Choi EB, Lee WH, Kim YS, et al. Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy 2012;67:1271-81.
54. Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet 2005;21:399-404.
55. Kang SM, Choi JW, Lee Y, Hong SH, Lee HJ. Identification of microRNA-size, small RNAs in Escherichia coli. Curr Microbiol 2013;67:609-13.
56. Lee HJ, Hong SH. Analysis of microRNA-size, small RNAs in Streptococcus mutans by deep sequencing. FEMS Microbiol Lett 2012;326:131-6.
57. Choi JW, Kwon TY, Hong SH, Lee HJ. Isolation and characterization of a microRNA-size secretable small RNA in streptococcus sanguinis. Cell Biochem Biophys 2018;76:293-301.
58. Han L, Lam EW, Sun Y. Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol Cancer 2019;18:59.
59. Nauts HC, Swift WE, Coley BL. The treatment of malignant tumors by bacterial toxins as developed by the late William B. Coley, M.D., reviewed in the light of modern research. Cancer Res 1946;6:205-16.
60. Zbar B, Bernstein I, Tanaka T, Rapp HJ. Tumor immunity produced by the intradermal inoculation of living tumor cells and living Mycobacterium bovis (strain BCG). Science 1970;170:1217-8.
61. Aso Y, Akazan H. Prophylactic effect of a Lactobacillus casei preparation on the recurrence of superficial bladder cancer. BLP Study Group. Urol Int 1992;49:125-9.
62. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013;342:967-70.
63. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079-84.
64. Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res 2012;18:2039-47.
65. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084-9.
66. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91-7.
67. Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol 2007;73:7767-70.
68. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104-8.
69. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359:97-103.
70. Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014;6:220ra11.
72. Franchi L, Warner N, Viani K, Nuñez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev 2009;227:106-28.
73. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest 2007;117:2197-204.
74. Sun J, Shi YH, Le GW, Ma XY. Distinct immune response induced by peptidoglycan derived from Lactobacillus sp. World J Gastroenterol 2005;11:6330-7.
75. Hajam IA, Dar PA, Shahnawaz I, Jaume JC, Lee JH. Bacterial flagellin-a potent immunomodulatory agent. Exp Mol Med 2017;49:e373.
76. Leleux JA, Pradhan P, Roy K. Biophysical attributes of CpG presentation control TLR9 signaling to differentially polarize systemic immune responses. Cell Rep 2017;18:700-10.
77. Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008;29:637-49.
78. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 2010;107:12204-9.
79. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016;16:341-52.
81. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013;341:569-73.
82. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013;504:451-5.
83. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol 2015;8:80-93.
84. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013;504:446-50.
85. Berman D, Parker SM, Siegel J, Chasalow SD, Weber J, et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun 2010;10:11.
86. Denning TL, Kim G, Kronenberg M. Cutting edge: CD4+CD25+ regulatory T cells impaired for intestinal homing can prevent colitis. J Immunol 2005;174:7487-91.
87. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 2016;7:10391.
88. Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol 1998;25:4-12.
89. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003;22:7265-79.
90. Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol 2009;6:638-47.
91. Strauss G, Westhoff MA, Fischer-Posovszky P, Fulda S, Schanbacher M, et al. 4-hydroperoxy-cyclophosphamide mediates caspase-independent T-cell apoptosis involving oxidative stress-induced nuclear relocation of mitochondrial apoptogenic factors AIF and EndoG. Cell Death Differ 2008;15:332-43.
92. Schwartz PS, Waxman DJ. Cyclophosphamide induces caspase 9-dependent apoptosis in 9L tumor cells. Mol Pharmacol 2001;60:1268-79.
93. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013;342:971-6.
94. Viaud S, Flament C, Zoubir M, Pautier P, LeCesne A, et al. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res 2011;71:661-5.
95. Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 2016;45:931-43.
96. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017;357:1156-60.
97. Masotti A. Interplays between gut microbiota and gene expression regulation by miRNAs. Front Cell Infect Microbiol 2012;2:137.
98. Maudet C, Mano M, Eulalio A. MicroRNAs in the interaction between host and bacterial pathogens. FEBS Lett 2014;588:4140-7.
99. Dalmasso G, Nguyen HT, Yan Y, Laroui H, Charania MA, et al. Microbiota modulate host gene expression via microRNAs. PLoS One 2011;6:e19293.
100. Kobayashi M, Funayama R, Ohnuma S, Unno M, Nakayama K. Wnt-β-catenin signaling regulates ABCC3 (MRP3) transporter expression in colorectal cancer. Cancer Sci 2016;107:1776-84.
101. Xue X, Feng T, Yao S, Wolf KJ, Liu CG, et al. Microbiota downregulates dendritic cell expression of miR-10a, which targets IL-12/IL-23p40. J Immunol 2011;187:5879-86.
102. Mjelle R, Sjursen W, Thommesen L, Sætrom P, Hofsli E. Small RNA expression from viruses, bacteria and human miRNAs in colon cancer tissue and its association with microsatellite instability and tumor location. BMC Cancer 2019;19:161.
103. Yu T, Guo F, Yu Y, Sun T, Ma D, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017;170:548-63.e16.
104. Dorward DW, Garon CF, Judd RC. Export and intercellular transfer of DNA via membrane blebs of Neisseria gonorrhoeae. J Bacteriol 1989;171:2499-505.
105. Brown L, Wolf JM, Prados-Rosales R, Casadevall A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol 2015;13:620-30.
106. Kim JH, Lee J, Park J, Gho YS. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin Cell Dev Biol 2015;40:97-104.
107. Yu YJ, Wang XH, Fan GC. Versatile effects of bacterium-released membrane vesicles on mammalian cells and infectious/inflammatory diseases. Acta Pharmacol Sin 2018;39:514-33.
108. Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol 2015;13:605-19.
109. Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 2010;64:163-84.
110. Haurat MF, Aduse-Opoku J, Rangarajan M, Dorobantu L, Gray MR, et al. Selective sorting of cargo proteins into bacterial membrane vesicles. J Biol Chem 2011;286:1269-76.
111. Ghosal A, Upadhyaya BB, Fritz JV, Heintz-Buschart A, Desai MS, et al. The extracellular RNA complement of Escherichia coli. Microbiologyopen 2015;4:252-66.
112. Resch U, Tsatsaronis JA, Le Rhun A, Stübiger G, Rohde M, et al. A Two-component regulatory system impacts extracellular membrane-derived vesicle production in group A streptococcus. MBio 2016;7:e00207-16.
113. Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, et al. The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 2016;19:32-43.
114. Azizian A, Epping I, Kramer F, Jo P, Bernhardt M, et al. Prognostic value of microRNAs in preoperative treated rectal cancer. Int J Mol Sci 2016;17:568.
115. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res 2012;72:4875-82.
116. Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013;339:543-8.
117. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26.
118. McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013;12:217-28.
119. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205-14.
120. van Vliet MJ, Harmsen HJ, de Bont ES, Tissing WJ. The role of intestinal microbiota in the development and severity of chemotherapy-induced mucositis. PLoS Pathog 2010;6:e1000879.
121. Marthey L, Mateus C, Mussini C, Nachury M, Nancey S, et al. Cancer immunotherapy with anti-CTLA-4 monoclonal antibodies induces an inflammatory bowel disease. J Crohns Colitis 2016;10:395-401.
122. Chen JH, Pezhouh MK, Lauwers GY, Masia R. Histopathologic features of colitis due to immunotherapy with anti-PD-1 antibodies. Am J Surg Pathol 2017;41:643-54.
123. Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 2010;120:4332-41.
124. Teillant A, Gandra S, Barter D, Morgan DJ, Laxminarayan R. Potential burden of antibiotic resistance on surgery and cancer chemotherapy antibiotic prophylaxis in the USA: a literature review and modelling study. Lancet Infect Dis 2015;15:1429-37.
125. Samet A, Sledzińska A, Krawczyk B, Hellmann A, Nowicki S, et al. Leukemia and risk of recurrent Escherichia coli bacteremia: genotyping implicates E. coli translocation from the colon to the bloodstream. Eur J Clin Microbiol Infect Dis 2013;32:1393-400.
126. Tulkens J, Vergauwen G, Van Deun J, Geeurickx E, Dhondt B, et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020;69:191-3.
127. Cavaillon JM. Exotoxins and endotoxins: inducers of inflammatory cytokines. Toxicon 2018;149:45-53.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Anfossi S, Calin GA. Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy. Cancer Drug Resist 2020;3:356-70. http://dx.doi.org/10.20517/cdr.2020.04
AMA Style
Anfossi S, Calin GA. Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy. Cancer Drug Resistance. 2020; 3(3): 356-70. http://dx.doi.org/10.20517/cdr.2020.04
Chicago/Turabian Style
Anfossi, Simone, George A. Calin. 2020. "Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy" Cancer Drug Resistance. 3, no.3: 356-70. http://dx.doi.org/10.20517/cdr.2020.04
ACS Style
Anfossi, S.; Calin GA. Gut microbiota: a new player in regulating immune- and chemo-therapy efficacy. Cancer Drug Resist. 2020, 3, 356-70. http://dx.doi.org/10.20517/cdr.2020.04
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 29
likes
Like This Article 29
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.