
MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution


Abstract
Neuroblastoma (NB) deriving from neural crest cells is the most common extra-cranial solid cancer at infancy. NB originates within the peripheral sympathetic ganglia in adrenal medulla and along the midline of the body. Clinically, NB exhibits significant heterogeneity stretching from spontaneous regression to rapid progression to therapy resistance. MicroRNAs (miRNAs, miRs) are small (19-22 nt in length) non-coding RNAs that regulate human gene expression at the post-transcriptional level and are known to regulate cellular signaling, growth, differentiation, death, stemness, and maintenance. Consequently, the function of miRs in tumorigenesis, progression and resistance is of utmost importance for the understanding of dysfunctional cellular pathways that lead to disease evolution, therapy resistance, and poor clinical outcomes. Over the last two decades, much attention has been devoted to understanding the functional roles of miRs in NB biology. This review focuses on highlighting the important implications of miRs within the context of NB disease progression, particularly miRs’ influences on NB disease evolution and therapy resistance. In this review, we discuss the functions of both the “oncomiRs” and “tumor suppressor miRs” in NB progression/therapy resistance. These are the critical components to be considered during the development of novel miR-based therapeutic strategies to counter therapy resistance.
Keywords
Introduction
Neuroblastoma (NB) is the prime extracranial solid tumor in infants[1,2]. It is a tremendously lethal malignancy that accounts for ~10% of all pediatric cancer deaths[3-5]. With 50% of the diagnosed cases designated as high-risk progressive disease, the overall survival (OS) rates remain around 40%, despite intensive multimodal clinical therapy (IMCT)[6]. Given the disease’s heterogeneity, resistance, and poor hematological reserve, cure for high-risk disease is rare, with < 10% 5-year OS and 2% 10-year survival (vs. 38%-71% for low-risk disease)[7,8]. NB is characterized by hematogenous metastasis[9]. Since NB is a highly heterogeneous disease and there is a continuous acquisition of genetic and molecular rearrangements in such heterogeneous clones that contribute to the therapy resistance, tumor relapse, and disease evolution, we must understand the acquired/adapted reorganizations if we are to successfully treat NB. Although epigenetic, genetic, and molecular rearrangements have been causally linked to disease evolution beyond IMCT, in this review we directed our focus on the role of microRNAs (miRs), particularly those involved in tumor dissemination and progression, generally designated as “metastamiRs”.
NB is derived from neural crest cells (NCCs) of sympathoadrenal lineage (SAPs)[6,10-13], when these cells cease to differentiate[14]. These SAPs undergo epithelial-to-mesenchymal transition (EMT), augmenting NCCs’ migratory abilities and with a complex interplay of sequential molecular events orchestrating functional response (e.g., catecholamine biosynthesis)[15] that dictates NB genesis. Since NB genesis could be drawn from SAPs destined for various lineages[16], NB is generally characterized with biological and clinical heterogeneity[17-19]. In the last two decades, significant advances have been made in staging, risk stratification, and treatment strategies. Briefly, for clinically stable stage I/II disease, surgery in which the tumor mass is excised is the prime treatment strategy, without any chemotherapy or other treatments[20]. However, for high-risk disease (stages III/IV), IMCT comprises the induction phase, with alternating regimens of high-dose chemotherapeutic drugs and load reduction surgery; followed by the consolidation phase with more intensive chemotherapy, along with radiotherapy and stem cell transplant; and finally the maintenance phase with retinoid drug treatment, immunotherapy, and immune-activating cytokine treatment. Despite IMCT, the disease evolves with frequent relapses in decreasing time intervals[21,22], reflecting the continuous acquisition of genetic and molecular rearrangements in the undifferentiated/poorly differentiated NB cells[23-25], and warrants a better understanding of the underlying genetic/molecular mechanisms. Defining such events would allow us to develop improved targeted therapeutic strategies for better control of the disease evolution and, subsequently, to achieve the desired clinical outcomes.
Discovered in 1993[26], miRs are a group of small non-coding RNAs of approximately 22 nucleotides in length that regulate gene expression at the post-transcriptional level. Thus far, a total of 1917 miRs have been identified in humans (CRCh38, miR Base). Studies have documented cell-/tissue-specific expression of select sets of miRs with roles in shaping cellular identity and cell-specific function[27,28]. miRs exert multi-dimensional control on gene expression, through direct DNA interaction, translational repression, or by routing mRNA degradation[29]. In general, miRs play crucial roles in maintaining homeostasis in cellular systems. Rearrangements in miR profiles could derail balance in development, differentiation, and growth, leading to diseases such as cancer. A plethora of studies have documented the functional role of miRs in regulating oncogenes, tumor suppressors, and genes involved in the cell cycle, apoptosis, cell migration, and angiogenesis[30-37]. Changes in miR expression associated with cancer development can occur through a manifold of mechanisms, each of which is a critical event that could greatly alter early development and give rise to the pathogenesis of childhood cancers. In this regard, researchers have indicated the benefit of non-invasive screening for serum miRs in the diagnosis and risk stratification of childhood solid tumors[38].
Functionally, the miR serves as a guide to the target mRNA through base-pairing and thereby negatively regulates the target expression. The complementarity level between miR and mRNA determines the mechanism of silencing, whether through cleavage of target mRNA with subsequent degradation or through translation inhibition. The biogenesis of miRs, mechanisms of action, and their roles in cancer were reviewed in detail elsewhere[39]. With about 60% of human protein-coding genes controlled by miRs, independent studies have consistently demonstrated the role of miRs in regulating cellular functions, including the cell cycle, proliferation, differentiation, apoptosis, and metabolism. Aberrant alterations (expression/suppression) in miRs have been shown to prompt carcinogenesis, tumor progression, cancer evolution, cancer stem cells, autophagy, multi-drug resistance (MDR), EMT, migration, invasion, metastasis, and others[40-42]. miRs can be designated as oncomiRs (inhibit tumor suppressor genes), metastamiRs (inhibit tumor stabilization genes), or tumor suppressor miRs (annul oncogenes), based on the targets regulated[43,44]. Studies have also shown tumor- (vs. normal tissue), tumor type-, and disease type- (primary or metastatic) specific miR signatures[45-47]. For instance, reduced levels of miR-Let-7 (targets proto-oncogene RAS)[48] and overexpression of polycistronic miR cluster miR-17-92[30,31] were shown in lung cancer, while miR-15 and miR-16 are frequently deleted in chronic lymphocytic leukemias[49]. However, attaining the cancer type-specific miRs blueprint is still in the early stages for various reasons, including continuously evolving signatures with disease progression, therapy resistance, cancer site, and others; exclusion of prognosis-relevant miRs with putative oncogenic functions based on their silent state or low expression; and inclusion of miRs that are capable of regulating targets of both arms (promotion and inhibition) of signaling where balance might shift with disease evolution. Furthermore, beyond the gene negative regulator function of miRs, studies have also indicated that miRs can moderate transcription[50,51] or activate translation[52].
Role of miRs in NB genesis
Researchers have shown that the tissues of early neural crest development are rich in miR diversity, and that select sets of miRs are explicitly expressed in NCCs[53], the primordial source from which NB originates. During development, miRs fine-tune protein levels, which contribute to the progressive changes in gene expression profiles of different cell types and/or maintain steady-state levels of genes for programmed processes, including NCCs induction, differentiation, delamination, and migration [Figure 1][54]. Furthermore, miRs have been shown to regulate EMT, the critical step leading from SAPs to NB genesis, in NCCs, dysregulation of which could contribute to neural crest disorders[55]. More importantly, the functional role of miRs in NB genesis is clearly documented in studies that showed preferential downregulation of miRs in an N-MYC-driven NB model appropriating the gene signature for tumor genesis, as well as to sustain N-MYC expression[56]. A recent miR-mRNA interactions study for network modeling in NB tumorigenesis designated miR-204 as a tumor suppressor. Expression of miR-204 directly bound with N-MYC mRNA, repressed its expression, and inhibited NB tumorigenesis[57]. Mostly, the function of miRs in NB has been indicated through high-content screening strategies coupled with sequential candidate function-finding strategy (using miR mimics or inhibitors) in terms of cell differentiation[58-60]. Du and colleagues documented the differentiation-inducing and oncosuppressive function of miR-2110, one among many neurite-inducing miRs identified by high-content screening[58], through targeted inhibition of TSKU[61].
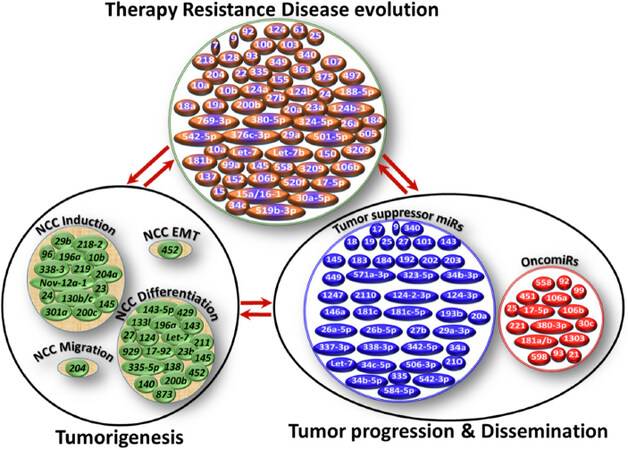
Figure 1. Schematic representation of miRs involved in neuroblastoma (NB) genesis, tumor progression, and therapy resistance. While cellular function- and NB process-specific miR modulation were realized, it was also clear that the functional roles of miRs (e.g., miR-9, miR-92) are conserved across the process, at least in the NB setting. In-depth analysis and crisscross comparisons of the documented functional roles of miRs across NB biological processes showed NB process (genesis, progression, therapy resistance) - specific miR(s) regulation/deregulation; within a process, function-specific miR(s) modulation; activation of function-specific miRs is complemented with compromised incompatible miR(s) (e.g., oncomiR activation with concomitant decrease of tumor suppressor miR or vice versa) and; the involvement of select miRs across the function within a process and also across the processes of NB evolution
Role of miRs in NB disease progression and metastasis
Deregulation of miRs may be an important mechanism that contributes to pathogenesis and heterogeneity of NB[62,63]. Clinical behaviors of NB may be considerably correlated with their specific genetic abnormalities[64] (e.g., amplifications, deletions, point mutations) and relatively rapid epigenetic changes (e.g., DNA methylations, histone modifications)[65]. For instance, studies have documented a strong inverse association between miR-2110 (low tumor levels) and its target, TSKU (high tumor mRNA levels), and further demonstrated a significant correlation of both low miR-2110 and high TSKU mRNA with poor patient survival[61]. Likewise, Chen and Stallings[66] showed underexpression of a specific subset of miRs in N-MYC-amplified NB cells, and targeting N-MYC restored these crucial miRs that play functional roles (e.g., miR-184 in inducing apoptosis). This data suggests that N-MYC could induce NB pathogenesis by regulating miRs that promote NB cell differentiation, apoptosis, and others.
Deregulation of miRs in malignant NB could be due to N-Myc amplifications, chromosomal deletion, or abnormal epigenetic regulation[62,67]. N-Myc binds to the promoter region of an array of miRs and regulates their expression. An independent study showed that at least 37 miRs were differentially expressed in NB with N-Myc amplification compared with N-Myc non-amplified NB[68]. While several oncomiRs (miR-17-5p, miR-92, miR-93, miR-99, miR-106a, and miR-221) are upregulated with N-Myc amplification in progressive NB, suppression of tumor suppressors like miR-34a is also observed with N-Myc amplification. In addition, N-Myc activates methyl-transferases and prompts methylation of target genes. To that note, deregulated epigenetic machinery can aberrantly modify the promoters of miRs in NB. Researchers have identified a panel of oncomiRs and tumor suppressors (let-7, miR-101, miR-202, miR-9, miR-34a, miR-340, miR-184, and miR-335) that are controlled under epigenetic regulation (aberrant DNA methylation or histone modification) in malignant NB[69,70]. For instance, Banelli and colleagues identified that miR-34b-3p, miR-34b-5p, miR-34c-5p, and miR-124-2-3p were significantly hypermethylated and consequently downregulated, particularly in the subset of NB patients at high risk of progression[71]. Conversely, miR-221 has been shown to increase the expression of N-MYC by directly targeting Nemo-like Kinase (NLK), regulating the cell cycle, and promoting the growth of NB cells[72].
Since evolving NB harbors a characteristic miR profile and given miRs’ biomarker designation for NB diagnosis, therapy response, and prognosis[62], stabilizing altered miR species could improve therapeutic strategies and yield a better outcome. An array of miRs functions as tumor suppressors as well as tumor evolution stabilization candidates, deregulation of which heavily contributes to NB progression [Table 1]. For example, stabilizing miR-203 directly targeted Sam68 translation and thereby inhibited NB cell proliferation, migration, and invasion[73]. Likewise, reinforcing miR-337-3p and miR-584-5p has been indicated to inhibit NB cell growth, invasion, metastasis, and angiogenesis by targeting MMP14[74,75]. In this line of tumor suppressor miRs, miR-449a has been extensively investigated for its beneficial functions in various tumors, including lung, liver, pancreatic, breast, bone, brain, prostate, head and neck, ovarian, endometrial, and bladder cancer[76-81]. In the context of NB, miR-449a has been shown to inhibit cell survival and growth through two converging mechanisms: inducing cell differentiation and cell cycle arrest[59].
Partial list of microRNAs involved in NB disease progression and therapy resistance
| miR | Type | HR-NB* | Target | Cellular function | NB process | Ref. |
|---|---|---|---|---|---|---|
| 204 | TSmiR | Inverse | N-MYC, BCL2, NTRK2 | ↑sensitivity to cisplatin | Chemo sensitization | [57] |
| 2110 | TSmiR | Inverse | TSKU | ↑differentiation, ↑neurite | Oncosuppression | [61] |
| 184 | TSmiR | Inverse | AKT | ↑cell death | Tumor suppression | [68,101] |
| 17-5p | OncomiR | Direct | p21, BIM, ERα, NHR-GR | ↑proliferation, ↓differentiation | Disease progression, therapy resistance | [134,136] |
| 18a | OncomiR | Direct | p21, BIM, ERα, NHR-GR | ↑proliferation, ↓differentiation | Disease progression, therapy resistance | [134,136] |
| 19a | OncomiR | Direct | p21, BIM, ERα, NHR-GR | ↑proliferation, ↓differentiation | Disease progression, therapy resistance | [134,136] |
| 20a | OncomiR | Direct | p21, BIM, ERα, NHR-GR | ↑proliferation, ↓differentiation | Disease progression, therapy resistance | [134,136] |
| 92 | OncomiR | Direct | p21, BIM, ERα, NHR-GR | ↑proliferation, ↓differentiation | Disease progression, therapy resistance | [134,136] |
| 221 | OncomiR | Direct | NLK | ↑cell cycle, ↑growth, ↑proliferation | ↑ n-MYC expression | [72] |
| 34a | TSmiR | Inverse | MYCN, E2F3, BCL2, CCND1, CDK6, TIMP-2 | ↑cell cycle arrest, ↑apoptosis. ↓angiogenesis | Tumor suppression | [100,102,138,139] |
| 9 | TSmiR | Inverse | MMP14 | ↓invasion, ↓metastasis, ↓angiogenesis | Tumor suppression | [107] |
| 340 | TSmiR | ↑ W/ RA | SOX2 | ↑differentiation, ↓stemness | Oncosuppression, chemosensitization | [69] |
| 34b-3p | TSmiR | Inverse | CCNE2, E2F3 | Regulates cell viability | Oncosuppression | [110] |
| 203 | TSmiR | Inverse | SAM68 | ↓proliferation, ↓invasion, ↓migration | Tumor suppression | [73] |
| 337-3p | TSmiR | Inverse | MMP14 | ↓proliferation,↑invasion, ↓migration ↓angiogenesis | Tumor suppression | [74] |
| 584-5p | TSmiR | Inverse | MMP14 | ↓proliferation, ↓invasion, ↓migration ↓angiogenesis | Tumor suppression | [75] |
| 449a | TSmiR | Inverse | MFAP4, PKP4, TSEN15, CDK6, LEF1 | ↓growth, ↓survival, ↑cell cycle arrest ↑differentiation | Tumorigenesis, differentiation | [59] |
| 506-3P | TSmiR | Inverse | CDK4 and STAT3 | ↑differentiation | Tumor suppression | [58] |
| 124-3p | TSmiR | Inverse | CDK4 and STAT4 | ↑differentiation | Tumor suppression | [58] |
| 193b | TSmiR | Inverse | MYCN, Cyclin D1, MCL1 | ↓viability, ↓proliferation, ↑cell cycle arrest, ↑cell death | Tumor suppression | [89] |
| 145 | TSmiR | Inverse | HIF 2α | ↓growth, ↓metastasis, ↓angiogenesis | Tumor suppression | [90] |
| 27b | TSmiR | Inverse | PPARγ | ↓growth, ↓proliferation | Tumor suppression | [98] |
| 542-3p | TSmiR | Inverse | Survivin | ↓proliferation, ↑apoptosis | Tumor suppression | [99] |
| 542-5p | TSmiR | Inverse | Survivin | proliferation, ↑apoptosis | Tumor suppression | [99] |
| 335 | TSmiR | Inverse | ROCK1, MAPK1, LRG1 | ↓invasion, ↓metastasis | Tumor suppression | [106] |
| 210 | TSmiR | Inverse | BCL2 | ↑apoptosis | Tumor suppression | [108] |
| 181c | TSmiR | Inverse | MAD7 | ↓invasion, ↓metastasis, ↓angiogenesis | Tumor suppression | [109] |
| 29a-3p | TSmiR | Inverse | CDK6, DNMT3A, DNMT3B | ↓cell viability | Tumor suppression | [110] |
| 517a -3p | TSmiR | Inverse | OLFM3, IFNAR1 | Regulates cell viability | Tumor suppression | [110] |
| 183 | TSmiR | Inverse | 85 targets, MCM 2-7 | ↓DNA replication | Tumor suppression | [112] |
| 323a-5p | TSmiR | Inverse | CHAF1A, KIF11, E2F2, INCENP, CDC25A, CCND1, FADD | ↓cell cycle arrest, ↑apoptosis | Tumor suppression | [116] |
| 342-5p | TSmiR | Inverse | AKT2, CCND1, MKNK2, BCL-X | ↓cell cycle arrest, ↑apoptosis | Tumor suppression | [116] |
| 26A-5P | TSmiR | Inverse | LIN28B | ↓oncogene | Oncosuppression | [117] |
| 26B-5P | TSmiR | Inverse | LIN28B | ↓oncogene | Oncosuppression | [117] |
| 338-3p | TSmiR | Inverse | PREX2A | ↓survival, ↓growth, ↑cell cycle arrest | Tumor suppression | [118] |
| 1247 | TSmiR | Inverse | ZNF346 | ↓proliferation, ↑cell-cycle arrest, ↑cell death | Tumor suppression | [121] |
| 146a | TSmiR | Inverse | BCL11A | Inhibits cell growth and promotes apoptosis | Tumor suppression | [122] |
| 558 | OncomiR | Direct | HPSE, VEGF, AGO2, EIF4E | ↑growth, ↑invasion, ↑metastasis, ↑angiogenesis | Tumor progression | [123,124] |
| 451 | OncomiR | Direct | MIF | ↑growth, ↑invasion, ↑migration | Tumor progression | [125] |
| 192 | TSmiR | Inverse | Dicer1 | ↓proliferation, ↓migration | Tumor suppression | [126] |
| 1303 | OncomiR | Direct | GSK3β, SFRP1 | ↑proliferation | Disease progression | [127] |
| 181 a/b | OncomiR | Direct | ABL1 | ↑growth, ↑invasion | Disease progression | [129] |
| 380-3p | OncomiR | Direct | p53 | ↓cell death | Disease progression | [130] |
| 21 | OncomiR | Direct | PTEN, APL, FOXO3A | ↑proliferation | Disease progression | [132] |
| 137 | ↓CR cells | CAR | ↑sensitivity to doxorubicin | Chemosensitization | [137] | |
| 15a/16-1 | ↓ CR | BMI1 | ↓BMI1 and GSH | Chemosensitization | [140] | |
| 61 | TSmiR | ↑W/ cisplatin | BDNF | ↓survival, ↑differentiation | Chemosensitization | [142,143] |
| 497 | TSmiR | Inverse | WEE1 | ↓cell viability, ↑cell death | Tumor suppression | [144] |
| 376c-3p | TSmiR | ↓W/IMCT | Cyclin D1 | ↑cell death | Tumor suppression | [145] |
| 155 | TSmiR | Inverse | TERF1 | ↓telomerase activity, ↓telomere length | Tumor suppression | [151] |
| 10A/10B | TSmiR | ↑W/RA | SRFS1 | ↑differentiation, ↓migration, ↓invasion | Tumor suppression | [157] |
| 128 | TSmiR | ↑W/RA | REELIN, DCX, NTRK3 | ↓motility,↑invasion, ↓growth | Tumor suppression | [158,159] |
| 129 | TSmiR | MYO10 | ↓growth, ↑sensitization to Cytoxan | Chemosensitization | [168] |
While miR-449a dictates NB cell differentiation by selectively targeting MFAP4, PKP4, and TSEN15, in parallel it also affects cell cycle arrest through downregulation of CDK6 and LEF1[59]. Previously, utilizing a high-content morphological screening approach, the same group identified three miR seed families (seed region is 5’ end of the mature miRNA consisting of similar eight nucleotides that are involved in inducing NB cell differentiation[58]. In this study, they showed that miR-506-3p and miR-124-3p have unparalleled differentiation-inducing capability and they exert their tumor-suppressive function in part by downregulating their targets, CDK4 and STAT3. miR-124 is most specifically expressed in the nervous system, plays a prominent role in neuronal differentiation, and is increased during neural development[82,83]. They also recognized that miR-506-3p is dramatically upregulated in differentiated NB cells, indicating its crucial role in differentiation and tumorigenesis. Similar to miR-449a, miR-193b is a well-documented and designated tumor suppressor in various human cancers[84-88]. miR-193b showed significantly lower expression in NB and corresponded to increased cell viability and proliferation[89]. Functionally, miR-193b induces G1 cell cycle arrest and NB cell death by selectively inhibiting the expression of N-MYC, Cyclin D1, and MCL1.
In parallel, an independent study identified the NB tumor suppressor function of miR-145, a miR that is enriched in germ-line and mesoderm-derived tissues[90]. Since its first record of downregulation and its role in the tumorigenesis of early stage colorectal cancer, its tumor suppressor function has been widely recognized in many human cancers[91-96], including nervous system tumors such as glioblastoma and CNS lymphoma[97]. Functionally, in NB, it has been shown that miR-145 targets HIF-2α and inhibits the hallmark physiognomies (growth, invasion, metastasis, and angiogenesis) of tumor progression and metastasis[90]. Another study indicated that miR-27b directly targets the 3’UTR of PPARγ and regulates NFκB, NHE1, and tumor growth and progression[98]. miR-542-3p and 5p, the miRs that inversely associate with poor prognosis in NB, directly target survivin and dictate tumor-suppressive functions[99].
Studies assessing the tumor suppressor functions of miRs in NB setting have shown that miR-34a and miR-184 are significantly underexpressed in NB[100,101]. Functionally, miR-34a targets N-MYC[100], while miR-184 targets AKT and orchestrates their tumor-suppressive function[102], miR-34a induces cell cycle arrest and apoptosis activation in NB cells[103]. Furthermore, it has been shown that CDK1 regulates miR-34a in NB cells[104]. Inhibition of CDK1 dictates increased expression of miR-34a, which in turn downregulates N-MYC and mediates the Survivin loss associated with cell death. Comparing the metastatic NB tissues, many studies identified the deregulation of miRs[105], however, only a few in-depth reports are available on the mechanism of regulation and the function of miRs in disease evolution. Studies that attempted to define the mechanism(s) of action (cell death, inhibition of invasion, metastasis) of miRs in disease evolution identified: miR-335, which targets many gene targets in the non-canonical TGFβ signaling pathway[106]; miR-9, which targets MMP14[107]; miR-210, which targets BCl2[108]; and miR-181c, which targets Smad7[109].
Consistently, it has been shown that miR-181c was significantly downregulated in metastatic tissues compared with primary NB tissues[109]. Likewise, Purello and colleagues showed that downregulation of miR-29a-3p, miR-34b-3p, miR-181c-5p, and miR-517a-3p in NB tissues (compared with normal adrenal gland) was primarily due to the hypermethylation of their promoters[110]. Since these tumor suppressors are disallowed in NB, uncontrolled expression of their targets CDK6, DNMT3a, DNMT3B, CCNA2, and E2F3 dictates disease progression. Deubzer and colleagues reported that transcription of miR-183 is significantly repressed by N-MYC and is highly induced in N-MYC-amplified NB in response to pan-HDAC inhibitor and cyclic tetrapeptide[111]. miR 183 expression studies by this group later identified 85 targets, including all 6 members of the minichromosome maintenance complex (MCM2, MCM3, MCM4, MCM5, MCM6, MCM7) that is critical for initiation and elongation during DNA replication[112].
The transcriptional regulator LMO1 has been shown to promote NB cell proliferation and has been designated as an NB susceptibility gene by GWAS[113]. Later, this group identified that LMO1 indirectly downregulates at least 18 miRs, including seven miRs of the Let7 family (7a-5p, 7b-5p, 7c, 7e-5p, 7f-5p, 7g-5p,7i-5p)[114]. The proliferation inhibition function of these seven members of the Let7 family was extensively documented. The regulation of these candidates by LMO1 in NB indicates well-orchestrated, cluster-focused control of tumor suppressor miRs in disease evolution. A number of mechanisms that could deregulate the Let-7 family in NB has been reported and was recently reviewed in detail by Daley and colleagues[115].
Many studies have identified and characterized tumor-suppressive miRs in NB and defined their mechanistic signaling flow-through in orchestrating tumor progression and disease resistance. A very recent observation with functional high-throughput screening in both in vitro and in vivo models identified the NB tumor-suppressive function of miR-323-5p and miR-342-5p, and indicated that the effect occurs through directly targeting CCND1, CHAF1A, INCENP, and Bcl-Xl[116]. Interestingly, two tumor suppressor miRs, miR-26a-5p and miR-26b-5p, which directly regulate LIN28B, are in turn affected by N-MYC expression, leading to N-MYC-driven LIN28B-mediated oncogenic processes[117]. Chen and colleagues identified PREX2a as a direct target of miR-338-3p[118]. PREX2a can directly interact with PTEN to inactivate its lipid phosphatase activity, leading to accumulation of PIP3 and, as a consequence, increased AKT phosphorylation, which in turn promotes NB cell survival, cell cycle progression, and tumor growth. miR-338-3p-dependent inhibition of PREX2a affects PTEN/AKT pathway-dependent tumor progression. miR-338-3p is transcribed from the intron 8 of the apoptosis-associated tyrosine kinase (AATK) gene, located on chromosome 17q25, playing a critical role in promoting cell death, neuronal differentiation, and neurite extension[119,120]. Assessing the biological function of miR-1247 in NB, it was realized that ZNF346 is the direct target of miR-1247 and its targeted inhibition of ZNF346 suppresses cell proliferation and induces cell cycle arrest and NB cell death[121]. Similarly, miR-146a has been shown to inhibit NB cell growth and promote apoptosis by directly down-modulating its target, BCL11A[122]. Similarly, miR-192 has been reported as an independent prognostic marker for relapse in NB patients; miR-192 directly targets Dicer1 and was functionally designated as a tumor suppressor miR[123].
Understanding the deregulation of designated oncomiRs in NB cells and recognizing their driving role in disease progression, dissemination, and clinical outcomes is of great importance [Table 1]. For instance, oncomiR-558 was shown to directly facilitate the transactivation and translation of HPSE and its downstream target VEGF, and contribute to NB tumor progression[124]. Studies have also demonstrated that miR-558 directly facilitates Ago2- and eIF4E-dependent HIF2α expression and dictates NB growth, invasion, metastasis, and angiogenesis[125]. Likewise, lower levels of miR-451 were inversely correlated with NB growth, invasion, and migration. It has been shown that miR-451 directly inhibits macrophage inhibitory factor, which critically mediates the biological effect of miR-451 in NB[126]. Stem-loop sequence miR-1303, on the other hand, has been shown to promote NB cell proliferation by selectively targeting GSK3β and SFRP1[127]. While GSK3β is critical for the phosphorylation of Cyclin D1, SFRP1 directly binds with WNTs and inhibits the WNT/β-catenin pathway, indicating that miR-1303-directed inhibition of these candidates could endorse disease progression. NB patients with high levels of SFRP1 have shown good prognosis.
In the highly tumorigenic sub-population of CD114+ cells, research/investigators have identified differential expression of 25 miRs, including those that control embryonic stem cells transition to neuronal precursors (miR-106b, miR-21, miR-25, miR-30e, miR-598, miR-93)[128]. However, miRs, including miR-25, miR-17, miR-18, miR-19, miR-20a, miR-143, and miR-27, are repressed during neuronal lineage differentiation of somatic stem cells. Although most of the miRs are validated transcriptional targets of N-MYC, most are also known to target p53 and its downstream targets, and correlate with poor prognosis in NB[128]. While miR-181c is designated as a tumor suppressor, miR-181a/b is recognized as an oncomiR. Expression of miR181a/b was positively associated with N-MYC amplification and NB progression. It has been shown that miR-181a/b directly affects ABl1 expression and thereby dictates NB disease evolution[129]. Also, miR-380-3p has been recognized as an oncomiR, as it has been shown to promote tumor growth by repressing p53, and is associated with poor outcome in N-MYC-amplified NB[130]. Consistent with these observations, miR profiling studies clearly recognized that differential regulation of specific subsets of miRs could be beneficial for risk stratification in NB[131].
Role of miRs in NB therapy resistance
NB-focused investigations indicated unique patterns of mutation(s) and specific miR dysregulation that could lead to therapy resistance[66,68]. In-depth analysis and crisscross comparisons of the documented functional roles of miRs across NB biological processes, including genesis, progression, therapy resistance, and evolution, clearly portrayed: (1) NB process-specific miR(s) regulation/deregulation; (2) within a process, function-specific miR(s) modulation; (3) that activation of function-specific miRs is complemented with compromised incompatible miR(s) (e.g., oncomiR activation with concomitant decrease of tumor suppressor miR or vice versa); and (4) the involvement of select miRs (e.g., miR-17-5p) across the function within a process and also across the processes of NB evolution [Figure 1, Table 1]. In particular, the miRs that dictate NB pathogenesis and tumor progression overlap to a large extent with those that orchestrate therapy resistance or sensitization. Differential modulation of at least seven miRs was reported in N-MYC-amplified NB cell lines (vs. N-MYC non-amplified) and further defined their role in orchestrated chromosomal imbalances[68]. A study in which primary NB tumors were screened revealed the underexpression of miRs in tumors with N-MYC amplification and demonstrated a substantial reversal with retinoic acid (RA) treatment, a compound that is known to reduce N-MYC expression and induce NB cell differentiation[67]. This study also showed that targeting N-MYC restores expression of miRs, indicating that N-MYC may mediate therapy resistance, in part, through directly or indirectly regulating the expression of miRs that are involved in cell differentiation, hence designating these miRs as potential therapeutic targets[67].
In addition to the induced therapy resistance through well-directed underexpression of miRs that play a role in apoptosis and differentiation, studies have also shown that the function of miRs also heavily contribute to therapy resistance. Researchers have demonstrated that miR-21 could target PTEN and thereby increase NB cell proliferation and endorse chemo-resistance[132]. miR-21 promoter contains highly conserved regions with consensus binding sites for transcriptional regulator AP1 and forkhead family protein FOXO3a. Findings from one study indicated high expression of miR-21 in cisplatin-resistant NB cells, and its selective silencing prompted chemo-sensitization[132]. Interestingly, miR-21, the designated oncomiR, has been shown to be strongly upregulated under N-MYC knockdown conditions, and did not prompt any significant differentiation or proliferation with forced repression, indicating the probable requirement for miR-21 and N-MYC parallel activation for a causal effect[133].
Five miRs within the polycistronic cluster (miR-17-5p, miR-18a, miR-19a, miR-20a, miR-92) were greatly expressed in NB cells with high N-MYC expression[134]. N-MYC binds directly to several sites of the 5’ and 3’ regions of this cluster candidate and hence could contribute to disease progression and therapy resistance[134]. While ectopic expression of miR-17-5p-92 has been shown to dramatically induce NB cell proliferation, miR-17-5p directly downregulates the tumor suppressor gene P21 by binding to its 3’ UTR region. Although cluster candidates miR-19 and miR-92 are known to modulate levels of the apoptotic BIM gene, it is now clear that miR-17-5p also targets BIM in NB cells. Targeting miR-17-5p with antagomiR profoundly inhibited NB growth in vivo and produced a reciprocal increase of both P21 and BIM[134]. In this regard, stabilizing miRs could serve as an all-inclusive strategy to overcome MDR, particularly for NB, since numerous miRs (miR-21, -128, -380-5P, -558, -375, -9, -15, -103, -107, -124, -29A, -152, -125b, -204, -363, -335, -17-92, and let 7) deregulate a panel of gene targets (discussed elsewhere in detail[135]) that orchestrate MDR.
It has been shown that the N-MYC-regulated miR-17-92 cluster inhibits NB cell differentiation through ERα repression, targeting nuclear hormone receptors, and suppression of glucocorticoid receptor expression associated with undifferentiated phenotype and decreased survival[136]. Chen and colleagues recognized an inverse expressional relationship between miR-137 and Constitute Androstane Receptor (CAR), a xenosensor and a key regulator of MDR[137]. This study showed that miR-137 is selectively downregulated in doxorubicin (Dox) -resistant NB cells; the stabilization (reinforcement) of miR-137 resulted in CAR protein suppression (effected through mRNA degradation) and promoted chemotherapy (Dox) sensitization. More importantly, the findings indicated the presence of a mechanistic negative feedback loop between miR-137 and CAR. Functionally, promoter hypermethylation and negative regulation of miR-137 by CAR contribute to the reduced miR-137 availability and increased CAR and MDR1, which contribute to Dox resistance[137].
Loss of tumor suppressor miR-34a in progressive NB also contributes to therapy resistance. N-MYC is the direct target of miR-34a, with two functional target sites in the N-MYC 3’ UTR[100]. In addition to targeting N-MYC, mir-34a is known to target other crucial players of therapy resistance, including E2F3, BCl2, CCND1, and CDK6[138]. NB-targeted delivery of miR-34a not only decreased tumor growth, but also significantly reduced tumor vascularization by targeting TIMP2, a critical component of MDR[139]. In a similar study, Domenicotti and colleagues[140]. reported the function of miR-15a/16-1 in acquired drug resistance for NB. Interestingly, this study recognized a mono-allelic deletion of the 13q14.3 locus in p53, the location of p53-inducible miR15a/16-1 in etoposide-resistant cells. Acquired loss of miR-15a/16-1 leads to induced expression of BMI1 oncoprotein and activation of GSH-dependent responses, leading to etoposide resistance[140]. Ryan and colleagues[141] reported that reinforcing the acquired loss of miR-204 targeted BCl2 and NTRK2, and thereby increased the sensitivity of NB cells to cisplatin. miR-204 is a tumor suppressor miR and is a designated predictor of outcome in NB.
Understanding how chemotherapy drugs inhibit cancer growth would place us in a better position to assess and/or prevent acquired resistance. Brain-derived neurotropic factor (BDNF) is a crucial protein that prompts neuronal cell survival, differentiation, and axon wiring through its interaction with tyrosine kinase receptor B (TrkB)[142]. BDNF-TrkB signaling has also been shown to affect the development, invasion, and outcome of many human tumors, including lung, bladder, pancreatic, and breast cancer. It has been shown that BDNF is the direct target of miR-61, and cisplatin treatment could significantly reinforce miR-61 and thereby inhibit BDNF in NB cells[143]. Similarly, miR-497 loss is associated with worse EFS and OS in N-MYC-amplified high-risk NB patients, and also corresponds with increased cell viability and decreased cell death. It has been shown that miR-497 directly targets WEE1, a key cell cycle regulator that orchestrates drug resistance. More importantly, ectopic expression of miR-497 or the inhibition of WEE1 resulted in significant chemotherapy sensitization to cisplatin, affirming that acquired loss of miR-497-dependent increased expression of WEE1 dictates drug resistance in NB cells[144]. Another study indicated a reduced expression of the tumor suppressor miR-376c-3p in the majority of the cell lines isolated from patients after IMCT[145]. miR-376c-3p directly targets Cyclin D1, an oncogene critical for NB pathogenesis. The therapy-associated acquired loss of miR-376c-3p in surviving NB cells illustrates its function in therapy resistance.
In agreement with the hypothesis that both oncomiRs vs. tumor suppressor miRs drives therapy resistance, a high-throughput profiling study comparing miR deregulation across three independent chemo-resistant models consistently identified upregulation of four miRs (miR-150, miR-188-5p, miR-501-5p, miR-519B-3p) and downregulation of another three miRs (miR-125b-1, miR-204, miR-769-3p)[146]. Such emerging roles of miRs in exacerbating MDR highlight its criticality in clinical management of drug-resistant NB. Some of the key players involved in NB therapy resistance and their functional targets, the modulation of those that dictate therapy response are provided in Figure 2. One of the prime strategies in this setting is to block upregulated oncomiR by using antisense oligonucleotides. As identified earlier, targeted delivery of miR-34a using GD2-coated nanoparticles showed significant reduction in tumor growth, increased apoptosis, and reduced vascularization[139]. Conversely, the other method is to reinforce and to stabilize the downregulated tumor suppressor miR(s) using mimics. For instance, independent studies reinforcing tumor suppressor miRs (e.g. miR-9, miR-27b, miR-34a, miR-145, miR-184, etc.) showed a profound benefit reducing NB progression, and clearly indicated that these miRs could serve as novel therapeutic targets[90,98,103,107,130,147,148]. Chemotherapy in combination with miR-based treatment is currently recognized as a better strategy for clinical management of progressive NB. Ray and colleagues[149] indicated that synthetic retinoid (4HPR) in combination with EGCG resulted in significant inhibition of oncomiRs miR-92, miR-93, and 106b, and induced the expression of tumor suppressors miR-7-1, miR-34a, and miR-99a. These findings suggest that treatments targeting oncomiRs and/or reinforcing tumor suppressor miRs would be highly beneficial in reverting induced therapy resistance in progressive NB.
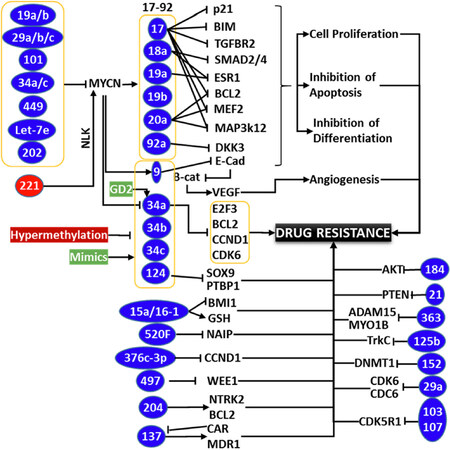
Figure 2. Schema showing the partial list of miRs involved in neuroblastoma (NB) therapy resistance. While MYCN regulated miRs have been documented to play crucial role in the therapy resistance through orchestrated clonal expansion and defying differentiation, acquired modulation of upstream miRs those regulate MYCN also plays crucial role in coordinating drug resistance and disease evolution. Hypermethylation of tumor suppressor miRs and hypomethylation of oncomiRs with clinical therapy in surviving cancer cells is regarded as one of the major mechanism for acquired loss of TS miRs and gain of oncomiRs, those dictate drug-resistance. Rearrangements on the levels of many key miRs inflicting therapy resistance through unique signaling flow-through are documented. Conversely, regulation/deregulation of many miRs converge on a signaling or functional event (e.g., Bcl2) to effect resistance. It is clearly evident that miRs play definitive roles in therapy resistance and miR-targeted approach could be an effective strategy for the treatment of resistant NB
Although it has been well recognized that MYCN amplification dramatically influences MDR by regulating an array of functional targets, including the genes involved in drug reflux, studies are highly limited in understanding the role of miRs in other high-risk subtypes that lack N-MYC amplification. In a recent study of NB chemo-resistance (resistance to cisplatin and cross-resistant to etoposide), it was shown that chromosomal rearrangement dictated significant overexpression of neural apoptosis inhibitory protein (NAIP) in drug-resistant cells[150]. Remarkably, this study identified significant loss of miR-520f in post-chemotherapy tumors (vs. pre-chemotherapy) and causally linked the loss of miR-520f to the induced expression of NAIP, which mediates drug resistance in this setting[150]. In parallel, an interesting study by Fabbri and colleagues[151] indicated that the paracrine exchange of exosomic miRs between NB cells and neighboring human monocytes could critically affect drug resistance. NB cells secreting exosomic miR-21 led to monocyte-dependent miR-155 upregulation in a TLR8- and NFkB-dependent manner. Expressed miR-155 directly targets telomerase inhibitor TERF1 and affects telomerase activity and telomere length. This study also asserted the exosomic transfer of miR-155 from surrounding tumor-associated macrophages[151]. Researchers have also reported that exosomes carry as cargo various miRs, including miR-16, miR-125b, miR-21, miR-23a, miR-24, miR-25, miR-27b, miR-218, miR-320a, and miR-92a, which act as oncomiRs, affecting the sequence of many gene targets (e.g., NFkB, STAT3. P53, TLR8), and contributing to drug resistance[152].
Due to the criticality of induced apoptosis or differentiation in NB cure, RA treatment is currently used as part of the NB treatment regimen[153]. Although RA treatment affects N-MYC regulation[154] and other critical targets[155] very early, it is generally accepted that these induced alterations could translate to differentiation or death. Since N-MYC directly regulates an array of miRs in NB, it is highly likely that RA treatment that inhibits N-MYC could inflict rearrangements. Assessing 70 miRs during RA-induced growth arrest identified upregulation of 14 miRs (miR-9, miR-124a, miR-125a, miR-125b, let-7a, let-7b, miR-7, miR-22, miR-23a, miR-24, miR-26a, miR-30a-5p, miR-100, miR-103) that are down-modulated in primary NB[156]. Functional analysis indicated the oncosuppressing potential of at least three miRs (miR-9, miR-125a, and b) and designated their possible use as diagnostic markers for tumorigenesis. An independent investigation by Stallings and Chen assessing the expression of 34 miRs (that exhibited significant variations between favorable vs. unfavorable NB) in NB cells exposed to RA treatment showed definitive upregulation of 17 miRs and downregulation of four miRs[66]. Interestingly, their outcomes indicated robust induction of miR-184, which functionally induced G1 cell cycle arrest, cell death, and complete suppression of miR181b and miR-92 (component of polycistronic cluster, highly expressed in N-MYC amplified NB[134] and known to modulate apoptotic gene BIM)[66]. Independently, miR-184 has been shown to inhibit NB cell survival by directly targeting AKT and killing N-MYC-amplified cells[101]. Further, miR-10a and miR-10b have been causally linked to RA treatment-induced NB cell differentiation[157]. Of the 42 differentially expressed miRs in NB cells treated with RA, the authors indicated profound expression of miRs-10a and b and functionally characterized their role in RA-induced differentiation, RA-inhibited migration, invasion, and in vivo metastasis. Moreover, it has been recognized that miRs-10a and b directly target SR-family splicing factor (SRFS1), inhibit SRFS1-dependent alternative splicing and translational functions, and thereby orchestrate RA treatment-induced NB cell differentiation[157]. Another study indicated an up-modulation of miR-128, a brain-enriched miR, in RA-differentiated NB cells. Induced expression of miR-128 suppressed the expression of Reelin and DCX and reduced NB cell motility, invasiveness, and growth[158]. Reinforcing miR-128, which regulates the truncated isoform of NTRK3, has been shown to cause morphological changes in NB cells and alter the expression of genes involved in cytoskeletal organization, apoptosis, and cell proliferation[159]. miR-340 induces a cell-context-dependent differentiation or death, and has been shown to be upregulated through the demethylation process with RA-treatment[69]. Induced expression of miR-340 has been shown to inhibit Sox2, a critical stemness maintenance factor.
More importantly, a recent high-throughput NGS study comparing miR profiles in isogenic cell lines derived from NB patients at diagnosis and at relapse after IMCT identified 42 (8 upregulated and 34 downregulated) differentially expressed miRs[160]. Uniquely, 22 of 34 downregulated miRs were encoded from 14q32 miR cluster. Reduced expression of miRs from 14p32 cluster was significantly associated with poor prognosis in a cohort of 226 NB patients. Downstream gene target analysis of the roles of this cluster of miRs in signaling and biological functions recognized their association with disease progression and drug resistance[160]. Recent studies are now appropriately focused on elucidating the potential of targeted miR(s) delivery in the treatment of progressive tumors, including high-risk human NB[161,162]. Essand and colleagues demonstrated the benefit of miR-124, miR-125, and miR-134 insertion in SFV-4 virus in reducing the neuro-virulence and increased oncolytic capacity with an NB cure rate of about 50% in mouse model[163]. Similarly, it has been shown that enterovirus EV71 stimulates miR-Let-7b and directly targets CCND1 in therapy-resistant SH-SY5Y cells, identifying EV71 as a potential candidate for miR-based therapy in NB[164]. Another study indicated that fluoxetine, a serotonin reuptake inhibitor, upregulated miR-572 and miR-663a consistently in NB cells and thereby inhibited their crucial targets (mir-572 - Cdkn1, Dicer1, Wnt7a; miR-663a - TGFβ1, PTEN, VEGFA) associated with NB cell differentiation and tumor evolution[165]. The role of miRs in NB cell differentiation and the possibilities of targeting miRs for differentiation therapy is discussed in detail elsewhere[166].
Novel strategies were adopted to investigate the benefit of delivering tumor suppressor miR-3245p, the independent predictor for NB[167]. Direct evidence demonstrating the benefit of a miR-based strategy for therapy-resistant NB was first sought by Fruci and colleagues. They showed that targeting miR-17-5p with antagomiR dramatically inhibited NB growth in vivo and showed a reciprocal increase of both P21 and BIM[134]. Later, an investigation by Li and colleagues recognized that miR-129 could inhibit tumor growth and, more importantly, promote NB cell sensitization to Cytoxan (chemo-drug often used in clinical treatment of NB) by selectively targeting MYO10[168]. Pal-Bhadra and colleagues identified novel conjugate [anthranilamide-pyrazolo (1,5-α) pyrimidine] -targeted miR-34-a, mir-34-c, mir-200b, mir-107, mir-542-5p, and mir-605, which led to the up-modulation of pro-apoptotic p21, Bax, and caspases, with concomitant downregulation of pro-survival Akt, E2F1, and Bcl2 in therapy-resistant human NB cells[169]. With the quantitative nuclear proteomics approach, studies have identified the mechanism of resveratrol, a natural phytoalexin-induced NB cell apoptosis. Resveratrol-induced miR-137 prompted the inhibition of polycomb protein histone methyltransferase EZH2, and reverted the EZH2-dependent regulation of tumor suppressors CLU and NGFR[170].
With the evolution of the new concept that, “embryonic stem cell (ESC) gene regulatory mechanisms in tumors with high pluripotency capacity could contribute to the increased risk of therapy failure”, studies are now focused on understanding the realm of ESC miRs and their function in therapy resistance. Although this focus is still in its infancy, researchers are attempting to understand the basics. On that note, with an in silico approach, studies have listed a panel of ESC miRs along with mRNA signatures that are associated with poor NB patient outcomes, specifically for the N-MYC-amplified and N-MYC non-amplified high-risk subsets[171]. In addition, the findings indicated that the ESC signature is majorly driven by FOXM1 and could be useful to inhibit tumor progression and therapy resistance. Adding to the complexity of miR biology in NB disease evolution and therapy resistance, dose-associated differential modulation of miRs was also reported in NB cells. For instance, while low doses of morphine reduced the expression of miR-133b and prompted cell proliferation, higher doses inhibited cell proliferation, with reduced levels of miR-133b and miR-128[172]. In addition, new miRs residing within frequently altered chromosomal regions in NB tumors that could play significant functional roles in NB biology and therapy resistance are consistently being cloned[173].
Conclusion and perspectives
Despite tremendous clinical efforts with IMCT over the last two decades, a cure for high-risk NB is rare, with < 10% 5-year OS and < 2% long-term survival. These rates are mainly attributed to the continuous ongoing acquisition of genetic and molecular rearrangements in the NB cells that dictates therapy resistance and disease evolution. In the past decade, studies have been appropriately focused and many molecular targets have been proposed. Most of these proposed approaches have not reached clinical trials for multiple reasons, including that aiming single target or signaling in one of the NB evolution processes has been shown to result in activation of alternative and compensatory pathways, leading to additive resistance; cell-type-dependent expression and function; drug (treatment) type; and dose-dependent expression and function. Hence, it is essential to identify new and improved therapeutic strategies for high-risk NB, particularly through all-inclusive targeting that could counter acquired therapy resistance and disease progression. In this regard, miRs are an astonishing new class of gene regulators, and the discovery of the role of miRs in molecular pathogenesis of NB opened up their possible applications in NB diagnosis, prognosis, and miR-based therapy. The current wealth of information on contextual (diagnosis, prognosis, resistance) miR profiles; the direct roles of miRs in NB biology, cellular function, and signaling flow-through; and converging and/or compromising miR rearrangements recognizes that the miR-based therapy is around the corner. As discussed here, attempts were made and are also in progress to target oncomiR(s), reinforce tumor suppressor miR(s), or a combination [Figure 3]. Emerging evidence will further allow us to aim for (1) unique or converging molecular pathways; (2) specific cellular process; or (3) a combination of processes. Although translation of these preliminary research data into clinical application is in its infancy, these findings will provide a strong basis for translation and clinical use in the treatment of NB. In summary, we strongly opine that miRs that contribute to disease progression also have critical roles in the acquired molecular rearrangements and orchestrated therapy resistance of NB. Overall, the presented evidence supports the enormous clinical potential of miRs in countering therapy resistance and disease evolution, and warrants further rigorous investigation.
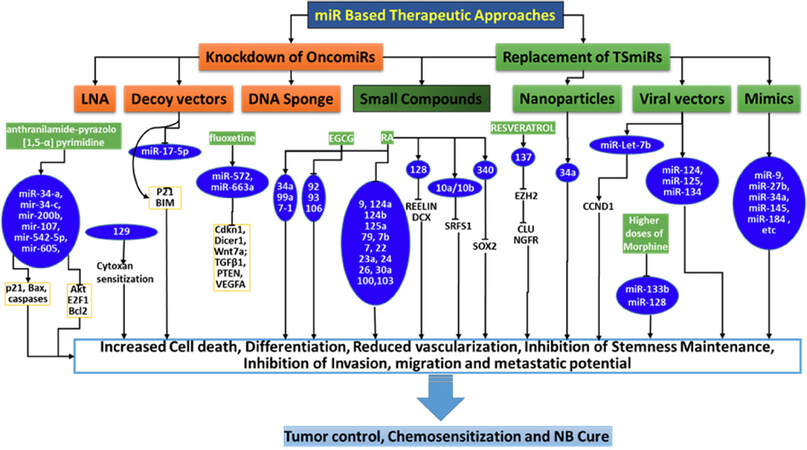
Figure 3. Schema showing the outline of the currently documented miR based treatment strategies for neuroblastoma (NB) cure. Independent studies validated various miR based strategies to increase cell death, inhibit clonal expansion, reduce vascularization, inhibit stem-like cell clonal selection and other crucial functions and, those effect NB cells for chemosensitization and NB cure
Declarations
AcknowledgmentsThe authors acknowledge the OUHSC Staff Editor (Ms. Kathy Kyler) for the help in critically reviewing this manuscript.
Authors’ contributionsWrote the manuscript: Aravindan N
Performed literature collection: Subramanian K
Performed literature analysis: Aravindan N, Subramanian K, Somasundaram DB
Performed literature interpretation: Aravindan N, Aravindan S
Technical support: Somasundaram DB
Conception and design of the study: Aravindan N, Herman TS, Aravindan S
Critically reviewed the manuscript: Somasundaram DB, Herman TS, Aravindan S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe authors are supported by research funding from the National Institutes of Health (NIH 1P20GM103639-01) from the COBRE Program of NIH; and OUHSC Department of Radiation Oncology Research Development Funds.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Marc TG, Gurney JG, Smith MA, Olshan AF. Sympathetic nervous system tumors. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995, National Cancer Institute. Bethesda, MD: NIH Pub.; 1999. pp. 65-72.
2. Gurney JG, Smith MA, Ross JA. Cancer among infants. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995, National Cancer Institute. Bethesda, MD: NIH Pub.; 1999. pp. 149-56.
3. Morgenstern DA, Baruchel S, Irwin MS. Current and future strategies for relapsed neuroblastoma: challenges on the road to precision therapy. J Pediatr Hematol Oncol 2013;35:337-47.
4. American-Cancer-Society. Cancer Facts & Figures. Atlanta: American Cancer Society; 2013. pp. 1-64.
5. Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 2010;28:2625-34.
6. De Bernardi B, Nicolas B, Boni L, Indolfi P, Carli M, et al. Disseminated neuroblastoma in children older than one year at diagnosis: comparable results with three consecutive high-dose protocols adopted by the Italian Co-Operative Group for Neuroblastoma. J Clin Oncol 2003;21:1592-601.
7. Garaventa A, Parodi S, De Bernardi B, Dau D, Manzitti C, et al. Outcome of children with neuroblastoma after progression or relapse. A retrospective study of the Italian neuroblastoma registry. Eur J Cancer 2009;45:2835-42.
8. London WB, Castel V, Monclair T, Ambros PF, Pearson AD, et al. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 2011;29:3286-92.
10. Grau E, Oltra S, Orellana C, Hernandez-Marti M, Castel V, et al. There is no evidence that the SDHB gene is involved in neuroblastoma development. Oncol Res 2005;15:393-8.
11. Castel V, Villamon E, Canete A, Navarro S, Ruiz A, et al. Neuroblastoma in adolescents: genetic and clinical characterisation. Clin Transl Oncol 2010;12:49-54.
12. Fulda S. The PI3K/Akt/mTOR pathway as therapeutic target in neuroblastoma. Curr Cancer Drug Targets 2009;9:729-37.
13. Jrebi NY, Iqbal CW, Joliat GR, Sebo TJ, Farley DR. Review of our experience with neuroblastoma and ganglioneuroblastoma in adults. World J Surg 2014;38:2871-4.
14. Magro G, Grasso S. The glial cell in the ontogenesis of the human peripheral sympathetic nervous system and in neuroblastoma. Pathologica 2001;93:505-16.
15. Crane JF, Trainor PA. Neural crest stem and progenitor cells. Annu Rev Cell Dev Biol 2006;22:267-86.
16. Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, et al. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004;6:838-45.
17. Schwab M, Evans A. Neuroblastoma - developmental and molecular biology meet therapy design. Cancer Lett 2003;197:1.
19. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 2013;13:397-411.
20. Saad DF, Gow KW, Milas Z, Wulkan ML. Laparoscopic adrenalectomy for neuroblastoma in children: a report of 6 cases. J Pediatr Surg 2005;40:1948-50.
21. Santana VM, Furman WL, McGregor LM, Billups CA. Disease control intervals in high-risk neuroblastoma. Cancer 2008;112:2796-801.
22. Simon T, Berthold F, Borkhardt A, Kremens B, De Carolis B, et al. Treatment and outcomes of patients with relapsed, high-risk neuroblastoma: results of German trials. Pediatr Blood Cancer 2011;56:578-83.
23. Molenaar JJ, Ebus ME, Koster J, van Sluis P, van Noesel CJ, et al. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res 2008;68:2599-609.
24. Barbieri E, De Preter K, Capasso M, Chen Z, Hsu DM, et al. Histone chaperone CHAF1A inhibits differentiation and promotes aggressive neuroblastoma. Cancer Res 2014;74:765-74.
25. Schleiermacher G, Janoueix-Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. Int J Cancer 2014;135:2249-61.
26. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993;75:843-54.
27. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, et al. Identification of tissue-specific microRNAs from mouse. Curr Biol 2002;12:735-9.
28. Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, et al. MicroRNA expression in zebrafish embryonic development. Science 2005;309:310-1.
30. Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005;65:9628-32.
31. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005;435:839-43.
32. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 2006;38:228-33.
33. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 2006;38:1060-5.
34. Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 2006;26:8191-201.
35. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006;103:2257-61.
36. Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem 2007;282:2135-43.
37. Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, et al. Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 2007;109:4399-405.
38. Murray MJ, Bell E, Raby KL, Rijlaarsdam MA, Gillis AJ, et al. A pipeline to quantify serum and cerebrospinal fluid microRNAs for diagnosis and detection of relapse in paediatric malignant germ-cell tumours. Br J Cancer 2016;114:151-62.
39. Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics 2010;11:537-61.
40. Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, et al. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 2010;299:L861-71.
42. Xia H, Hui KM. MicroRNAs involved in regulating epithelial-mesenchymal transition and cancer stem cells as molecular targets for cancer therapeutics. Cancer Gene Ther 2012;19:723-30.
43. Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, et al. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res 2007;67:1419-23.
44. Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, et al. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem 2007;282:1479-86.
45. Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A 2004;101:11755-60.
46. Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, et al. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer 2006;5:24.
47. Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer 2007;120:1046-54.
48. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, et al. RAS is regulated by the let-7 microRNA family. Cell 2005;120:635-47.
49. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002;99:15524-9.
50. Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 2008;105:1608-13.
51. Khraiwesh B, Arif MA, Seumel GI, Ossowski S, Weigel D, et al. Transcriptional control of gene expression by microRNAs. Cell 2010;140:111-22.
52. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007;318:1931-4.
53. Ward NJ, Green D, Higgins J, Dalmay T, Munsterberg A, et al. MicroRNAs associated with early neural crest development in Xenopus laevis. BMC Genomics 2018;19:59.
54. Weiner AMJ. MicroRNAs and the neural crest: from induction to differentiation. Mech Dev 2018;154:98-106.
55. Sheehy NT, Cordes KR, White MP, Ivey KN, Srivastava D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010;137:4307-16.
56. Beckers A, Van Peer G, Carter DR, Mets E, Althoff K, et al. MYCN-targeting miRNAs are predominantly downregulated during MYCNdriven neuroblastoma tumor formation. Oncotarget 2015;6:5204-16.
57. Ooi CY, Carter DR, Liu B, Mayoh C, Beckers A, et al. Network modeling of microRNA-mRNA interactions in neuroblastoma tumorigenesis identifies miR-204 as a direct inhibitor of MYCN. Cancer Res 2018;78:3122-34.
58. Zhao Z, Ma X, Hsiao TH, Lin G, Kosti A, et al. A high-content morphological screen identifies novel microRNAs that regulate neuroblastoma cell differentiation. Oncotarget 2014;5:2499-512.
59. Zhao Z, Ma X, Sung D, Li M, Kosti A, et al. MicroRNA-449a functions as a tumor suppressor in neuroblastoma through inducing cell differentiation and cell cycle arrest. RNA Biol 2015;12:538-54.
60. Zhao Z, Ma X, Shelton SD, Sung DC, Li M, et al. A combined gene expression and functional study reveals the crosstalk between N-Myc and differentiation-inducing microRNAs in neuroblastoma cells. Oncotarget 2016;7:79372-87.
61. Zhao Z, Partridge V, Sousares M, Shelton SD, Holland CL, et al. MicroRNA-2110 functions as an onco-suppressor in neuroblastoma by directly targeting Tsukushi. PLoS One 2018;13:e0208777.
62. Stallings RL. MicroRNA involvement in the pathogenesis of neuroblastoma: potential for microRNA mediated therapeutics. Curr Pharm Des 2009;15:456-62.
63. Schulte JH, Horn S, Schlierf S, Schramm A, Heukamp LC, et al. MicroRNAs in the pathogenesis of neuroblastoma. Cancer Lett 2009;274:10-5.
64. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003;3:203-16.
65. Tonini GP, Romani M. Genetic and epigenetic alterations in neuroblastoma. Cancer Lett 2003;197:69-73.
66. Chen Y, Stallings RL. Differential patterns of microRNA expression in neuroblastoma are correlated with prognosis, differentiation, and apoptosis. Cancer Res 2007;67:976-83.
67. Buechner J, Einvik C. N-myc and noncoding RNAs in neuroblastoma. Mol Cancer Res 2012;10:1243-53.
68. Bray I, Bryan K, Prenter S, Buckley PG, Foley NH, et al. Widespread dysregulation of MiRNAs by MYCN amplification and chromosomal imbalances in neuroblastoma: association of miRNA expression with survival. PLoS One 2009;4:e7850.
69. Das S, Bryan K, Buckley PG, Piskareva O, Bray IM, et al. Modulation of neuroblastoma disease pathogenesis by an extensive network of epigenetically regulated microRNAs. Oncogene 2013;32:2927-36.
70. Romania P, Bertaina A, Bracaglia G, Locatelli F, Fruci D, et al. Epigenetic deregulation of microRNAs in rhabdomyosarcoma and neuroblastoma and translational perspectives. Int J Mol Sci 2012;13:16554-79.
71. Parodi F, Carosio R, Ragusa M, Di Pietro C, Maugeri M, et al. Epigenetic dysregulation in neuroblastoma: a tale of miRNAs and DNA methylation. Biochim Biophys Acta 2016;1859:1502-14.
72. He XY, Tan ZL, Mou Q, Liu FJ, Liu S, et al. MicroRNA-221 enhances MYCN via targeting nemo-like kinase and functions as an oncogene related to poor prognosis in neuroblastoma. Clin Cancer Res 2017;23:2905-18.
73. Zhao D, Tian Y, Li P, Wang L, Xiao A, et al. MicroRNA-203 inhibits the malignant progression of neuroblastoma by targeting Sam68. Mol Med Rep 2015;12:5554-60.
74. Xiang X, Mei H, Zhao X, Pu J, Li D, et al. miRNA-337-3p suppresses neuroblastoma progression by repressing the transcription of matrix metalloproteinase 14. Oncotarget 2015;6:22452-66.
75. Xiang X, Mei H, Qu H, Zhao X, Li D, et al. miRNA-584-5p exerts tumor suppressive functions in human neuroblastoma through repressing transcription of matrix metalloproteinase 14. Biochim Biophys Acta 2015;1852:1743-54.
76. Meng H, Huang Q, Zhang X, Huang J, Shen R, et al. MiR-449a regulates the cell migration and invasion of human non-small cell lung carcinoma by targeting ADAM10. Onco Targets Ther 2019;12:3829-38.
77. Cheng J, Wu LM, Deng XS, Wu J, Lv Z, et al. MicroRNA-449a suppresses hepatocellular carcinoma cell growth via G1 phase arrest and the HGF/MET c-Met pathway. Hepatobiliary Pancreat Dis Int 2018;17:336-44.
78. Li F, Liang J, Bai L. MicroRNA-449a functions as a tumor suppressor in pancreatic cancer by the epigenetic regulation of ATDC expression. Biomed Pharmacother 2018;103:782-9.
79. Xu B, Zhang X, Wang S, Shi B. MiR-449a suppresses cell migration and invasion by targeting PLAGL2 in breast cancer. Pathol Res Pract 2018;214:790-5.
80. Li Y, Jiang T, Shao L, Liu Y, Zheng C, et al. Mir-449a, a potential diagnostic biomarker for WNT group of medulloblastoma. J Neurooncol 2016;129:423-31.
81. Mao A, Liu Y, Wang Y, Zhao Q, Zhou X, et al. miR-449a enhances radiosensitivity through modulating pRb/E2F1 in prostate cancer cells. Tumour Biol 2016;37:4831-40.
82. Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 2008;6:14.
83. Ko HY, Lee J, Lee YS, Choi Y, Ali BA, et al. Bioimaging of transcriptional activity of microRNA124a during neurogenesis. Biotechnol Lett 2015;37:2333-40.
84. Choi KH, Shin CH, Lee WJ, Ji H, Kim HH. Dual-strand tumor suppressor miR-193b-3p and -5p inhibit malignant phenotypes of lung cancer by suppressing their common targets. Biosci Rep 2019;39:BSR20190634.
85. Hashemi ZS, Forouzandeh Moghadam M, Khalili S, Ghavami M, Salimi F, et al. Additive effect of metastamiR-193b and breast cancer metastasis suppressor 1 as an anti-metastatic strategy. Breast Cancer 2019;26:215-28.
86. Bhayadia R, Krowiorz K, Haetscher N, Jammal R, Emmrich S, et al. Endogenous tumor suppressor microRNA-193b: therapeutic and prognostic value in acute myeloid leukemia. J Clin Oncol 2018;36:1007-16.
87. Mitra AK, Chiang CY, Tiwari P, Tomar S, Watters KM, et al. Microenvironment-induced downregulation of miR-193b drives ovarian cancer metastasis. Oncogene 2015;34:5923-32.
88. Chen J, Zhang X, Lentz C, Abi-Daoud M, Pare GC, et al. miR-193b regulates Mcl-1 in melanoma. Am J Pathol 2011;179:2162-8.
89. Roth SA, Hald OH, Fuchs S, Lokke C, Mikkola I, et al. MicroRNA-193b-3p represses neuroblastoma cell growth via downregulation of Cyclin D1, MCL-1 and MYCN. Oncotarget 2018;9:18160-79.
90. Zhang H, Pu J, Qi T, Qi M, Yang C, et al. MicroRNA-145 inhibits the growth, invasion, metastasis and angiogenesis of neuroblastoma cells through targeting hypoxia-inducible factor 2 alpha. Oncogene 2014;33:387-97.
91. Ye D, Zhou C, Deng H, Lin L, Zhou S. MicroRNA-145 inhibits growth of laryngeal squamous cell carcinoma by targeting the PI3K/Akt signaling pathway. Cancer Manag Res 2019;11:3801-12.
92. Zhang XF, Zhang XQ, Chang ZX, Wu CC, Guo H. MicroRNA145 modulates migration and invasion of bladder cancer cells by targeting Ncadherin. Mol Med Rep 2018;17:8450-6.
93. Lin Y, Ge X, Wen Y, Shi ZM, Chen QD, et al. MiRNA-145 increases therapeutic sensibility to gemcitabine treatment of pancreatic adenocarcinoma cells. Oncotarget 2016;7:70857-68.
94. Chen Y, Sun Y, Rao Q, Xu H, Li L, et al. Androgen receptor (AR) suppresses miRNA-145 to promote renal cell carcinoma (RCC) progression independent of VHL status. Oncotarget 2015;6:31203-15.
95. Noguchi S, Mori T, Hoshino Y, Yamada N, Nakagawa T, et al. Comparative study of anti-oncogenic microRNA-145 in canine and human malignant melanoma. J Vet Med Sci 2012;74:1-8.
96. Chen Z, Zeng H, Guo Y, Liu P, Pan H, et al. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res 2010;29:151.
97. Wan X, Cheng Q, Peng R, Ma Z, Chen Z, et al. ROCK1, a novel target of miR-145, promotes glioma cell invasion. Mol Med Rep 2014;9:1877-82.
98. Lee JJ, Drakaki A, Iliopoulos D, Struhl K. MiR-27b targets PPARgamma to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene 2012;31:3818-25.
99. Althoff K, Lindner S, Odersky A, Mestdagh P, Beckers A, et al. miR-542-3p exerts tumor suppressive functions in neuroblastoma by downregulating Survivin. Int J Cancer 2015;136:1308-20.
100. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008;27:5204-13.
101. Foley NH, Bray IM, Tivnan A, Bryan K, Murphy DM, et al. MicroRNA-184 inhibits neuroblastoma cell survival through targeting the serine/threonine kinase AKT2. Mol Cancer 2010;9:83.
102. Stallings RL, Foley NH, Bryan K, Buckley PG, Bray I. Therapeutic targeting of miRNAs in neuroblastoma. Expert Opin Ther Targets 2010;14:951-62.
103. Tivnan A, Tracey L, Buckley PG, Alcock LC, Davidoff AM, et al. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer 2011;11:33.
104. Chen Y, Tsai YH, Tseng SH. Inhibition of cyclin-dependent kinase 1-induced cell death in neuroblastoma cells through the microRNA-34a-MYCN-survivin pathway. Surgery 2013;153:4-16.
105. Terrile M, Bryan K, Vaughan L, Hallsworth A, Webber H, et al. miRNA expression profiling of the murine TH-MYCN neuroblastoma model reveals similarities with human tumors and identifies novel candidate miRNAs. PLoS One 2011;6:e28356.
106. Lynch J, Fay J, Meehan M, Bryan K, Watters KM, et al. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-beta signalling pathway. Carcinogenesis 2012;33:976-85.
107. Zhang H, Qi M, Li S, Qi T, Mei H, et al. MicroRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol Cancer Ther 2012;11:1454-66.
108. Chio CC, Lin JW, Cheng HA, Chiu WT, Wang YH, et al. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch Toxicol 2013;87:459-68.
109. Li Y, Wang H, Li J, Yue W. MiR-181c modulates the proliferation, migration, and invasion of neuroblastoma cells by targeting Smad7. Acta Biochim Biophys Sin (Shanghai) 2014;46:48-55.
110. Maugeri M, Barbagallo D, Barbagallo C, Banelli B, Di Mauro S, et al. Altered expression of miRNAs and methylation of their promoters are correlated in neuroblastoma. Oncotarget 2016;7:83330-41.
111. Lodrini M, Oehme I, Schroeder C, Milde T, Schier MC, et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res 2013;41:6018-33.
112. Lodrini M, Poschmann G, Schmidt V, Wunschel J, Dreidax D, et al. Minichromosome maintenance complex is a critical node in the miR-183 signaling network of MYCN-amplified neuroblastoma cells. J Proteome Res 2016;15:2178-86.
113. Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 2011;469:216-20.
114. Saeki N, Saito A, Sugaya Y, Amemiya M, Sasaki H. Indirect down-regulation of tumor-suppressive let-7 family microRNAs by LMO1 in neuroblastoma. Cancer Genomics Proteomics 2018;15:413-20.
115. Powers JT, Tsanov KM, Pearson DS, Roels F, Spina CS, et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 2016;535:246-51.
116. Soriano A, Masanas M, Boloix A, Masia N, Paris-Coderch L, et al. Functional high-throughput screening reveals miR-323a-5p and miR-342-5p as new tumor-suppressive microRNA for neuroblastoma. Cell Mol Life Sci 2019;76:2231-43.
117. Beckers A, Van Peer G, Carter DR, Gartlgruber M, Herrmann C, et al. MYCN-driven regulatory mechanisms controlling LIN28B in neuroblastoma. Cancer Lett 2015;366:123-32.
118. Chen X, Pan M, Han L, Lu H, Hao X, et al. miR-338-3p suppresses neuroblastoma proliferation, invasion and migration through targeting PREX2a. FEBS Lett 2013;587:3729-37.
119. Raghunath M, Patti R, Bannerman P, Lee CM, Baker S, et al. A novel kinase, AATYK induces and promotes neuronal differentiation in a human neuroblastoma (SH-SY5Y) cell line. Brain Res Mol Brain Res 2000;77:151-62.
120. Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res 2004;14:1902-10.
121. Wu T, Lin Y, Xie Z. MicroRNA-1247 inhibits cell proliferation by directly targeting ZNF346 in childhood neuroblastoma. Biol Res 2018;51:13.
122. Li SH, Li JP, Chen L, Liu JL. miR-146a induces apoptosis in neuroblastoma cells by targeting BCL11A. Med Hypotheses 2018;117:21-7.
123. Feinberg-Gorenshtein G, Guedj A, Shichrur K, Jeison M, Luria D, et al. MiR-192 directly binds and regulates Dicer1 expression in neuroblastoma. PLoS One 2013;8:e78713.
124. Qu H, Zheng L, Pu J, Mei H, Xiang X, et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Hum Mol Genet 2015;24:2539-51.
125. Qu H, Zheng L, Song H, Jiao W, Li D, et al. MicroRNA-558 facilitates the expression of hypoxia-inducible factor 2 alpha through binding to 5’-untranslated region in neuroblastoma. Oncotarget 2016;7:40657-73.
126. Liu G, Xu Z, Hao D. MicroRNA451 inhibits neuroblastoma proliferation, invasion and migration by targeting macrophage migration inhibitory factor. Mol Med Rep 2016;13:2253-60.
127. Li Z, Xu Z, Xie Q, Gao W, Xie J, et al. miR-1303 promotes the proliferation of neuroblastoma cell SH-SY5Y by targeting GSK3beta and SFRP1. Biomed Pharmacother 2016;83:508-13.
128. Hsu DM, Agarwal S, Benham A, Coarfa C, Trahan DN, et al. G-CSF receptor positive neuroblastoma subpopulations are enriched in chemotherapy-resistant or relapsed tumors and are highly tumorigenic. Cancer Res 2013;73:4134-46.
129. Liu X, Peng H, Liao W, Luo A, Cai M, et al. MiR-181a/b induce the growth, invasion, and metastasis of neuroblastoma cells through targeting ABI1. Mol Carcinog 2018;57:1237-50.
130. Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med 2010;16:1134-40.
131. De Preter K, Mestdagh P, Vermeulen J, Zeka F, Naranjo A, et al. miRNA expression profiling enables risk stratification in archived and fresh neuroblastoma tumor samples. Clin Cancer Res 2011;17:7684-92.
132. Chen Y, Tsai YH, Fang Y, Tseng SH. Micro-RNA-21 regulates the sensitivity to cisplatin in human neuroblastoma cells. J Pediatr Surg 2012;47:1797-805.
133. Buechner J, Henriksen JR, Haug BH, Tomte E, Flaegstad T, et al. Inhibition of mir-21, which is up-regulated during MYCN knockdown-mediated differentiation, does not prevent differentiation of neuroblastoma cells. Differentiation 2011;81:25-34.
134. Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 2008;3:e2236.
135. Buhagiar A, Ayers D. Chemoresistance, cancer stem cells, and miRNA influences: the case for neuroblastoma. Anal Cell Pathol (Amst) 2015;2015:150634.
136. Ribeiro D, Klarqvist MDR, Westermark UK, Oliynyk G, Dzieran J, et al. Regulation of nuclear hormone receptors by MYCN-driven miRNAs impacts neural differentiation and survival in neuroblastoma patients. Cell Rep 2016;16:979-93.
137. Takwi AA, Wang YM, Wu J, Michaelis M, Cinatl J, et al. miR-137 regulates the constitutive androstane receptor and modulates doxorubicin sensitivity in parental and doxorubicin-resistant neuroblastoma cells. Oncogene 2014;33:3717-29.
138. Sun F, Fu H, Liu Q, Tie Y, Zhu J, et al. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett 2008;582:1564-8.
139. Tivnan A, Orr WS, Gubala V, Nooney R, Williams DE, et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS One 2012;7:e38129.
140. Marengo B, Monti P, Miele M, Menichini P, Ottaggio L, et al. Etoposide-resistance in a neuroblastoma model cell line is associated with 13q14.3 mono-allelic deletion and miRNA-15a/16-1 down-regulation. Sci Rep 2018;8:13762.
141. Ryan J, Tivnan A, Fay J, Bryan K, Meehan M, et al. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br J Cancer 2012;107:967-76.
142. Arevalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci 2006;63:1523-37.
143. Sun YX, Yang J, Wang PY, Li YJ, Xie SY, et al. Cisplatin regulates SH-SY5Y cell growth through downregulation of BDNF via miR-16. Oncol Rep 2013;30:2343-9.
144. Creevey L, Ryan J, Harvey H, Bray IM, Meehan M, et al. MicroRNA-497 increases apoptosis in MYCN amplified neuroblastoma cells by targeting the key cell cycle regulator WEE1. Mol Cancer 2013;12:23.
145. Bhavsar SP, Lokke C, Flaegstad T, Einvik C. Hsa-miR-376c-3p targets Cyclin D1 and induces G1-cell cycle arrest in neuroblastoma cells. Oncol Lett 2018;16:6786-94.
146. Ayers D, Mestdagh P, Van Maerken T, Vandesompele J. Identification of miRNAs contributing to neuroblastoma chemoresistance. Comput Struct Biotechnol J 2015;13:307-19.
147. Zhi F, Wang R, Wang Q, Xue L, Deng D, et al. MicroRNAs in neuroblastoma: small-sized players with a large impact. Neurochem Res 2014;39:613-23.
148. Tivnan A, Foley NH, Tracey L, Davidoff AM, Stallings RL. MicroRNA-184-mediated inhibition of tumour growth in an orthotopic murine model of neuroblastoma. Anticancer Res 2010;30:4391-5.
149. Chakrabarti M, Khandkar M, Banik NL, Ray SK. Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res 2012;1454:1-13.
150. Harvey H, Piskareva O, Creevey L, Alcock LC, Buckley PG, et al. Modulation of chemotherapeutic drug resistance in neuroblastoma SK-N-AS cells by the neural apoptosis inhibitory protein and miR-520f. Int J Cancer 2015;136:1579-88.
151. Challagundla KB, Wise PM, Neviani P, Chava H, Murtadha M, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst 2015;107:djv135.
152. Haug BH, Hald OH, Utnes P, Roth SA, Lokke C, et al. Exosome-like extracellular vesicles from MYCN-amplified neuroblastoma cells contain oncogenic miRNAs. Anticancer Res 2015;35:2521-30.
153. Reynolds CP, Lemons RS. Retinoid therapy of childhood cancer. Hematol Oncol Clin North Am 2001;15:867-910.
154. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985;313:404-6.
155. Truckenmiller ME, Vawter MP, Cheadle C, Coggiano M, Donovan DM, et al. Gene expression profile in early stage of retinoic acid-induced differentiation of human SH-SY5Y neuroblastoma cells. Restor Neurol Neurosci 2001;18:67-80.
156. Laneve P, Di Marcotullio L, Gioia U, Fiori ME, Ferretti E, et al. The interplay between microRNAs and the neurotrophin receptor tropomyosin-related kinase C controls proliferation of human neuroblastoma cells. Proc Natl Acad Sci U S A 2007;104:7957-62.
157. Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J Biol Chem 2011;286:4150-64.
158. Evangelisti C, Florian MC, Massimi I, Dominici C, Giannini G, et al. MiR-128 up-regulation inhibits Reelin and DCX expression and reduces neuroblastoma cell motility and invasiveness. FASEB J 2009;23:4276-87.
159. Guidi M, Muinos-Gimeno M, Kagerbauer B, Marti E, Estivill X, et al. Overexpression of miR-128 specifically inhibits the truncated isoform of NTRK3 and upregulates BCL2 in SH-SY5Y neuroblastoma cells. BMC Mol Biol 2010;11:95.
160. Roth SA, Knutsen E, Fiskaa T, Utnes P, Bhavsar S, et al. Next generation sequencing of microRNAs from isogenic neuroblastoma cell lines isolated before and after treatment. Cancer Lett 2016;372:128-36.
161. Nallasamy P, Chava S, Verma SS, Mishra S, Gorantla S, et al. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: immuno-oncology perspective. Semin Cancer Biol 2018;52:53-65.
162. Verissimo CS, Molenaar JJ, Fitzsimons CP, Vreugdenhil E. Neuroblastoma therapy: what is in the pipeline? Endocr Relat Cancer 2011;18:R213-31.
163. Ramachandran M, Yu D, Dyczynski M, Baskaran S, Zhang L, et al. Safe and effective treatment of experimental neuroblastoma and glioblastoma using systemically delivered triple microRNA-detargeted oncolytic semliki forest virus. Clin Cancer Res 2017;23:1519-30.
164. Du X, Wang H, Xu F, Huang Y, Liu Z, et al. Enterovirus 71 induces apoptosis of SHSY5Y human neuroblastoma cells through stimulation of endogenous microRNA let-7b expression. Mol Med Rep 2015;12:953-9.
165. Mundalil Vasu M, Anitha A, Takahashi T, Thanseem I, Iwata K, et al. Fluoxetine increases the expression of miR-572 and miR-663a in human neuroblastoma cell lines. PLoS One 2016;11:e0164425.
166. Sousares M, Partridge V, Weigum S, Du L. MicroRNAs in neuroblastoma differentiation and differentiation therapy. Adv Mod Oncol Res 2017;3:213-22.
167. Curtin C, Nolan JC, Conlon R, Deneweth L, Gallagher C, et al. A physiologically relevant 3D collagen-based scaffold-neuroblastoma cell system exhibits chemosensitivity similar to orthotopic xenograft models. Acta Biomater 2018;70:84-97.
168. Wang X, Li J, Xu X, Zheng J, Li Q. miR-129 inhibits tumor growth and potentiates chemosensitivity of neuroblastoma by targeting MYO10. Biomed Pharmacother 2018;103:1312-8.
169. Ramaiah MJ, Pushpavalli SN, Lavanya A, Bhadra K, Haritha V, et al. Novel anthranilamide-pyrazolo[1,5-a]pyrimidine conjugates modulate the expression of p53-MYCN associated micro RNAs in neuroblastoma cells and cause cell cycle arrest and apoptosis. Bioorg Med Chem Lett 2013;23:5699-706.
170. Ren X, Bai X, Zhang X, Li Z, Tang L, et al. Quantitative nuclear proteomics identifies that miR-137-mediated EZH2 reduction regulates resveratrol-induced apoptosis of neuroblastoma cells. Mol Cell Proteomics 2015;14:316-28.
171. Vanhauwaert S, Decaesteker B, De Brouwer S, Leonelli C, Durinck K, et al. In silico discovery of a FOXM1 driven embryonal signaling pathway in therapy resistant neuroblastoma tumors. Sci Rep 2018;8:17468.
172. Gonzalez-Nunez V, Noriega-Prieto JA, Rodriguez RE. Morphine modulates cell proliferation through mir133b &mir128 in the neuroblastoma SH-SY5Y cell line. Biochim Biophys Acta 2014;1842:566-72.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Aravindan N, Subramanian K, Somasundaram DB, Herman TS, Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist 2019;2:1086-105. http://dx.doi.org/10.20517/cdr.2019.68
AMA Style
Aravindan N, Subramanian K, Somasundaram DB, Herman TS, Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resistance. 2019; 2(4): 1086-105. http://dx.doi.org/10.20517/cdr.2019.68
Chicago/Turabian Style
Aravindan, Natarajan, Karthikeyan Subramanian, Dinesh Babu Somasundaram, Terence S. Herman, Sheeja Aravindan. 2019. "MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution" Cancer Drug Resistance. 2, no.4: 1086-105. http://dx.doi.org/10.20517/cdr.2019.68
ACS Style
Aravindan, N.; Subramanian K.; Somasundaram DB.; Herman TS.; Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist. 2019, 2, 1086-105. http://dx.doi.org/10.20517/cdr.2019.68
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 32
likes
Like This Article 32
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.