
New insights into acquired endocrine resistance of breast cancer

Abstract
The translational research strategy of targeting estrogen receptor α (ERα) positive breast cancer and then using long term anti-hormone adjuvant therapy (5-10 years) has reduced recurrences and mortality. However, resistance continues to occur and improvements are required to build on the success of tamoxifen and aromatase inhibitors (AIs) established over the past 40 years. Further translational research has described the evolution of acquired resistance of breast cancer cell lines to long term estrogen deprivation that parallels clinical experience over years. Additionally, recent reports have identified mutations in the ERα obtained from the recurrences of AI treated patients. These mutations allow the ERα to activate without ligands and auto stimulate metastatic tumor growth. Furthermore, the new biology of estrogen-induced apoptosis in acquired resistant models in vitro and in vivo has been interrogated and applied to clinical trials. Inflammation and stress are emerging concepts occurring in the process of acquired resistance and estrogen-induced apoptosis with different mechanisms. In this review, we will present progress in the understanding of acquired resistance, focus on stress and inflammatory responses in the development of acquired resistance, and consider approaches to create new treatments to improve the treatment of breast cancer with endocrine resistance.
Keywords
Introduction
Estrogen receptor α (ERα) is a critical nuclear transcription factor to mediate cell proliferation and metabolism through binding to its ligand estrogen (E2) in breast cancer. Approximately 70% of breast cancer patients are ER-positive. Thus, targeting ER with tamoxifen or inhibition of E2 synthesis via aromatase inhibitors (AIs) are standard treatments for ER-positive breast cancer[1,2]. The translational strategy of long term anti-hormone adjuvant therapy targeted to ER has saved millions of women’s life due to effectively control the growth of breast cancer[3]. Results from clinical trials demonstrate that the risk of breast cancer relapse is reduced by extending adjuvant tamoxifen therapy from 5 to 10 years[4,5]. Consistently, increasing the duration of adjuvant therapy with an AI letrozole to 10 years decreases recurrence rates and contralateral breast cancer[6], whereas Mamounas et al.[7] observe that 5 years of letrozole therapy does not significantly prolong disease-free survival compared with placebo to patients with early-stage breast cancer. Unfortunately, acquired resistance to anti-hormone therapy is an inevitable challenge in the clinic after prolonged therapies[8,9]. Understanding the mechanisms that underlie this resistance provides an opportunity to develop strategies for overcoming it.
There are three types of resistance to selective estrogen receptor modulators (SERMs): metabolic resistance, de novo resistance, and acquired resistance[8]. Metabolic[10] and de novo[8] resistance have been extensively reviewed and will not be considered further. Acquired resistance to tamoxifen has different biological characteristics according to the treatment time on xenografted mouse model [Figure 1]. Initially, tamoxifen acts as E2 antagonist in breast cancer to inhibit growth during the successful treatment of breast cancer, but then causes tamoxifen-stimulated breast cancer growth after a year or two of treatment which is a unique form of acquired drug resistance[3]. The AIs and fulvestrant can prevent tumor growth in tamoxifen-resistant disease. This phase of drug resistance is referred to as Phase I resistance. In phase II, constant exposure to tamoxifen more than 5 years results in continued tamoxifen-stimulated growth, but E2 induces apoptosis at this stage. Eventually, autonomous growth (Phase III) occurs after indefinite treatment that is unresponsive to fulvestrant or AIs, but E2 still induces apoptosis at this stage [Figure 1]. The distinct features at different phases of acquired resistance suggest that cell populations are clearly being selected over years of therapy so that those cells can adapt and grow in a stressful environment[11].
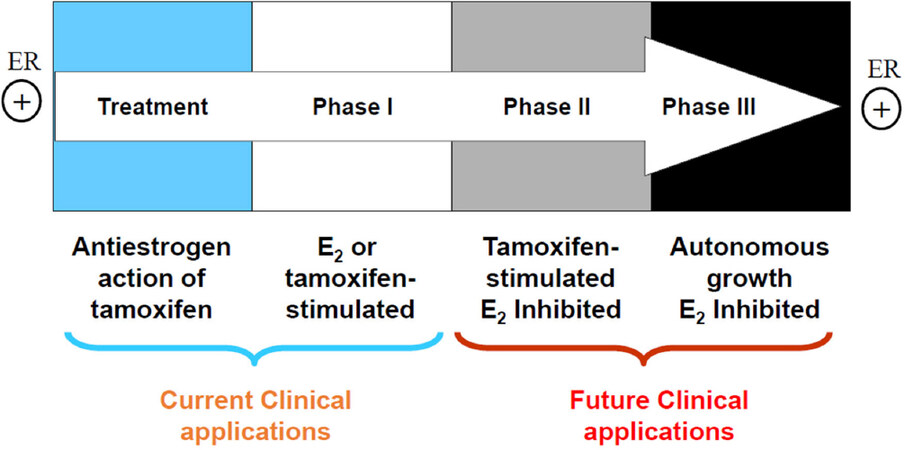
Figure 1. The evolution of resistance to tamoxifen after long term therapy. Phase I acquired resistance develops after a year or two of therapy of estrogen receptor (ER)-positive metastatic breast cancer. Tamoxifen stimulates cell growth, but aromatase inhibitors (AIs) and fulvestrant inhibit cell growth at this stage. Phase II acquired resistance occurs after 5 years of tamoxifen treatment. Tamoxifen continues to stimulate cell growth, but E2 induces apoptosis. Eventually, autonomous growth occurs after indefinite tamoxifen therapy for ER-positive breast cancer, which is referred as Phase III acquired resistance. Cells are unresponsive to AIs or fulvestrant, but E2 still induces apoptosis[11]
Several mechanisms have been proposed to contribute to the acquired endocrine resistance, including activation of growth factor receptor and kinase pathways (e.g., HER-2 and MAPK)[12-14], amplification of transcriptional co-activator proteins (e.g., SRC3)[15], mutations in the ligand-binding domain of ERα[16], and constitutive activation of other inflammation-associated transcription factors such as nuclear factor κB (NF-κB)[17,18]. Particularly, inflammation is now considered a hallmark of cancer and plays a key role in all aspects of tumor biology, including initiation, angiogenesis, resistance, and metastasis[19-21]. Moreover, inflammatory response suppresses the function of ERα, thereby affecting the response to SERMs[21]. Importantly, long term anti-hormone therapy alters the interactions between ERα and other inflammation- and stress-associated transcription factors such as NF-κB and peroxisome proliferator-activated receptor γ (PPARγ)[18,22,23], which ultimately switch the cellular response to E2 from proliferation to apoptosis in vitro[24,25] and in vivo[26,27]. This laboratory discovery has been translated into the clinical trial[28,29].
We will review how stressful anti-hormone therapy alters the function of ERα, including its interactions with membrane-associated molecules, mutations, and crosstalk with nuclear inflammation-associated transcription factors, thereby leading to the endocrine resistance. Furthermore, we will focus on how these inflammation-associated transcription factors modulate subsequent E2-induced apoptosis and find approaches to improve therapeutic effects of E2-induced apoptosis on endocrine resistant breast cancer.
Crosstalk between growth factor receptors and ERα in acquired endocrine resistance
Compelling evidence has demonstrated that several tyrosine kinase receptors, including HER-2[12,13], epithelial growth factor receptor (EGFR)[30,31], and insulin-like growth factor-1 receptor (IGF-1R)[32,33] participate in acquired endocrine resistance in breast cancer. Therefore, the most common strategy for overcoming endocrine resistant breast cancer is inhibition of these tyrosine kinase receptors in breast cancer patients[34,35]. In addition to activating downstream signaling pathways such as PI3K/Akt and MAPK, these tyrosine kinase receptors cross talk with ERα and evade the anti-hormone therapy in breast cancer[12,30-33]. The interaction between ERα and growth factor receptors results in the redistribution of ERα from nucleus to extra nuclear areas and increasing the non-genomic pathway of ERα, which will further activate PI3K/Akt and MAPK pathways[30,32]. In another aspect, ERα can be phosphorylated by activated MAPK and Akt, resulting in ligand-independent transcriptional activity[36,37]. Particularly, aberrant activation of PI3K/Akt by PI3KCA mutations has been implicated in endocrine resistance of breast cancer[38,39]. Additionally, more investigations disclose that activity of non-receptor tyrosine kinase c-Src is increased[40,41] and plays a key role in the mediation of interaction between ERα and growth factor receptor in endocrine resistant breast cancer cells[30]. Thus, targeting c-Src can overcome acquired endocrine resistance[30,42]. Moreover, multiple membrane-associated molecules including focal adhesion molecules, adapter proteins, and growth factor receptors are identified to be activated in SERM-resistant breast cancer cells[32,43][Figure 2]. These active membrane-associated molecules integrally activate downstream signal pathways and lead to be unresponsive to tamoxifen [Figure 2].
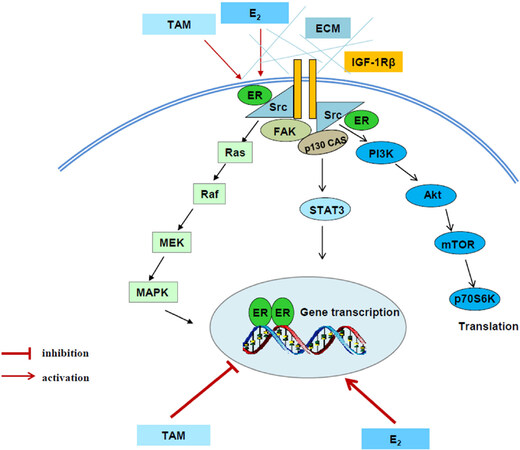
Figure 2. Signal transduction pathways differentially regulated by E2 and tamoxifen in tamoxifen-resistant model. E2 and tamoxifen (TAM) exert differential functions on nuclear estrogen receptor α (ERα). E2 activates classical ER-target genes but TAM acts to block gene activation. Both E2 and TAM increase the non-genomic activity of ERα through membrane-associated molecules such as extracellular matrix (ECM), c-Src, insulin-like growth factor-1 receptor (IGF-1R), and focal adhesion kinase (FAK) to enhance downstream signaling cascades, leading to acquired resistance[32]
Despite activation of multiple membrane-associated molecules, considerable results implicate that ERα is still a major drive of growth utilized by both E2 and SERMs in resistant models in vivo[44,45] and in vitro[46]. In contrast to E2 that activates classical ER-target genes, SERMs continue to act as effective antiestrogens to inhibit classical ER-target genes, even at the time of growth stimulation[32]. This conclusion is supported by our previous results that growth of tumors by tamoxifen or fulvestrant lacks the induction of E2-responsive genes[47]. Other groups also reported similar results that tamoxifen suppresses classical ERE-regulated genes during occurrence of acquired resistance in vitro[48] and in vivo[49]. Consistently, expression of IGF-1R is up-regulated by E2 through ERα but downregulated by tamoxifen[32], which leads to the loss of IGF-1R in the tamoxifen resistant breast cancer[50]. However, phosphorylation of IGF-1R is increased by tamoxifen due to the non-genomic activity of ERα[32]. All of these findings suggest that SERMs consistently inhibit classical ERα transcriptional activity regardless of being SERM sensitive or resistant breast cancer cells. In contrast, ERα tethering pathway such as activator protein-1 (AP-1) family members are activated in tamoxifen resistant breast cancer[51-54]. Apart from interaction with other transcription factors, stress-related kinases such as c-Jun NH2-terminus kinase (JNK) has been documented to activate AP-1 proteins through phosphorylation[53-55]. In line with these results, JNK and p38 have been found to promote acquired resistance in breast cancer[56]. Notably, our findings demonstrate that both JNK and Akt are commonly regulated by IGF-1R in SERM resistant breast cancer cells[57]. All together, these factors including growth factor receptors, stress-associated kinases, and AP-1 family members are activated after anti-hormone therapy, which contribute to SERM-resistance in ER-positive breast cancer.
ERα mutation occurrence in acquired resistant breast cancer
ERα continues to be expressed in the majority of cases with acquired endocrine resistance. However, the function of ERα has been altered after long term endocrine therapies. Recent genetic studies have shown that high frequency of ERα gene (ESR1) mutations (around 20%) occurs in acquired resistant tumors[58], mainly in AI resistant tumors. In contrast, it is extremely rare to find ERα mutations in primary breast cancer[16,58,59], including those matched primary tumors from patients in which ERα mutations are found after progress of endocrine resistance[58]. These results suggest that selective pressures from endocrine therapies are critical for the acquisition of ESR1 mutations[16,58,60]. Remarkably, several ESR1 point mutations identified in acquired resistant breast cancer occur in ER ligand binding domain (LBD)[16,58-63], just a few amino acids within or near the helix 12 region of the LBD. This region has function for undergoing conformational changes during ERα activation[16,58-64]. Amino acid 351 allele mutation was first found in MCF-7 xenografted tumors after long term tamoxifen treatment[64,65]. The majority of other mutation sites are detected in metastatic breast cancer tumors or cell lines after long term endocrine therapies[58,61,62,66-68]. Based on these findings, Tyr537 and Asp538 are the hot spots of ESR1 mutations[58,61,62,66-68]. These single allele mutations do not affect the dimerization of ERα, but they continuously increase the transcriptional activity of ERα[67,68], which result in the loss of response to tamoxifen and fulvestrant. Of note, ERα mutations are also enriched in PI3KCA mutant tumors and most of these point mutations are ERα phosphorylation sites[69]. Tyr537 is a unique site phosphorylated by c-Src[70], which is implicated in hormone binding, dimerization, and hormone-dependent transcriptional activity. Other mutation sites are phosphorylated at serine residues through RAS/MAPK[36] or downstream signal of growth factor receptors[71-73]. It remains unclear whether ERα mutation is related with over activation of kinases after acquired endocrine resistance. Recently, Mao et al.[74] reported that Y537S mutation constitutively increases the unfolded protein response (UPR) with high expression of XBP1 and Bip/GRP78, which are associated with tamoxifen resistance. However, ER biomodulator, BHPI further elicits UPR in breast cancer cells with ERα mutations[74]. This persistent activation of UPR converts cell responses from protection to death, leading to completely inhibit proliferation of breast cancer cells with ERα mutations[74,75]. Moreover, some novel antiestrogens or selective estrogen receptor down-regulators are developed to overcome acquired resistance caused by ERα mutations[76-78]. All of these results highlight the importance and functional consequence of ERα mutations and provide an important resource for studying endocrine resistance of breast cancer.
Alteration of interaction between ERα and inflammation-associated transcription factors after acquired resistance
In addition to the critical role in female reproduction, E2 directly modulates lipid metabolism and the function of mitochondria, thereby influencing adipocyte differentiation and energy homeostasis[79-82]. Thus, E2 deficiency caused by menopause or anti-hormone therapies results in metabolic stress, demonstrating fat redistribution and insulin resistance[80-83]. Specifically, fatty acid and cholesterol metabolism are increased after endocrine therapy or menopause in breast cancer cells, along with abnormal activation of cytokines locally and distally[83,84]. Both inflammatory factors and lipid metabolism regulators [such as PPARγ, sterol regulatory element-binding protein 1(SREBP1), and CCAAT/enhancer binding protein β (C/EBP β)] have been identified to result in acquired resistance in breast cancer[84-88]. In particular, PPARγ is a master adipocyte modulator to affect the lipid and energy metabolism[89,90], which function is closely related with the levels of E2[23,86,91]. Many observations have demonstrated that a bidirectional crosstalk exists between ERα and PPARγ in the regulation of proliferation, differentiation, metabolism, and inflammation in breast cancer[92-95]. Additionally, NF-κB is another key transcription factor in responsible for inflammation and acquired resistance in ER-positive breast cancer[96-98]. Long term endocrine therapy is inclined to create an inflammatory microenvironment in breast cancer[99]. It has been reported that cytokines and chemokines released in the inflammatory environment activate NF-κB-associated pathways that desensitize cell response to SERMs[99]. Therefore, repression of NF-κB activity can restore sensitivity to ERα antagonists[100]. An inverse relationship between ERα and NF-κB has been observed in the development of endocrine resistant breast cancer[101,102]. E2 has a potential to suppress the activation of NF-κB[18]. However, long term anti-hormone therapy alters the function of ERα in the regulation of metabolism and inflammation[103] that results in the constitutive activation of NF-κB[18]. Apart from interaction with ERα, the activity of NF-κB is suppressed by C/EBPβ and PPARγ[18,23,104,105]. Particularly, trans-suppression of NF-κB by the PPARγ agonist is a major mechanism underlying inhibition of inflammation and acceleration of insulin sensitivity in the clinic[106,107]. Furthermore, the inflammatory factor is able to reprogram the motifs on ERα binding sites on chromatin which is closely associated with the endocrine resistance in breast cancer[108,109]. These results demonstrate that long term anti-hormone therapy alters the function of ERα and its interaction with inflammation-associated transcription factors that results in the endocrine resistance in breast cancer.
E2-induced apoptosis in acquired resistant breast cancer
The primary purpose of anti-hormone therapy is to prevent E2 from binding to ERα (SERMs) or inhibit synthesis of E2 (AIs) in ER-positive breast cancer, thereby blocking the proliferation of breast cancer cells. Paradoxically, administration of physiological concentrations of E2 can induce apoptosis in acquired resistant breast cancer in vitro[24,25] and in vivo[26,27]. This scientific discovery has been used in the clinical trial for the treatment of aromatase inhibitor resistant breast cancer patients with 30% of benefit[29]. This rationale is also been used to interpret why E2 alone hormone replacement therapy (HRT) can reduce the incidence of breast cancer in hysterectomized postmenopausal women[110]. Our further clinically relevant findings disclose that the synthetic progestin medroxyprogesterone acetate for the classical HRT in combination with E2 has glucocorticoid activity that is able to block E2-induced apoptosis[111], implicating that inflammation and stress are involved in the apoptosis induced by E2[112-115]. In support with this view, it has been reported that long term anti-hormone therapy is stressful for breast cancer, along with activation of multiple stress- and inflammation-associated transcription factors and pathways[18,23,57,116-119]. Furthermore, E2 treatment widely activates stress responses including endoplasmic reticulum stress, oxidative stress, and inflammatory stress in long term estrogen deprivation (LTED) breast cancer cells[117,118]. Among these stress responses, the endoplasmic reticulum is a critical regulatory site for conveying signals between the nucleus and cytoplasm to induce apoptosis[18,118]. Three sensors of endoplasmic reticulum stress are activated but perform different functions after E2 treatment. One of these sensors, protein kinase RNA-like endoplasmic reticulum kinase (PERK) is responsible for homeostasis of unfolded proteins and plays a critical role in E2-induced apoptosis[57,118]. The other two sensors, inositol-requiring protein 1 alpha (IRE1α) and ATF-6, mainly mediate endoplasmic reticulum-associated degradation of phospholipids[57]. Differential functions of the endoplasmic reticulum stress sensors suggest that abnormal protein folding and lipid metabolism occur after exposure to E2 in LTED breast cancer cells[23,57].
How ERα triggers stress responses remain unclear. Although E2-induced stress utilizes c-Src tyrosine kinase[46,118], a well-known molecule to mediate non-genomic pathway of ERα[30,32], it is confirmed that nuclear ERα mediates E2-induced stress and apoptosis, but not non-genomic effects of ERα[118]. The fact that PERK activation is crucial for E2-induced apoptosis suggests an accumulation of unfolded proteins in the endoplasmic reticulum after E2 treatment[57,118]. Most likely, E2 increases the expression of some short half-life nuclear proteins such as AP-1 family member c-Fos[120], which is increased by E2 and leads to rapid increase the misfolded protein in the endoplasmic reticulum[121]. Our recent findings demonstrate that PERK kinase increases DNA-binding activity of signal transducer and activator of transcription 3 which promotes NF-κB translocation to nucleus and activation of NF-κB-dependent tumor necrosis factor α (TNFα)[18,22]. The PERK/NF-κB/TNFα axis is identified as the key drive to induce apoptosis after E2 treatment of LTED breast cancer cells[18,22][Figure 3]. In addition of regulating apoptosis, NF-κB is a critical molecule to mediate metabolism, stress, inflammation, and proliferation, depending on the context of cells[122]. It also has a close crosstalk with other transcription factors to regulate inflammatory responses[102]. For example, nuclear receptor PPARγ suppresses the function of NF-κB in a variety of cells[103,105]. Our results show that PPARγ agonist suppresses the NF-κB DNA-binding activity and blocks E2-induced apoptosis in LTED breast cancer cells[23]. In contrast, antagonist of PPARγ inhibits breast cancer cell growth[23,123,124] and increases E2-induced apoptosis via regulation of oxidative stress and NF-κB-dependent TNFα expression[23]. Furthermore, the mechanism underlying glucocorticoids blockade of E2-induced apoptosis is also mediated by selective suppression of NF-κB DNA-binding activity and subsequent inhibition of TNFα expression in LTED breast cancer cells[121]. Thus, nuclear activity of NF-κB can be modulated by multiple other transcription factors to affect therapeutic effects of E2-induced apoptosis in anti-hormone resistant breast cancer [Figure 4]. These findings also imply that biological function of ERα is affected by several other transcription factors, depending on the ligands present in the nucleus.
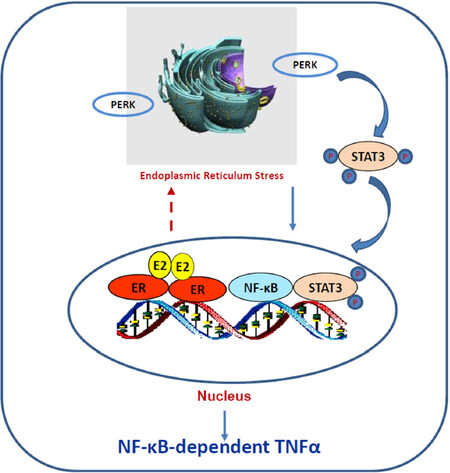
Figure 3. The protein kinase RNA-like endoplasmic reticulum kinase (PERK)/nuclear factor κB (NF-κB)/tumor necrosis factor α (TNFα) axis is activated by E2 to induce apoptosis in long term estrogen deprivation breast cancer cells. E2 activates nuclear estrogen receptor α (ERα) and accumulates unfolded proteins in the endoplasmic reticulum, which activates PERK in response to the misfolded proteins. This stress kinase phosphorylates signal transducer and activator of transcription 3 (STAT3) and increases its DNA-binding activity. Subsequently, activated STAT3 promotes NF-κB DNA binding and induction of TNFα expression[22]
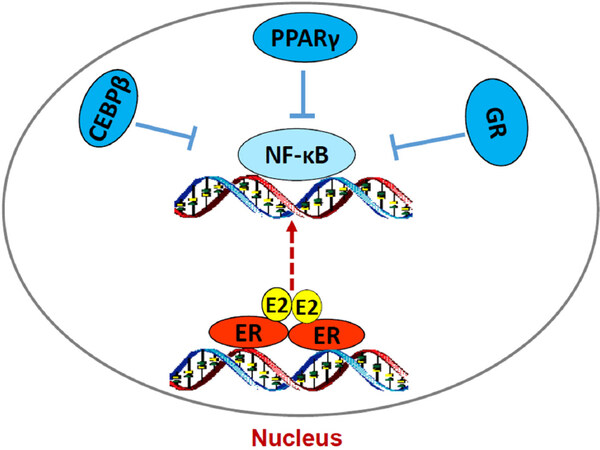
Figure 4. Regulation of nuclear factor κB (NF-κB) DNA-binding activity by other transcription factors in acquired resistant breast cancer cells. E2/estrogen receptor α (ERα) activates endoplasmic reticulum stress and subsequently increases NF-κB DNA-binding activity. However, lipid metabolism-associated transcription factors CCAAT/enhancer binding protein β (C/EBP β) and peroxisome proliferator-activated receptor γ (PPARγ) and inflammation modulator GR all suppress the DNA-binding activity of NF-κB, thereby inhibition of E2-induced apoptosis in long term estrogen deprivation breast cancer cells
Conclusion
In summary, long term anti-hormone therapy is a stress pressure for ER-positive breast cancer cells that selects cell populations for the adaptation to the treatment[11]. During adaptation, multiple stress- and inflammation-associated transcription factors and pathways are activated and participate in the promotion of acquired endocrine resistance. Simultaneously, these stress and inflammatory responses create a microenvironment facilitating E2-induced apoptosis in the acquired resistant breast cancer cells. Remarkably, ERα is the initial target for the endocrine therapy whereas it is also the initial site to trigger apoptosis. Although ERα continues to be expressed, the interactions between ERα and stress- and inflammation-associated transcription factors such as NF-κB and PPARγ are altered when resistance occurs[18,23]. The ultimate effects of stress and inflammation can either promote proliferation or induce apoptosis, depending on the context of tumor cells[96,125,126]. Generally, persistent stress makes cell susceptible to apoptosis[127]. Therefore, many novel compounds are designed to manipulate stress responses for the therapy of diseases[74,127]. Collectively, the discovery of E2-induced apoptosis not only has clinical relevance to treat aromatase inhibitor-resistant breast cancer and reduce breast cancer incidence in postmenopausal women[29,110], but also a general principal has emerged to understand sex steroid-induced apoptosis in long-term androgen deprived prostate cancer[128]. Ongoing strategic studies in our laboratory are addressing the mechanisms underlying sex steroid-induced apoptosis in a range of models of normal and cancer cells.
Declarations
Authors’ contributionsGenerated conception: Jordan VC, Fan P
Wrote the manuscript: Fan P
Revised the manuscript: Jordan VC
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the NIH/NCI (P30-CA016672) (Peter Pisters), Susan G. Komen for the Cure Foundation (SAC100009) to Jordan VC, Cancer Prevention Research Institute of Texas (CPRIT) for the STARs and STARs plus Awards to Jordan VC, the George and Barbara Bush Foundation for Innovative Cancer Research to Jordan VC, and the benefactors of the Dallas/Fort Worth Living Legend Chair of Cancer Research to Jordan VC.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 2007;72:7-25.
2. Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res 2003;9:1980-9.
3. Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell 2004;5:207-13.
4. Davies C, Pan H, Godwin J, Gray R, Arriagada R, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805-16.
5. Pan H, Gray R, Braybrooke J, Davies C, Taylor C, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med 2017;377:1836-46.
6. Goss PE, Ingle JN, Pritchard KI, Robert NJ, Muss H, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209-19.
7. Mamounas EP, Bandos H, Lembersky BC, Jeong JH, Geyer CE Jr, et al. Use of letrozole after aromatase inhibitor-based therapy in postmenopausal breast cancer (NRG Oncology/NSABP B-42): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019;20:88-99.
8. Jordan VC, O’Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol 2007;25:5815-24.
9. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 2009;9:631-43.
10. Brauch H, Schwab M. Prediction of tamoxifen outcome by genetic variation of CYP2D6 in post-menopausal women with early breast cancer. Br J Clin Pharmacol 2014;77:695-703.
11. Fan P, Jordan VC. Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations. Steroids 2014;90:44-52.
12. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 2004;96:926-35.
13. Sabnis GJ, Jelovac D, Long B, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res 2005;65:3903-10.
14. Martin LA, Ghazoui Z, Weigel MT, Pancholi S, Dunbier A, et al. An in vitro model showing adaptation to long-term oestrogen deprivation highlights the clinical potential for targeting kinase pathways in combination with aromatase inhibition. Steroids 2011;76:772-6.
15. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003;95:353-61.
16. Jordan VC, Curpan R, Maximov PY. Estrogen receptor mutations found in breast cancer metastases integrated with the molecular pharmacology of selective ER modulators. J Natl Cancer Inst 2015;107:djv075.
17. Frasor J, El-Shennawy L, Stender JD, Kastrati I. NF-κB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol Cell Endocrinol 2015;418:235-9.
18. Fan P, Tyagi AK, Agboke FA, Mathur R, Pokharel N, et al. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov 2018;4:15.
21. Murray JI, West NR, Murphy LC, Watson PH. Intratumoural inflammation and endocrine resistance in breast cancer. Endocr Relat Cancer 2015;22:R51-67.
22. Fan P, Jordan VC. How PERK kinase conveys stress signals to nuclear factor-κB to mediate estrogen-induced apoptosis in breast cancer cells? Cell Death Dis 2018;9:842.
23. Fan P, Abderrahman B, Chai TS, Yerrum S, Jordan VC. Targeting peroxisome proliferator-activated receptor γ to increase estrogen-induced apoptosis in estrogen-deprived breast cancer cells. Mol Cancer Ther 2018;17:2732-45.
24. Song RX, Mor G, Naftolin F, McPherson RA, Song J, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17 beta-estradiol. J Natl Cancer Inst 2001;93:1714-23.
25. Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst 2005;97:1746-59.
26. Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res 2000;6:2028-36.
27. Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst 2003;95:1586-97.
28. Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer 2015;22:R1-31.
29. Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, et al. Lower-dose vs. high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA 2009;302:774-80.
30. Fan P, Wang J, Santen RJ, Yue W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res 2007;67:1352-60.
31. Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, et al. Bidirectional cross talk between ERalpha and EGFR signaling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat 2006;96:131-46.
32. Fan P, Agboke FA, Cunliffe HE, Ramos P, Jordan VC. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur J Cancer 2014;50:2866-76.
33. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERα through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol 2011;25:516-28.
34. Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res 2011;17:1147-59.
35. Verma S, Miles D, Gianni L, Krop IE, Welslau M, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783-91.
36. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995;270:1491-4.
37. Li SY, Rong M, Grieu F, Iacopetta B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Res Treat 2006;96:91-5.
38. Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120:2406-13.
39. Szijgyarto Z, Flach KD, Opdam M, Palmieri C, Linn SC, et al. Dissecting the predictive value of MAPK/AKT/estrogen-receptor phosphorylation axis in primary breast cancer to treatment response for tamoxifen over exemestane: a Translational Report of the Intergroup Exemestane Study (IES)-PathIES. Breast Cancer Res Treat 2019;175:149-63.
40. Hiscox S, Morgan L, Green TP, Barrow D, Gee J, et al. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat 2006;97:263-74.
41. Zhao Y, Planas-Silva MD. Mislocalizaion of cell-cell adhesion complexes in tamoxifen-resistant breast cancer cells with elevated c-Src tyrosine kinase activity. Cancer Lett 2009;275:204-12.
42. Hiscox S, Jordan NJ, Smith C, James M, Morgan L, et al. Dual targeting of SRC and ER prevents acquired antihormone resistance in breast cancer cells. Breast Cancer Res Treat 2009;115:57-67.
43. Fan P, Cunliffe HE, Griffith OL, Agboke FA, Ramos P, et al. Identification of gene regulation patterns underlying both oestrogen- and tamoxifen-stimulated cell growth through global gene expression profiling in breast cancer cells. Eur J Cancer 2014;50:2877-86.
44. Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res 1988;48:5183-7.
45. Gottardis MM, Jiang SY, Jeng MH, Jordan VC. Inhibition of tamoxifen-stimulated growth of an MCF-7 tumor variant in athymic mice by novel steroidal antiestrogens. Cancer Res 1989;49:4090-3.
46. Fan P, Agboke FA, McDaniel RE, Sweeney EE, Zou X, et al. Inhibition of c-Src blocks oestrogen-induced apoptosis and restores oestrogen-stimulated growth in long-term oestrogen-deprived breast cancer cells. Eur J Cancer 2014;50:457-68.
47. Osipo C, Meeke K, Cheng D, Weichel A, Bertucci A, et al. Role for HER2/neu and HER3 in fulvestrant-resistant breast cancer. Int J Oncol 2007;30:509-20.
48. Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, et al. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat 2003;81:81-93.
49. Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 2008;68:826-33.
50. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res 2012;72:3372-80.
51. Malorni L, Giuliano M, Migliaccio I, Wang T, Creighton CJ, et al. Blockade of AP-1 potentiates endocrine therapy and overcomes resistance. Mol Cancer Res 2016;14:470-81.
52. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007;7:59.
53. Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst 2000;92:1926-34.
54. Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, et al. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res 1999;5:251-6.
56. Aesoy R, Sanchez BC, Norum JH, Lewensohn R, Viktorsson K, et al. An autocrine VEGF/VEGFR2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol Cancer Res 2008;6:1630-8.
57. Fan P, Cunliffe HE, Maximov PY, Agboke FA, McDaniel RE, et al. Integration of downstream signals of insulin-like growth factor-1 receptor by endoplasmic reticulum stress for estrogen-induced growth or apoptosis in breast cancer cells. Mol Cancer Res 2015;13:1367-76.
58. Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017;36:2286-96.
59. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61-70.
60. Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions and cell free DNA of breast cancer patients. Clin Cancer Res 2015;22:1130-7.
61. Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun 2017;8:1865.
62. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446-51.
63. Toy W, Shen Y, Won H, Green B, Sakr RA, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439-45.
64. Catherino WH, Wolf DM, Jordan VC. A naturally occurring estrogen receptor mutation results in increased estrogenicity of a tamoxifen analog. Mol Endocrinol 1995;9:1053-63.
65. Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res Treat 1994;31:129-38.
66. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 2013;73:6856-64.
67. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 2014;20:1757-67.
68. Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer research 1997;57:1244-9.
69. Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016;7:11579.
70. Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol 1995;9:24-33.
71. Jin K, Park S, Teo WW, Korangath P, Cho SS, et al. HOXB7 is an ERα cofactor in the activation of HER2 and multiple ER target genes leading to endocrine resistance. Cancer Discov 2015;5:944-59.
72. Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120:2406-13.
74. Mao C, Livezey M, Kim JE, Shapiro DJ. Antiestrogen resistant cell lines expressing estrogen receptor α mutations upregulate the unfolded protein response and are killed by BHPI. Sci Rep 2016;6:34753.
75. Andruska ND, Zheng X, Yang X, Mao C, Cherian MM, et al. Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc Natl Acad Sci U S A 2015;112:4737-42.
76. Zhao Y, Laws MJ, Guillen VS, Ziegler Y, Min J, et al. Structurally novel antiestrogens elicit differential responses from constitutively active mutant estrogen receptors in breast cancer cells and tumors. Cancer Res 2017;77:5602-13.
77. Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem 2015;58:4888-904.
78. De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, et al. Optimization of a novel binding motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetra hydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J Med Chem 2015;58:8128-40.
79. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 1999;340:1801-11.
80. Torres MJ, Kew KA, Ryan TE, Pennington ER, Lin CT, et al. 17β-Estradiol directly lowers mitochondrial membrane microviscosity and improves bioenergetic function in skeletal muscle. Cell Metab 2018;27:167-79.
81. Gupte AA, Pownall HJ, Hamilton DJ. Estrogen: an emerging regulator of insulin action and mitochondrial function. J Diabetes Res 2015;2015:916585.
82. Maniu A, Aberdeen GW, Lynch TJ, Nadler JL, Kim SO, et al. Estrogen deprivation in primate pregnancy leads to insulin resistance in offspring. J Endocrinol 2016;230:171-83.
83. Misso ML, Jang C, Adams J, Tran J, Murata Y, et al. Differential expression of factors involved in fat metabolism with age and the menopause transition. Maturitas 2005;51:299-306.
84. Ribas R, Ghazoui Z, Gao Q, Pancholi S, Rani A, et al. Identification of chemokine receptors as potential modulators of endocrine resistance in oestrogen receptor-positive breast cancers. Breast Cancer Res 2014;16:447.
85. Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell 2017;65:1122-35.
86. Du T, Sikora MJ, Levine KM, Tasdemir N, Riggins RB, et al. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res 2018;20:106.
87. Skildum A, Dornfeld K, Wallace K. Mitochondrial amplification selectively increases doxorubicin sensitivity in breast cancer cells with acquired antiestrogen resistance. Breast Cancer Res Treat 2011;129:785-97.
88. Choi HK, Yang JW, Roh SH, Han CY, Kang KW. Induction of multidrug resistance associated protein 2 in tamoxifen-resistant breast cancer cells. Endocr Relat Cancer 2007;14:293-303.
90. Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest 2015;125:3461-76.
91. Weigt C, Hertrampf T, Kluxen FM, Flenker U, Hülsemann F, et al. Molecular effects of ER alpha- and beta-selective agonists on regulation of energy homeostasis in obese female Wistar rats. Mol Cell Endocrinol 2013;377:147-58.
92. Suzuki T, Hayashi S, Miki Y, Nakamura Y, Moriya T, et al. Peroxisome proliferator-activated receptor gamma in human breast carcinoma: a modulator of estrogenic actions. Endocr Relat Cancer 2006;13:233-50.
93. Yin Y, Yuan H, Zeng X, Kopelovich L, Glazer RI. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res 2009;69:687-94.
94. Talbert DR, Allred CD, Zaytseva YY, Kilgore MW. Transactivation of ERalpha by rosiglitazone induces proliferation in breast cancer cells. Breast Cancer Res Treat 2008;108:23-33.
95. Llopis J, Westin S, Ricote M, Wang Z, Cho CY, et al. Ligand-dependent interactions of coactivators steroid receptor coactivator-1 and peroxisome proliferator-activated receptor binding protein with nuclear hormone receptors can be imaged in live cells and are required for transcription. Proc Natl Acad Sci USA 2000;97:4363-8.
96. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol 2011;12:715-23.
97. Shah KN, Wilson EA, Malla R, Elford HL, Faridi JS. Targeting ribonucleotide reductase M2 and NF-κB activation with Didox to circumvent tamoxifen resistance in breast cancer. Mol Cancer Ther 2015;14:2411-21.
98. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, et al. Enhanced NF-κB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007;14:59-74.
99. Baumgarten SC, Frasor J. Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol 2012;26:360-71.
100. Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006;124:615-29.
101. Sas L, Lardon F, Vermeulen PB, Hauspy J, Van Dam P, et al. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res 2012;14:212.
102. De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 2006;25:6868-86.
103. Cvoro A, Tzagarakis-Foster C, Tatomer D, Paruthiyil S, Fox MS, et al. Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol Cell 2006;21:555-64.
104. Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998;391:82-6.
105. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79-82.
106. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 2014;20:573-91.
107. Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A 2009;106:22504-9.
108. Franco HL, Nagari A, Kraus WL. TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell 2015;58:21-34.
109. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481:389-93.
110. Anderson GL, Chlebowski RT, Aragaki AK, Kuller LH, Manson JE, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal hysterectomy: extended follow-up of the Women’s Health Initiative Randomised Trial. Lancet Oncol 2012;13:476-86.
111. Sweeney EE, Fan P, Jordan VC. Molecular modulation of estrogen-induced apoptosis by synthetic progestins in hormone replacement therapy: an insight into the women’s health initiative study. Cancer Res 2014;74:7060-8.
112. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005;353:1711-23.
113. De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol 2010;10:497-504.
114. Oh KS, Patel H, Gottschalk RA, Lee WS, Baek S, et al. Anti-inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity 2017;47:298-309.
115. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest 2012;122:2130-40.
116. Miller WR, Larionov A. Molecular effects of oestrogen deprivation in breast cancer. Mol Cell Endocrinol 2011;340:127-36.
117. Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A 2011;108:18879-86.
118. Fan P, Griffith OL, Agboke FA, Anur P, Zou X, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer Res 2013;73:4510-20.
119. Sweeney EE, Fan P, Jordan VC. Mechanisms underlying differential response to estrogen-induced apoptosis in long-term estrogen-deprived breast cancer cells. Int J Oncol 2014;44:1529-38.
120. Ferrara P, Andermarcher E, Bossis G, Acquaviva C, Brockly F, et al. The structural determinants responsible for c-Fos protein proteasomal degradation differ according to the conditions of expression. Oncogene 2003;22:1461-74.
121. Fan P, Siwak DR, Abderrahman B, Agboke FA, Yerrum S, et al. Suppression of Nuclear Factor-κB by Glucocorticoid Receptor Blocks Estrogen-induced Apoptosis in Estrogen-deprived Breast Cancer Cells. Mol Cancer Ther. Forthcoming 2019.
122. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008;454:455-62.
123. Zaytseva YY, Wang X, Southard RC, Wallis NK, Kilgore MW. Down-regulation of PPARgamma1 suppresses cell growth and induces apoptosis in MCF-7 breast cancer cells. Mol Cancer 2008;7:90.
124. Wang X, Sun Y, Wong J, Conklin DS. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene 2013;32:5512-21.
125. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 2013;339:286-91.
126. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010;107:8788-93.
127. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov 2013;12:703-19.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fan P, Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist 2019;2:198-209. http://dx.doi.org/10.20517/cdr.2019.13
AMA Style
Fan P, Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resistance. 2019; 2(2): 198-209. http://dx.doi.org/10.20517/cdr.2019.13
Chicago/Turabian Style
Fan, Ping, V. Craig Jordan. 2019. "New insights into acquired endocrine resistance of breast cancer" Cancer Drug Resistance. 2, no.2: 198-209. http://dx.doi.org/10.20517/cdr.2019.13
ACS Style
Fan, P.; Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019, 2, 198-209. http://dx.doi.org/10.20517/cdr.2019.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 4
likes
Like This Article 4
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.