
Pharmacogenetics of thiopurines

Abstract
Polychemotherapeutic protocols for the treatment of pediatric acute lymphoblastic leukemia (ALL) always include thiopurines. Specific approaches vary in terms of drugs, dosages and combinations. Such therapeutic schemes, including risk-adapted intensity, have been extremely successful for children with ALL who have reached an outstanding 5-year survival of greater than 90% in developed countries. Innovative drugs such as the proteasome inhibitor bortezomib and the bi-specific T cell engager blinatumomab are available to further improve therapeutic outcomes. Nevertheless, daily oral thiopurines remain the backbone maintenance or continuation therapy. Pharmacogenetics allows the personalization of thiopurine therapy in pediatric ALL and clinical guidelines to tailor therapy on the basis of genetic variants in TPMT and NUDT15 genes are already available. Other genes of interest, such as ITPA and PACSIN2, have been implicated in interindividual variability in thiopurines efficacy and adverse effects and need additional research to be implemented in clinical protocols. In this review we will discuss current literature and clinical guidelines available to implement pharmacogenetics for tailoring therapy with thiopurines in pediatric ALL.
Keywords
Introduction
Thiopurines such as mercaptopurine and thioguanine (MP and TG, Figure 1A and B respectively) are lympholytic drugs used in all phases of therapy for acute lymphoblastic leukemia (ALL), with MP being part of the mainstay maintenance therapy[1]; instead, the MP prodrug azathioprine (AZA, Figure 1C) is employed in non-malignant conditions such as inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC) during the maintenance phase of treatment[2].
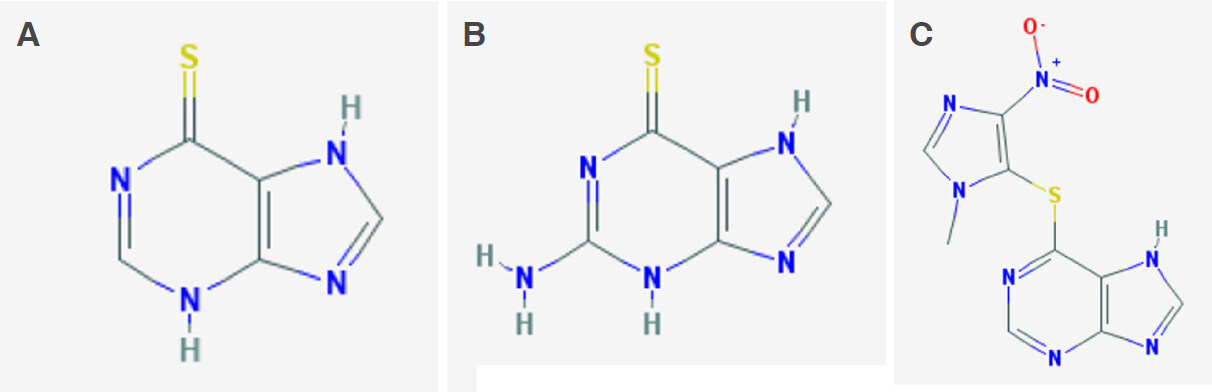
Figure 1. Thiopurines chemical structure. A: thioguanine; B: mercaptopurine; C: azathioprine
Thiopurines are antimetabolites similar in structure to purines: in particular MP is an analogue of hypoxanthine and TG of guanine. In cells, thiopurines undergo complex anabolic and catabolic processes. The anabolic pathway produces thionucleotides (TGN), including thioguanosine mono-, di-, tri-phosphate (tGMP, tGDP, tGTP) and deoxythioguanosine mono-, di-, tri-phosphate (tdGMP, tdGDP, tdGTP), associated with therapeutic efficacy. The conversion of MP to tdGMP/tGMP involves the consecutive action of the enzymes of salvage pathway of nucleotides biosynthesis, in particular hypoxanthine phosphoribosyltransferase 1 (HPRT1), inositol monophosphate dehydrogenase (IMPDH) and guanosine monophosphate synthetase (GMPS), whereas TG is directly converted by HPRT1 in a single step reaction [Figure 2]. The resulting tdGTP/tGTP thionucleotides antagonize the incorporation of canonical dGTP and GTP into DNA and RNA, thus impairing DNA and RNA polymerases and subsequently inducing cell-cycle arrest and apoptosis because of altered DNA, RNA and protein synthesis[3]. Furthermore, the cytotoxic action of these drugs in lymphoid cells is implemented by additional mechanisms of action such as the inhibition of the purine de novo synthesis pathway and tGTP-mediated inhibition of Ras-related C3 botulinum toxin substrate 1 (Rac-1), a GTPase of the Rho family[3]. Catabolic pathways of thiopurines are mediated by the enzymes xanthine oxidase (XO) and thiopurine methyltransferase (TPMT). The extensive first-pass metabolism of the drug by XO in the liver and intestinal mucosa is responsible for the low bioavailability of oral MP (less than 20%) and generates the main inactive metabolite, 6-thiouric acid, excreted in the urine[4]. Indeed, it is known that allopurinol, a structural isomer of hypoxanthine and a XO inhibitor, influences thiopurine pharmacokinetics and promotes TGN production[4]. MP and TG are also converted into methylmercaptopurine (MMP) derivatives by TPMT. This reaction can occur also in cells different from hepatocytes, since TPMT is ubiquitously expressed. MMP is not converted to nucleotides, as it is a poor HPRT substrate and has no antileukemic activity[5]. The synthesis of MMP therefore is in competition with the anabolic pathway of thiopurines. TPMT catalyses the S-methylation also of intermediate thionucleotides leading to TGN, producing secondary methylated nucleotides (MMPN), with potential cytotoxic activity through the inhibition of de novo purine synthesis. The balance between TGN and MMPN has been related to thiopurines response and cytotoxicity[6]. In IBD, TGN concentrations between 230 and 450 pmol/8 × 108 red blood cells (RBC) are associated with the therapeutic index and clinical efficacy, while higher TGN concentrations have been related to myelosuppression and other severe complications such as infections[7]; concentrations of MMPN above 5,700 pmol/8 × 108 RBC have been associated with hepatotoxicity[8]. To authors’ knowledge, there is no general consensus for the range of TGN/MMPN plasma concentrations that should be achieved for drug efficacy and toxicity in ALL: some clinical protocols suggest MP dose reduction with TGN concentration above 1000 pmol/8 × 108 RBC in order to avoid excessive toxicity[9]. Thiopurine active metabolites persist in the blood for a long time in contrast to MP plasma concentration that rapidly declines after oral administration (MP serum peak is achieved within 2 h, half-life ranges from 20 to 120 min according to patients age and drug formulation): TGN are detected in RBC by the 3rd day of treatment and reach the steady state around the 3rd week; in IBD patients, clinical response to thiopurines is further delayed and appears between weeks 8 and 17 of therapy making these drugs unsuitable to achieve early disease remission but useful in long-term maintenance treatments[10]. TGN are routinely detected in RBC rather than in leukocytes, although RBC lack IMPDH and have therefore a thiopurine metabolism different from with that of principal target cells of these drugs. However, erythrocyte TGN levels have been found to correlate to leukocyte TGN levels in pediatric patients[11,12].
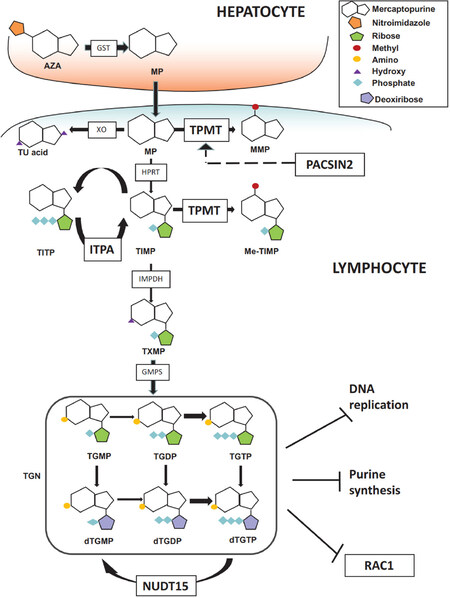
Figure 2. Thiopurine pathway. MP: mercaptopurine; MMP: methyl-mercaptopurine; TIMP: thioinosine monophosphate; Me-TIMP: methyl-thioinosine monophosphate; TGDP: thioguanine diphosphate; TGMP: thioguanine monophosphate; TGTP: thioguanine triphosphate; TITP: thioinosinetriphosphate; Me-TITP: methylthioinosinetriphosphate; TGN: thioguanine nucleotide; TU acid: thiouric acid; TXMP: thioxantine monophosphate; AZA: azathioprine; IMPDH: Inosine-5′-monophosphate dehydrogenase; ITPA: inosine triphosphate pyrophosphatase; GMPS: GMP synthase; GST: glutathione-transferase; NUDT15: nucleotide triphosphate diphosphatase gene; PACSIN2: Protein kinase C and casein kinase substrate in neurons protein 2; TPMT: thiopurine methyltransferase; XO: xanthine oxidase. Hatched arrow indicates the infleunce of PACSIN2 on TPMT gene expression and TPMT activity
In ALL, thiopurines are always used in the context of a polychemotherapeutic protocol that comprises glucocorticoids, vincristine, asparaginase, cytarabine, anthracyclines, cyclophosphamide and tyrosine-kinase inhibitors such as imatinib; in particular, MP is used in combination with methotrexate[13-15]. Specific ALL polychemotherapeutic approaches differ across worldwide protocols in terms of drugs, dosages and combinations; however, they consistently share the therapeutic scheme with an initial remission-induction phase to eradicate the burden of tumoral cells and restore normal hematopoiesis, followed by a remission-consolidation therapy and then by a long-term maintenance regimen (up to 24 months after diagnosis) to eliminate minimal residual leukemic cells. Such therapeutic scheme, combined with a risk-adapted intensive polychemotherapy, has been extremely successful for children with ALL who have reached an outstanding 5-year survival greater than 90% in developed countries. Two of the currently running main clinical trials in childhood ALL, the European AIEOP-BFM (Associazione Italiana di Ematologia ed Oncologia Pediatrica-Berlin-Frankfurt-Munster) 2017 protocol and the Total Therapy XVII (TOTAL XVII) used at the St. Jude Children’s Research Hospital (SJCRH) in Memphis, USA (ClinicalTrials.gov identifiers: NCT03643276 and NCT03117751, respectively http://clinicaltrials.gov) are aimed at further improving cure rate and quality of life of patients by exploring the effect of innovative drugs such as the proteasome inhibitor bortezomib and the bi-specific T cell engager blinatumomab. Nevertheless, thiopurines continue to be of interest: indeed, being administered daily throughout the duration of the entire therapy, although introduced 4/5 weeks after diagnosis and therapy start (i.e., after the induction phase), they remain the backbone of the polychemotherapy in both clinical trials. A detailed assessment of thiopurine metabolism and, in particular, of MP tolerance, toxicity and treatment outcome is one of the explorative objectives of the SJCRH TOTAL XVII protocol.
The most common drug related adverse effect of thiopurines is bone marrow suppression: leukopenia is a well-recognized effect occurring in more than 50% of ALL patients and 15%-20% in IBD patients[16], leading to dose reductions or therapy interruption, with consequent increase of disease recurrence and infection rate. Gastrointestinal complication are also very common in terms of anorexia, diarrhea, nausea, vomiting and stomatitis; the use of thiopurines may be limited also by the development of pancreatitis, although this complication is very rare in ALL patients (< 5%)[17]. Among other drug related adverse effects, there are hepatic toxicity and flu-like symptoms.
Pharmacogenetics
Pharmacogenetics of thiopurines in pediatric ALL is very well studied. Table 1 summarizes the genetic factors involved in thiopurine toxicities and further discussed in this review: TPMT variants have the major established role in clinics; clinical guidelines are now also available for NUDT15 polymorphisms, whereas the pharmacogenetic contributions of other mentioned genes are either smaller or not yet steadily confirmed and therefore require further studies for their clinical implementation.
Putative pharmacogenetic indicator for thiopurines
| Gene | Protein function | Genetic variants | Influence on mercaptopurine response in patients | Ref. |
|---|---|---|---|---|
| TPMT | S-methylation* | rs1800462
(*2, 238C>G, pAla80Pro) rs1800460 (*3B, 460G>A, p.Ala154Thr) rs1142345 (*3C, 719A>G, p.Tyr240Cys) | Severe leukopenia and hematological toxicity in carriers of the variant alleles | [36] |
| NUDT15 | Diphosphatase, catalyses the hydrolysis of nucleoside triphosphates and their oxidized forms | rs116855232
(415C>T, p.Arg139Cys) rs147390019 (416G>A, p.Arg139His) rs186364861 (52G>A, p.Val18Ile) rs746071566 (Insertion 36_37insGGAGTC p.Val18_Val19insGlyVal) | Thiopurine intolerance and adverse drug reaction in carriers of the loss-of-function NUDT15 genetic polymorphisms | [41]
[42] [43] [36] |
| PACSIN2 | Vesicle formation, endocytosis and caveole biogenesis | rs2413739 (T>C) | Severe gastrointestinal and haematological toxicities in TT genotype patients | [70]
[71] |
| ITPA | Pyrophosphatase that hydrolyzes the non-canonical purine nucleotides inosine triphosphate | rs1127354 (94C>A, p.Pro32Thr)
rs7270101 (IV2 +21A>C) | Severe febrile neutropenia and hepatotoxicity in carriers of the variant alleles | [59]
[6] |
TPMT
TPMT (EC 2.1.1.67) is ~30 kDa cytosolic enzyme expressed ubiquitously in cells. Its function on xenobiotics is very well established: as already mentioned above, it is responsible for the formation of methylated thiopurines derivatives and its activity is inversely related to the cytoplasmic accumulation of TGN[18]. In contrast, the endogenous function of TPMT is unknown: however, a role in the methylation of endogenous selenoproteins has been hypothesized and a very recent study confirmed in vitro a role for TPMT in the biosynthesis of selenocysteine[19]. Additionally, also a role in the biosynthesis of the molibdenum-binding co-factor molybdopterin has been postulated but not fully demonstrated yet[20]. TPMT is encoded by a 27 kb gene, composed of 10 exons, located in the short arm of chromosome 6[21]. Its expression varies and the patient genetic background is responsible for the observed interindividual differences in clinical efficacy and hematologic toxicity of thiopurine therapy[22]. Recent genome-wide association studies (GWAS) have provided evidences of TPMT as the only monogenic trait that influences the TPMT phenotype, finding that only variants in TPMT gene are significantly associated with TPMT activity in ALL children at genome-wide level (n = 1,026; P = 8.6 × 10-61) and in a mixed-population (two cohorts of adult healthy volunteers and one of pediatric ALL patients, n = 1,212; P = 1.2 × 10-72)[23,24]. TPMT activity is inherited as an autosomal codominant trait and approximately 1 in 300 individuals has very low TPMT activity, 10% have intermediate activity and 89% show normal/high activity[25]. Indeed 10% of Europeans are positive for a genetic variant in TPMT and 0.5% are TPMT completely defective[26]. Studies that correlate TPMT genotype and phenotype in different populations show that there are more than 38 allelic variants responsible for a possible impairment of the enzyme activity[27]. However, only few alleles, such as TPMT*2 and TPMT*3, comprise 95% of the loss of function variants and therefore are clinically relevant. TPMT*2 allele (Single Nucleotide Polymorphism (SNP) rs1800462) is defined by the 238G>C transversion and the p.Ala80Pro amino acid substitution, whereas the TPMT*3 family alleles are defined by the SNPs 460G>A (rs1800460, p.Ala154Thr) and 719A>G (rs1142345, p.Tyr240Cys). In particular, TPMT*3A is the haplotype characterized by both 460G>A and 719A>G transitions, TPMT*3B corresponds to the SNP rs1800460 alone and TPMT*3C to the single SNP rs1142345[27]. Genetic variants affecting TPMT activity are different among the main ethnic groups: indeed, the TPMT*3A is the variant most present in Europeans, while TPMT*3C is the most frequent in African and South-east Asian populations (allele frequency of 3%)[28]. In Africans, an additional allele putatively presenting reduced enzymatic activity, TPMT*8, identified by the polymorphism rs56161402, is present[29].
All these genetic variations lead to TPMT activity alterations: several studies have shown that the mechanism for decreased levels of TPMT protein and catalytic activity is the enhanced degradation of TPMT allozymes encoded by the TPMT*2 and TPMT*3A alleles[30-32]. These TPMT activity alterations are related to a high risk of severe side effects during thiopurine treatment. Many studies have demonstrated that standard thiopurine doses administration is correlated to a higher risk of developing severe and even fatal hematologic toxicity in individuals with low TPMT activity compared to individuals without genetic variations in this gene. In particular, this side effect is due to the accumulation of TGN[33]. Murine models knock-out for Tpmt do not show any apparent phenotype in the absence of drug challenge, whereas they present differences in TGN concentrations, MMPN concentrations and toxicity among Tpmt(-/-), Tpmt(+/-), and Tpmt(+/+) genotypes when thiopurines are administered, resembling the same phenotype of human individuals carrying TPMT defective polymorphisms[34].
Clinical guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC) are currently available and recommend to assess TPMT status in term of either genetic variations or activity in ALL and IBD patients before starting thiopurine therapy, in order to optimize dosages and thus drug efficacy and tolerance[35,36]. Nonetheless, there are many cases of patients without TPMT defective variants who are intolerant to a full dose of thiopurines and present leukopenia or other adverse effects, suggesting that also other genes or environmental factors can contribute to the risk of these side effects during thiopurine therapy[37].
NUDT15
Recent independent agnostic pharmacogenomic studies have identified a novel pharmacogene important for thiopurines: the nudix hydrolase 15 gene (NUDT15, also known as MTH2), is ~10 kb long, and is located on q region of chromosome 13[38]. NUDT15 (EC 3.6.1.9) is an ubiquitously expressed nucleotide triphosphate diphosphatase that converts oxidized GTP to its monophosphate form, preventing the integration of the damaged purine nucleotides into DNA and subsequent mismatch repair[39]. The triphosphate thionucleotides are potential substrate for NUDT15 that can hydrolyse and inactivate them. In patients with defective NUDT15, treatment with a standard dose of MP leads to an excessive accumulation of tGTP/tdGTP and consequent extensive DNA damage and cytotoxicity[40].
Yang et al.[38] performed an Immunochip-based association study in 978 Korean CD patients treated with thiopurines, investigating the association of 196524 genetic variants previously shown to be genetic susceptibility loci for drug-induced leukopenia in autoimmune or inflammatory diseases. The missense SNP 415C>T of NUDT15 gene (rs116855232), that induces p.Arg139Cys change, was associated with early leukopenia in the combined analysis of the discovery and replication samples (OR = 35.6; P combined = 4.88 × 10-94). Similarly, a GWAS study was conducted, using the HumanExomeBeadChip array (244770 SNPs), on a discovery cohort including 657 ALL children and a replication cohort of 371 ALL patients. The association with MP dose intensity (defined as the ratio between clinician-prescribed MP dose and protocol dose) was investigated, finding rs1142345 in TPMT and rs116855232 in NUDT15 as significantly associated loci (P ~9 × 10-9) with independent replication. Patients that presented a homozygous TT genotype for NUDT15 rs116855232 were very sensitive to MP and they could tolerate only 8.3% of the usual MP dose; this polymorphism explains 22% of variance in thiopurine-tolerance[41]. Moriyama et al.[42] reported that NUDT15 variant p.Arg139Cys did not affect enzymatic activity but rather protein stability, likely due to a loss of supportive intramolecular bonds that caused rapid proteasomal degradation in cells.
Moriyama et al.[47] sequenced all exons of NUDT15 in 3 ALL cohorts including 270 children from Guatemala, Singapore and Japan and identified 4 coding SNPs located in exons 1 and 3 associated with a loss of enzymatic activity (from 74.4% to 100%): the missense SNP rs116855232, the 416G>A SNP (rs147390019) that induces p.Arg139His and two other variants that affect the Val18 residue, the 52G>A resulting in a conversion of valine into isoleucine (p.Val18Ile) and the 36_37insGGAGTC insertion that leads to an in frame addition of a glycine and a valine residue (p.Val18_Val19insGlyVal). They also identified 5 NUDT15 haplotypes (*1-*5) with distinct combinations of these 4 variants and they classified patients into 3 diplotypic groups: normal activity (*1/*1), intermediate activity (*1/*2, *1/*3, *1/*4 and *1/*5), and low activity (*2/*3, *3/*3 and *3/*5). Loss-of-function NUDT15 diplotypes were consistently associated with thiopurine intolerance. In vitro studies on human lymphoid cell line showed an increased level of active TGTP in the NUDT15 knock-down cells compared to control cells, with the elevation of intracellular incorporated DNA thionucleotides and consequently higher thiopurines induced apoptosis[42].
A meta-analysis study of NUDT15 variants investigating 1,752 patients from 7 independent cohorts (both ALL and IBD) showed a significant association between NUDT15 rs116855232 T allele and myelotoxicity: indeed, the presence of the T allele increases ~8 times the probability to have leukopenia in comparison to the C allele (P < 0.00001, OR = 7.86, 95% CI: 6.13, 10.08). Analysing 2745 patients from 13 cohorts, the authors also found that T carriers tolerated lower mean daily thiopurines dose (P < 0.00001)[43]. A second meta-analysis, including 16 studies on rs116855232 (p.Arg139Cys), confirmed its role as clinically relevant predictor of thiopurine-induced leukopenia[44]. This SNP is the most common NUDT15 genetic variant, particularly frequent in East Asians (10.4%) and Hispanics (7.1%), but almost absent in Europeans (0.46%) and Africans[45]. Distribution of other NUDT15 genetic variants are also ethnic specific: rs147390019 is common only in Hispanics (1.7%)[43] and rs186364861 in Asians (1.6%)[43]. Recently, a NUDT15 variant with a significant prevalence in Caucasian patients was discovered in a cohort of patients with IBD, rs746071566, (p.Gly17_Val18del) and was associated with myelosuppression during thiopurine therapy[46]. The relevance of this variant in Caucasian patients with ALL has still to be fully understood.
International guidelines of thiopurines treatment suggest to consider NUDT15 together with TPMT genotypes before drug administration. Patients who present the above-mentioned genetic variants need to adjust drug dosage. CPIC suggests to use the normal starting dose for NUDT15 normal metabolizers (e.g., NUDT15 *1/*1, MP 75 mg/m2/day in ALL) and a reduced one in intermediate metabolizers (e.g., NUDT15 *1/*3, MP 30%-80% normal starting dose) and poor metabolizers (e.g., NUDT15 *3/*3; MP 10 mg/m2/day in ALL)[36].
Moriyama et al.[47] evaluated the effects of NUDT15 on thiopurines metabolism and identified DNA-incorporated thioguanine (DNA-TG) and TGN as pharmacologic markers for NUDT15 genotype-guided thiopurines dosing. They analyzed the level of these metabolites in a cohort of 55 Japanese ALL pediatric patients: those with NUDT15 deficiencies accumulated DNA-TG more efficiently than patients without NUDT15 variants (P = 4.0 × 10-9). They also showed that cytosolic TGN and nuclear DNA-TG were positively correlated with each other (P = 6.5 × 10-4), but the ratio of DNA-TG to TGN was significantly higher in NUDT15 deficient patients (P = 3.6 × 10-9), suggesting that DNA-TG is a more relevant MP metabolite than TGN to inform NUDT15 genotype-guided dose adjustments[47].
ITPA
Human inosine triphosphate pyrophosphatase (ITPA, EC 3.6.1.19) is a ~21 kDa enzyme that is ubiquitously expressed in the cytoplasm of cells and acts in homodimer form[48]. ITPA catalyses the hydrolysis of the triphosphate moieties from noncanonical (deoxy-) purine triphosphate, such as inosine triphosphate, deoxy-inosine triphosphate, xanthosine triphosphate, deoxy-xantosine triphosphate (ITP, dITP, XTP and dXTP respectively), thus recovering the monophosphate derivatives and releasing the pyrophosphate group. IMP/dIMP are fundamental intermediates in purine metabolism and can be converted into canonical adenosine monophosphate (AMP/dAMP, and therefore ATP/dATP) and guanosine monophosphate (GMP/dGMP, hence GTP/dGTP). It has been estimated that nearly one third of the human population has a genetically determined decreased ITPase activity[49]. The resulting unusual accumulation of noncanonical purine triphosphate can be toxic for the cells because of their interference with polymerase enzymes, and their incorporation into DNA or RNA. The 19 kb ITPA gene is located on the short arm of chromosome 20 and includes 8 exons[50]. Genetic knock-out (Itpa-/-) mice present abnormal features of growth retardation and cardiac myofiber disarray and die within 2 weeks after birth, likely because of the accumulation of ITP in the nucleotide pool found in erythrocytes[51]. Some reports have associated ITPA variants to human diseases, such as psychiatric disorders, encephalopathy, young-onset tuberculosis and infertility[49,52-54]. The human ITPA gene carries several polymorphisms associated with a reduction of enzymatic activity among which ITPA SNPs rs1127354 and rs7270101. SNP rs1127354 consists in the C94A transversion at the exon 2 level, leading to the amino acid substitution p.Pro32Thr. This substitution determines complete depletion of ITPA activity in homozygous individuals and a reduction to about 25% in heterozygous individuals[55]. The frequency of this polymorphism is around 5%-7% in the Caucasian population, but varies greatly depending on the ethnicity ranging from 2% in Hispanic populations up to 19% in Asians[56]. rs7270101 instead is the intronic transversion IVS2 +21A>C, that alters a very conserved adenine at the level of a splicing site, thus leading to an aberrant mRNA. This variant has a less severe effect than the C94A alteration: indeed, patients heterozygous for this SNP show an activity of about 60% compared to wild-type[55]. Genetic variants in ITPA are of major interest in pharmacogenetic research being associated with altered outcomes for patients undergoing antimetabolite therapy, in particular antileukemic agents such as thiopurines and methotrexate or antivirals such ribavirin or zidovudine[57,58].
The occurrence of toxicities during the ALL maintenance therapy was previously associated with ITPA variants: in the context of the SJCRH Total XIIIB protocol, Stocco et al.[59] found that ALL patients with a variant ITPA allele had a higher probability of developing severe febrile neutropenia. Increased accumulation of MMPN nucleotides has been described both in patients’ blasts, during a mercaptopurine-based window therapy, and erythrocytes, during maintenance therapy with MP doses adjusted for TPMT genotype. Adam de Beaumais et al.[6] found that wild-type TPMT/variant ITPA ALL patients treated with the EORTC (European Organization for Research and Treatment of Cancer)-58951 protocol had higher MMPN concentration in erythrocytes in comparison to wild-type TPMT/wild-type ITPA and that a MMPN threshold above 5000 pmol/8 × 108 RBC was associated to an increased risk of hepatotoxicity. Controversial results were reported in Asian populations: Kim et al.[60] did not find a difference in the cumulative incidence of grade III/IV febrile neutropenia according to ITPA genotypes in Korean ALL pediatric patients whereas Malaysian patients with ITPA 94A allele seemed more prone to develop fever and liver toxicity in therapeutic protocols not individualized for TPMT[61]. An Italian pharmacogenetic multicentric study was performed on 508 pediatric ALL patients treated with the AIEOP-BFM 2000 protocol: four children were homozygous variant for rs1127354 (94 AA genotype) and had a significant 13-fold increase in the risk of developing severe (grade III-IV) neurological toxicities during the 2 months induction phase, when compared with wild-type subjects, and showed also an higher risk of developing severe gastrointestinal complications (8-fold increase in risk)[62]. However, in this study, MP were introduced only four weeks after the beginning of the treatment, thus the incidence of these adverse effects could not be uniquely ascribable to thiopurines and the contribution of other drugs, such methotrexate, could be considered.
PACSIN2
Protein kinase C and casein kinase substrate in neurons protein 2 (PACSIN2) is a member of the PACSIN protein family[63]. PACSIN2 is an ubiquitously expressed protein of ~56 kDa and is localized in physical proximity to membranes. Structurally, PACSIN2 contains a membrane-sculpting Bin-Amphiphysin-Rvs (BAR) domain at N-terminal, a central region with a proline rich motif and a C-terminal Src homology 3 (SH3) domain[64-65]. The F-BAR domain regulates membrane curvatures and invaginations formation involved in vesicle budding, endocytosis and caveole biogenesis[66-68], whereas the SH3 domain mediates the interaction with many membrane proteins. PACSIN2 is encoded by a 180kbp gene located in the long arm of chromosome 22 which includes 15 exons. Human and murine PACSIN2 cDNA and protein show significant homology with 79.8% and 93.6% identity, respectively. The PACSIN2 knock-out mouse presents a decreased mean corpuscular volume, decreased mean corpuscular hemoglobin concentration, decreased leukocyte cell number, increased heart weight and improved glucose tolerance[69]. A recent genome wide analysis on a panel of 30 human HapMap cell lines trios demonstrated that the intronic SNP rs2413739 (T>C) of PACSIN2 can influence TPMT activity. In ALL patients enrolled in the Total XIIIB/XV protocols at SJCRH, the TT genotype was associated with lower TPMT activity in patients’ RBC and with an higher rate of gastrointestinal toxicity during thiopurines therapy. This latter association was also validated in an independent cohort of ALL patients treated with the AIEOP-BFM ALL 2000 protocol[70]. A retrospective study in pediatric ALL patients treated with thiopurines suggested that the presence of rs2413739 in PACSIN2 gene is a significant risk factor for mercaptopurine induced hematological toxicity in patients with wild-type TPMT[71]. It could be hypothesized that the increased toxicities observed in rs2413739 TT carriers are due to lower TPMT activity. However, no contribution of rs2413739 on thiopurine metabolites (TGN and MMPN) was reported, thus the mechanistic link between the PACSIN2 SNP and thiopurine adverse effects is not yet clearly established.
The PACSIN2 SH3 domain can interact with other proteins, such as Ras-related C3 botulinum toxin substrate 1 (Rac-1)[67]. Several evidences suggest a possible interplay among PACSIN2, Rac-1 and thiopurines. The physical interaction between the proline-rich hypervariable region of Rac-1 and the SH3 domain of PACSIN2 is well known. Due to its role in intracellular vesicle-mediated transport, PACSIN2 could serve as a molecular brake on Rac-1 signaling, by mediating the Rac-1-GTP internalization toward early endosomes, thus slowing down cell processes, such as growth and differentiation, regulated by the small GTPase[72]. The thiopurine metabolite tGTP binds to Rac-1 as a competitive antagonist of GTP: this binding suppresses the activation of Rac-1 and leads to apoptosis. Indeed, in IBD patients, effective MP therapy led to decreasing concentrations of Rac-1-GTP and Rac-1 expression[73]. PACSIN2 moreover may affect TPMT activity: indeed, the knock-down of PACSIN2 mRNA in human leukemia cells NALM6 resulted in significantly lower TPMT expression and enzymatic activity[70] that can in turn result in a higher tGTP availability and Rac-1 inhibition. Although not clear enough, these evidences suggest that PACSIN2, Rac-1 and their expression level could be additional potential biomarkers of thiopurines response and should be further investigated.
Rac-1
Rac proteins play a major role in T cell development, differentiation, and proliferation[74] and Rac-1 knock-outs are embryonic lethal in mice[75]. Rho GTPase proteins cycle between a GDP-bound and a GTP-bound state through the action of guanine nucleotide exchange factors (GEFs) and the opposing GTPase activating proteins (GAP); the GTP-bound state is generally considered as the active conformation, regulating signaling pathways in cells. The binding of tGTP instead of GTP on Rac-1 suppresses its activation, and thus the activation of its downstream targets such as mitogen-activated protein kinase, NF-κB and the antiapoptotic protein Bcl-xL. In vivo and in vitro studies suggested that Rac-1 plays a key role in thiopurines mechanism of action[24]. Tiede and collaborators have demonstrated that binding of AZA-generated tGTP to Rac-1 induced the mitochondrial pathway of apoptosis in primary CD4+ T lymphocytes lamina propria cells of healthy volunteers and CD patients, by downregulating Bcl-xL mRNA and protein[24]. An in vitro study on primary T cells isolated from healthy donors clarified the molecular mechanism beneath the tGTP-block of Rac-1, showing that the accumulation of inactive tGDP-Rac-1 proteins over time depends on the inhibition of the GEF factor Vav[76]. IBD patients treated with these drugs present a reduced median Rac-1 expression compared with patients without immunosuppressive therapy, and among MP treated patients, non-responders showed an increased median active Rac-1 expression[73]. In animal models, neutrophil and macrophage conditional Rac1 knock-out show normal bowel histology and do not present any obvious colonic phenotype; nonetheless they are protected against dextran sulphate sodium-induced colitis and show a reduced level of the proinflammatory cytokine IL-1β and of the neutrophil chemokine KC compared to wild-type mice[77]. The pharmacogenetics of RAC1, a ~30 kDa gene located on p arm of chromosome 7, has been investigated in IBD, but not in ALL patients. Two intronic RAC1 polymorphisms (rs10951982 and rs4720672) have been associated with UC susceptibility in a discovery and in 2 independent replication cohorts; IBD patients who had the rs10951982 G risk allele had increased expression of RAC1 compared to those without this allele[77]. The same SNPs, together with RAC1 SNP rs34932801, was genotyped in 59 IBD pediatric patients and no association was found with thiopurine effectiveness after 12 months of therapy[78]. In contrast, a retrospective report on a cohort of 156 thiopurine-treated CD adults has shown an association between the GC heterozygous genotype in SNP rs34932801 and poorer response to thiopurines, in comparison to wild-type GG[79].
Epigenetics
Epigenetic factors, in particular considering molecular factors that can be inherited from one generation to the other but that do not depend on the DNA sequence, such as methylation of cytosines on DNA and non-coding RNA profiles, such as microRNAs (miRNAs) and long non-coding RNAs, have been recently considered as innovative pharmacogenomic determinants of interindividual variability in drug efficacy and toxicity[80].
For thiopurines, studies on the effect of pharmacoepigenetic factors are still limited. Thiopurines have been shown to reduce global DNA methylation[81]. This effect was demonstrated in vitro in human leukemia cells and has been related to the reduction of de novo purine synthesis (DNPS) induced by thionucleotides, which determines reduction in ATP levels and depletion of S-adenosylmethionine (SAM) concentration, an important co-factor of methyltransferases, including DNA methyltransferases, enzymes involved in the regulation of DNA methylation[82]. Also TPMT activity, by modulating the capability of thionucleotides to inhibit DNPS, has been shown to influence the reduction of DNA methylation induced by mercaptopurine and thioguanine[83]. However, how this effect of thiopurines on DNA methylation may be related to interindividual variability in efficacy and adverse effects of these drugs or on the expression of candidate genes involved in thiopurines pharmacokinetics and pharmacodynamics is still unknown.
In the treatment of pediatric ALL, mercaptopurine is generally associated with methotrexate, both during consolidation and maintenance therapies. The epigenetic factors influencing interindividual variability in response to these phases of therapy may be of relevance also for thiopurine effects. For examples, a major adverse effect of consolidation therapy for pediatric ALL is oral mucositis, which is generally ascribed to methotrexate. However, as already mentioned, in some protocols, such as SJCRH Total XIIIB protocol, this adverse effect has been related also to TPMT activity[70], which indicated that also interindividual variability in mercaptopurine disposition may influence this adverse effect. A recent study evaluated DNA methylation profiles associated with oral mucositis during consolidation therapy in children with ALL. This study confirmed also in patients a significant reduction in SAM levels during methotrexate/mercaptopurine therapy (doses respectively 5 g/m2 every 2 weeks with leucovorin rescue 15 mg/m2/dose and daily mercaptopurine 25 mg/m2). In this study, DNA methylation was measured considering the percentage of 12 CpG islands in the LINE1 gene, that is considered a surrogate for DNA global methylation[84]. Intriguingly, a small increase in this DNA methylation marker was observed during therapy, with no correlation with SAM levels and incidence of oral mucositis. One limitation of this study is that DNA was extracted from peripheral blood mononuclear cells and since DNA methylation is cell specific, different effects could be observed in tissues more directly involved in methotrexate/mercaptopurine, effects such as residual lymphoblasts and epithelial cells of the gastrointestinal tract.
Considering non coding RNAs, studies are also limited, in particular for the association with the clinical effects of thiopurines. One study considered miRNA profiles after treatment with mercaptopurine in rat placenta, as a model to study toxicity of this drug in fetal development. Differentially expressed miRNAs could be identified and were involved in process such as apoptosis, inflammation and ischemia[85].
Non coding RNAs affecting the expression of pharmacogenes in human liver have been identified and associated with age. Among these genes, also those relevant for thiopurines pharmacokinetics have been considered[86]. Results showed that some miRNAs were associated with both age and TPMT expression; in particular, hsa-miR-34a-5p and has-miR-125b-5p showed the largest significant negative correlation with TPMT expression. Interestingly, TPMT activity measured in erythrocytes is negatively associated with age in children with ALL[23] and age has been shown to modulate thiopurine pharmacokinetics in pediatric patients[87]. The molecular mechanisms by which age influences TPMT activity could involve epigenetic factors, however more research is needed to clarify this phenomenon.
While the contribution of epigenetic factors to mercaptopurine induced adverse effects in pediatric ALL is still uncertain and speculative, further studies on this topic may bring important insights to improve therapy of patients taking mercaptopurine.
Genotyping vs. phenotyping
Many genes involved in thiopurine pharmacogenetics (i.e., TPMT, NUDT15, ITPA) encode for enzymes whose activity can be measured in patients, for examples in erythrocytes. For TPMT there are many studies considering both phenotyping and genotyping, while for the other enzymes the information available is still limited.
Both TPMT genotyping or phenotyping before starting thiopurines treatment allows to identify patients with reduced enzymatic activity and increased risk of drug induced adverse effects and to adjust starting dose accordingly or to use an alternative therapy. There is a risk of TPMT misclassification when only genotyping or phenotyping is used. Genotype-phenotype concordances are usually set between 76% and 99%[22]. Indeed, the conduction of both tests may give clinicians a more complete picture about patient’s risks during therapy, but there is no consensus on the evaluation of both in all patients, also for the associated costs. The Food and Drug Administration (FDA) still indicates genotyping or phenotyping before drug administration to identify TPMT status[88], while the American College of Gastroenterology indicates phenotyping as the first choice for patients using thiopurines for IBD because phenotyping reports a quantitative level of TPMT enzyme activity[89]. Genotyping certainly detects variations useful to predict the enzyme activity and the possible development of adverse effects directly associated with enzyme deficiency. One advantage of genetic tests is that they can be performed before starting therapy and the information obtained will be unchanged for the entire patient lifespan. Despite the relatively simple and rapid procedure, genetic tests provide data about key variants generally detecting the most common genetic variants associated with enzyme decrease but is not able to give a full prediction because they do not verify the presence of rare or previously undiscovered variants that will not be detected by variant-specific genotyping methods[90]. The advent of genome wide and next-generation sequencing (NGS) approaches and the continued reduction in the costs of these technologies will partly overcome this problem. Moreover, the genotype is not solely responsible for patient’s susceptibility of a drug. Genotyping tests will always provide a partial point of view since they do not consider variability of enzyme activity due to the presence of drug interactions, presence of others pathologies or other epigenetic factors. Phenotyping consists in a more direct observation of how the drug is metabolized and considers also inter-individual differences between patients with the same genetic arrangement. For this purpose, the activity of TPMT in lysed erythrocytes can be directly and successfully determined using reversed-phase high performance liquid chromatography (HPLC)[91]. It is important to measure TPMT activity on a blood sample taken before starting thiopurine administration and during therapy, because its activity can increase during treatment and thus can interfere with test results[36]. Moreover, phenotyping test shall be performed at least 3 months after a transfusion of erythrocytes[92]: during ALL chemotherapy many patients receive transfusions therefore the utility of phenotyping in this setting may be limited.
In conclusion, TPMT genotyping and phenotyping provide complementary information. Despite the usually quite high levels of concordance between the analysis, the combination of both genotype and phenotype tests insures a higher precision in the treatment personalization.
Conclusion
With the development of highly efficient sequencing techniques and the expected more diffuse availability of patient specific genotyping data, particularly for severe life threatening disease such as pediatric ALL, thiopurines pharmacogenetics may significantly contribute to reduce adverse effects and improve efficacy, by considering multilocus genotyping of TPMT and NUDT15. More research is needed to further improve pharmacogenetics of thiopurines by including additional gene variants, such as ITPA and PACSIN2, to obtain further clinical and mechanistic evidence.
Declarations
Authors’ contributionsDesigned the review, performed literature search, wrote and revised the manuscript: Franca R
Performed literature search, wrote and revised the manuscript: Zudeh G
Performed literature search, wrote and revised the manuscript: Pagarin S
Designed the review and revised the manuscript: Rabusin M
Performed literature search, wrote and revised the manuscript: Lucafò M
Designed the review, performed literature search, wrote and revised the manuscript: Stocco G
Designed the review, wrote and revised the manuscript: Decorti G
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis project is supported by the Italian Ministry of Health (Progetto Ricerca Corrente 5/2012).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Adamson PC, Poplack DG, Balis FM. The cytotoxicity of thioguanine vs mercaptopurine in acute lymphoblastic leukemia. Leuk Res 1994;18:805-10.
2. Lucafò M, Franca R, Selvestrel D, Curci D, Pugnetti L, et al. Pharmacogenetics of treatments for inflammatory bowel disease. Expert Opin Drug Metab Toxicol 2018;14:1209-23.
3. Cara CJ, Pena AS, Sans M, Rodrigo L, Guerrero-Esteo M, et al. Reviewing the mechanism of action of thiopurine drugs: towards a new paradigm in clinical practice. Med Sci Monit 2004;10:RA247-54.
4. Berends SE, Strik AS, Löwenberg M, D’Haens GR, Mathôt RAA. Clinical pharmacokinetic and pharmacodynamic considerations in the treatment of ulcerative colitis. Clin Pharmacokinet 2019;58:15-37.
5. Krenitsky TA, Papaioannou R, Elion GB. Human hypoxanthine phosphoribosyltransferase. I. Purification, properties, and specificity. J Biol Chem 1969;244:1263-70.
6. Adam de Beaumais T, Fakhoury M, Medard Y, Azougagh S, Zhang D, et al. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br J Clin Pharmacol 2011;71:575-84.
7. Nguyen CM, Mendes MA, Ma JD. Thiopurine methyltransferase (TPMT) genotyping to predict myelosuppression risk. PLoS Curr 2011;3:RRN1236.
8. Dubinsky MC, Lamothe S, Yang HY, Targan SR, Sinnett D, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology 2000;118:705-13.
9. Ben-Horin S, Van Assche G, Chowers Y, Fudim E, Ungar B, et al. Pharmacokinetics and immune reconstitution following discontinuation of thiopurine analogues: implications for drug withdrawal strategies. J Crohns Colitis 2018;12:1410-7.
10. Derijks LJ, Gilissen LP, Engels LG, Bos LP, Bus PJ, et al. Pharmacokinetics of 6-mercaptopurine in patients with inflammatory bowel disease: implications for therapy. Ther Drug Monit 2004;26:311-8.
11. Lancaster DL, Patel N, Lennard L, Lilleyman JS. Leucocyte versus erythrocyte thioguanine nucleotide concentrations in children taking thiopurines for acute lymphoblastic leukaemia. Cancer Chemother Pharmacol 2002;50:33-6.
12. Cuffari C, Seidman EG, Latour S, Théorêt Y. Quantitation of 6-thioguanine in peripheral blood leukocyte DNA in Crohn’s disease patients on maintenance 6-mercaptopurine therapy. Can J Physiol Pharmacol 1996;74:580-5.
13. Stocco G, Franca R, Verzegnassi F, Londero M, Rabusin M, et al. Pharmacogenomic approaches for tailored anti-leukemic therapy in children. Curr Med Chem 2013;20:2237-53.
14. Franca R, Kuzelicki NK, Sorio C, Toffoletti E, Montecchini O, et al. Targeting kinase-activating genetic lesions to improve therapy of pediatric acute lymphoblastic leukemia. Curr Med Chem 2018;25:2811-25.
15. Ling Y, Xie Q, Zhang Z, Zhang H. Protein kinase inhibitors for acute leukemia. Biomark Res 2018;6:8.
16. Avallone EV, Pica R, Cassieri C, Zippi M, Paoluzi P, et al. Azathioprine treatment in inflammatory bowel disease patients: type and time of onset of side effects. Eur Rev Med Pharmacol Sci 2014;18:165-70.
17. Ledder O, Lemberg DA, Day AS. Thiopurine-induced pancreatitis in inflammatory bowel diseases. Expert Rev Gastroenterol Hepatol 2015;9:399-403.
18. Citterio-Quentin A, El Mahmoudi A, Perret T, Conway A, Ryan A, et al. Red Blood cell IMPDH activity in adults and children with or without azathioprine: relationship between thiopurine metabolites, ITPA and TPMT activities. Basic Clin Pharmacol Toxicol 2018.
19. Urbančič D, Kotar A, Šmid A, Jukič M, Gobec S, et al. Methylation of selenocysteine catalysed by thiopurine S-methyltransferase. Biochim Biophys Acta Gen Subj 2019;1863:182-90.
20. Schwarz G, Csaszar J, Schaeffeler E, Schwab, M. Diagnostic in vitro method, 2013 [Application number: US20160160264A1]. Available from: https://patents.google.com/patent/US20160160264A1/en. [Last accessed on 15 Apr 2019].
21. Collie-Duguid ES, Pritchard SC, Powrie RH, Sludden J, Collier DA, et al. The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics 1999;9:37-42.
22. Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 2004;14:407-17.
23. Liu C, Yang W, Pei D, Cheng C, Smith C, et al. Genomewide approach validates thiopurine methyltransferase activity is a monogenic pharmacogenomic trait. Clin Pharmacol Ther 2017;101:373-81.
24. Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, et al. CD28-dependent Rac-1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest 2003;111:1133-45.
25. Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 1997;126:608-14.
26. Hindorf U, Appell ML. Genotyping should be considered the primary choice for pre-treatment evaluation of thiopurine methyltransferase function. J Crohns Colitis 2012;6:655-9.
27. Chadli Z, Kerkeni E, Hannachi I, Chouchene S, Ben Fredj N, et al. Distribution of genetic polymorphisms of genes implicated in thiopurine drugs metabolism. Ther Drug Monit 2018;40:655-9.
28. Saiz-Rodríguez M, Ochoa D, Belmonte C, Román M, Martínez-Ingelmo C, et al. Influence of thiopurine S-methyltransferase polymorphisms in mercaptopurine pharmacokinetics in healthy volunteers. Basic Clin Pharmacol Toxicol 2019;124:449-55.
29. Oliveira E, Quental S, Alves S, Amorim A, Prata MJ. Do the distribution patterns of polymorphisms at the thiopurine S-methyltransferase locus in sub-Saharan populations need revision? Hints from Cabinda and Mozambique. Eur J Clin Pharmacol 2007;63:703-6.
30. Tai HL, Krynetski EY, Schuetz EG, Yanishevski Y, Evans WE. Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): mechanisms for the genetic polymorphism of TPMT activity. Proc Natl Acad Sci U S A 1997;94:6444-9.
31. Wang L, Sullivan W, Toft D, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: chaperone protein association and allozyme degradation. Pharmacogenetics 2003;13:555-64.
32. Wang L, Nguyen TV, McLaughlin RW, Sikkink LA, Ramirez-Alvarado M, et al. Human thiopurine S-methyltransferase pharmacogenetics: variant allozyme misfolding and aggresome formation. Proc Natl Acad Sci U S A 2005;102:9394-9.
33. Coenen MJ, de Jong DJ, van Marrewijk CJ, Derijks LJ, Vermeulen SH, et al. Identification of patients with variants in tpmt and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology 2015;149:907-17.e7.
34. Hartford C, Vasquez E, Schwab M, Edick MJ, Rehg JE, et al. Differential effects of targeted disruption of thiopurine methyltransferase on mercaptopurine and thioguanine pharmacodynamics. Cancer Res 2007;67:4965-72.
35. Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011;89:387-91.
36. Relling MV, Schwab M, Whirl-Carrillo M, Suarez-Kurtz G, Pui CH, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther 2018.
37. Meijer B, Kreijne JE, van Moorsel SAW, Derijks LJJ, Bouma G, et al. 6-methylmercaptopurine-induced leukocytopenia during thiopurine therapy in inflammatory bowel disease patients. J Gastroenterol Hepatol 2017;32:1183-90.
38. Yang SK, Hong M, Baek J, Choi H, Zhao W, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet 2014;46:1017-20.
39. Yang JJ, Whirl-Carrillo M, Scott SA, Turner AJ, Schwab M, et al. Pharmacogene variation consortium gene introduction: NUDT15. Clin Pharmacol Ther 2018.
40. Kakuta Y, Kinouchi Y, Shimosegawa T. Pharmacogenetics of thiopurines for inflammatory bowel disease in East Asia: prospects for clinical application of NUDT15 genotyping. J Gastroenterol 2018;53:172-80.
41. Yang JJ, Landier W, Yang W, Liu C, Hageman L, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 2015;33:1235-42.
42. Moriyama T, Nishii R, Perez-Andreu V, Yang W, Klussmann FA, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 2016;48:367-73.
43. Yin D, Xia X, Zhang J, Zhang S, Liao F, et al. Impact of NUDT15 polymorphisms on thiopurines-induced myelotoxicity and thiopurines tolerance dose. Oncotarget 2017;8:13575-85.
44. Cargnin S, Genazzani AA, Canonico PL, Terrazzino S. Diagnostic accuracy of NUDT15 gene variants for thiopurine-induced leukopenia: a systematic review and meta-analysis. Pharmacol Res 2018;135:102-11.
45. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285-91.
46. Walker G, Harrison J, Voskuil M, Heap G, Heerasing N. NUDT15 variants contribute to thiopurine-induced myelosuppression in European populations. 2018. Available from: https://www.ecco-ibd.eu/publications/congress-abstract-s/abstracts-2018/item/op035-nudt15-variants-contribute-to-thiopurine-induced-myelosuppression-in-european-populations.html. [Last accessed on 15 Apr 2019].
47. Moriyama T, Nishii R, Lin TN, Kihira K, Toyoda H, et al. The effects of inherited NUDT15 polymorphisms on thiopurine active metabolites in Japanese children with acute lymphoblastic leukemia. Pharmacogenet Genomics 2017;27:236-9.
48. Lin S, McLennan AG, Ying K, Wang Z, Gu S, et al. Cloning, expression, and characterization of a human inosine triphosphate pyrophosphatase encoded by the itpa gene. J Biol Chem 2001;276:18695-701.
49. Burgis NE. A disease spectrum for ITPA variation: advances in biochemical and clinical research. J Biomed Sci 2016;23:73.
50. Sumi S, Marinaki AM, Arenas M, Fairbanks L, Shobowale-Bakre M, et al. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet 2002;111:360-7.
51. Behmanesh M, Sakumi K, Abolhassani N, Toyokuni S, Oka S, et al. ITPase-deficient mice show growth retardation and die before weaning. Cell Death Differ 2009;16:1315-22.
52. Vanderheiden BS. Possible implication of an inosinetriphosphate metabolic error and glutamic acid decarboxylase in paranoid schizophrenia. Biochem Med 1979;21:22-32.
53. Nakauchi A, Wong JH, Mahasirimongkol S, Yanai H, Yuliwulandari R, et al. Identification of ITPA on chromosome 20 as a susceptibility gene for young-onset tuberculosis. Hum Genome Var 2016;3:15067.
54. Mollaahmadi F, Moini A, Salman Yazdi R, Behmanesh M. The rs1127354 polymorphism in itpa is associated with susceptibility to infertility. Cell J 2018;20:73-7.
55. Stenmark P, Kursula P, Flodin S, Gräslund S, Landry R, et al. Crystal structure of human inosine triphosphatase. Substrate binding and implication of the inosine triphosphatase deficiency mutation P32T. J Biol Chem 2007;282:3182-7.
56. Marsh S, King CR, Ahluwalia R, McLeod HL. Distribution of ITPA P32T alleles in multiple world populations. J Hum Genet 2004;49:579-81.
57. Fellay J, Thompson AJ, Ge D, Gumbs CE, Urban TJ, Shianna KV, et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature 2010;464:405-8.
58. Coelho AV, Silva SP, Zandonà L, Stocco G, Decorti G, et al. Role of inosine triphosphate pyrophosphatase gene variant on fever incidence during zidovudine antiretroviral therapy. Genet Mol Res 2017;16.
59. Stocco G, Cheok MH, Crews KR, Dervieux T, French D, et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther 2009;85:164-72.
60. Kim H, Kang HJ, Kim HJ, Jang MK, Kim NH, et al. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: a possible association between survival rate and ITPA polymorphism. PLoS One 2012;7:e45558.
61. Wan Rosalina WR, Teh LK, Mohamad N, Nasir A, Yusoff R, et al. Polymorphism of ITPA 94C>A and risk of adverse effects among patients with acute lymphoblastic leukaemia treated with 6-mercaptopurine. J Clin Pharm Ther 2012;37:237-41.
62. Franca R, Rebora P, Bertorello N, Fagioli F, Conter V, et al. Pharmacogenetics and induction/consolidation therapy toxicities in acute lymphoblastic leukemia patients treated with AIEOP-BFM ALL 2000 protocol. Pharmacogenomics J 2017;17:4-10.
63. Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci 2000;113:4511-21.
64. Frost A, Unger VM, De Camilli P. The BAR domain superfamily: membrane-molding macromolecules. Cell 2009;137:191-6.
65. Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 2004;303:495-9.
66. Senju Y, Rosenbaum E, Shah C, Hamada-Nakahara S, Itoh Y, et al. Phosphorylation of PACSIN2 by protein kinase C triggers the removal of caveolae from the plasma membrane. J Cell Sci 2015;128:2766-80.
67. de Kreuk BJ, Anthony EC, Geerts D, Hordijk PL. The F-BAR protein PACSIN2 regulates epidermal growth factor receptor internalization. J Biol Chem 2012;287:43438-53.
68. Hansen CG, Howard G, Nichols BJ. Pacsin 2 is recruited to caveolae and functions in caveolar biogenesis. J Cell Sci 2011;124:2777-85.
69. Ritter B, Modregger J, Paulsson M, Plomann M. PACSIN 2, a novel member of the PACSIN family of cytoplasmic adapter proteins. FEBS Lett 1999;454:356-62.
70. Stocco G, Yang W, Crews KR, Thierfelder WE, Decorti G, et al. PACSIN2 polymorphism influences TPMT activity and mercaptopurine-related gastrointestinal toxicity. Hum Mol Genet 2012;21:4793-804.
71. Smid A, Karas-Kuzelicki N, Jazbec J, Mlinaric-Rascan I. PACSIN2 polymorphism is associated with thiopurine-induced hematological toxicity in children with acute lymphoblastic leukaemia undergoing maintenance therapy. Sci Rep 2016;6:30244.
72. Lam BD, Hordijk PL. The Rac-1 hypervariable region in targeting and signaling: a tail of many stories. Small GTPases 2013;4:78-89.
73. Seinen ML, van Nieuw Amerongen GP, de Boer NK, Mulder CJ, van Bezu J, et al. Rac-1 as a potential pharmacodynamic biomarker for thiopurine therapy in inflammatory bowel disease. Ther Drug Monit 2016;38:621-7.
74. Shin JY, Wey M, Umutesi HG, Sun X, Simecka J, et al. Thiopurine prodrugs mediate immunosuppressive effects by interfering with Rac-1 protein function. J Biol Chem 2016;291:13699-714.
75. Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, et al. Rac-1 is required for the formation of three germ layers during gastrulation. Oncogene 1998;17:3427-33.
76. Poppe D, Tiede I, Fritz G, Becker C, Bartsch B, et al. Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J Immunol 2006;176:640-51.
77. Muise AM, Walters T, Xu W, Shen-Tu G, Guo CH, et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase Rac-1 are associated with ulcerative colitis. Gastroenterology 2011;141:633-41.
78. Lev-Tzion R, Renbaum P, Beeri R, Ledder O, Mevorach R, et al. Rac-1 polymorphisms and thiopurine efficacy in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2015;61:404-7.
79. Koifman E, Karban A, Mazor Y, Chermesh I, Waterman M, et al. Thiopurine effectiveness in patients with Crohn’s disease: a study of genetic and clinical predictive factors. Inflamm Bowel Dis 2013;19:1639-44.
80. Fisel P, Schaeffeler E, Schwab M. DNA methylation of ADME genes. Clin Pharmacol Ther 2016;99:512-27.
81. De Abreu R, Lambooy L, Stet E, Vogels-Mentink T, Van den Heuvel L. Thiopurine induced disturbance of DNA methylation in human malignant cells. Adv Enzyme Regul 1995;35:251-63.
82. Lambooy LH, Leegwater PA, van den Heuvel LP, Bökkerink JP, De Abreu RA. Inhibition of DNA methylation in malignant MOLT F4 lymphoblasts by 6-mercaptopurine. Clin Chem 1998;44:556-9.
83. Hogarth LA, Redfern CP, Teodoridis JM, Hall AG, Anderson H, et al. The effect of thiopurine drugs on DNA methylation in relation to TPMT expression. Biochem Pharmacol 2008;76:1024-35.
84. Lisanti S, Omar WA, Tomaszewski B, De Prins S, Jacobs G, et al. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS One 2013;8:e79044.
85. Taki K, Fukushima T, Ise R, Horii I, Yoshida T. Microarray analysis of 6-mercaptopurine-induced-toxicity-related genes and microRNAs in the rat placenta. J Toxicol Sci 2013;38:159-67.
86. Burgess KS, Philips S, Benson EA, Desta Z, Gaedigk A, et al. Age-Related Changes in MicroRNA Expression and Pharmacogenes in Human Liver. Clin Pharmacol Ther 2015;98:205-15.
87. Stocco G, Martelossi S, Arrigo S, Barabino A, Aloi M, et al. Multicentric case-control study on azathioprine dose and pharmacokinetics in early-onset pediatric inflammatory bowel disease. Inflamm Bowel Dis 2017;23:628-34.
88. Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 2012;92:414-7.
89. Kornbluth A, Sachar DB, Gastroenterology PPCotACo. Ulcerative colitis practice guidelines in adults: American college of gastroenterology, practice parameters committee. Am J Gastroenterol 2010;105:501-23.
91. Stocco G, Martelossi S, Barabino A, Fontana M, Lionetti P, Decorti G, et al. TPMT genotype and the use of thiopurines in paediatric inflammatory bowel disease. Dig Liver Dis 2005;37:940-5.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Franca R, Zudeh G, Pagarin S, Rabusin M, Lucafò M, Stocco G, Decorti G. Pharmacogenetics of thiopurines. Cancer Drug Resist 2019;2:256-70. http://dx.doi.org/10.20517/cdr.2019.004
AMA Style
Franca R, Zudeh G, Pagarin S, Rabusin M, Lucafò M, Stocco G, Decorti G. Pharmacogenetics of thiopurines. Cancer Drug Resistance. 2019; 2(2): 256-70. http://dx.doi.org/10.20517/cdr.2019.004
Chicago/Turabian Style
Franca, Raffaella, Giulia Zudeh, Sofia Pagarin, Marco Rabusin, Marianna Lucafò, Gabriele Stocco, Giuliana Decorti. 2019. "Pharmacogenetics of thiopurines" Cancer Drug Resistance. 2, no.2: 256-70. http://dx.doi.org/10.20517/cdr.2019.004
ACS Style
Franca, R.; Zudeh G.; Pagarin S.; Rabusin M.; Lucafò M.; Stocco G.; Decorti G. Pharmacogenetics of thiopurines. Cancer Drug Resist. 2019, 2, 256-70. http://dx.doi.org/10.20517/cdr.2019.004
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 0
likes
Like This Article 0
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.