
Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors

Abstract
Testicular germ cell tumors (TGCTs) are a cancer pharmacology success story with a majority of patients cured even in the highly advanced and metastatic setting. Successful treatment of TGCTs is primarily due to the exquisite responsiveness of this solid tumor to cisplatin-based therapy. However, a significant percentage of patients are, or become, refractory to cisplatin and die from progressive disease. Mechanisms for both clinical hypersensitivity and resistance have largely remained a mystery despite the promise of applying lessons to the majority of solid tumors that are not curable in the metastatic setting. Recently, this promise has been heightened by the realization that distinct (and perhaps pharmacologically replicable) epigenetic states, rather than fixed genetic alterations, may play dominant roles in not only TGCT etiology and progression but also their curability with conventional chemotherapies. In this review, it discusses potential mechanisms of TGCT cisplatin sensitivity and resistance to conventional chemotherapeutics.
Keywords
Introduction
Testicular germ cell tumors (TGCTs) are the most common neoplasm in men age 15 to 44 years old with increasing incidence in the last 40 years[1]. Metastatic TGCTs have been a model for transforming a once fatal metastatic solid tumor into one that is curable. The discovery in the mid-70s that TGCTs are sensitive to conventional cisplatin-based combination chemotherapy eventually resulted in an improvement in 5-year survival from less than 10% to over 80% even in patients with disseminated disease[2,3]. However, 15%-20% of all patients and 50% of poor risk patients are refractory to treatment and only 50% of patients can be cured after relapse with salvage therapy consisting of high dose platinum followed by stem cell transplant or traditional chemotherapies[4-6]. Most patients progressing after high dose therapy will succumb to their disease as will those patients who undergo late relapse[6,7]. Attempts to incorporate newer targeted therapies to treat refractory TGCTs have been unsuccessful. An average of nearly 40 years of life are lost when a patient dies from testicular cancer, over a decade more than any other adult malignancy[8,9]. Further, TGCT patients successfully treated with cisplatin-based therapies suffer from acute and life-long toxicities including infertility, hypogonadism, androgen deficiency, decreased lung and kidney function and neurotoxicity and have a greater risk of developing cardiovascular disease and secondary malignancies[10]. Hence, there is a pressing clinical need to devise new strategies to treat cisplatin refractory TGCTs and a rationale to devise targeted, cisplatin-sparing therapies.
TGCTs remain the only solid malignancy curable with chemotherapy. A greater understanding of the hypersensitivity and resistance of TGCTs has the potential to not only impact refractory patients but also may inform strategies to sensitize other solid tumors to conventional chemotherapies[11,12]. TGCTs are classified based on histology into two distinct subtypes seminomas and nonseminomas. Both seminomas and nonseminomas likely arise from precursor cells called germ cell neoplasia in situ (GCNIS)[13-15]. Nonseminomas can be further classified as embryonal carcinoma (EC), teratoma, yolk sac tumor and choriocarcinoma. Pluripotent EC are the stem cell-like component of nonseminoma. TGCTs and EC have an intrinsic hypersensitivity to drug-induced cell death[16,17]. The basis of this hypersensitivity and mechanisms to account for chemotherapy resistance remain elusive. TGCTs likely represent transformed germ cells and may have inherited unique mechanisms of sensitivity to DNA damage and other stress to prevent germline mutations. Alterations in traditional mechanisms of cisplatin sensitivity and resistance in other solid tumors have generally not been accepted as the reasons for the hypersensitivity of TGCTs[3,12,18].
TGCTs have one of the lowest overall somatic mutation rates of all solid tumors and possess unique epigenetic states that are likely a reflection of the epigenetics of their primordial germ cell (PGC) origins[19,20]. Further EC and other pluripotent cells are known to undergo extensive plasticity in tumorigenicity depending on the environment[21,22]. These findings suggest that epigenetics may play a larger role in the chemotherapeutic hypersensitivity and resistance of TGCTs. Recently genetic susceptibility, biological signaling, and genetic and environmental factors have been examined to explain the mechanisms responsible for TGCTs curability, pathogenesis and development of treatment resistance. This review will focus on potential mechanisms of cisplatin resistance in TGCTs that may relate to the exquisite sensitivity of this solid tumor to conventional chemotherapeutics that may provide strategies to overcome acquired cisplatin resistance.
Etiology and pathology of TGCTs
The incidence of TGCTs has been steadily rising in young males[9]. According to the 2018 WHO global cancer observatory, TGCTs are estimated to have the largest number of new cases among all cancers for males under the age of 34 years in the USA and second largest worldwide [Figure 1][23]. The incidence of TGCTs varies widely with geographic location with the highest incidence in northern European countries and the lowest in Africa nations [Figure 2]. The reasons for these differences is not known but is hypothesized to be a combination of inheritable and environmental factors including exposure to endocrine disruptors in utero. Common risks for TGCTs include endocrine disruptors, cryptorchidism[24], Down’s syndrome[25], family history of cancer[26], low birth weight, premature birth, birth order, bleeding during pregnancy, hormone exposure during pregnancy, high maternal age and neonatal jaundice[27,28]. Late age at puberty, male infertility and testicular dysgenesis syndrome are also associated with TGCTs[29-33].
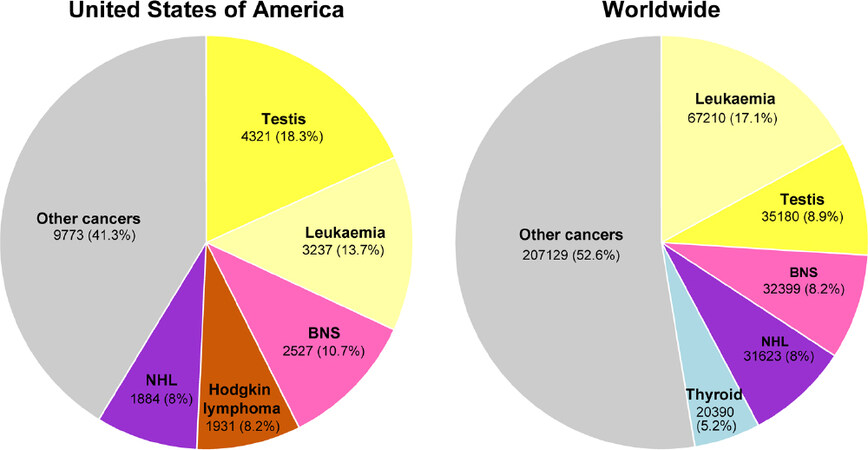
Figure 1. Estimated new cancers in males aged 0-34 in the USA and worldwide for 2018. Pie charts represent the distribution of new cancer cases in the United States and worldwide for males 0-34 years of age. Data source: World Health Organization global cancer observatory.
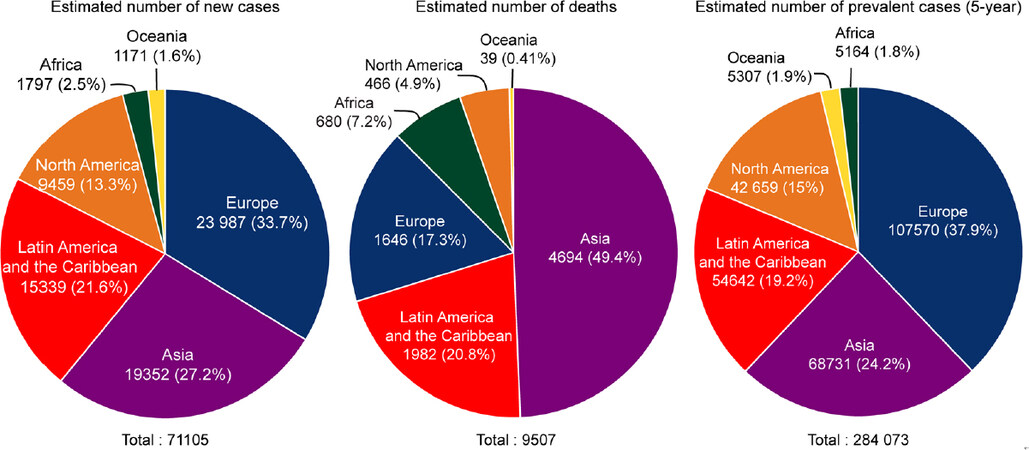
Figure 2. Estimated number of new TGCT cases, deaths and prevalence worldwide for 2018. Pie charts represent the distribution of new TGCT cases, TGCT deaths and TGCT prevalence worldwide for all aged males. Data source: WHO global cancer observatory. TGCTs: testicular germ cell tumors
EC cells resemble embryonic stem (ES) cells[22]. Both cell types possess pluripotency and have close similarities in gene expression[34,35]. Interestingly, despite similarities, EC cells are malignant, while ES cells are not. GCNIS-derived TGCTs have cytogenetic and molecular anomalies that mainly include aneuploidy and gain and loss of distinct chromosomal regions coupled with somatic mutation rates that are low[19,20]. Chromosome 12p amplification, such as isochromosome 12p and chromosome 12p overrepresentation are nearly universally present and pathognomonic for TGCTs[36,37]. Other chromosomal anomalies associated with TGCTs include gain of genetic material on chromosome 1, 2p, 7, 8, 12, 14q, 15q, 17q, 21q, and X and the deletion of genetic material from chromosome 4, 5, 11q, 13q, and 18q2[38]. Similarly, multiple passaging of ES cells has been shown to result in acquired alterations similar to those found in TGCTs and EC, including chromosome 12, 17, and X gain[39]. A recent analysis performed on a 150 TGCT cohort from the TCGA[40] indicates gain/amplification of 12p in the majority of patients with gain in 12q, 8q, 22q and deletion/loss of 11q, 18q, 18p, 9p, 4q, 10q, 5q, 16q and 19q also occurring with much less frequency [Figure 3].
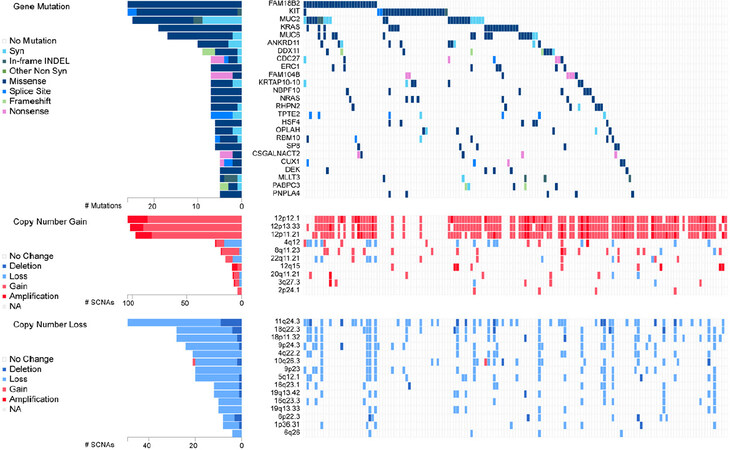
Figure 3. Genetic alteration in TGCTs. Analysis of 150 patient TGCT cohort generated through FireBrowse. Top genetic alterations are arranged by frequency. Data source: TCGA. TGCTs: testicular germ cell tumors
The GCNIS legion is considered a precursor for all TGCT subtypes. GCNIS has been hypothesized to emerge from PGCs or early gonocytes that have transformed in utero[41-43]. Evidence that TGCTs begin during development include similarities in expression of the markers KIT, OCT4, and PLAP, cell morphology, genomic imprinting and transcriptomics between GCNIS and PGCs[44,45]. Further, abnormalities of germ cell development are associated with TGCT incidence[46]. In addition extragonadal TGCTs are found at the midline, which is where PGCs migrate during development[47,48]. Taken together the data support that TGCTs come from arrested PGCs or gonocytes that lie dormant but further progress with gonadal hormones during sexual maturation[41].
Genetic drivers of TGCTs are poorly understood compared to other cancers. Several genes have been implicated in the pathogenesis of TGCTs, but their exact roles are still uncertain and underscore that TGCT etiology is likely widely polygenic in nature. Genome-wide association studies have been useful in identifying variants that contribute to risk of TGCTs. Allelic variation within the kit-ligand gene 12q22 is the strongest genetic risk factor for TGCTs[49]. Several other allelic variants have been identified that correlate with TGCTs susceptibility[45]. Somatic mutation in TGCTs are relatively rare and of relatively low frequency and occur mostly in seminoma. Gain-of function mutations in KIT are most frequent at 18%[19,20]. Other mutations in seminoma include KRAS (14%) and NRAS (4%) [Figure 3].
Epigenetics of TGCTs
There is considerable evidence that epigenetics may play a role in TGCT biology. As stated above, environmental exposures in utero and testicular dysgenesis syndromes are major risk factors for TGCTs. Further, normal ES cells are very similar in gene expression and biology to EC cells. Transformation of both of these pluripotent cell types are influenced by environmental factors exemplified by findings that ES cells form teratoma and teratocarcinomas when implanted ectopically in mice, while EC cells in certain situations can participate in normal development when placed in the early embryo[21,50]. Recent studies have characterized the pattern of TGCT DNA methylation. It is important to remember that PGCs normally undergo erasure of imprinting during development[51]. Thus, TGCTs may have arisen from a germ cell lacking imprinting. Evidence to support this comes from the finding that IGF2 and H19 are expressed from both alleles in TGCTs but are otherwise exclusively expressed from one parental allele in adult tissues[52]. TGCT genomic DNA is hypomethylated in comparison to most solid tumors. Seminomas possess very little DNA methylation while EC have an intermediate level of DNA methylation compared to somatic cancers and EC-derived teratoma are hypermethylated[20,52-55]. The level of DNA methylation roughly correlates with cisplatin curability with seminoma being the most curable followed by EC and other nonseminoma components besides teratoma. Teratoma is resistant to cisplatin treatment.
Interestingly, pluripotent cells including EC cells possess high levels of non-CpG methylation called mCpH (H = A, C, T) along gene bodies of genes that are highly expressed[52,56]. In EC up to 25% of all cytosine methylation is mCpH while over 99% of all cytosine methylation in somatic cells is CpG. Also, mCpH does not occur in the mature components of nonseminoma or in seminoma, suggesting mCpH is closely associated with the pluripotent and reprogrammed state of EC[52]. The role of non-CpG methylation in EC is currently unclear.
Histone modifications are well-established epigenetic regulators. Polycomb complexes modify chromatin to repress homeotic genes which have an important role in stem cell dynamics[57,58]. Gene repression is initiated by polycomb repression complex 2 which contains histone methyltransferase activity resulting in histone H3 trimethylation on lysine 27 (H3K27me3). The H3K27me3 then recruits PRC complex 1 to further mediate repression of gene expression. Alterations in BMI1 and EZH2 and other polycomb components frequently occurs in cancer[59,60]. Little is known concerning alterations in histone modifiers in TGCTs[61,62]. The H3K4me demethylase, LSD1 has been shown to be a key regulator of pluripotency in ES and EC cells. LSD1 has been reported to be highly expressed in EC and seminoma which correlates with LSD1 and HDAC1 inhibitor sensitivity[63,64].
Another form of epigenetic regulation recently studied in TGCTs is mediated by miRNAs. Voorhoeve et al.[65], showed that in TGCT cells miR-372 and miR-373 neutralized p53 function through inhibition of the tumor suppressor LATS2. Several miRNAs appear to be specifically expressed in TGCTs and have been investigated as potential new serum biomarkers for TGCT patients[66]. For example, miR-371a-3p was reported to have high sensitivity and specificity for detecting TGCT disease burden and was not elevated in control patients or patients with other cancer types[66,67]. Serum levels of miR-371a-3p increased with TGCT progression and decline after surgery[66,67]. Since TGCTs appear to have distinct epigenetics compared to normal tissues and other cancers, the use of epigenetic biomarkers may be particularly useful for disease management of TGCT patients.
Mechanism of cisplatin resistance in TGCTs
Several principles of solid tumor chemotherapy were validated during the optimization of curative TGCTs regimens. Seminomas are very sensitive to cisplatin-based therapy and are highly curable regardless of stage[2]. Nonseminoma are somewhat less sensitive to chemotherapy especially for tumors of more advanced grade and stage[2]. For advanced TGCTs, bleomycin, etoposide and cisplatin or etoposide, ifosfamide and cisplatin (VIP) are the first line standards of care. The International Germ Cell Cancer Collaborative Group devised a risk classification based on serum markers and primary and metastatic sites that divided metastatic nonseminoma patients into good, intermediate, and poor risk with cure rates of 90%, 75% and 50%, respectively[68]. Currently there are no predictive biomarkers to identify which poor-risk patients will die from disease. There is a clinical need to better identify which high risk patients will fail conventional therapy. Salvage high-dose chemotherapy with stem cell rescue or conventional dose chemotherapy can induce remissions in approximately 25% of relapse cases[69]. The treatment of refractory multi-relapsing disease is very challenging and most cases cannot be cured. Targeted approaches including tyrosine-kinase inhibitors and immunotherapy have failed to demonstrate activity in these refractory cases. Mechanisms of hypersensitivity and acquired resistance in TGCTs are likely multi-factorial and could include cellular detoxification, altered platinum accumulation, DNA repair, and alterations in apoptotic pathways[12,18,70]. However, these mechanisms are shared by many solid tumors that are not curable. Further TGCTs are highly sensitive to a variety of agents that do not share common import and export pathways and mediate different forms of DNA damage repaired by distinct mechanisms. Hence, mechanisms responsible for TGCT sensitivity and resistance may be related to how these tumors respond to damaged DNA mediated in part by their unique cellular context[3,71]. In the next section, we will review suggested mechanisms of therapy resistance in TGCTs.
Cisplatin resistance mechanisms can be classified as pre-target, on-target, and post-target[70]. Pre-target involve alterations preceding the binding of cisplatin to DNA, on-target resistance involves alterations directly relate to DNA-cisplatin adducts, post-target resistance involves mechanisms downstream of cisplatin-mediated DNA damage [Figure 4]. In this section we will discuss these cisplatin resistance mechanisms.
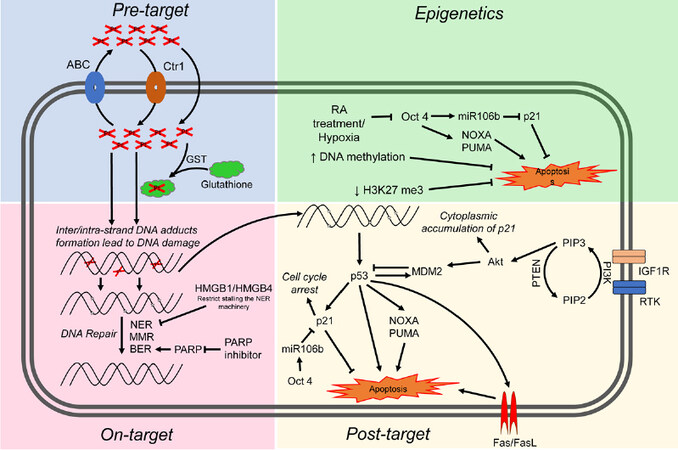
Figure 4. Proposed mechanisms mediating cisplatin sensitivity and resistance in TGCTs. Cisplatin resistance mechanisms are classified as pre-target, on-target, post-target and epigenetic mechanisms. ABC: atp-binding cassette transporters; GST: glutathione s-transferase; NER: nucleotide excision repair; MMR: mismatch repair; BER: base excision repair; RA: retinoic acid; RTK: receptor tyrosine kinase; TGCTs: testicular germ cell tumors
Pre-target resistance
Before cisplatin binds DNA, cancer cells can avoid cytotoxicity by two main mechanisms: first, decreased cellular accumulation of cisplatin and second, enhanced cisplatin detoxification by glutathione, metallothioneins and nucleophilic cytoplasmic “scavengers”[70]. Cellular uptake of cisplatin is typically mediated by passive diffusion or facilitated transport, although the copper transporter SLC31A1 has also been implicated as a cisplatin transport mechanism[72]. Cisplatin uptake rates in TGCT and other cancer cells have been reported to be similar[73]. A number of ATP-binding cassette transporters including MDR1 have been linked to cisplatin efflux[70]. However, no cisplatin resistance mechanism related to drug transporters has been reported for TGCTs.
Resistance to cisplatin is often associated with increased levels of thiol-containing proteins such as glutathione and metallothionein which detoxify cisplatin through conjugation. In general it appears that the level of these detoxifiers are low in TGCTs compared to some other tumor types[74]. Interestingly, one study showed high levels of a glutathione-S-transferase, an enzyme that conjugates GSH in resistant teratoma[75]. These finding suggest that low levels of exporters and detoxifiers may play at least a contributory role in TGCT sensitivity to cisplatin. However, in general pre-target mechanisms have not been widely recognized as significant sources of cisplatin resistance in TGCTs.
On-target resistance
Inter/intra-strand DNA adducts and subsequent apoptosis can be defective in cisplatin-resistant cancer cells due to a variety of mechanisms. TGCTs have been shown to have similar rates of cisplatin DNA adduct formation upon initial exposure compared to other tumor types[71,74,76]. However, other reports have implied that TGCTs possess a diminished capacity to repair cisplatin adducts[77,78]. Correspondingly, cisplatin-resistant cells may have acquired the ability to repair adducts at an enhanced rate or gained the ability to tolerate unrepaired lesions. The majority of cisplatin DNA lesions are repaired by the nucleotide excision repair (NER) system[79,80]. One proposed mechanism for cisplatin resistance involves high expression of the high-mobility group box protein 1 (HMGB1) in TGCT cells[81]. HMGB1 appears to bind selectively to cisplatin DNA crosslinks and interferes with NER[82-85]. Similar to HMGB1, HMGB4 is also highly expressed in TGCTs and likewise recognizes cisplatin-DNA intrastrand cross-links, stalling the NER machinery which otherwise would excise and repair the damage[81]. Hence, one proposed mechanism to explain the chemosensitivity of TGCT is decreased capacity to repair interstrand platinum-DNA adducts, the most toxic from of cisplatin induced adducts by either excision repair or homologous recombination. We refer the reader to an excellent recent review on this topic[86].
Several studies have linked defective mismatch repair (MMR), microsatellite instability and BRAF mutations with relapse and treatment failure in TGCT patients[87-90]. Refractory TGCTs have been shown to have decreased expression of MMR genes including MLH1, MLH2, or MSH6, suggesting that decreased MMR may be a mechanism of TGCT resistance[89]. However, the precise molecular mechanisms are unclear. Future studies are needed to solidify the exact role of MMR in cisplatin resistance in TGCTs. Poly (ADP-ribose) polymerase (PARP) is involved in the repair of DNA single-strand breaks via base excision repair (BER), a key cellular DNA repair pathway. Mego and colleagues observed overexpression of PARP in TGCTs compared to normal testicular tissue and PARP inhibitors were shown to reverse cisplatin resistance[91,92].
Post-target resistance
Post-target resistance to cisplatin can result from alterations in signal transduction pathways that mediate apoptosis in response to DNA damage. Non-repairable cisplatin-induced DNA damage leads to the activation of a multi-branched signaling cascade with proapoptotic outcomes. A distinguishing feature of TGCTs is that they are devoid of p53 mutations compared to almost all other solid tumors[19,20]. Several groups have provided evidence that hyperactivation of p53 is a key mechanism for the hypersensitivity of TGCTs to cisplatin[93-97]. However, the role of p53 mutations in cisplatin resistance of TGCTs has been controversial[3,96]. Mutations in p53 and overexpression of MDM2 do appear to occur in a proportion of cisplatin refractory TGCTs but the extent of these mutations is unclear and will require studies with larger numbers of cisplatin refractory cases[98,99]. High abundance of MDM2 has been shown to correlate with cisplatin resistance in TGCT cell lines and disruption of the MDM2-p53 interaction by nutlin-3 strongly potentiated p53-dependent apoptosis via the Fas/FasL pathway[100]. NOXA and PUMA, downstream targets of p53 and p53 family members p63 and p73, have been linked to the cisplatin sensitivity of TGCTs[95,101,102]. Overexpression of the cell cycle regulator CCND1 has been described as a mechanism of cisplatin resistance[103]. Upregulation of IGF1R expression and signaling was also found to contribute to acquired cisplatin resistance in an in vitro nonseminoma model[104].
Di Vizio et al.[105] provided evidence that the PDGFR/PI3K/AKT pathway plays a role in the development of TGCTs. Dysregulation was mainly due to loss of PTEN in TGCTs. Other studies demonstrated a role for this pathway in cisplatin resistance in TGCTs patients and cell lines[106,107]. Inhibition of PDGFRβ mediated phosphorylation of AKT by Ly294002 reversed cisplatin resistance in TGCT cell lines[107]. Furthermore, activation of the PI3K/AKT pathway phosphorylated MDM2 and p21 which led to cytoplasmic accumulation of p21 and inhibition of p53 mediated apoptosis in response to cisplatin[106,107]. Somatic mutations within PI3KCA, AKT and FGFR3 were also reported in cisplatin resistant TGCTs[108]. Taken together, pre-clinical studies indicate that targeting this pathway may reverse cisplatin resistance in TGCTs[107,109,110]. However, Mego et al.[111] showed limited efficacy of the mTOR inhibitor everolimus against unselected heavily pretreated refractory TGCTs.
The cisplatin-resistant phenotype can also be sustained by alterations in signaling pathways that are not directly engaged by cisplatin, so called “off-target” effects[112-114]. It is well recognized that epigenetic alterations occurring upon differentiation increase cisplatin resistance in TGCT cells[115]. TGCTs have been shown to undergo differentiation after retinoic acid treatment and molecular changes that occur in these cells after treatment have been well characterized[114,116]. Gutekunst et al.[95] demonstrated short-term differentiation of EC cells by retinoic acid significantly decreases the expression of IER3, NOXA and PUMA and caused a loss in cisplatin hypersensitivity. Other studies have reported that loss of OCT4 expression leads to cisplatin resistance in EC cells[112,113]. Events in addition to differentiation that can lead to downregulation of OCT4 include hypoxia[117] and cisplatin treatment[118]. OCT4 is also known to induce the expression of miR106b which in turn suppresses p21 expression[106]. Similarly, increased OCT4 expression was also significantly correlated with cisplatin response in xenograft TGCTs models, cell lines, and tumors[112,116,119,120]. Altogether, these finding suggest a connection between OCT4, NOXA and the cytoplasmic expression of p21 in cisplatin resistance TGCTs.
Epigenetics
Epigenetic silencing, for example linked to hypermethylation, could mediate TGCT cisplatin resistance by both on-target and post-target/off target mechanisms. Accumulating evidence suggests that an increase in DNA methylation may be associated with cisplatin resistance[61,121,122]. Seminomas are generally considered more sensitive to cisplatin than nonseminoma and are severely hypomethylated. EC cells are also sensitive to cisplatin but have a higher incidence of resistance and have an intermediate level of methylation while more difficult to treat teratomas, yolk sac tumors and choriocarcinoma have the highest level of DNA methylation[37,52]. Methylation of several specific gene promoters have been associated with inherent and acquired cisplatin resistance in TGCTs and cell lines including RASSF1A, HIC1, MGMT and CALCA[123-125]. Additionally, cisplatin treatment has been shown to be associated with increased DNA methylation in vivo[123]. We have shown that a series of testicular cancer derived EC cell lines are hypersensitive to the DNA methytransferase inhibitors decitabine and quadecitabine in vitro and in vivo compared to somatic tumors[126-128]. This hypersensitivity extended to cisplatin refractory EC and was dependent on high levels of the DNA methyltransferase, DNMT3B. Importantly, pretreatment of cisplatin refractory cells in vitro and in vivo could resensitize them to cisplatin and this activity was associated with activation of p53 and immune-related pathways[126-128]. These findings have been validated by a number of additional studies[124,129,130]. This data provided the rationale to combine cisplatin and guadecitabine in a phase I study at Indiana University [NCT02429466]. Significant responses were observed including two complete and two partial remissions in heavily pretreated platinum refractory TGCT patients (Spinella et al., in preparation). In a new study with an extended series of cisplatin resistant EC cells we unexpectedly found a tight association between cisplatin resistance and decreased H3K27 methylation of polycomb target genes that was associated with a decrease in expression of BMI1 and EZH2[131] (Spinella et al., in preparation). How the decrease in H3K27 methylation of polycomb targets relates to DNA methylation is currently being investigated. Interestingly, decreased H3K27 methylation has been previously associated with increased DNA promoter methylation in ES cells[132-134]. Another promising approach to overcome cisplatin resistance in a number of cancer types including TGCTs is the used of the bromodomain inhibitor JQ1 that target the epigenetic reader bromo and extra-terminal domain proteins[135-137].
We speculate that epigenetic context is a key driver of cisplatin resistance and sensitivity in TGCTs and specifically the DNA hypomethylation may be associated with cisplatin hypersensitivity [Figure 5]. This could be achieved by at least two, mutually-nonexclusive, mechanisms. In the first (on-target) more “open”, pluripotent-like chromatin may be more accessible to cisplatin binding and adduct formation. In the second (post-target/off target) the open, pluripotent chromatin of TGCTs may possess an inherent transcriptional plasticity not shared with other solid cancers. This may allow for a more robust acute transcriptional response to DNA damage, for example mediated by p53. In this model, epigenetic reprogramming associated with loss of pluripotency would be a source of cisplatin resistance in teratomas and perhaps other TGCT subtypes than are or become refractory to cisplatin [Figure 5]. A more complete understanding of the epigenetic state of sensitive and resistant TGCTs, especially in the clinical setting is needed to fully test this model.
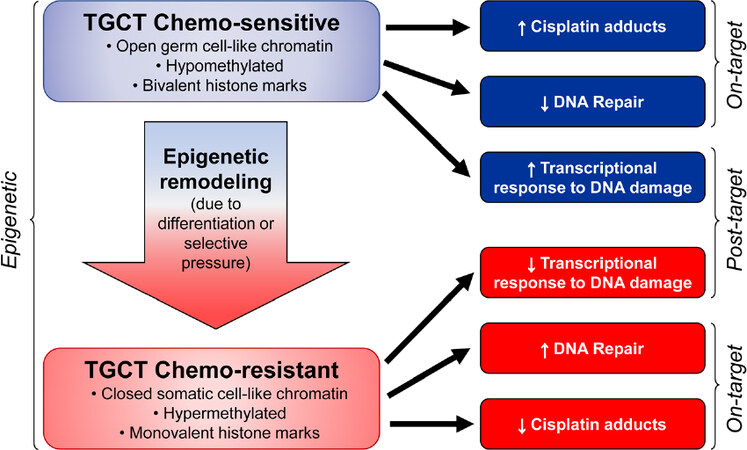
Figure 5. Epigenetic context as a key determinant of cisplatin sensitivity and resistance in TGCTs. TGCTs: testicular germ cell tumors
Conclusions
Mechanisms of cisplatin hypersensitivity and acquired resistance in TGCTs are likely multi-factorial and associated with the germ cell origins of these solid tumors. Epigenetics may play a more prominent role in the biology of TGCTs compared to somatic solid tumors that possess higher mutation rates and more frequent and obvious driver mutations. This likely extends to one of the important clinical features of TGCT, their curability. Considering that epigenetic states are malleable, one can envision reinstating “sensitive” epigenetics to cisplatin refractory TGCTs and theoretically even to common somatic solid tumors. Of course and as always, how these strategies alter toxicity to normal tissues will be an important issue.
Declarations
Authors’ contributionsGenerated conception: Singh R, Fazal Z, Freemantle SJ, Spinella MJ
Wrote manuscript: Singh R, Fazal Z, Spinella MJ
Revised manuscript: Singh R, Freemantle SJ, Spinella MJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by NIH; National Cancer Institute grant (R01CA211875), (R03CA223709); a Reach Grant from the Alex’s Lemonade Stand Foundation (MJS).
Conflicts of interestThe author declares that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Ghazarian AA, Kelly SP, Altekruse SF, Rosenberg PS, McGlynn KA. Future of testicular germ cell tumor incidence in the United States: Forecast through 2026. Cancer 2017;123:2320-8.
3. Giuliano CJ, Freemantle SJ, Spinella MJ. Testicular germ cell tumors: A paradigm for the successful treatment of solid tumor stem cells. Curr Cancer Ther Rev 2006;2:255-70.
4. Allen JC, Kirschner A, Scarpato KR, Morgans AK. Current management of refractory germ cell tumors and future directions. Curr Oncol Rep 2017;19:8.
5. Einhorn LH, Williams SD, Chamness A, Brames MJ, Perkins SM, et al. High-dose chemotherapy and stem-cell rescue for metastatic germ-cell tumors. N Engl J Med 2007;357:340-8.
6. Feldman DR, Sheinfeld J, Bajorin DF, Fischer P, Turkula S, et al. TI-CE high-dose chemotherapy for patients with previously treated germ cell tumors: results and prognostic factor analysis. J Clin Oncol 2010;28:1706-13.
7. Oing C, Alsdorf WH, von Amsberg G, Oechsle K, Bokemeyer C. Platinum-refractory germ cell tumors: an update on current treatment options and developments. World J Urol 2017;35:1167-75.
8. Albers P, Albrecht W, Algaba F, Bokemeyer C, Cohn-Cedermark G, et al. Guidelines on testicular cancer: 2015 update. Eur Urol 2015;68:1054-68.
9. Znaor A, Lortet-Tieulent J, Jemal A, Bray F. International variations and trends in testicular cancer incidence and mortality. Eur Urol 2014;65:1095-106.
10. Abouassaly R, Fossa SD, Giwercman A, Kollmannsberger C, Motzer RJ, et al. Sequelae of treatment in long-term survivors of testis cancer. Eur Urol 2011;60:516-26.
11. Tandstad T, Kollmannsberger CK, Roth BJ, Jeldres C, Gillessen S, et al. Practice Makes Perfect: The Rest of the Story in Testicular Cancer as a Model Curable Neoplasm. J Clin Oncol 2017;35:3525-28.
12. Schmidtova S, Kalavska K, Kucerova L. Molecular mechanisms of cisplatin chemoresistance and its circumventing in testicular germ cell tumors. Curr Oncol Rep 2018;20:88.
13. Berney DM, Looijenga LH, Idrees M, Oosterhuis JW, Rajpert-De Meyts E, et al. Germ cell neoplasia in situ (GCNIS): evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016;69:7-10.
14. Spiller CM, Bowles J. Germ cell neoplasia in situ: The precursor cell for invasive germ cell tumors of the testis. Int J Biochem Cell Biol 2017;86:22-5.
15. Mitchell RT, Camacho-Moll M, Macdonald J, Anderson RA, Kelnar CJ, et al. Intratubular germ cell neoplasia of the human testis: heterogeneous protein expression and relation to invasive potential. Mod Pathol 2014;27:1255-66.
16. Kristensen DM, Sonne SB, Ottesen AM, Perrett RM, Nielsen JE, et al. Origin of pluripotent germ cell tumours: the role of microenvironment during embryonic development. Mol Cell Endocrinol 2008;288:111-8.
17. Spierings DC, de Vries EG, Vellenga E, de Jong S. The attractive Achilles heel of germ cell tumours: an inherent sensitivity to apoptosis-inducing stimuli. J Pathol 2003;200:137-48.
18. Kalavska K, Conteduca V, De Giorgi U, Mego M. Molecular mechanisms of resistance in testicular germ cell tumors - clinical implications. Curr Cancer Drug Targets 2018;18:967-78.
19. Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun 2015;6:5973.
20. Shen H, Shih J, Hollern DP, Wang L, Bowlby R, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep 2018;23:3392-406.
21. Bulic-Jakus F, Katusic Bojanac A, Juric-Lekic G, Vlahovic M, Sincic N. Teratoma: from spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip Rev Dev Biol 2016;5:186-209.
22. Andrews PW, Matin MM, Bahrami AR, Damjanov I, Gokhale P, et al. Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem Soc Trans 2005;33:1526-30.
23. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424.
24. Ferguson L, Agoulnik AI. Testicular cancer and cryptorchidism. Front Endocrinol (Lausanne) 2013;4:32.
25. Dieckmann KP, Rube C, Henke RP. Association of Down’s syndrome and testicular cancer. J Urol 1997;157:1701-4.
26. Soto AM, Sonnenschein C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol 2010;6:363-70.
27. Piltoft JS, Larsen SB, Dalton SO, Johansen C, Baker JL, et al. Early life risk factors for testicular cancer: a case-cohort study based on the Copenhagen School Health Records Register. Acta Oncol 2017;56:220-4.
28. Cook MB, Akre O, Forman D, Madigan MP, Richiardi L, et al. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer experiences of the son. Int J Epidemiol 2010;39:1605-18.
29. Maule M, Malavassi JL, Richiardi L. Age at puberty and risk of testicular cancer: a meta-analysis. Int J Androl 2012;35:828-34.
30. Moller H, Skakkebaek NE. Risk of testicular cancer in subfertile men: case-control study. BMJ 1999;318:559-62.
31. Jacobsen R, Bostofte E, Engholm G, Hansen J, Olsen JH, et al. Risk of testicular cancer in men with abnormal semen characteristics: cohort study. BMJ 2000;321:789-92.
32. Hanson HA, Anderson RE, Aston KI, Carrell DT, Smith KR, et al. Subfertility increases risk of testicular cancer: evidence from population-based semen samples. Fertil Steril 2016;105:322-8.
33. Zequi Sde C, da Costa WH, Santana TB, Favaretto RL, Sacomani CA, et al. Bilateral testicular germ cell tumours: a systematic review. BJU Int 2012;110:1102-9.
34. Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, et al. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A 2003;100:13350-5.
35. Josephson R, Ording CJ, Liu Y, Shin S, Lakshmipathy U, et al. Qualification of embryonal carcinoma 2102Ep as a reference for human embryonic stem cell research. Stem Cells 2007;25:437-46.
36. Atkin NB, Baker MC. Specific chromosome change, i(12p), in testicular tumours? Lancet 1982;2:1349.
37. Facchini G, Rossetti S, Cavaliere C, D’Aniello C, Di Franco R, et al. Exploring the molecular aspects associated with testicular germ cell tumors: a review. Oncotarget 2018;9:1365-79.
38. Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer 2005;5:210-22.
39. Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol 2007;25:207-15.
40. FireBrowse portal. Available from https://firebrowse.org/.
41. Skakkebaek NE, Berthelsen JG, Giwercman A, Muller J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl 1987;10:19-28.
42. Rijlaarsdam MA, Looijenga LH. An oncofetal and developmental perspective on testicular germ cell cancer. Semin Cancer Biol 2014;29:59-74.
43. Litchfield K, Levy M, Huddart RA, Shipley J, Turnbull C. The genomic landscape of testicular germ cell tumours: from susceptibility to treatment. Nat Rev Urol 2016;13:409-19.
44. Honecker F, Stoop H, de Krijger RR, Chris Lau YF, Bokemeyer C, et al. Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J Pathol 2004;203:849-57.
45. Batool A, Karimi N, Wu XN, Chen SR, Liu YX. Testicular germ cell tumor: a comprehensive review. Cell Mol Life Sci 2019;76:1713-27.
46. Skakkebaek NE, Rajpert-De Meyts E, Jorgensen N, Carlsen E, Petersen PM, et al. Germ cell cancer and disorders of spermatogenesis: an environmental connection? APMIS 1998;106:3-11.
47. Oosterhuis JW, Stoop H, Honecker F, Looijenga LH. Why human extragonadal germ cell tumours occur in the midline of the body: old concepts, new perspectives. Int J Androl 2007;30:256-63.
48. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol 2018;2018:9059382.
49. Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet 2009;41:811-5.
50. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A 1975;72:3585-9.
51. Lobo J, Gillis AJM, Jerónimo C, Henrique R, Looijenga LH. Human germ cell tumors are developmental cancers: Impact of epigenetics on pathobiology and clinic. Int J Mol Sci 2019;20:pii: E258.
52. Killian JK, Dorssers LC, Trabert B, Gillis AJ, Cook MB, Oosterhuis JW, Meltzer PS, McGlynn KA, Looijenga LH, et al. Imprints and DPPA3 are bypassed during pluripotency- and differentiation-coupled methylation reprogramming in testicular germ cell tumors. Genome Res 2016;26:1490-504.
53. Rijlaarsdam MA, Tax DM, Gillis AJ, Dorssers LC, Koestler DC, et al. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One 2015;10:e0122146.
54. van der Zwan YG, Rijlaarsdam MA, Rossello FJ, Notini AJ, et al. Seminoma and embryonal carcinoma footprints identified by analysis of integrated genome-wide epigenetic and expression profiles of germ cell cancer cell lines. PLoS One 2014;9:e98330.
55. Smiraglia DJ, Szymanska J, Kraggerud SM, Lothe RA, Peltomaki P, et al. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene 2002;21:3909-16.
57. Grossniklaus U, Paro R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harb Perspect Biol 2014;6:a019331.
58. Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell 2017;171:34-57.
59. Sashida G, Oshima M, Iwama A. Deregulated Polycomb functions in myeloproliferative neoplasms. Int J Hematol 2019; doi: 10.1007/s12185-019-02600-6.
60. Wang W, Qin JJ, Voruganti S, Nag S, Zhou J, Zhang R. Polycomb group (PcG) proteins and human cancers: Multifaceted Functions and therapeutic implications. Med Res Rev 2015;35:1220-67.
61. Jostes S, Nettersheim D, Schorle H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat Rev Urol 2019;16:245-59.
62. Almstrup K, Nielsen JE, Mlynarska O, Jansen MT, Jorgensen A, et al. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer 2010;103:1269-76.
63. Hoang N, Zhang X, Zhang C, Vo V, Leng F, et al. New histone demethylase LSD1 inhibitor selectively targets teratocarcinoma and embryonic carcinoma cells. Bioorg Med Chem 2018;26:1523-37.
64. Yin F, Lan R, Zhang X, Zhu L, Chen F, et al. LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol Cell Biol 2014;34:158-79.
65. Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006;124:1169-81.
66. Syring I, Bartels J, Holdenrieder S, Kristiansen G, Muller SC, et al. Circulating serum miRNA (miR-367-3p, miR-371a-3p, miR-372-3p and miR-373-3p) as biomarkers in patients with testicular germ cell cancer. J Urol 2015;193:331-7.
67. Dieckmann KP, Radtke A, Geczi L, Matthies C, Anheuser P, et al. Serum levels of microRNA-371a-3p (M371 test) as a new biomarker of testicular germ cell tumors: results of a prospective multicentric study. J Clin Oncol 2019;37:1412-23.
68. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol 1997;15:594-603.
69. Rick O, Bokemeyer C, Beyer J, Hartmann JT, Schwella N, et al. Salvage treatment with paclitaxel, ifosfamide, and cisplatin plus high-dose carboplatin, etoposide, and thiotepa followed by autologous stem-cell rescue in patients with relapsed or refractory germ cell cancer. J Clin Oncol 2001;19:81-8.
70. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012;31:1869-83.
71. Fenske AE, Glaesener S, Bokemeyer C, Thomale J, Dahm-Daphi J, et al. Cisplatin resistance induced in germ cell tumour cells is due to reduced susceptibility towards cell death but not to altered DNA damage induction or repair. Cancer Lett 2012;324:171-8.
72. Safaei R, Howell SB. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hematol 2005;53:13-23.
73. Sark MW, Timmer-Bosscha H, Meijer C, Uges DR, Sluiter WJ, et al. Cellular basis for differential sensitivity to cisplatin in human germ cell tumour and colon carcinoma cell lines. Br J Cancer 1995;71:684-90.
74. Masters JR, Thomas R, Hall AG, Hogarth L, Matheson EC, et al. Sensitivity of testis tumour cells to chemotherapeutic drugs: role of detoxifying pathways. Eur J Cancer 1996;32A:1248-53.
75. Mayer F, Stoop H, Scheffer GL, Scheper R, Oosterhuis JW, et al. Molecular determinants of treatment response in human germ cell tumors. Clin Cancer Res 2003;9:767-73.
76. Pera MF, Friedlos F, Mills J, Roberts JJ. Inherent sensitivity of cultured human embryonal carcinoma cells to adducts of cis-diamminedichloroplatinum(II) on DNA. Cancer Res 1987;47:6810-3.
77. Bedford P, Fichtinger-Schepman AM, Shellard SA, Walker MC, Masters JR, et al. Differential repair of platinum-DNA adducts in human bladder and testicular tumor continuous cell lines. Cancer Res 1988;48:3019-24.
78. Hill BT, Scanlon KJ, Hansson J, Harstrick A, Pera M, et al. Deficient repair of cisplatin-DNA adducts identified in human testicular teratoma cell lines established from tumours from untreated patients. Eur J Cancer 1994;30A:832-7.
79. Wood RD, Araujo SJ, Ariza RR, Batty DP, Biggerstaff M, et al. DNA damage recognition and nucleotide excision repair in mammalian cells. Cold Spring Harb Symp Quant Biol 2000;65:173-82.
80. Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res 2008;18:64-72.
81. Awuah SG, Riddell IA, Lippard SJ. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc Natl Acad Sci U S A 2017;114:950-5.
82. Kartalou M, Essigmann JM. Recognition of cisplatin adducts by cellular proteins. Mutat Res 2001;478:1-21.
83. Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev 2007;107:1387-407.
84. Yamada M, O’Regan E, Brown R, Karran P. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res 1997;25:491-6.
85. Shu X, Xiong X, Song J, He C, Yi C. Base-Resolution Analysis of Cisplatin-DNA Adducts at the Genome Scale. Angew Chem Int Ed Engl 2016;55:14246-9.
86. Bloom JC, Loehr AR, Schimenti JC, Weiss RS. Germline genome protection: implications for gamete quality and germ cell tumorigenesis. Andrology 2019; doi: 10.1111/andr.12651.
87. Honecker F, Wermann H, Mayer F, Gillis AJ, Stoop H, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol 2009;27:2129-36.
88. Rudolph C, Melau C, Nielsen JE, Vile Jensen K, et al. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell Oncol (Dordr) 2017;40:341-55.
89. Mayer F, Gillis AJ, Dinjens W, Oosterhuis JW, Bokemeyer C, et al. Microsatellite instability of germ cell tumors is associated with resistance to systemic treatment. Cancer Res 2002;62:2758-60.
90. Velasco A, Riquelme E, Schultz M, Wistuba II, Villarroel L, et al. Microsatellite instability and loss of heterozygosity have distinct prognostic value for testicular germ cell tumor recurrence. Cancer Biol Ther 2004;3:1152-8.
91. Mego M, Cierna Z, Svetlovska D, Macak D, Machalekova K, Miskovska V, Chovanec M, Usakova V, Obertova J, Babal P, Mardiak J. PARP expression in germ cell tumours. J Clin Pathol 2013;66:607-12.
92. Cavallo F, Graziani G, Antinozzi C, Feldman DR, Houldsworth J, et al. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS One 2012;7:e51563.
93. Kersemaekers AM, Mayer F, Molier M, van Weeren PC, Oosterhuis JW, et al. Role of P53 and MDM2 in treatment response of human germ cell tumors. J Clin Oncol 2002;20:1551-61.
94. Kerley-Hamilton JS, Pike AM, Li N, DiRenzo J, Spinella MJ. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embryonal carcinoma. Oncogene 2005;24:6090-100.
95. Gutekunst M, Oren M, Weilbacher A, Dengler MA, Markwardt C, et al. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS One 2011;6:e19198.
96. Romano FJ, Rossetti S, Conteduca V, Schepisi G, Cavaliere C, et al. Role of DNA repair machinery and p53 in the testicular germ cell cancer: a review. Oncotarget 2016;7:85641-9.
97. Duale N, Lindeman B, Komada M, Olsen AK, Andreassen A, et al. Molecular portrait of cisplatin induced response in human testis cancer cell lines based on gene expression profiles. Mol Cancer 2007;6:53.
98. Bagrodia A, Lee BH, Lee W, Cha EK, Sfakianos JP, et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J Clin Oncol 2016;34:4000-7.
99. Taylor-Weiner A, Zack T, O’Donnell E, Guerriero JL, Bernard B, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016;540:114-8.
100. Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA, de Jong S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis 2011;2:e148.
101. Grande L, Bretones G, Rosa-Garrido M, Garrido-Martin EM, Hernandez T, et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J Biol Chem 2012;287:26495-505.
102. Beyer U, Moll-Rocek J, Moll UM, Dobbelstein M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc Natl Acad Sci U S A 2011;108:3624-9.
103. Noel EE, Yeste-Velasco M, Mao X, Perry J, Kudahetti SC, et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am J Pathol 2010;176:2607-15.
104. Selfe J, Goddard NC, McIntyre A, Taylor KR, Renshaw J, et al. IGF1R signalling in testicular germ cell tumour cells impacts on cell survival and acquired cisplatin resistance. J Pathol 2018;244:242-53.
105. Di Vizio D, Cito L, Boccia A, Chieffi P, Insabato L, et al. Loss of the tumor suppressor gene PTEN marks the transition from intratubular germ cell neoplasias (ITGCN) to invasive germ cell tumors. Oncogene 2005;24:1882-94.
106. Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest 2010;120:3594-605.
107. Juliachs M, Munoz C, Moutinho CA, Vidal A, Condom E, et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin Cancer Res 2014;20:658-67.
108. Feldman DR, Iyer G, Van Alstine L, Patil S, Al-Ahmadie H, et al. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin Cancer Res 2014;20:3712-20.
109. Castillo-Avila W, Piulats JM, Garcia Del Muro X, Vidal A, Condom E, et al. Sunitinib inhibits tumor growth and synergizes with cisplatin in orthotopic models of cisplatin-sensitive and cisplatin-resistant human testicular germ cell tumors. Clin Cancer Res 2009;15:3384-95.
110. Juliachs M, Vidal A, Del Muro XG, Piulats JM, Condom E, et al. Effectivity of pazopanib treatment in orthotopic models of human testicular germ cell tumors. BMC Cancer 2013;13:382.
111. Mego M, Svetlovska D, Miskovska V, Obertova J, Palacka P, et al. Phase II study of everolimus in refractory testicular germ cell tumors. Urol Oncol 2016;34:122 e17-22.
112. Mueller T, Mueller LP, Holzhausen HJ, Witthuhn R, Albers P, et al. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem Cell Biol 2010;134:197-204.
113. Mueller T, Mueller LP, Luetzkendorf J, Voigt W, Simon H, et al. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour Biol 2006;27:71-83.
114. Snow GE, Kasper AC, Busch AM, Schwarz E, Ewings KE, et al. Wnt pathway reprogramming during human embryonal carcinoma differentiation and potential for therapeutic targeting. BMC Cancer 2009;9:383.
115. Timmer-Bosscha H, de Vries EG, Meijer C, Oosterhuis JW, Mulder NH. Differential effects of all-trans-retinoic acid, docosahexaenoic acid, and hexadecylphosphocholine on cisplatin-induced cytotoxicity and apoptosis in a cisplantin-sensitive and resistant human embryonal carcinoma cell line. Cancer Chemother Pharmacol 1998;41:469-76.
116. Honecker F, Rohlfing T, Harder S, Braig M, Gillis AJ, Balabanov S, et al. Proteome analysis of the effects of all-trans retinoic acid on human germ cell tumor cell lines. J Proteomics 2014;96:300-13.
117. Wu YC, Ling TY, Lu SH, Kuo HC, Ho HN, Yeh SD, Shen CN, Huang YH. Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res 2012;72:4963-73.
118. Abada PB, Howell SB. Cisplatin induces resistance by triggering differentiation of testicular embryonal carcinoma cells. PLoS One 2014;9:e87444.
119. Gutekunst M, Mueller T, Weilbacher A, Dengler MA, Bedke J, et al. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res 2013;73:1460-9.
120. Koster R, de Jong S. Lessons learned from testicular cancer: identification of cytoplasmic p21 as an Achilles’ heel of cisplatin resistance. Cell Cycle 2010;9:4776-7.
121. Buljubašić R, Buljubašić M, Bojanac AK, Ulamec M, Vlahović M, et al. Epigenetics and testicular germ cell tumors. Gene 2018;661:22-33.
122. Sonnenburg D, Spinella MJ, Albany C. Epigenetic Targeting of Platinum Resistant Testicular Cancer. Curr Cancer Drug Targets 2016;16:789-95.
123. Koul S, McKiernan JM, Narayan G, Houldsworth J, Bacik J, et al. Role of promoter hypermethylation in cisplatin treatment response of male germ cell tumors. Mol Cancer 2004;3:16.
124. Wermann H, Stoop H, Gillis AJ, Honecker F, van Gurp RJ, et al. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol 2010;221:433-42.
125. Martinelli CMDS, Lengert AVH, Cárcano FM, Silva ECA, Brait M, et al. MGMT and CALCA promoter methylation are associated with poor prognosis intesticular germ cell tumor patients. Oncotarget 2016;8:50608-17.
126. Beyrouthy MJ, Garner KM, Hever MP, Freemantle SJ, Eastman A, et al. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res 2009;69:9360-6.
127. Biswal BK, Beyrouthy MJ, Hever-Jardine MP, Armstrong D, Tomlinson CR, et al. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA Damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLoS One 2012;7:e53003.
128. Albany C, Hever-Jardine MP, von Herrmann KM, Yim CY, Tam J, et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017;8:2949-59.
129. Wongtrakoongate P, Li J, Andrews PW. Aza-deoxycytidine induces apoptosis or differentiation via DNMT3B and targets embryonal carcinoma cells but not their differentiated derivatives. Br J Cancer 2014;110:2131-8.
130. Oing C, Verem I, Mansour WY, Bokemeyer C, Dyshlovoy S, et al. F. 5-azacitidine exerts prolonged pro-apoptotic effects and overcomes cisplatin-resistance in non-seminomatous germ cell tumor cells. Int J Mol Sci 2018;20:pii: E21.
132. Singh R, Fazal Z, Corbet AK, Bikorimana E, Rodriguez JC, et al. Epigenetic remodeling through downregulation of polycomb repressive complex 2 mediates chemotherapy resistance in testicular germ cell tumors. Cancers (Basel) 2019;11:pii: E796.
132. Brinkman AB, Gu H, Bartels SJ, Zhang Y, Matarese F, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 2012;22:1128-38.
133. Saksouk N, Barth TK, Ziegler-Birling C, Olova N, Nowak A, et al. Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol Cell 2014;56:580-94.
134. van Mierlo G, Dirks RAM, De Clerck L, Brinkman AB, Huth M, et al. Integrative Proteomic Profiling Reveals PRC2-Dependent Epigenetic Crosstalk Maintains Ground-State Pluripotency. Cell Stem Cell 2019;24:123-37e8.
135. Momeny M, Eyvani H, Barghi F, Ghaffari SH, Javadikooshesh S, et al. Inhibition of bromodomain and extraterminal domain reduces growth and invasive characteristics of chemoresistant ovarian carcinoma cells. Anticancer Drugs 2018;29:1011-20.
136. Zanellato I, Colangelo D, Osella D. JQ1, a BET inhibitor, synergizes with cisplatin and induces apoptosis in highly chemoresistant malignant pleural mesothelioma cells. Curr Cancer Drug Targets 2018;18:816-28.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Singh R, Fazal Z, Freemantle SJ, Spinella MJ. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist 2019;2:580-94. http://dx.doi.org/10.20517/cdr.2019.19
AMA Style
Singh R, Fazal Z, Freemantle SJ, Spinella MJ. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resistance. 2019; 2(3): 580-94. http://dx.doi.org/10.20517/cdr.2019.19
Chicago/Turabian Style
Singh, Ratnakar, Zeeshan Fazal, Sarah J. Freemantle, Michael J. Spinella. 2019. "Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors" Cancer Drug Resistance. 2, no.3: 580-94. http://dx.doi.org/10.20517/cdr.2019.19
ACS Style
Singh, R.; Fazal Z.; Freemantle SJ.; Spinella MJ. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580-94. http://dx.doi.org/10.20517/cdr.2019.19
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 30
likes
Like This Article 30
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.