
Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer


Abstract
Breast cancer is one of the leading causes of death in women in the United States. In general, patients with breast cancer undergo surgical resection of the tumor and/or receive drug treatment to kill or suppress the growth of cancer cells. In this regard, small molecule kinase inhibitors serve as an important class of drugs used in clinical and research settings. However, the development of resistance to these compounds, in particular HER2 and CDK4/6 inhibitors, often limits durable clinical responses to therapy. Emerging evidence indicates that PI3K/AKT/mTOR pathway hyperactivation is one of the most prominent mechanisms of resistance to many small molecule inhibitors as it bypasses upstream growth factor receptor inhibition. Importantly, the PI3K/AKT/mTOR pathway also plays a pertinent role in regulating various aspects of cancer metabolism. Recent studies from our lab and others have demonstrated that altered lipid metabolism mediates the development of acquired drug resistance to HER2-targeted therapies in breast cancer, raising an interesting link between reprogrammed kinase signaling and lipid metabolism. It appears that, upon development of resistance to HER2 inhibitors, breast cancer cells rewire lipid metabolism to somehow circumvent the inhibition of kinase signaling. Here, we review various mechanisms of resistance observed for kinase inhibitors and discuss lipid metabolism as a potential therapeutic target to overcome acquired drug resistance.
Keywords
Introduction
Following the breakthrough success of Gleevec, small molecule kinase inhibitors have become a paradigm-shifting class of cancer therapeutics over the past two decades. As opposed to function blocking antibodies, kinase inhibitors are small molecules permeable to the blood-brain barrier and are therefore more efficacious in treating brain tumors. Furthermore, the ability to selectively target oncoproteins of interest offers a clinical benefit in mitigating off-target toxicities often seen with radiation therapy and conventional chemotherapy, such as doxorubicin and cisplatin. In the pre-clinical research setting, kinase inhibitors serve as valuable tool compounds that are able to rapidly inhibit kinase activity in tissue culture cell lines across species. Moreover, kinase inhibitors allow researchers to dissociate the role of kinase activity from protein scaffolding, a distinct advantage over RNAi- and CRISPR-mediated approaches. It is estimated that 25% of all ongoing drug development is dedicated to this class of drugs[1]. Since 2001, over 10,000 patent applications have been filed in the U.S. alone for kinase inhibitors[1]. As of June 2019, the FDA has approved 48 kinase inhibitors, 43 of which are indicated for the treatment of various cancers[2]. As ubiquitous and efficacious as these drugs have been, the common problem that continues to plague the clinic is the development of acquired drug resistance. Common mechanisms of resistance to kinase inhibitor resistance include kinase reprogramming[3,4], overexpression of drug efflux pumps[5], and drug resistant mutations[6] among others.
Breast cancer is the most commonly diagnosed malignancy in women worldwide, with an estimated incidence of nearly two million cases annually[7]. Breast cancers can be categorized into three molecular subtypes based on expression of hormone receptors (HR), such as estrogen receptor (ER) or progesterone receptor (PR), and amplification/overexpression of the human epidermal growth factor receptor 2 (HER2). Roughly 70% breast cancers fall into the ER+ category, 20% fall into the HER2+ category, and the remaining 10%, which do not exhibit ER/PR expression or HER2 amplification/overexpression, are referred to as triple negative. Molecular subtype is the major determining factor which guides treatment strategies in breast cancer. As with other cancer types, treatment regimens often include kinase inhibitors. The HER2 receptor tyrosine kinase and the cell cycle regulators cyclin-dependent kinases 4 and 6 (CDK4/6) are targeted in HER2+ and ER+ breast cancer, respectively. However, development of drug resistance often limits therapeutic efficacy of these drugs to transient clinical benefits. Mechanisms of resistance to these inhibitors often revolve around PI3K/AKT/mTOR pathway hyperactivation and targeting this compensatory pathway has been shown to lead to re-sensitization[8-11].
In this review, we highlight altered lipid metabolism as an emerging mechanism of resistance to small molecule kinase inhibitor therapy in breast cancer cells. Lipid metabolic rewiring is becoming increasingly recognized as a critical mechanism that promotes the growth and survival of cancer cells[12-15] and is often associated with maintenance of cancer cell stemness[16,17] and development of chemoresistance[17,18]. It is becoming clear that resistance mechanisms to kinase inhibitors in breast cancer often involve PI3K/AKT/mTOR pathway hyperactivation[8-11,19]. Notably, PI3K/AKT/mTOR pathway plays a critical role in the regulation of several lipid metabolic processes such as FA synthesis[20], FA uptake[21], FA storage into lipid droplets[22,23] and FAO[24,25]. Recent reports have demonstrated that targeting lipid metabolic pathways may be a valuable therapeutic strategy to re-sensitize cells resistant to HER2-targeted therapies in breast cancer[18,26,27]. We discuss the rationale behind targeting lipid metabolic rewiring in the treatment of drug resistant breast cancer cells as a therapeutic approach that requires further investigation.
Kinases targeted in breast cancer
HER family receptor tyrosine kinases in HER2+ breast cancer
Twenty percent of breast cancers exhibit overexpression or genetic amplification of the HER2 receptor and are classified as the HER2+ subtype[28]. HER2 is a member of the HER family of receptor tyrosine kinases, which include EGFR (HER1), HER2, HER3, and HER4. Upon binding to exogenous ligands, these receptors undergo homo- and heterodimerization, transphosphorylation, and activate downstream PI3K/AKT and MAPK pathways to promote cancer cell growth and survival[29]. Unlike other HER family members, HER2 is constitutively active and does not require ligand-induced activation[30]. HER2 is always in an active conformation and is known to be the preferred dimeric partner of all HER family members, with a preference for HER2-HER3 heterodimer formation[29,30].
Although standard of care for HER2+ breast cancers involves treatment regimens centered around the HER2-targeted antibody trastuzumab, there are several small molecule inhibitors used in the clinic that also target HER family receptors [Table 1]. Lapatinib, a dual inhibitor of EGFR and HER2, was the first small molecule inhibitor FDA approved for the treatment of advanced HER2+ breast cancer. Displaying promise in pre-clinical studies, lapatinib was shown to inhibit the growth of trastuzumab-resistant cancer cells[31] and was shown to have additive effects with HER2-targeted antibodies[32,33], offering hope for the treatment of breast cancer patients who previously relapsed on trastuzumab-based therapies. Although lapatinib exhibited only modest clinical success as a single agent in humans[34], it has been shown to be effective in certain settings. Addition of lapatinib to capecitabine displayed increased efficacy over capecitabine alone in women who progressed after treatment with an anthracycline + taxane + trastuzumab based therapy[35,36]. In the neoadjuvant setting, addition of lapatinib to trastuzumab was also shown to elicit superior clinical responses as compared to trastuzumab alone[37]. Importantly, lapatinib also demonstrated clinical activity in HER2+ breast cancer patients with brain metastases[38], offering a distinct advantage over trastuzumab in managing CNS metastatic disease.
Kinase inhibitors for the treatment of breast cancer
| Kinase inhibitor | Drug target | FDA approval status* |
|---|---|---|
| Lapatinib (Tykerb) | EGFR, HER2 | Initially approved in 2007 and currently indicated for (1) use in combination with capecitabine for the treatment of patients with advanced or metastatic HER2+ breast cancer and who have received prior therapy including an anthracycline, a taxane, and trastuzumab or (2) in combination with letrozole for the treatment of postmenopausal women with ER+HER2+ for whom hormonal therapy is indicated |
| Neratinib (Nerlynx) | EGFR, HER2, HER4 | Approved in 2017 for extended adjuvant treatment of early stage HER2+ breast cancer following adjuvant trastuzumab-based therapy and granted Orphan Drug Designation in 2019 for the treatment of breast cancer patients with brain metastases |
| Pyrotinib | EGFR, HER2, HER4 | Approved for clinical use in China in combination with capecitabine for the treatment of advanced or metastatic HER2+ breast cancer. Currently in Phase III clinical trials in the U.S. (NCT03980054, NCT03080805, NCT03863223, NCT02973737, NCT03588091) |
| Palbociclib (Ibrance) | CDK4, CDK6 | Granted accelerated approval in 2015 and currently indicated for the treatment of ER-HER2+ metastatic breast cancer in combination with (1) an aromatase inhibitor as initial endocrine-based therapy or (2) fulvestrant in patients who progressed on previous endocrine therapy |
| Ribociclib (Kisqali) | CDK4, CDK6 | Approved in 2017 and currently indicated for the treatment of ER-HER2+ metastatic breast cancer in combination with (1) an aromatase inhibitor as initial endocrine-based therapy or (2) fulvestrant as initial endocrine-based therapy or following progression on endocrine therapy |
| Abemaciclib (Verzenio) | CDK4, CDK6 | Granted priority review in 2015 and regular approval in 2017. Currently indicated for the treatment of ER+HER2- metastatic breast cancer in combination with (1) an aromatase inhibitor as initial endocrine-based therapy or (2) fulvestrant following progression on endocrine therapy or (3) as monotherapy in patients exhibiting disease progression following endocrine therapy and prior chemotherpy in the metastatic setting |
| Alpelisib (Piqray) | PI3K | Approved in 2019 for use in combination with fulvestrant for the treatment of HR+HER2-PIK3CA mutated advanced or metastatic breast cancer following progression on endocrine therapy |
| Everolimus (Afinitor) | mTOR | First approved in 2009 for the treatment of renal cell carcinoma and expanded to include treatment of ER+HER2- metastatic breast cancer in combination with exemestane following progression on letrozole or anastrozole |
Mechanisms of lapatinib resistance have been widely studied in the literature. Common mechanisms include HER3[3] and HER4[4,27] amplification, ligand-mediated HER family hyperactivation[39,40], PI3K hyperactivation[10,11,19], and metabolic reprogramming of glucose[41,42] and glutamine[43] utilization pathways. Additionally, many of these also manifest as mechanisms of resistance to trastuzumab as well[44,45].
In 2017, the FDA approved a second HER family-targeted inhibitor, neratinib, an irreversible inhibitor of EGFR, HER2, and HER4. HER3 is not targeted due to the lack of intrinsic kinase activity. Importantly, neratinib therapy was not shown to be associated with an increased incidence of cardiotoxicity[46] as opposed to trastuzumab. However, the major reported side effect is high grade nausea and diarrhea[46]. In 2019, neratinib was granted Orphan Drug Designation and its indication was expanded to include the treatment of breast cancer-related brain metastases. Mechanisms of resistance to neratinib are still under investigation but targeting Src[47] and mTORC1[48] have shown promise. In addition to lapatinib and neratinib, gefitinib and afatinib are two other FDA approved kinase inhibitors that have shown clinical efficacy in breast cancer but are currently used only for the treatment of non-small cell lung cancer (NSCLC)[49,50]. More recently, addition of a new pan-HER inhibitor pyrotinib to capecitabine demonstrated increased clinical efficacy as compared to lapatinib and capecitabine in HER2+ patients who have progressed on taxanes, anthracyclines, and/or trastuzumab (NCT02422199). Although the drug is already approved for use in China[51], it currently remains in several Phase III clinical trials in the U.S. (NCT03980054, NCT03080805, NCT03863223, NCT02973737, and NCT03588091).
CDK4/6 inhibitors in ER+ breast cancer
Dysregulation of the cell cycle is a well-known hallmark of cancer[52]. Progression through the cell cycle is mediated by the formation of several heteromeric complexes consisting of a cyclin family protein and a serine/threonine cyclin-dependent kinase (CDK). In particular, the Cyclin D1-CDK4/6 complex plays a critical role in the progression from G1/S transition, a checkpoint regulated by the retinoblastoma-associated protein (Rb1). Under normal circumstances, mitogenic signaling induces the expression of D-type cyclins, Cyclin D1, 2, and 3[53]. Cyclin D then binds and activates CDK4 or CDK6, resulting in the phospho-inactivation of the tumor suppressors, Rb1 and Rb1-like proteins p130 and p107. In the hypophosphorylated state, Rb1 represses the transcription of genes mediating cell cycle progression by binding to the transactivation domain of E2F. Upon phosphorylation by the CDK4/6-Cyclin D complex, Rb1 is inactivated, releases E2F, and allows for induction of genes controlling cell cycle progression (Cyclin E and Cyclin A), DNA replication (MCM7 and PCNA), and mitotic progression (Cyclin B1 and CDK1)[54]. Induction of E2F subsequently promotes the formation of a positive feedback loop by inducing expression of E-type cyclins, which mediate S phase transition and DNA synthesis[55]. Antagonizing CDK4/6 phosphorylation promotes the preservation of hypophosphorylated Rb, thereby restoring the inhibition of E2F-mediated induction of downstream target genes.
There are currently three FDA approved CDK4/6 targeting drugs on the market: palbociclib, ribociclib, and abemaciclib [Table 1]. In 2015, palbociclib became the first CDK4/6 inhibitor to be approved by the FDA and was granted accelerated approval in combination with the nonsteroidal aromatase inhibitor letrozole as first-line therapy for treating advanced ER+HER2- breast cancer following results from the Phase II PALOMA-1 study (NCT00721409), which showed that the addition of palbociclib to letrozole therapy in advanced ER+HER2- breast cancer nearly doubled progression free survival from 10.2 months to 20.2 months[56,57]. The Phase II PALOMA-2 study (NCT01740427) demonstrated similar findings, with the addition of palbociclib to letrozole extending median progression free survival from 14.5 months to 24.8 months in patients with advanced ER+HER2- breast cancer[58]. In 2016, the FDA expanded the indication of palbociclib to include combination therapy with fulvestrant for the treatment of advanced ER+HER2- breast cancer that progressed on endocrine therapy following results from the Phase III PALOMA-3 study (NCT01942135), which demonstrated that addition of palbociclib to fulvestrant for the treatment of ER+HER2- metastatic breast cancer that progressed on endocrine therapy extended median progression free survival from 4.6 months to 9.5 months[56,59]. Since then, the FDA has approved two additional CDK4/6 inhibitors: ribociclib in 2017 and abemaciclib in 2018. The combination of a CDK4/6 inhibitor with an aromatase inhibitor is now standard of care as first-line therapy for the treatment of ER+HER2- breast cancer in the U.S.
Although dispensable for normal development of mammary tissue, Cyclin D is essential for breast tumor development and progression, rendering breast cancer cells selectively sensitive to CDK4/6 kinase inhibitors. Indeed, CDK4/6 inhibition sensitized ER+ and HER2+ breast cancer cells to tamoxifen and trastuzumab, respectively[60]. Recent work has highlighted the tight coupling between the ER and the Cyclin D1-CDK4/6 pathways, thereby rendering ER+ breast cancers especially dependent upon CDK4/6 for proliferation[60]. Cyclin D1 expression has long been known to be elevated and predictive of poor clinical outcome in ER+ breast cancer[61]. ER is known to promote induction of Cyclin D1 expression[62]. Moreover, Cyclin D1 (but not D2 or D3) has been shown to directly interact with ER to promote its transcriptional activity, thereby further establishing Cyclin D1 dependency in hormone receptor positive breast cancer[63]. Lastly, CDK4/6 inhibitors were also shown to be effective in sensitizing PIK3CA mutant breast cancer cells to PI3K inhibitors, demonstrating efficacy in overcoming intrinsic and acquired resistance to PI3K inhibition[64].
As with HER2-targeted therapies, activation of PI3K/AKT/mTOR signaling is becoming increasingly recognized as an important mechanism in the development of resistance to CDK4/6 inhibitors[56,65-68]. Herrera-Abreu demonstrated that AKT phosphorylation is increased following chronic treatment with palbociclib and that PI3K/AKT signaling is responsible for sustaining E2F-induced expression of Cyclin E and CDK2[65]. Notably, antagonizing PI3K also inhibited downstream CDK2 activation, offering an additional mechanism of suppressing proliferation[65]. Indeed, PTEN loss was also demonstrated to mediate resistance to the CDK4/6 inhibitor ribociclib as well as PI3Kα inhibitors in the clinical setting[69]. Furthermore, the combination of ER, CDK4/6, and PI3K triple targeting demonstrated greater efficacy than single or double therapies in in vitro and in vivo models of ER+ breast cancer[65,70]. Notably, inhibiting Rb phosphorylation by antagonizing CDK4/6 was shown to induce AKT activation by releasing Rb-mediated mTORC2 suppression[66] and facilitate metabolic rewiring via mTORC1 to promote glycolysis and oxidative metabolism in pancreatic ductal adenocarcinoma cells[68]. Therefore, it would be valuable to determine whether mTOR hyperactivation could also promote loss of sensitivity to CDK4/6-targeted therapy. As such, in vitro studies have also shown that mTORC1/2 inhibition can decrease Cyclin D1, Rb phosphorylation, and E2F-mediated transcription[71]. Additionally, ER, CDK4/6, and mTOR triple targeting has also yielded promising results in pre-clinical models[71]. It is believed that upregulation of Cyclin D underlies many of these mechanisms of resistance and, importantly, Cyclin D and Cyclin E upregulation can be blocked by PI3K/mTOR inhibition[67,72]. Additional mechanisms of CDK4/6 resistance include CDK6 amplification[73] and Cyclin E overexpression/amplification[65,72,74]. Lastly, Rb null cells are inherently resistant to CDK4/6 inhibition[75]. Furthermore, Rb1 loss and mutations were shown to be associated with CDK4/6 inhibitor resistance in patient-derived xenograft (PDX) models[65] and in patients with metastatic breast cancer[69,76].
Role of PI3K/AKT/mTOR pathway in lipid metabolism and drug resistance
The PI3K family is composed of lipid kinases that phosphorylate the 3ʹOH group of phosphatidylinositols[77]. Class 1A PI3K family members are the most widely implicated in cancer and promote the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) on the plasma membrane to generate the second messenger, phosphatidylinositol-3,4,5-trisphosphate (PIP3)[77]. AKT and phosphoinositide-dependent protein kinase 1 (PDK1) directly bind to PIP3 and are recruited to the plasma membrane. PDK1 phosphorylation activates AKT and allows for activation of downstream target proteins that promote growth and survival in cancer cells. Mutation in PIK3CA is one of the most common events in breast cancer[78,79] and hyperactivation of the PI3K/AKT pathway has long been thought to be a major downstream mechanism of resistance to HER2-targeted therapies[3,80] as well as escape from hormone dependence in ER+ breast cancer[81]. While pharmacological inhibitors of PI3K and AKT have long been pursued as therapeutics in breast cancer, inhibitors have historically performed poorly in clinical trials due to off target effects/toxicity, such as autoimmune disorders, hypertension, dysregulation of blood glucose levels, and hepatotoxicity, among others[82,83]. However, in May 2019, alpelisib, a first in class PI3K inhibitor, was FDA approved for use in combination with fulvestrant for the treatment of men and postmenopausal women with HR+, HER2-, PIK3CA-mutated, advanced or metastatic breast cancer following progression on or after an endocrine-based regimen. Unfortunately, several pre-clinical models of PI3K inhibitors have revealed compensatory mechanisms of resistance, such as loss of PTEN[84] or PI3K amplification or mutation[85,86]. Moreover, HER3 amplification can also occur, thereby facilitating PI3K hyperactivation as well[87].
mTOR is the catalytic component of two functionally distinct protein complexes, mTOR complexes 1 (mTORC1) and 2 (mTORC2), whose activities are regulated by different cofactors. mTOR is a master regulator of cell growth and metabolism and is often dysregulated in a variety of cancers. mTOR regulates the expression and activity of several pertinent metabolic enzymes involved in amino acid, glucose, nucleotide, and lipid metabolism[88]. Furthermore, mTOR-mediated metabolic reprogramming has been shown to promote resistance to a variety of chemotherapies such as lapatinib[9] and neratinib[48]. There currently are two mTOR inhibitors that are FDA approved. Temsirolimus was approved in 2007 for the treatment of advanced kidney cancer and everolimus was approved in 2012 for use in combination with the aromatase inhibitor exemestane for the treatment of HR+HER2- metastatic breast cancer after progression on letrozole or anastrozole therapy. Everolimus is also indicated for a variety of other cancers such as kidney cancer, subependymal giant cell astrocytoma, and renal angiomyolipomas associated with tuberous sclerosis, as well as neuroendocrine tumors of pancreatic origin, gastrointestinal origin, or lung origin.
While the PI3K/AKT/mTOR pathway is extensively studied for its role in proliferative signaling in breast cancer, it is important to note that this central signaling axis also plays a critical role in regulating various aspects of metabolism including glycolysis[24,88], glutaminolysis[89], fatty acid (FA) synthesis[20,90], and FA oxidation (FAO)[24].
Glucose and glutamine metabolism
PI3K/AKT/mTOR signaling promotes glycolysis in a number of ways, such as GLUT1- and GLUT4-mediated[91,92] glucose uptake into cells. Notably, mTORC2 promotes feedforward activation of AKT, which further activates hexokinase 2 to promote the entry of glucose into the glycolytic cascade[88]. Moreover, mTOR signaling is also thought to induce GLUT1 and HK2 expression via activation of HIF1a and MYC[88,93,94]. Pyruvate, the final product of glycolysis, can then be converted to lactate or acetyl coA for use in the TCA cycle. An additional mechanism of feeding the TCA that is regulated by AKT/mTOR is glutamate anaplerosis[89]. Glutamate can be converted to glutamine, which can then be converted to the TCA intermediate α-ketoglutarate (α-KG). Importantly, α-KG can have two distinct metabolic fates in the cell. α-KG can either be used as fuel for the TCA cycle or converted back into citrate, which can then be exported from the mitochondria and converted to acetyl coA by ATP citrate lyase (ACLY)[89]. Acetyl coA can then be converted to malonyl coA by acetyl coA carboxylase 1 (ACC1) and these two coA products can be used as substrates for FA synthesis.
FA synthesis
FAs are critical biomolecules utilized by all cells in membrane biogenesis, energy production, and signal transduction. Cancer cell growth and proliferation require an abundance of FAs to accommodate rapid membrane synthesis, a process classically thought to depend solely upon endogenous FA synthesis in cancer cells[95,96]. Interestingly, cancer cells almost universally exhibit a “lipogenic phenotype” characterized by exacerbated levels of FA biogenesis, even in the presence of abundant circulating exogenous FAs[95,97]. It is estimated that ~90% FAs in cancer cells are synthesized by the enzyme FA synthase (FASN)[95,98,99]. FASN is known to be overexpressed across several cancer types, including breast cancer, with expression increasing with tumor stage and predictive of poor prognosis[97,100-103]. Importantly, under physiological conditions, normal cells exhibit a sole preference for acquiring FAs from the circulation rather than depending on de novo lipogenesis[104]. Furthermore, pharmacological inhibition of FASN has been shown to be lethal to cancer cells in vitro and in vivo but not to normal cells[105], highlighting the importance of FAs in cancer biology while simultaneously revealing a promising therapeutic window. As such, FASN has been extensively investigated for use as a potential therapeutic target for decades.
FASN has emerged as an important enzyme in HER2+ breast cancers, in particular since HER2 directly phospho-activates FASN protein[106] as well as regulating transcription of FASN mRNA[107]. FASN inhibitors have yielded promising results in animal studies[108-110] but have achieved limited success in clinical trials due to poor pharmacokinetics[111] and off target toxicities, such as anorexia[112]. To date, the only FASN inhibitor to advance to clinical studies is TVB-2640, which has been shown to elicit promising responses across a variety of tumor types, including KRASMUT NSCLC, ovarian, and breast cancers[113]. TVB-2640 is currently being investigated in a Phase II clinical study (NCT03179904), combining FASN inhibition with trastuzumab and paclitaxel for the treatment of late stage HER2+ breast cancers. Additionally, dysregulation of de novo lipogenesis can also involve overexpression of other FA synthesis pathway proteins, such as acetyl coA carboxylase (ACC) and ACLY[114,115]. Inhibitors of these proteins are also currently undergoing pre-clinical development[116,117]. Importantly, AKT has been shown to activate the master regulator of sterol regulatory element binding protein 1 (SREBP1) at the levels of gene expression and protein expression via mTORC1 signaling[20,118,119]. Activated SREBP1 is cleaved and acts as a transcription factor that induces the expression of a variety of FA synthesis genes, including FASN, ACACA (which encodes ACC1), ACLY, and CD36, which encodes a transmembrane FA uptake transporter.
FA uptake
While the therapeutic efficacy of FASN synthesis has been largely studied, the role of deregulated FA uptake and β-oxidation pathways are gaining attention for their roles in promoting cancer cell growth and metastasis[120-124]. Indeed, the role of FA uptake and “opportunistic nutrient acquisition” has even been proposed as an emerging hallmark of cancer[12]. Recent work has shown that cancer cells are able to instruct neighboring adipocytes to release FAs, which in turn the cancer cell can acquire and utilize, implying a critical role for tumor-stromal crosstalk in the regulation of tumor growth[120,121]. Furthermore, it was reported that cancer cells can also acquire FAs from the circulation via lipoprotein lipolysis catalyzed by lipoprotein lipase (LPL)[122]. Importantly, LPL-mediated lipolysis of FAs requires FA channel-mediated uptake into cells. The canonical FA transporter downstream of LPL is CD36[123]. While CD36 (also known as platelet glycoprotein 4) is a multifunctional protein that regulates angiogenesis and cellular adhesion in addition to FA uptake, CD36 as a FA transporter has emerged as an important player in the progression of many cancer types[124,125]. Pascual et al.[124] showed that inhibiting CD36 attenuated metastasis of oral squamous cell carcinoma[124]. Ladanyi et al.[125] showed that adipocytes induce the expression of CD36 in ovarian cancer cells, thereby promoting metastasis[125]. Importantly, work from our lab has also identified a critical role of CD36 in the development of resistance to HER2-targeted therapies in breast cancer[18]. Wang et al.[21] demonstrated that rapamycin attenuates murine hepatic steatosis by reducing CD36 expression via inhibiting transcriptional efficiency, offering a potential mechanism of reducing CD36 expression in vivo[21]. Overall, the role of FA uptake has been a previously understudied area that may have therapeutic potential and requires further investigation.
FA β-oxidation
Cancer cells canonically exhibit preferential usage of the glycolysis pathway even in the presence of excess oxygen, a process referred to as the “Warburg Effect”[126]. However, cancer cells also often utilize the β-oxidative pathway to meet bioenergetics demands as well. In fact, β-oxidation is even known to be the dominant bioenergetic pathway in prostate cancer[127] and has been demonstrated to be a critical metabolic pathway in triple negative breast cancer[128,129]. Furthermore, recent work has shown that FAO is a major source of acetyl coA for histone acetylation[130] and that acidosis reprograms cancer cell FA metabolism via changes in histone acetylation[131]. These findings suggest that, in hypoxic regions of tumors where glycolysis likely fuels ATP production, cancer cells could adapt flexibility to use FAO to meet bioenergetic demands. Moreover, lipid droplet dynamics are known to play an essential role in regulating FA partitioning in cancer cells. Triglyceride (TG) synthesis and storage of excess FAs in lipid droplets protects cells from overabundance of free fatty acids and lipotoxicity. Interestingly, different breast cancer subtypes have been shown to exhibit distinct preferences in FA handling, such as the source of FAs used for TG synthesis. Although triple negative MDA-MB-231 cells exhibit greater levels of TGs as compared to ER+HER2- MCF7 cells[129], the former tend to divert exogenous FAs towards TG synthesis[132] while the latter tend to rely upon de novo lipogenesis to support this process[129]. Two unique isoforms of diacylglycerol transferase (DGAT) mediate the synthesis of TGs in cells from FA and diacylglycerol, where DGAT1 promotes TGs synthesis from exogenous FAs and DGAT2 from endogenous FAs. As such, MDA-MB-231 cells express elevated levels of DGAT1, whereas MCF7 cells express elevated levels of DGAT2[129]. Lipid droplets are dynamic organelles and the TGs they carry can also be hydrolyzed by intracellular lipases to release FAs during times of need, such as nutrient stress. Notably, lipid droplets can serve as a pro-tumorigenic source of FAs. Indeed, monoacylglycerol lipase (MAGL) mediated FA lipolysis has been shown to promote migration, invasion, survival, and tumor growth[133] and antagonizing MAGL was shown to attenuate this phenotype. Therefore, targeting FA esterification and lipid droplet hydrolysis have been proposed as potential therapeutic strategies that require further investigation[133,134].
While the role of AKT/mTOR signaling on FA synthesis is well understood, its role in regulating β-oxidation is recognized but remains poorly characterized. mTOR inhibition by rapamycin was shown to reduced glucose transport capacity, glycogen synthesis, and glycolysis by approximately 40% but also induced β-oxidation by 60% in skeletal muscle cells[24]. In rat hepatocytes, rapamycin induced oxidation of exogenous FAs by 46%-100% and reduced esterification of exogenous FAs and de novo lipogenesis by 40%-60%[25]. Because rapamycin is a selective inhibitor for mTORC1, these effects are thought to be mediated by the mTORC1 complex.
Targeting lipid metabolic rewiring in drug resistant cancer cells
Metabolic rewiring has long been recognized as a bona fide hallmark of cancer[52] and the importance of lipid metabolism in cancer biology is gaining salience[12-15]. Apart from serving bioenergetics purposes, FAs exhibit a number of fates within cells including production of phospholipids for membrane biosynthesis and alteration of lipid raft composition to reconfigure growth factor receptor signaling[95,135]. Altered lipid metabolism in cancer cells has also been shown to promote growth and survival[12-15], cancer cell stemness[16,17], and chemoresistance[17,18]. Given the intricate crosstalk between the PI3K/AKT/mTOR pathway on regulation of FA uptake, synthesis, and oxidation, we believe that the hyperactivation of this pathway that often plays a driving role in the development of drug resistance may also coincide with concomitant changes in regulation of lipid metabolism [Figure 1].
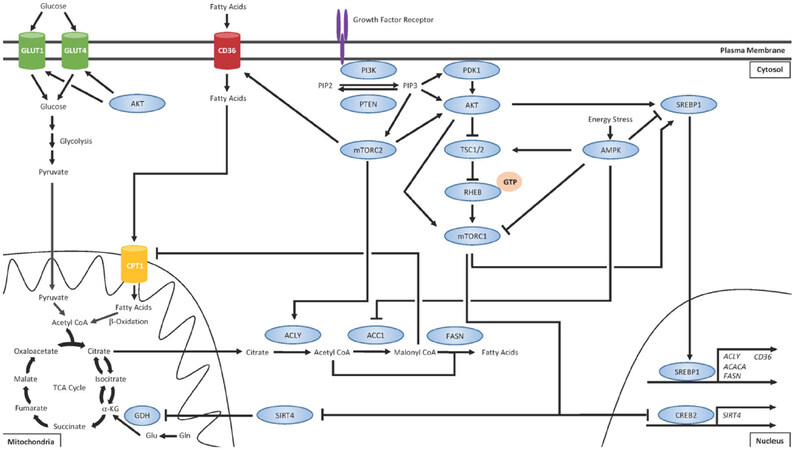
Figure 1. PI3K/AKT/mTOR regulates various aspects of lipid metabolism
Upon activation of growth factor receptors, such as the HER family receptor tyrosine kinases, the PI3K/AKT/mTOR signaling cascade is often the major effector pathway. The lipid kinase PI3K catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) on the plasma membrane to generate the second messenger, phosphatidylinositol-3,4,5-trisphosphate (PIP3)[77]. PIP3 recruits phosphoinositide-dependent protein kinase 1 (PDK1) and AKT to the plasma membrane, where PDK1 activates AKT. Tuberous sclerosis complex 1 (TSC1) and TSC2 form a heterodimer and negatively regulate mTORC1. TSC2 possesses GTPase-activating protein activity and, under normal circumstances, stimulates RHEB GTPase activity to hydrolyze bound GTP and convert RHEB into its inactive state[132]. Activated AKT phosphorylates TSC2 and prevents TSC1/TSC2 heterodimerization, which allows for GTP loading onto RHEB[132]. GTP-bound RHEB can then directly activate mTORC1[133,134]. Furthermore, the mTORC1 component PRAS40 is also a direct substrate of AKT, allowing for an additional mechanism of mTOR pathway activation[135]. One of the major mechanisms of mTORC1-mediated regulation of lipid metabolism is the activation of the SREBP1 transcription factor, which promotes the transcription of several genes that promote FA synthesis, such as ACLY, ACACA (which encodes ACC1), and FASN, as well as FA uptake[92,119]. mTORC1 also stimulates glutamine (Gln) anaplerosis by activating glutamine dehydrogenase (GDH) via destabilizing CREB2 and suppressing expression of SIRT4[89]. Through this mechanism, Gln can be converted into glutamate (Glu) and then into α-KG. α-KG can then either be used as fuel for the TCA cycle or converted back into citrate, which can be exported from the mitochondria and converted to acetyl coA by ATP citrate lyase (ACLY). Acetyl coA can then be converted to malonyl coA by acetyl coA carboxylase 1 (ACC1) and these two coA products can be used as substrates for FA synthesis catalyzed by FASN. The mTORC2 complex serves several functions distinct from the mTORC1 complex. mTORC2 has been shown to activate AKT[88] and ACLY[136] as well as promote the expression of the FA uptake channel CD36[137]. In addition to regulating lipid metabolism, AKT can also promote membrane localization of GLUT transporters and increased glucose uptake and glycolysis[91,92]. Importantly, the master regulator of energy homeostasis AMP-activated protein kinase (AMPK) is well recognized as having opposing effects on mTOR signaling[138-140]. AMPK negatively regulates mTOR by phospho-inactivating TSC2 and the mTORC1 subunit Raptor[141]. While mTOR is generally active during conditions of high energy states, AMPK is activated during nutrient stress. AMPK restores energy balance by promoting signaling for catabolic processes such as glycolysis, autophagy, and FAO while concomitantly inhibiting energetically costly anabolic processes such as FA synthesis[142].
Transcriptomic analysis of trastuzumab resistant HER2+ cell lines revealed genes involved in the lipid metabolism, glycolysis, and vitamin A metabolism as the most commonly differentially expressed as compared to parental cells, suggesting that metabolic reprogramming is associated with acquired resistance[136]. Several groups have also noted that endocrine resistance is associated with lipid metabolic reprogramming[137,138]. Hultsch et al.[137] demonstrated that tamoxifen resistance is associated with increased lysosomal cholesterol accumulation[137] and Du et al.[138] demonstrated that MDA-MB-134 endocrine resistant cells exhibit exacerbated levels of SREBP1, the master regulator of FA, and cholesterol synthesis, and were hypersensitive to SREBP1 and FAO inhibition as compared to parental cells[138]. In addition, many mechanisms of resistance to CDK4/6 inhibitors, such as Rb loss[65,69,76] and Cyclin D upregulation[67], often result in increased activation of CDK4 signaling. Jin et al.[139] identified C/EBPa as a critical target of the Cyclin D3-CDK4 complex[139]. C/EBPa is a major transcription factor that regulates adipocyte differentiation and controls the expression of genes such as CD36 and DGAT. Jin et al.[139] found increased activation of Cdk4 in mouse models of nonalcoholic fatty liver disease (NAFLD) and human NAFLD patients[139]. They demonstrated that inhibition of Cdk4 can reverse hepatic steatosis in NAFLD mice, providing evidence that aberrant Cdk4 signaling can promote dysregulation of lipid metabolism in certain contexts[139]. Supporting this idea, Kitajima et al.[140] demonstrated that Rb1 inactivation in MCF7 cells results in metabolic reprogramming and increased dependency on FAO[140]. Moreover, Franco et al.[68] observed mTOR-mediated metabolic reprogramming in response to palbociclib treatment in pancreatic ductal adenocarcinoma cells that promoted increased glycolysis and oxidative metabolism[68]. They observed that mTOR inhibition antagonized the metabolic changes and sensitized tumors to CDK4/6 inhibition[68]. Furthermore, Tarrado-Castellarnau et al.[141] observed similar findings and identified MYC as a critical factor upstream of mTOR-mediated metabolic reprograming in response to CDK4/6 inhibition[141]. MYC was demonstrated to play a central role in modulating glutamine metabolism by upregulating glutamine transporters and transcriptionally repressing microRNA-23a/b to promote glutaminase (GLS1) expression[141]. GLS1 can promote mitochondrial respiration by converting glutamine to glutamate, which can then be converted to α-ketoglutarate (α-KG) and be used as fuel for the TCA cycle or as a substrate for de novo FA synthesis [Figure 1]. Importantly, GLS1 inhibition was shown to restore sensitivity to CDK4/6 inhibition[141]. Although the mechanism of lipid metabolic rewiring has yet to be fully elucidated in the arena of acquired resistance to CDK4/6 inhibitors, we encourage investigation into this area as we anticipate its importance. These observations raise the intriguing possibility of targeting lipid metabolism as an alternative therapeutic strategy in endocrine-resistant cells that have also stopped responding to CDK4/6 inhibition. Therefore, targeting these changes may allow for a promising therapeutic strategy to overcome acquired drug resistance to HER2-targeted and CDK4/6-targeted therapies in HER2+ and ER+ breast cancers, respectively.
Several groups recently demonstrated that breast cancer cells resistant to lapatinib and trastuzumab both exhibit increased sensitivity to FASN inhibition as opposed to parental cells and indicating that targeting FASN activity may be a valuable therapeutic approach to circumventing resistance to targeted therapy[26,142]. Additionally, it was also shown that FASN inhibition reduced HER2 levels in trastuzumab resistant cells and sensitized them to trastuzumab[143]. Lastly, Blancafort et al.[27] found that the combination of anti-FASN polyphenolic compounds with an mTOR inhibitor (temsirolimus) and a HER2 dimerization domain-targeted antibody (pertuzumab) had the greatest combinatorial effect in suppressing the growth of lapatinib and trastuzumab dual resistant cells[27].
Work from our lab has also identified CD36 as an important mediator of resistance to HER2-targeted therapies. We found that pharmacologic inhibition of CD36 with the small molecule inhibitor sulfosuccinimidyl oleate (SSO) or a function blocking antibody could re-sensitize lapatinib resistant cells to targeted therapy in vitro as well as in vivo[18]. Furthermore, RNA-seq analysis of human breast tumors pre- and post-treatment with trastuzumab and lapatinib (either alone or in combination) revealed that CD36 expression increased following treatment with targeted therapy. Furthermore, high CD36 expression exhibited by tumors of this cohort were also shown to be associated with poorer patient prognosis. Unfortunately, there are currently no FDA approved CD36 inhibitors that are available for use in humans. Therefore, it remains to be seen whether combination of CD36 inhibition can recapitulate the re-sensitization to HER2-targeted therapy observed in our preclinical studies.
Carnitine palmitoyl transferase (CPT1) is the rate limiting enzyme that catalyzes FAO. There are three isoforms of this enzyme, namely CPT1A, CPT1B, and CPT1C, which exhibit differences in tissue expression. All three isoforms have been reported to play an important role in the context of drug resistance. Samudio et al.[144] showed that leukemia cells can exhibit mitochondrial uncoupling and promote a metabolic preference for FAO over pyruvate oxidation. Exploiting this dependency, they demonstrated that inhibition of carnitine palmitoyl transferase 1A (CPT1A) could sensitize leukemia cells to the BH3-mimetic ABT-747[144]. Wang et al.[16] demonstrated that adipocyte derived leptin promotes JAK/STAT3 pathway activation in breast cancer cell to promote carnitine palmitoyl transferase B (CPT1B)-mediated dependency upon FAO and that this metabolic switch promotes cancer stemness and paclitaxel chemoresistance[16]. Importantly, inhibiting FAO re-sensitized resistant cells to chemotherapy[16]. Moreover, carnitine palmitoyl transferase 1C (CPT1C), the remaining isoform of CPT1, is commonly upregulated in lung tumors and promotes increased FAO, rapamycin resistance, as well as adaptation to metabolic stress such as glucose deprivation and hypoxia[145]. Inhibition of FAO by mercaptoacetate and etomoxir has also been shown to sensitize paclitaxel-resistant lung adenocarcinoma cells[146]. Lastly, Wu et al.[147] identified long-chain fatty acyl coA synthetase (ACSL4), which facilitates FAO, as an important factor that promotes hormone and lapatinib resistance[147]. Therefore, several instances have demonstrated the therapeutic potential of targeting FAO to sensitize cancer cells to chemotherapies.
The energy sensor AMP-activated protein kinase (AMPK) is the master regulator of cellular energy homeostasis and is well recognized as having opposing effects on mTOR signaling by activating TSC2[148-156]. While mTOR is active during conditions of high energy states, AMPK is activated by elevated AMP:ATP ratio during nutrient stress and signals for catabolic pathways to restore energy balance[157]. AMPK negatively regulates mTOR by phospho-inactivating TSC2 and the mTORC1 subunit Raptor[158]. In doing so, AMPK promotes bioenergetics processes such as glycolysis, autophagy, and FAO, while concomitantly inhibiting energetically costly anabolic processes such as FA synthesis[157]. Importantly, palbociclib was recently shown to induce activation of AMPK[159] and CDK4 has been shown to suppress FAO by directly antagonizing AMPKα2[160]. Therefore, AMPK activation may be a valuable therapeutic target to antagonize mTOR-mediated lipid metabolic reprogramming. The interconnectedness of signaling pathways that control lipid metabolism in cancer cells is summarized in Figure 1.
Conclusion
Lipid metabolic reprogramming is an emerging mechanism of resistance to kinase inhibitor therapy in breast cancer. Given that mTOR hyperactivation is often implicated in development of acquired resistance to targeted therapy and that the AKT/mTOR signaling regulates various aspects of metabolism, exploiting rewired metabolic dependencies may allow for therapeutic re-sensitization of drug resistant cells. This would also allow for the possibility of repositioning existing lipid metabolic inhibitors into treatment regimens for breast cancer. Future work should be dedicated to identifying critically altered players in lipid metabolism that promote development of drug resistance and to assess whether targeting these pathways hold any therapeutic promise.
Declarations
AcknowledgmentsWe would like to thank Dr. William B. Kinlaw (Dartmouth College), Dr. R. Brooks Robey (Dartmouth College), and members of the Kurokawa lab for various discussions.
Authors’ contributionsWrote the manuscript: Feng WW, Kurokawa M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by an NCI Career Development Award (R00 CA140948), an NIH (R03 CA208384), and a Mary Kay Foundation research grant (to Kurokawa M).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17:48.
2. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res 2019;144:19-50.
3. Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Pro Natl Acad Sci USA 2011;108:5021-6.
4. Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 2015;14:648-55.
5. Polli JW, Humphreys JE, Harmon KA, Castellino S, O’mara MJ, et al. The role of efflux and uptake transporters in N-{3-chloro-4-[(3-fluorobenzyl) oxy] phenyl}-6-[5-({[2-(methylsulfonyl) ethyl] amino} methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 2008;36:695-701.
6. Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, et al. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol 2009;21:255-62.
7. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941-53.
8. Brünner-Kubath C, Shabbir W, Saferding V, Wagner R, Singer CF, et al. The PI3 kinase/mTOR blocker NVP-BEZ235 overrides resistance against irreversible ErbB inhibitors in breast cancer cells. Breast Cancer Res Treat 2011;129:387-400.
9. Joly MM, Hicks DJ, Jones B, Sanchez V, Estrada MV, et al. Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res 2016;76:4752-64.
10. Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008;68:9221-30.
11. Brady SW, Zhang J, Seok D, Wang H, Yu D. Enhanced PI3K p110α signaling confers acquired lapatinib resistance that can be effectively reversed by a p110α-selective PI3K inhibitor. Mol Cancer Ther 2014;13:60-70.
13. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016;5:e189.
14. Liu Q, Luo Q, Halim A, Song G. Targeting lipid metabolism of cancer cells: a promising therapeutic strategy for cancer. Cancer Lett 2017;401:39-45.
15. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, et al. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 2013;52:585-9.
16. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab 2018;27:136-50.
17. Chen CL, Kumar DB, Punj V, Xu J, Sher L, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab 2016;23:206-19.
18. Feng WW, Wilkins O, Bang S, Ung M, Li J, et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep 2019;29:3405-20.
19. Wang Q, Liu P, Spangle JM, Von T, Roberts TM, et al. PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 2016;35:3607.
20. Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab 2010;21:268-76.
21. Wang C, Yan Y, Hu L, Zhao L, Yang P, et al. Rapamycin-mediated CD36 translational suppression contributes to alleviation of hepatic steatosis. Biochem Biophys Res Commun 2014;447:57-63.
22. Yue S, Li J, Lee SY, Lee HJ, Shao T, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014;19:393-406.
23. Fazolini NP, Cruz AL, Werneck MB, Viola JP, Maya-Monteiro CM, et al. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015;14:2667-76.
24. Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism 2006;55:1637-44.
25. Brown NF, Stefanovic-Racic M, Sipula IJ, Perdomo G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism 2007;56:1500-7.
26. Puig T, Aguilar H, Cufí S, Oliveras G, Turrado C, et al. A novel inhibitor of fatty acid synthase shows activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res 2011;13:R131.
27. Blancafort A, Giró-Perafita A, Oliveras G, Palomeras S, Turrado C, et al. Dual fatty acid synthase and HER2 signaling blockade shows marked antitumor activity against breast cancer models resistant to anti-HER2 drugs. PLoS One 2015;10:e0131241.
28. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987;235:177-82.
29. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 2012;9:16.
30. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol 1996;16:5276-87.
31. Xia W, Liu LH, Ho P, Spector NL. Truncated ErbB2 receptor (p95 ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene 2004;23:646.
32. Xia W, Gerard CM, Liu L, Baudson NM, Ory TL, et al. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 2005;24:6213.
33. O’Donovan N, Byrne AT, O’Connor AE, McGee S, Gallagher WM, et al. Synergistic interaction between trastuzumab and EGFR/HER-2 tyrosine kinase inhibitors in HER-2 positive breast cancer cells. Invest New Drugs 2011;29:752-9.
34. Blackwell KL, Pegram MD, Tan-Chiu E, Schwartzberg LS, Arbushites MC, et al. Single-agent lapatinib for HER2-overexpressing advanced or metastatic breast cancer that progressed on first-or second-line trastuzumab-containing regimens. Ann Oncol 2009;20:1026-31.
35. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006;355:2733-43.
36. Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 2008;112:533-43.
37. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, De Azambuja E, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 2012;379:633-40.
38. Lin NU, Diéras V, Paul D, Lossignol D, Christodoulou C, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res 2009;15:1452-9.
39. Sato Y, Yashiro M, Takakura N. Heregulin induces resistance to lapatinib-mediated growth inhibition of HER 2-amplified cancer cells. Cancer Sci 2013;104:1618-25.
40. Leung WY, Roxanis I, Sheldon H, Buffa FM, Li JL, et al. Combining lapatinib and pertuzumab to overcome lapatinib resistance due to NRG1-mediated signalling in HER2-amplified breast cancer. Oncotarget 2015;6:5678.
41. Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol 2012;8:596.
42. Ruprecht B, Zaal EA, Zecha J, Wu W, Berkers CR, et al. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res 2017;77:1842-53.
43. Deblois G, Smith HW, Tam IS, Gravel SP, Caron M, et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat Commun 2016;7:12156.
44. Yang L, Li Y, Shen E, Cao F, Li L, et al. NRG1-dependent activation of HER3 induces primary resistance to trastuzumab in HER2-overexpressing breast cancer cells. Int J Oncol 2017;51:1553-62.
45. Zhao Y, Liu H, Liu Z, Ding Y, LeDoux SP, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res 2011;71:4585-97.
46. Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol 2010;28:1301-7.
47. Conlon N, Browne A, Breen L, Lowry M, O’Driscoll L, et al. 64P The potential of neratinib plus dasatinib in overcoming and preventing neratinib resistance in HER2-positive breast cancer models. Ann Oncol 2019;30:mdz238-062.
48. Sudhan DR, Guerrero-Zotano A, Won H, Ericsson PG, Servetto A, et al. Hyperactivation of Torc1 Drives Resistance to the Pan-Her Tyrosine Kinase Inhibitor Neratinib in Her2-Mutant Cancers. Cancer Cell ; doi: 10.2139/ssrn.3362256.
49. Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res 2011;17:1147-59.
50. Ring A, Wheatley D, Hatcher H, Laing R, Plummer R, et al. Phase I study to assess the combination of afatinib with trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Clin Cancer Res 2015;21:2737-44.
53. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501-12.
54. Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010;29:4018.
55. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016;13:417.
56. Portman N, Alexandrou S, Carson E, Wang S, Lim E, et al. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr Relat Cancer 2019;26:R15-30.
57. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25-35.
58. Finn RS, Martin M, Rugo HS, Jones S, Im SA, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016;375:1925-36.
59. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17:425-39.
60. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009;11:R77.
61. Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res 1999;5:2069-76.
62. Sabbah M, Courilleau D, Mester J, Redeuilh G. Estrogen induction of the cyclin D1 promoter: involvement of a cAMP response-like element. Proc Natl Acad Sci U S A 1999;96:11217-22.
63. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011;11:558.
64. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014;26:136-49.
65. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res 2016;76:2301-13.
66. Zhang J, Xu K, Liu P, Geng Y, Wang B, et al. Inhibition of Rb phosphorylation leads to mTORC2-mediated activation of Akt. Molecular Cell 2016;62:929-42.
67. Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 2017;3:39-55.
68. Franco J, Balaji U, Freinkman E, Witkiewicz AK, Knudsen ES. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep 2016;14:979-90.
69. Costa C, Wang Y, Ly A, Hosono Y, Ellen M, et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov 2019; doi: 10.1158/2159-8290.CD-18-0830.
71. Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol Cancer Ther 2018;17:908-20.
72. Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014;5:6512.
73. Yang C, Li Z, Bhatt T, Dickler M, Giri D, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017;36:2255.
74. Caldon CE, Sergio CM, Kang J, Muthukaruppan A, Boersma MN, et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol Cancer Ther 2012;11:1488-99.
75. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004;3:1427-38.
76. Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2017;29:640-5.
77. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009;9:550.
78. Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 2004;3:772-5.
79. Lee JW, Soung YH, Kim SY, Lee HW, Park WS, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 2005;24:1477.
80. Nahta R, O’Regan RM. Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway. Clin breast cancer 2010;10:S72-8.
81. Miller TW, Hennessy BT, González-Angulo AM, Fox E, Mills GB, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120:2406-13.
82. Esposito A, Viale G, Curigliano G. Safety, tolerability, and management of toxic effects of phosphatidylinositol 3-kinase inhibitor treatment in patients with cancer: a review. JAMA Oncol 2019; doi: 10.1001/jamaoncol.2019.0034.
83. Greenwell IB, Ip A, Cohen JB. PI3K inhibitors: understanding toxicity mechanisms and management. Oncology (Williston Park) 2017;31:821-8.
84. Juric D, Castel P, Griffith M, Griffith OL, Won HH, et al. Convergent loss of PTEN leads to clinical resistance to a PI (3) Kα inhibitor. Nature 2015;518:240.
85. Nakanishi Y, Walter K, Spoerke JM, O’Brien C, Huw LY, et al. Activating mutations in PIK3CB confer resistance to PI3K inhibition and define a novel oncogenic role for p110β. Cancer Res 2016;76:1193-203.
86. Huw LY, O’Brien C, Pandita A, Mohan S, Spoerke JM, et al. Acquired PIK3CA amplification causes resistance to selective phosphoinositide 3-kinase inhibitors in breast cancer. Oncogenesis 2013;2:e83.
87. Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA 2012;109:2718-23.
88. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer 2018;18:744-57.
89. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013;153:840-54.
90. Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res 2008;68:1003-11.
91. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 2007;18:1437-46.
92. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol 2009;19:R1046-52.
93. Tran Q, Lee H, Park J, Kim SH, Park J. Targeting cancer metabolism-revisiting the Warburg effects. Toxicol Res 2016;32:177.
94. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, et al. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22:7004-14.
95. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7:763-77.
96. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nature Reviews Cancer 2016;16:732.
97. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 2006;66:5977-80.
98. Ookhtens MU, Kannan RA, Lyon IR, Baker NO. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol Regul Integr Comp Physiol 1984;247:R146-53.
99. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA 2013;110:8882-7.
100. Alo PL, Visca P, Marci A, Mangoni A, Botti C, et al. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996;77:474-82.
101. Wang YY, Kuhajda FP, Li JN, Pizer ES, Han WF, et al. Fatty acid synthase (FAS) expression in human breast cancer cell culture supernatants and in breast cancer patients. Cancer Lett 2001;167:99-104.
102. Alo PL, Amini M, Piro F, Pizzuti L, Sebastiani V, et al. Immnunohistochemical expression and prognostic significance of fatty acid synthase in pancreatic carcinoma. Anticancer Res 2007;27:2523-7.
103. Cai Y, Wang J, Zhang L, Wu D, Yu D, et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol 2015;32:391.
104. Weiss L, Hoffman GE, Schreiber R, Andrea H, Fuchs E, et al. Fatty-acid biosynthesis in man, a pathway of minor importance. Purification, optimal assay conditions, and organ distribution of fatty-acid synthase. Biol Chem Hoppe-Seyler 1986;367:905-12.
105. Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, et al. Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine 2015;2:808-24.
106. Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res 2010;12:R96.
107. Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, Chinnaiyan AM. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res 2007;63:132-9.
108. Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene 2005;24:39-46.
109. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res 2004;64:2070-5.
110. Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, et al. Fasnall, a selective FASN inhibitor, shows potent anti-tumor activity in the MMTV-Neu model of HER2+ breast cancer. Cell Chem Biol 2016;23:678-88.
111. Zhi J, Melia AT, Funk C, Viger-Chougnet A, Hopfgartner G, et al. Metabolic profiles of minimally absorbed orlistat in obese/overweight volunteers. J Clin Pharmacol 1996;36:1006-11.
112. Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000;288:2379-81.
113. Brenner AJ, Von Hoff DD, Infante JR, Patel MR, Jones SF, et al. First-in-human investigation of the oral first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. J Clin Oncol 2015;33:TPS2615.
114. Yoon S, Lee MY, Park SW, Moon JS, Ko YK, et al. Up-regulation of acetyl-CoA carboxylase α and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem 2007;282:26122-31.
115. Wang D, Yin L, Wei J, Yang Z, Jiang G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumor Biol 2017;39:1010428317698338.
116. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 2016;22:1108.
117. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311-21.
118. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005;24:6465.
119. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008;8:224-36.
120. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17:1498.
121. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab 2017;5:1.
122. Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther 2011;10:427-36.
123. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase-and CD36-mediated pathways. J Lipid Res 2009;50:S86-90.
124. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41.
125. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018;37:2285.
127. Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 2006;9:230.
128. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 2016;22:427.
129. Giudetti AM, De Domenico S, Ragusa A, Lunetti P, Gaballo A, et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim Biophys Acta 2019;1864:344-57.
130. McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, et al. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep 2016;17:1463-72.
131. Corbet C, Pinto A, Martherus R, de Jesus JP, Polet F, et al. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab 2016;24:311-23.
132. Balaban S, Lee LS, Varney B, Aishah A, Gao Q, et al. Heterogeneity of fatty acid metabolism in breast cancer cells underlies differential sensitivity to palmitate-induced apoptosis. Mol Oncol 2018;12:1623-38.
133. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010;140:49-61.
134. Jarc E, Kump A, Malavašič P, Eichmann TO, Zimmermann R, et al. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim Biophys Acta 2018;1863:247-65.
135. Bi J, Ichu TA, Zanca C, Yang H, Zhang W, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab 2019;30:525-38.
136. Dubianski R, Sarnowska E, Leszczynski M, Kubala S, Olszewski W, et al. 17P HER2-positive breast cancer resistance to trastuzumab is associated with metabolic switch. Ann of Oncol 2018;29.
137. Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 2018;18:850.
138. Du T, Sikora MJ, Levine KM, Tasdemir N, Riggins RB, et al. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res 2018;20:106.
139. Jin J, Valanejad L, Nguyen TP, Lewis K, Wright M, et al. Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Rep 2016;16:744-56.
140. Kitajima S, Yoshida A, Kohno S, Li F, Suzuki S, et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017;36:5145.
141. Tarrado-Castellarnau M, de Atauri P, Tarragó-Celada J, Perarnau J, Yuneva M, et al. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol Syst Biol 2017;13.
143. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin™) by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol 2007;31:769-76.
144. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 2010;120:142-56.
145. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev 2011;25:1041-51.
146. Li J, Zhao S, Zhou X, Zhang T, Zhao L, et al. Inhibition of lipolysis by mercaptoacetate and etomoxir specifically sensitize drug-resistant lung adenocarcinoma cell to paclitaxel. PLoS One 2013;8:e74623.
147. Wu X, Li Y, Wang J, Wen X, Marcus MT, et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One 2013;8:e77060.
148. Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009;37:217-22.
149. Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol 2005;15:702-13.
150. Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, et al. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev 2008;22:2172-7.
151. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007;9:316.
152. Chen Y, Qian J, He Q, Zhao H, Toral-Barza L, et al. mTOR complex-2 stimulates acetyl-CoA and de novo lipogenesis through ATP citrate lyase in HER2/PIK3CA-hyperactive breast cancer. Oncotarget 2016;7:25224.
153. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017;32:807-23.
154. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577-90.
155. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012;32:2-11.
156. Hindupur SK, González A, Hall MN. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb Perspect Biol 2015;7:a019141.
157. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251.
158. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008;30:214-26.
159. Hsieh FS, Chen YL, Hung MH, Chu PY, Tsai MH, et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK 4/6-independent manner. Mol Oncol 2017;11:1035-49.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Feng WW, Kurokawa M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist 2020;3:1-17. http://dx.doi.org/10.20517/cdr.2019.100
AMA Style
Feng WW, Kurokawa M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resistance. 2020; 3(1): 1-17. http://dx.doi.org/10.20517/cdr.2019.100
Chicago/Turabian Style
Feng, William W., Manabu Kurokawa. 2020. "Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer" Cancer Drug Resistance. 3, no.1: 1-17. http://dx.doi.org/10.20517/cdr.2019.100
ACS Style
Feng, WW.; Kurokawa M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist. 2020, 3, 1-17. http://dx.doi.org/10.20517/cdr.2019.100
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 43
likes
Like This Article 43
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.