
Drug-adapted cancer cell lines as preclinical models of acquired resistance


Abstract
Acquired resistance formation limits the efficacy of anti-cancer therapies. Acquired and intrinsic resistance differ conceptually. Acquired resistance is the consequence of directed evolution, whereas intrinsic resistance depends on the (stochastic) presence of pre-existing resistance mechanisms. Preclinical model systems are needed to study acquired drug resistance because they enable: (1) in depth functional studies; (2) the investigation of non-standard treatments for a certain disease condition (which is necessary to identify small groups of responders); and (3) the comparison of multiple therapies in the same system. Hence, they complement data derived from clinical trials and clinical specimens, including liquid biopsies. Many groups have successfully used drug-adapted cancer cell lines to identify and elucidate clinically relevant resistance mechanisms to targeted and cytotoxic anti-cancer drugs. Hence, we argue that drug-adapted cancer cell lines represent a preclinical model system in their own right that is complementary to other preclinical model systems and clinical data.
Keywords
Introduction
Despite improvements in therapy outcomes in recent decades, except for a few exceptions (e.g., testicular cancer, Hodgkin’s lymphoma, childhood acute lymphoblastic leukaemia) cure rates remain low for advanced cancers that require systemic therapy, typically metastatic disease. In such advanced cases, the impetus typically lies on the prolongation of life and the improvement of quality of life[1-8].
The efficacy of systemic anti-cancer therapies is limited by the occurrence of resistance. Resistance can be “intrinsic” or “upfront”, i.e., cancer cells do not respond to therapy from the outset. Many cancer diseases, however, initially respond well to therapy, but after a temporary response resistant cancer cells emerge leading to “acquired” resistance, ultimately resulting in therapy failure and patient death[8-19]. Hence, cancer diseases that have become resistant to the available treatment options represent an unmet clinical need. New strategies including new biomarkers that indicate effective follow-up therapies (on an individualised basis) are needed for such patients for which no established therapy options are available anymore [Figure 1].
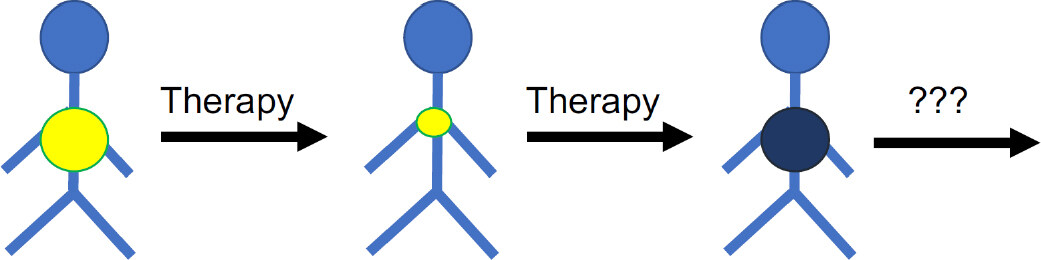
Figure 1. Many cancer diseases respond initially well to therapy but cancer cells become eventually resistant to therapy. An improved understanding of the mechanisms and processes underlying resistance formation is necessary to identify biomarkers that guide the use of efficient next-line therapies for tumours that have do not respond to the available standard therapies anymore
Intrinsic and acquired resistance mechanisms may differ
There is a conceptional difference between the mechanisms and processes underlying intrinsic and acquired drug resistance formation. Intrinsic resistance is the consequence of pre-existing, potentially stochastic changes that render cancer cells insensitive to the standard treatment. In contrast, acquired resistance is the consequence of selection and adaptation processes in response to therapy, i.e., of directed evolution induced by the therapy. In line with this, differences have been described between intrinsic and acquired resistance mechanisms[20-23]. Hence, acquired resistance needs to be studied in the context of the underlying (co)-evolutionary processes to establish a specific systems level understanding.
Preclinical model systems are needed to deliver biomarkers for the effective use of “liquid biopsies” for therapy monitoring
The systematic elucidation of resistance formation depends on the combined use of preclinical model systems in combination with clinical data and specimens. Preclinical model systems enable in-depth functional and systems level studies that are difficult or impossible to perform using primary cancer cells, tissues, and/or organoids. In addition, non-standard treatments can be systematically investigated in preclinical model systems. This is not possible in a clinical setting, where patients receive standard therapies that provide the highest probability of treatment success. Hence, biomarkers that: (1) identify (small) groups of patients that are unlikely to respond to standard therapies; and (2) guide the use of more promising therapies to such patients need to be derived from preclinical models. Finally, preclinical model systems enable the direct comparison of different therapies in the same system. Such comparisons are not possible in the clinics, where every patient can only be treated once.
So-called “liquid biopsies” including circulating tumour DNA and circulating tumour cells enable the monitoring of cancer evolution and therapy response in ever greater detail[24]. The clinical implementation of liquid biopsies still faces many technological and methodological challenges[24,25]. However, the first FDA-approved assays based on liquid biopsies are available and have been shown to improve therapy outcomes[24,26-32].
With the advancement of liquid biopsies for the monitoring of cancer cell evolution, a much more advanced understanding of the processes underlying therapy response and resistance formation will be required to make effective use of the wealth of omics data derived from liquid biopsies. Only an in-depth molecular understanding will enable the identification of biomarkers that indicate therapy failure early and inform the choice of effective next-line therapies. Such knowledge and the associated (putative) biomarkers will have to originate, at least in part, from research performed in preclinical model systems before they are tested in a clinical setting.
Drug-adapted cancer cell lines reflect clinical resistance mechanisms
Cancer cell lines are among the most commonly used pre-clinical models[33,34]. They are relatively easy to handle and enable high throughput analysis at relatively low cost and in a timely fashion. There is increasing agreement that the use of (larger) cell line panels improves the value of results[34,35]. The NCI60 panel of the National Cancer Institute is the oldest and best characterised cancer cell line panel, which has contributed to the discovery of many anti-cancer drugs[36,37]. If typical caveats such as cell line cross-contamination and misauthentication as well as mycoplasma contamination[38] are avoided, the investigation of cancer cell lines provides substantial information on cancer cell biology and drug sensitivity, as, for example, confirmed by large pharmacogenomic screens including the Genomics of Drug Sensitivity in Cancer, the Cancer Cell Line Encyclopedia, and the Cancer Therapeutics Response Portal[39-45]. Since most cancer cell lines have been derived from patients at diagnosis, however, they primarily reflect intrinsic resistance.
Drug-adapted cell lines better reflect the evolutionary processes leading to resistance formation. They have enabled the discovery of major drug resistance mechanisms and the identification and elucidation of clinically relevant acquired resistance mechanisms to targeted and cytotoxic anti-cancer drugs[33]. The ATP-binding cassette (ABC) transporters, arguably the most important mediators of drug resistance in cancer cells [Figure 2], were detected in drug-adapted cells. ABCB1 (also known as P-glycoprotein or MDR1) was discovered as the first member of the family of ABC transporters in colchicine-adapted Chinese hamster ovarian cells[46]. It is a promiscuous efflux pump that transports a wide range of structurally different substrates and provides resistance to a large number of anti-cancer drugs from various classes[47,48]. ABCC1 (also known as MRP1), another member of the ABC transporter family, is also of high importance as a cancer cell resistance mechanism[47,48] and was identified in a doxorubicin-adapted subline of the lung cancer cell line H69[49].
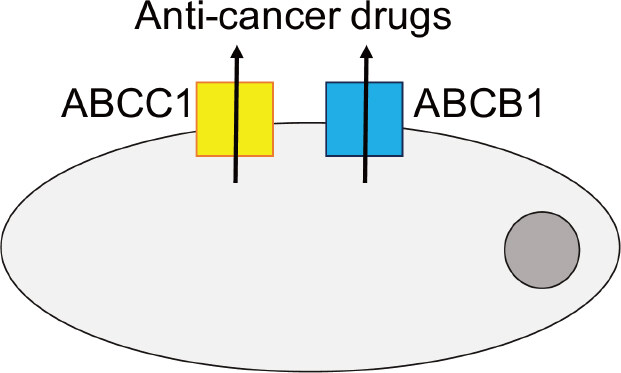
Figure 2. Members of the ATP-binding cassette (ABC) transporter family, including ABCB1 and ABCC1 as prominent members, belong to the most important mediators of drug resistance in cancer. Various members of the ABC transporter family function as efflux pumps that remove (often a wide range of structurally different anti-cancer drugs) from cancer cells and interfere with the achievement of effective intracellular drug concentrations
Without intending to provide a comprehensive overview, we have selected a few studies that illustrate the potential of drug-adapted cancer cell lines to reveal clinically relevant resistance mechanisms. Non-small cell lung cancer patients, who harbour cancer cells characterised by activating EGFR mutations, are treated with EGFR tyrosine kinase inhibitors[50]. In a landmark study, MET amplification was discovered as a resistance mechanism in a gefitinib-adapted subline of the EGFR exon 19 mutant non-small cell lung cancer cell line HCC827[51]. Further investigation of resistance formation to EGFR tyrosine kinase inhibitors using drug-adapted non-small cell lung cancer cell lines revealed that the origin of the resistance-mediating T790M EGFR mutation may differ in different cell line systems and patients[52]. Pre-existing T790M mutant subpopulations can either be selected, or de novo T790M mutations can be induced. The mode of resistance formation shaped the resistance phenotype of the resulting drug-resistant sublines. Induction of de novo T790M mutations, but not selection of pre-existing T790M mutant clones, was associated with an enhanced cellular resistance to apoptosis, which was caused by an increase in the cellular levels of anti-apoptotic bcl-2 proteins[52]. Furthermore, erlotinib-resistant colonies derived from non-small cell lung cancer cell lines reflected clinically observed resistance mechanisms[53].
Drug-adapted cancer cell lines have also been shown to reflect clinical resistance formation to other kinase inhibitors that target specific oncogenic driver events. Inhibitors that specifically target constitutively active oncogenic V600E-mutant BRAF, have improved the therapy of melanoma patients whose tumours consist of cells that harbour V600E BRAF mutations. Unfortunately, responses are short-lived, and resistance formation is inevitable[54]. Key acquired resistance mechanisms to V600E-specific BRAF inhibitors including NRAS mutation, BRAF amplification, dimerization of aberrantly spliced V600E-mutant BRAF, and PDGFRB upregulation were all identified in drug-adapted cancer cell lines[55-57]. Moreover, clinically 5 relevant resistance mechanisms were represented in EGFR, HER2, and ALK inhibitor-adapted cancer cell lines[58,59].
Drug-adapted cancer cell lines also reflect clinical resistance formation against various other “targeted” anti-cancer drugs that interfere with features that are exclusively or predominantly found in cancer cells, as demonstrated by the following examples. Prostate cancer cell lines adapted to the antiandrogen enzalutamide enabled the identification of F876L mutations in the androgen receptor as a clinically relevant resistance mechanism[60,61]. MDM2 inhibitors are under development as a novel class of anti-cancer drugs for the treatment of TP53 wild-type cancer cells from different cancer entities. TP53 encodes p53, a major tumour suppressor protein. MDM2 is a p53 target gene that encodes for MDM2, a major endogenous inhibitor of p53. MDM2 physically interacts with p53 and mediates its ubiquitination and proteasomal degradation. MDM2 inhibitors activate p53 signalling by interference with the MDM2/p53 interaction[62-64]. Adaptation of TP53 wild-type cancer cell lines has been associated with the formation of loss-of-function TP53 mutations in many model systems[65-70]. In agreement, MDM2 inhibitor treatment of liposarcoma patients was associated with the emergence of TP53 mutations[71].
Drug-adapted cancer cell lines are also used to elucidate resistance mechanisms to cytotoxic anti-cancer agents. A subfraction of cells that critically depend on notch- and hedgehog signalling have been shown to be critically involved in resistance formation to doxorubicin in castration-resistant prostate cancer cells[72]. A number of recent studies investigated resistance formation in acute myeloid leukaemia cells using drug-adapted cell lines and identified GLI1, EZH2, and SAMHD1 as clinically relevant resistance mechanisms to cytarabine-based therapies[73-75]. In addition, increased glucocorticoid sensitivity was detected in cytarabine-adapted acute myeloid leukaemia cell lines and patient samples[76]. The use of drug-adapted cell lines has also shown that acquired resistance to cytotoxic drugs can be associated with decreased sensitivity to kinase inhibitors[77,78]. The clinical impact of this is difficult to determine, however, because the baseline sensitivity of tumours to different anti-cancer therapies prior to the first-line treatment is not typically known.
Multiple resistance models are needed to reflect the heterogeneity of the processes associated with resistance formation
It is now generally accepted that cancer diseases are associated with tremendous intra-tumour heterogeneity[79-81]. Although therapy-induced heterogeneity has not been investigated to the same extent, there are indications that the processes underlying resistance formation are likely to be as complex[52,82-88].
The advantage of cancer cell lines as models is that they are relatively easy to handle and enable high throughput analysis at relatively low cost and in a timely fashion. Although they do not reflect the original heterogeneity of the tumour they have been derived from, they are not as homogenous or clonal as previously believed[34-37,89]. Resistance can occur by selection of pre-existing drug-resistant subpopulations or by adaptation of originally drug-sensitive cells to anti-cancer therapies. Both mechanisms have been shown to be represented in drug-adapted cancer cell lines[52,66-70,90-101].
In this context, we have adapted the TP53 wild-type acute myeloid leukaemia (AML) cell lines MV4-11, OCI-AML-2, OCI-AML-3, and SIG-M5 to the MDM2 inhibitor nutlin-3 in multiple independent experiments[102]. Nutlin-3-adapted sublines of the same AML cell lines displayed a substantial heterogeneity in the response to other anti-cancer drugs. Notably, the biggest fold change (11.4) was detected in the response of two nutlin-3-adapted MV4-11 sublines to doxorubicin, although nutlin-3 treatment selected a pre-existing TP53 mutant subpopulation in this cell line. This indicates that even the drug-induced selection of a defined pre-existing subpopulation in a cell line can result in phenotypically different sublines[102]. New technologies including single cell approaches will enable the elucidation of selection and adaptation processes during resistance formation in more detail[94,103,104].
Since many models will be needed to cover the complexity associated with acquired resistance formation, we have established the Resistant Cancer Cell Line collection by adapting initially chemosensitive cancer cell lines to clinical concentrations of targeted and cytotoxic anti-cancer drugs to enable the systematic investigation of acquired drug resistance mechanisms. It currently contains 1300 cancer cell lines based on 125 parental cell lines from 16 cancer entities and reflects acquired resistance to 67 drugs (https://research.kent.ac.uk/ibc/the-resistant-cancer-cell-line-rccl-collection). The DEN50-R platform is another project dedicated to the generation of drug-adapted cancer cell line panels (http://www.den50-r.org).
Conclusion
This perspective is focused on the use of drug-adapted cancer cell lines as models of acquired drug resistance in cancer. Drug-adapted cancer cell lines are, like every model system, associated with specific advantages and limitations. Models including primary cancer cell cultures, three-dimensional cell (co-)culture systems, tumour-derived organoids, and animal models better reflect certain aspects of tumour growth such as intra-tumour heterogeneity, three-dimensional architecture, cancer cell interaction with the cancer microenvironment, and/ or metastatic behaviour[105-114]. Such models can be used to study processes that cannot be studied in cell lines. In this context, acquired resistance models have been established based on cell line- and patient-derived xenografts, organoids, and transgenic tumour models[115-125]. However, cell lines enable the establishment of a substantially larger number of models within a given timeframe and at a given cost, which is critical for studying the drug-induced heterogeneity. Notably, data so far suggest that the drug adaptation of cancer cell lines reveals similar resistance mechanisms as cell line-derived xenografts and transgenic mouse models[116,118,123,125].
In conclusion, drug-adapted cancer cell lines reflect clinically relevant acquired drug resistance mechanisms and represent a preclinical model system in their own right, which is complementary to other preclinical models and clinical specimens. Drug-adapted cancer cell lines enable systems level studies and the direct comparison of different therapies in the same system that cannot be performed in the clinics. Hence, drug-adapted cancer cell lines offer potential for the identification of biomarkers that indicate resistance formation and, ideally, effective next-line therapies [Figure 3]. Many drug-adapted cancer cell lines will be needed to cover the complexity of the mechanisms underlying resistance formation.
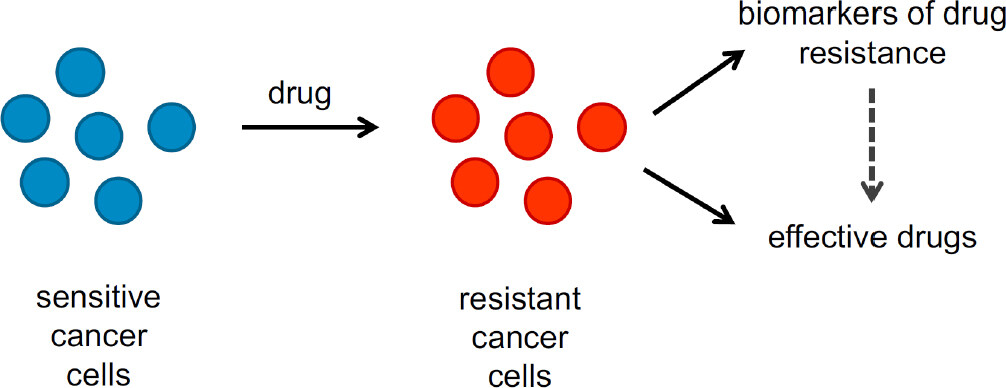
Figure 3. Drug-adapted cancer cell lines enable the identification of candidate biomarkers that enable the early detection of resistance formation and, in combination with drug screens and functional genomics approaches, the selection of effective next-line therapies
Declarations
Authors’ contributionsContributed to the writing and revision of the article and read and approved the final version.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström PU, et al. Bladder cancer. Lancet 2016;388:2796-810.
3. Iacobucci I, Mullighan CG. Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol 2017;35:975-83.
4. Litwin MS, Tan HJ. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017;317:2532-42.
5. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature 2018;553:446-54.
6. Lim SH, Johnson PWM. Optimizing therapy in advanced-stage Hodgkin lymphoma. Blood 2018;131:1679-88.
7. Pierorazio PM, Albers P, Black PC, Tandstad T, Heidenreich A, et al. Non-risk-adapted Surveillance for Stage I Testicular Cancer: Critical Review and Summary. Eur Urol 2018;73:899-907.
8. Fenton TR, Garrett MD, Wass MN, Michaelis M. What really matters - response and resistance in cancer therapy. Cancer Drug Resist 2018;1:200-3.
10. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26.
11. Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. Optimal Dosing for Targeted Therapies in Oncology: Drug Development Cases Leading by Example. Clin Cancer Res 2016;22:1318-24.
12. Wong KM, Capasso A, Eckhardt SG. The changing landscape of phase I trials in oncology. Nat Rev Clin Oncol 2016;13:106-17.
13. Gatzka MV. Targeted Tumor Therapy Remixed-An Update on the Use of Small-Molecule Drugs in Combination Therapies. Cancers (Basel) 2018;10:pii:E155.
14. Kavanagh S, Nee A, Lipton JH. Emerging alternatives to tyrosine kinase inhibitors for treating chronic myeloid leukemia. Expert Opin Emerg Drugs 2018;23:51-62.
15. Kleczko EK, Heasley LE. Mechanisms of rapid cancer cell reprogramming initiated by targeted receptor tyrosine kinase inhibitors and inherent therapeutic vulnerabilities. Mol Cancer 2018;17:60.
16. Larionov AA. Current Therapies for Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Patients. Front Oncol 2018;8:89.
17. Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, et al. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers (Basel) 2018;10:pii:E62.
18. Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer 2018;17:49.
19. Tomasello C, Baldessari C, Napolitano M, Orsi G, Grizzi G, et al. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit Rev Oncol Hematol 2018;123:149-61.
20. Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther 2013;14:1143-6.
21. Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res 2015;21:2157-66.
22. Miklos W, Pelivan K, Kowol CR, Pirker C, Dornetshuber-Fleiss R, et al. Triapine-mediated ABCB1 induction via PKC induces widespread therapy unresponsiveness but is not underlying acquired triapine resistance. Cancer Lett 2015;361:112-20.
23. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med 2017;23:114-9.
24. Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 2019;20:71-88.
25. Hayes DF. Precision Medicine and Testing for Tumor Biomarkers-Are All Tests Born Equal? JAMA Oncol 2018;4:773-4.
26. Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, et al. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med 2011;9:133.
27. Leighl NB, Rekhtman N, Biermann WA, Huang J, Mino-Kenudson M, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the study of lung cancer/association for molecular pathology guideline. J Clin Oncol 2014;32:3673-9.
28. Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol 2016;2:1014-22.
29. Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol Ther 2017;174:1-21.
30. Nian J, Sun X, Ming S, Yan C, Ma Y, et al. Diagnostic Accuracy of Methylated SEPT9 for Blood-based Colorectal Cancer Detection: A Systematic Review and Meta-Analysis. Clin Transl Gastroenterol 2017;8:e216.
31. Song L, Jia J, Peng X, Xiao W, Li Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci Rep 2017;7:3032.
32. Kalemkerian GP, Narula N, Kennedy EB, Biermann WA, Donington J, et al. Molecular Testing Guideline for the Selection of Patients With Lung Cancer for Treatment With Targeted Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J Clin Oncol 2018;36:911-9.
33. Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer 2010;10:241-53.
34. Gordon JL, Brown MA, Reynolds MM. Cell-Based Methods for Determination of Efficacy for Candidate Therapeutics in the Clinical Management of Cancer. Diseases 2018;6:pii:E85.
35. Dai X, Cheng H, Bai Z, Li J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J Cancer 2017;8:3131-41.
36. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813-23.
37. Goodspeed A, Heiser LM, Gray JW, Costello JC. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol Cancer Res 2016;14:3-13.
38. Geraghty RJ, Capes-Davis A, Davis JM, Downward J, Freshney RI, et al. Guidelines for the use of cell lines in biomedical research. Br J Cancer 2014;111:1021-46.
39. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603-7.
40. Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013;154:1151-61.
41. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955-61.
42. Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016;166:740-54.
43. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503-8.
44. Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019;176:1282-94.e20.
45. van der Meer D, Barthorpe S, Yang W, Lightfoot H, Hall C, et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res 2019;47:D923-29.
46. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152-62.
47. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219-34.
48. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer 2018;18:452-64.
49. Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992;258:1650-4.
50. Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res 2018;139:395-411.
51. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
52. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 2016;22:262-9.
53. Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun 2016;7:10690.
54. Torres-Collado AX, Knott J, Jazirehi AR. Reversal of Resistance in Targeted Therapy of Metastatic Melanoma: Lessons Learned from Vemurafenib (BRAFV600E-Specific Inhibitor). Cancers (Basel) 2018;10:E157.
55. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973-7.
56. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011;480:387-90.
57. Shi H, Moriceau G, Kong X, Lee MK, Lee H, Ket al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 2012;3:724.
58. Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011;471:523-6.
59. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346:1480-6.
60. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 2013;3:1020-9.
61. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov 2013;3:1030-43.
62. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013;13:83-96.
63. Cinatl J Jr, Speidel D, Hardcastle I, Michaelis M. Resistance acquisition to MDM2 inhibitors. Biochem Soc Trans 2014;42:752-7.
64. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 2017;10:133.
65. Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011;30:4678-86.
66. Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug resistant cancer cells. Cell Death Dis 2011;2:e243.
67. Jones RJ, Bjorklund CC, Baladandayuthapani V, Kuhn DJ, Orlowski RZ. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol Cancer Ther 2012;11:2243-53.
68. Michaelis M, Rothweiler F, Agha B, Barth S, Voges Y, et al. Human neuroblastoma cells with acquired resistance to the p53 activator RITA retain functional p53 and sensitivity to other p53 activating agents. Cell Death Dis 2012;3:e294.
69. Hoffman-Luca CG, Ziazadeh D, McEachern D, Zhao Y, Sun W, et al. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin Cancer Res 2015;21:2558-68.
70. Drummond CJ, Esfandiari A, Liu J, Lu X, Hutton C, et al. TP53 mutant MDM2-amplified cell lines selected for resistance to MDM2-p53 binding antagonists retain sensitivity to ionizing radiation. Oncotarget 2016;7:46203-18.
71. Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 2016;7:12609.
72. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012;22:373-88.
73. Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014;511:90-3.
74. Göllner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med 2017;23:69-78.
75. Schneider C, Oellerich T, Baldauf HM, Schwarz SM, Thomas D, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med 2017;23:250-5.
76. Malani D, Murumägi A, Yadav B, Kontro M, Eldfors S, et al. Enhanced sensitivity to glucocorticoids in cytarabine-resistant AML. Leukemia 2017;31:1187-95.
77. Benedetti V, Perego P, Luca Beretta G, Corna E, Tinelli S, et al. Modulation of survival pathways in ovarian carcinoma cell lines resistant to platinum compounds. Mol Cancer Ther 2008;7:679-87.
78. Cossa G, Lanzi C, Cassinelli G, Carenini N, Arrighetti N, et al. Differential outcome of MEK1/2 inhibitor-platinum combinations in platinum-sensitive and -resistant ovarian carcinoma cells. Cancer Lett 2014;347:212-24.
79. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017;168:613-28.
81. Stanta G, Bonin S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front Med (Lausanne) 2018;5:85.
82. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR Inhibitors. Sci Transl Med 2011;3:75ra26.
83. Jun HJ, Acquaviva J, Chi D, Lessard J, Zhu H, et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2012;31:3039-50.
84. Basile KJ, Abel EV, Dadpey N, Hartsough EJ, Fortina P, et al. In vivo MAPK reporting reveals the heterogeneity in tumoral selection of resistance to RAF inhibitors. Cancer Res 2013;73:7101-10.
85. Patel C, Stenke L, Varma S, Lindberg ML, Björkholm M, ey al. Multidrug resistance in relapsed acute myeloid leukemia: evidence of biological heterogeneity. Cancer 2013;119:3076-83.
86. Kemper K, Krijgsman O, Cornelissen-Steijger P, Shahrabi A, Weeber F, et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol Med 2015;7:1104-18.
87. Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, et al. Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res 2015;75:4372-83.
88. Quéré G, Descourt R, Robinet G, Autret S, Raguenes O, et al. Mutational status of synchronous and metachronous tumor samples in patients with metastatic non-small-cell lung cancer. BMC Cancer 2016;16:210.
89. Litzenburger UM, Buenrostro JD, Wu B, Shen Y, Sheffield NC, et al. Single-cell epigenomic variability reveals functional cancer heterogeneity. Genome Biol 2017;18:15.
90. Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One 2008;3:e3077.
91. Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011;30:4678-86.
92. Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal 2011;4:ra17.
93. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 2016;22:262-9.
94. Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun 2017;8:1231.
95. Hata AN, Rowley S, Archibald HL, Gomez-Caraballo M, Siddiqui FM, et al. Synergistic activity and heterogeneous acquired resistance of combined MDM2 and MEK inhibition in KRAS mutant cancers. Oncogene 2017;36:6581-91.
96. Kim C, Gao R, Sei E, Brandt R, Hartman J, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018;173:879-893.e13.
97. Liu Y, Li Y, Ou Q, Wu X, Wang X, et al. Acquired EGFR L718V mutation mediates resistance to osimertinib in non-small cell lung cancer but retains sensitivity to afatinib. Lung Cancer 2018;118:1-5.
98. Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med 2018;24:1167-77.
99. Schulte M, Köster J, Rahmann S, Schramm A. Cancer evolution, mutations, and clonal selection in relapse neuroblastoma. Cell Tissue Res 2018;372:263-8.
100. Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet 2019;51:207-16.
101. Sanchez-Vega F, Hechtman JF, Castel P, Ku GY, Tuvy Y, et al. EGFR and MET Amplifications Determine Response to HER2 Inhibition in ERBB2-Amplified Esophagogastric Cancer. Cancer Discov 2019;9:199-209.
102. Michaelis M, Schneider C, Rothweiler F, Rothenburger T, Mernberger M, et al. TP53 mutations and drug sensitivity in acute myeloid leukaemia cells with acquired MDM2 inhibitor resistance. biorXiv 2018; doi: 10.1101/404475.
103. Su Y, Wei W, Robert L, Xue M, Tsoi J, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A 2017;114:13679-84.
104. Sharma A, Cao EY, Kumar V, Zhang X, Leong HS, et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat Commun 2018;9:4931.
105. Behrens D, Walther W, Fichtner I. Pancreatic cancer models for translational research. Pharmacol Ther 2017;173:146-58.
106. Fichtner I, Klinghammer K, Behrens D, Flechsig S, Rolff J, et al. Animal models for personalized treatment options. Int J Clin Pharmacol Ther 2017;55:698-700.
107. Kodack DP, Farago AF, Dastur A, Held MA, Dardaei L, et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep 2017;21:3298-309.
108. Lampreht Tratar U, Horvat S, Cemazar M. Transgenic Mouse Models in Cancer Research. Front Oncol 2018;8:268.
109. Tellez-Gabriel M, Cochonneau D, Cadé M, Jubellin C, Heymann MF, et al. Circulating Tumor Cell-Derived Pre-Clinical Models for Personalized Medicine. Cancers (Basel) 2018;11:pii: E19.
110. Xu H, Lyu X, Yi M, Zhao W, Song Y, et al. Organoid technology and applications in cancer research. J Hematol Oncol 2018;11:116.
111. Koga Y, Ochiai A. Systematic Review of Patient-Derived Xenograft Models for Preclinical Studies of Anti-Cancer Drugs in Solid Tumors. Cells 2019;8:pii: E418.
112. Ramamoorthy P, Thomas SM, Kaushik G, Subramaniam D, Chastain KM, et al. Metastatic Tumor-in-a-Dish, a Novel Multicellular Organoid to Study Lung Colonization and Predict Therapeutic Response. Cancer Res 2019;79:1681-95.
113. Smith RC, Tabar V. Constructing and Deconstructing Cancers using Human Pluripotent Stem Cells and Organoids. Cell Stem Cell 2019;24:12-24.
114. Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer 2019;19:65-81.
115. Naundorf H, Becker M, Lykkesfeldt AE, Elbe B, Neumann C, et al. Development and characterization of a tamoxifen-resistant breast carcinoma xenograft. Br J Cancer 2000;82:1844-50.
116. Zhang Z, Lee JC, Lin L, Olivas V, Au V, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852-60.
117. Cottu P, Bièche I, Assayag F, El Botty R, Chateau-Joubert S, et al. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin Cancer Res 2014;20:4314-25.
118. Kopp F, Hermawan A, Oak PS, Ulaganathan VK, Herrmann A, et al. Sequential Salinomycin Treatment Results in Resistance Formation through Clonal Selection of Epithelial-Like Tumor Cells. Transl Oncol 2014;7:702-11.
119. Pirazzoli V, Nebhan C, Song X, Wurtz A, Walther Z, et al. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep 2014;7:999-1008.
120. Pyo KH, Lim SM, Kim HR, Sung YH, Yun MR, et al. Establishment of a Conditional Transgenic Mouse Model Recapitulating EML4-ALK-Positive Human Non-Small Cell Lung Cancer. J Thorac Oncol 2017;12:491-500.
121. Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, et al. An approach to suppress the evolution of resistance in BRAFV600E-mutant cancer. Nat Med 2017;23:929-37.
122. Buzzelli JN, Ouaret D, Brown G, Allen PD, Muschel RJ. Colorectal cancer liver metastases organoids retain characteristics of original tumor and acquire chemotherapy resistance. Stem Cell Res 2018;27:109-20.
123. Esteve-Arenys A, Valero JG, Chamorro-Jorganes A, Gonzalez D, Rodriguez V, Dlouhy I, et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018;37:1830-44.
124. Lau WM, Teng E, Huang KK, Tan JW, Das K, et al. Acquired Resistance to FGFR Inhibitor in Diffuse-Type Gastric Cancer through an AKT-Independent PKC-Mediated Phosphorylation of GSK3β. Mol Cancer Ther 2018;17:232-42.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Michaelis M, Wass MN, Cinatl J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist 2019;2:447-56. http://dx.doi.org/10.20517/cdr.2019.005
AMA Style
Michaelis M, Wass MN, Cinatl J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resistance. 2019; 2(3): 447-56. http://dx.doi.org/10.20517/cdr.2019.005
Chicago/Turabian Style
Michaelis, Martin, Mark N. Wass, Jindrich Cinatl. 2019. "Drug-adapted cancer cell lines as preclinical models of acquired resistance" Cancer Drug Resistance. 2, no.3: 447-56. http://dx.doi.org/10.20517/cdr.2019.005
ACS Style
Michaelis, M.; Wass MN.; Cinatl J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447-56. http://dx.doi.org/10.20517/cdr.2019.005
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 20 clicks
Cite This Article 20 clicks

 Like This Article 16
likes
Like This Article 16
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.