
Resistance to anti-tubulin agents: From vinca alkaloids to epothilones

Abstract
This review describes the mechanism of action - inhibition of microtubules - and the most important mechanisms of resistance for vinca alkaloids, taxanes and epothilones. Resistance is a major problem in vinca and taxane chemotherapy and arises in most cases from overexpression of efflux pumps that transport the drugs out of the cancer cells and from modifications of the target, the microtubules, by overexpression of tubulin isotypes or by attachment of proteins to the ends of the microtubules so that the target is no longer recognized by the drugs. In some cases, however, this process can have the opposite effect, leading to sensitization, e.g., for vinca alkaloids in cases where taxanes are not or no longer effective. The link between resistance due to efflux pumps and the pharmacokinetics and metabolism of the drugs is also covered. Other types of resistance that are addressed include detoxification of drugs within the cancer cell and blockade of apoptosis, post-translational modifications of microtubules and other protein pathways, micro-RNAs, induction of oncogenes, and cancer stem cells, which, taken together, offer particularly multifold possibilities for preventing drug activity. The use of biomarkers for the prediction of clinical outcome and for the direction of future therapy is also addressed.
Keywords
Introduction
Anti-tubulin agents (ATAs) represent a class of anti-cancer drugs that are targeting one of the most fundamental processes in the organism, mitosis and cell division[1]. This had double-edged consequences. On the one hand it provided a boost of anti-cancer activity compared to previously available drugs, and on the other hand, it may elicit a host of defense mechanisms, both in the cancer cells leading to resistance and in normal cells resulting in side effects. Since the target is so essential to the existence and well-functioning of the organism, the number of mechanisms that can take over if one of them is blocked, is very high and until today only a limited number have already been extensively investigated. In this review, I will address the most important ones of them.
ATAs are directed against tubulins. Tubulins are the building blocks of microtubules, which are part of the cytoskeleton and which are essential for many physiological processes such as formation of the mitotic spindle and chromosome segregation during cell division, for migration or intracellular transport and for the construction of special structures such as cilia or flagella and their movements.
Microtubules are tiny, very rigid, cylindrical, hollow tubes with a diameter of approximately 25 nm, composed of the two globular proteins, α- and β-tubulin with a total molecular weight of 50 kDa each [Figure 1]. In humans, eight isoforms of each α- and β-tubulin have been identified[2]. Some isotypes are specifically expressed in specialized cells and tissues. The predominant isotype is β-III in brain and testicular cells and β-I in other cell types. Isotype expression changes during development[3,4]. Post-translational modifications (PTMs) such as acetylation, phosphorylation, palmitoylation, sumoylation, S-nitrosylation, amination, glutamylation, glycylation, tyrosination, and detyrosination can lead to further diversification of microtubules[5-9]. Brain tubulin, which is a mixture of different tubulin isotypes with a majority of β-III, is heavily posttranslationally modified[4,10-12]. Most of the PTMs are reversible and their distribution is specific for the individual cell type[5]. The enzymes that introduce these modifications are essential to normal development[13-15]. Increased levels of tubulin modifications are a hallmark of cancer.
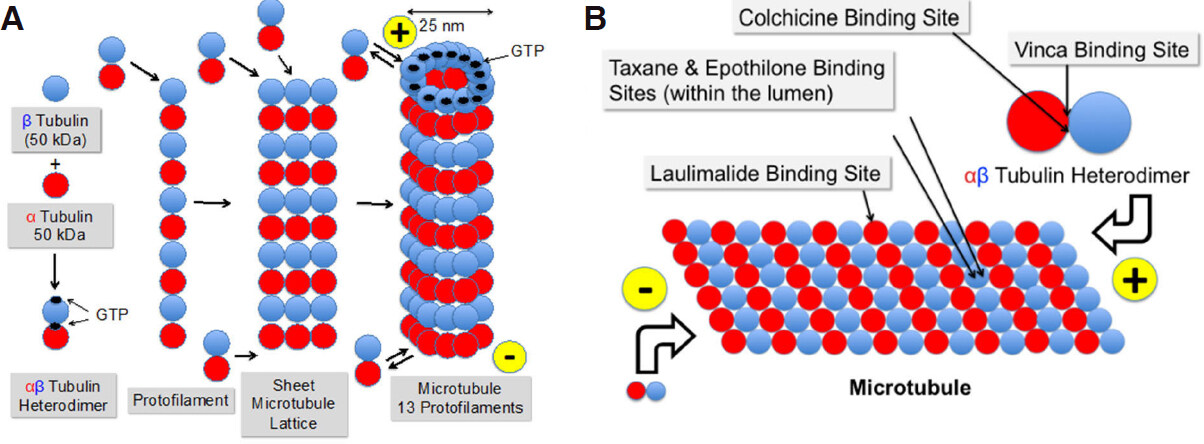
Figure 1. Formation of microtubules from α- and β-tubulin via αβ-heterodimers and protofilaments (A) and binding sites of colchicine, laulimalide, taxanes & epothilones, and vinca alkaloids (B)
In vitro, and in the presence of GTP, one α- and one β-tubulin molecule are spontaneously and non-convalently linked together to form a heterodimer as soon as their concentrations exceed a critical value[16]. The N-terminal regions of α- and β-tubulin are able to bind GTP, which provides the energy needed for polymerization by hydrolysis to GDP and phosphate. Whereas the GTP binding site of β-tubulin is freely accessible and therefore can be used for energy purposes, the GTP binding site of α-tubulin is hidden in the interior of the heterodimer [Figure 1]. The heterodimers are available in the cells at relatively high concentrations of 1-10 μmol/L, compared to the total tubulin concentration, which is in the range of 20 μmol/L[17,18].
Heterodimers are then lining up to form linear protofilaments and subsequently two-dimensional sheets. When 11-15 protofilaments have been linked together, they fold to form a tube, the microtubule[19-21]. The side with α-tubulin at the end is negatively charged, the other side, exposing β-tubulin, carries a positive charge. The negative end of the microtubule (α-tubulin) is attached to the microtubule-organizing center at the centrosomes and the positive end (β-tubulin) is growing into the cytoplasm of the cell by attaching new heterodimers[22]. The growth rate of microtubules varies between 5 and 20 μm/min[22]. However, there is a constant lengthening and shortening of the microtubules at both ends by adding and severing heterodimers[23,24] called “dynamic instability”. The addition and elimination of heterodimers is slower at the minus end of the microtubule. There are periods where overall shortening of the microtubule is dominant, termed “catastrophe” and periods where lengthening is ongoing, called “rescue”. When the number of GTP-bound heterodimers at the microtubule end drops below a threshold level, the microtubule undergoes catastrophe and switches to rapid depolymerization[21]. This behavior is characteristic for non-equilibrium processes and constitutes the dynamic instability. The kinetics of these processes is controlled by a whole bunch of proteins[1], termed microtubule-associated proteins (MAPs), such as for example γ-tubulin[16]. These proteins have a competing role for either stabilizing the microtubules, such as tau, MAP1, MAP2, MAP4, XMAP215 or destabilizing them, such as stathmin, XKCM1, XKIF2, and katanin[25,26]. They can address both sides of the microtubules and either promote or block microtubule polymerization or enhance or inhibit disassembly of the polymers. Reviews on this topic have been published by Janke et al.[6], Farache et al.[16], Ohi et al.[27], Akhmanova et al.[1], and Valiron et al.[28]. The dynamics of microtubules vary strongly during the cell cycle. Their turnover is slow in interphase cells. It can take minutes or hours depending on the cell type[29,30]. Mitosis accelerates this process up to 100-fold[31]. Anti-tubulin drugs with few exceptions decrease both the growing and shortening rate of the microtubules and the duration, increase the rescue frequency, and increase the time spent in a pause state[32].
Classes of anti-tubulin agents
Meanwhile, inhibition of tubulin is a well-established target in anticancer treatment[33]. Anti-tubulin agents come in two flavors, either stabilizing or destabilizing microtubules. Taxanes, and epothilones are microtubule-targeting agents that suppress microtubule dynamics by stabilizing microtubules and thereby interfering with the functioning of the mitotic spindle. As a consequence, the cell cycle comes to an arrest in rapidly proliferating cancer cells[34], finally leading to apoptosis. Colchicine and vinca alkaloids destabilize the microtubules, leading to disassembly and cell death.
Colchicine
The oldest drug that was investigated using the tubulin target is colchicine, which has been in use for more than 3000 years in the form of plant extracts. Colchicine has been approved by the FDA for the prophylaxis and treatment of gout flares and treatment of Mediterranean fever[35]. It is a natural product and was initially extracted from meadow saffron (Colchicum autumnale). Colchicine is able to block mitosis[36]. It tightly binds to the “colchicine binding site” [Figure 1] on heterodimers[37], but not on microtubules. Following colchicine binding, the dimers are no longer able to form protofilaments or to attach to microtubules due to a change in conformation[38]. However, the required dose for cancer treatment was so high that the therapeutic index was too low for use in cancer indications.
Vinca alkaloids
The discovery of the anticancer properties of vinca alkaloids in the late 1950s was a milestone in the development of cancer chemotherapy. Vinca alkaloids were originally obtained from the Madagascar periwinkle plant (Catharanthus roseus). They exhibit hypoglycemic and cytotoxic efficacy[39] and have been used for treating diabetes and high blood pressure and as disinfectants. Five compounds have been approved as anticancer agents, vinblastine, vinorelbine, vincristine, vindesine, and vinflunine. Additionally, a liposomal formulation of vincristine, with sphingomyelin and cholesterol-based nanoparticles, has been approved to overcome the toxicity limitations of standard vincristine, the dose of which had to be capped at 2 mg for safety reasons.
The vinca binding site on microtubules[40] is different from that of other anti-tubulin agents [Figure 1]. Vinca alkaloids induce tubulin to form alternate spiral polymers that will propagate into a microtubule GDP-containing lattice and dissociate microtubules by peeling spiral protofilaments[32]. These spirals are stable, equilibrium polymers that slowly dissociate. Vinca alkaloids are cell cycle phase-specific for the M-phase[41]. At high concentrations vinca alkaloids depolymerize microtubules and destroy mitotic spindles. The dividing cancer cells appear blocked in mitosis with condensed chromosomes. At low concentrations, they block mitosis more subtly, and cells die by apoptosis[42]. Similar to taxanes, vinca alkaloids exhibit antivascular and antiangiogenic effects, potently and rapidly inducing vascular disruption, thereby leading to tumor necrosis[43]. Vincristine, vinorelbine and vinblastine are on the WHO List of Essential Medicines.
Taxanes
Paclitaxel (Taxol) is a microtubule-stabilizing diterpenoid, which had been isolated from the bark of Pacific yew (Taxus brevifolia) in 1971. At the beginning, only minimal amounts were available from the NIH since the cost of paclitaxel supply was prohibitive. The initial production of paclitaxel in the 1990’s required ten tons of bark from the Pacific Yew tree to produce one kilogram of paclitaxel[44]. For this purpose approximately 2000-4000 trees had to be cut down. A typical dose for breast cancer treatment needs 175 mg/m2 IV over 3 h every 3 weeks for 4 courses, which means a total of approximately 1.3 g per patient. One kg of paclitaxel therefore could - theoretically - treat approximately 700 patients. In practice, the number is much lower due to losses during manufacture, analytical testing, packaging, etc. Meanwhile the production costs could significantly be reduced by fermentation techniques.
In vitro, paclitaxel binds reversibly to the microtubule polymer in a 1:1 stoichometry relative to the heterodimer[45,46] within the lumen of the microtubule, enhancing the polymerization of tubulin[45,47]. Initially, it was stated that the drug does not bind to the αβ-tubulin heterodimer[48]. Later, Field et al.[49] could show that zampanolide and dactylolide also reacted with the same residue in unassembled tubulin, thus providing the first direct evidence of the existence of the luminal site in dimeric tubulin. Microtubules formed in vitro with paclitaxel present are extraordinarily stable. They even do not depolymerize upon addition of CaCl2, cooling down or diluting the solution[50]. Paclitaxel is able to induce polymerization in the absence of GTP, which normally is absolutely necessary[51]. Docetaxel is using the same binding site as paclitaxel, though with higher affinity[52]. Cell killing by paclitaxel is bi-phasically concentration dependent starting at 5-50 nmol/L where paclitaxel stabilizes the mitotic spindle and arrests the cell at mitosis. Blockade of cell proliferation then leads to apoptosis. At 1000 times higher concentrations of 5-50 μmol/L, polymerization of microtubules and bundle formation during interphase are increased, thereby inhibiting the initiation of S phase and finally leading to necrosis[52,53]. Paclitaxel additionally induces apoptosis by binding to the apoptosis-stopping protein B-cell leukemia-2 (Bcl-2) and thereby arresting its function. The cause for this effect is the similarity in the binding site of the natural Bcl-2 inducer, Nur77, on Bcl-2 and the taxane-binding site on microtubules[54]. Ganguly et al.[55] proposed that paclitaxel rescues mutant cell division by inhibiting the detachment of microtubule minus-ends from centrosomes rather than by altering plus-end microtubule dynamics. Komlodi-Pasztor stated that the interference of paclitaxel with intracellular trafficking on microtubules is probably its most important function[56]. Additional effects of paclitaxel include the triggering of macrophages for TNFα and IL-1 production[57] and its antiangiogenic activity by downregulating VEGF and Ang-1 expression in tumor cells and by increasing the secretion of TSP-1 in the tumor microenvironment[58].
Taxanes with the approved drugs, paclitaxel (1991), docetaxel (1995), and cabazitaxel (2010) are a widely used class of anticancer agents. Galsky published a review for the latest approved drug, cabazitaxel[59]. Not approved but in clinical trials are tesetaxel and larotaxel[60]. Paclitaxel is also available as a protein-bound formulation, nanoparticle albumin-bound paclitaxel or nab-paclitaxel (Abraxane). Paclitaxel is on the WHO List of Essential Medicines. Approved cancer indications and dosing schedules are summarized in Table 1.
Available anti-tubulin agents with their years of approval, main indications, dose ranges and types of therapy (monotherapy, combination partners)
| Drug | Main indications | Dose | Combinations |
|---|---|---|---|
| Vinblastine 1961* | Hodgkin’s disease, non-Hodgkin lymphoma, histiocytic lymphoma, mycosis fungoides, testis, Kaposi’s sarcoma, choriocarcinoma, breast, kidney | 3.7 mg/m2-18.5 mg/m2 | Monotherapy, mechlorethamine, doxorubicin, vincristine, bleomycin, etoposide, dacarbazine, brentuximab, cisplatin, ifosfamide, methotrexate, mitomycine |
| Vincristine 1963* | Leukemias, lymphomas, myeloma, breast, lung, head & neck, sarcomas, Wilms’ tumor, neuroblastoma, retinoblastoma, medulloblastoma, | 0.8 mg/m2-2 mg | Monotherapy, doxorubicin, carboplatin mechlorethamine, vinblastine, bleomycin, etoposide, cyclophosphamide, procarbazine, topotecan, dactinomycin, leucovorin, actinomycin D |
| Vindesine 1982*** | ALL, CML, melanoma, breast | 3 mg/m2-4 mg/m2 | Monotherapy, cisplatin |
| Vinorelbine 1994* | NSCLC, Hodgkin’s disease, non-Hodgkin lymphoma, rhabdomyosarcoma, Wilm’s tumor, neuroblastoma | 25 mg/m2-30 mg/m2 | Monotherapy, cisplatin |
| Vinflunine 2009** | Urothelial carcinoma | 280 mg/m2-320 mg/m2 | Monotherapy |
| Vincristine Liposomal 2012* | Philadelphia chromosome-negative ALL | 2.25 mg/m2 | Monotherapy |
| Paclitaxel 1992* | Ovarian, breast, lung, gastric, Kaposi’s sarcoma | 100 mg/m2-210 mg/m2 | Monotherapy, cisplatin, doxorubicin |
| Docetaxel 1996* | Breast, lung, prostate, gastric, head & neck | 75 mg/m2-100 mg/m2 | Monotherapy, cyclophosphamide, cisplatin, 5-fluorouracil |
| Nab-Paclitaxel 2005* | Breast, lung, pancreas | 100 mg/m2-260 mg/m2 | Monotherapy, carboplatin, gemcitabine |
| Cabazitaxel 2010* | Prostate | 20 mg/m2-25 mg/m2 | Monotherapy |
| Ixabepilone 2007* | Breast | 40 mg/m2 | Capecitabine |
Epothilones
The latest drug class comprises the epothilones. The name was derived from their chemical structure as epoxide, thiazole and ketone. They initially raised high hopes due to their increased potency and by overcoming taxane and vinca resistance. Another advantage of the epothilones (excluding ixabepilone) over taxanes is their higher water solubility allowing formulations without additional solubilizers such as Cremophor EL, which might lead to hypersensitivity reactions. Epothilones were isolated from the myxobacterium Sorangium cellulosum in the 1990s[61]. According to their chemical structures, natural epothilones are characterized by the letters A-F. Only class B and D epothilones have been selected for development as anti-cancer drugs and as a basis for further chemical modification and optimization. Epothilone B is considerably more potent than epothilone A[61]. In general, the epothilones are more potent than paclitaxel by at least a factor of 10. The most potent epothilone is sagopilone with IC50 values in the sub-nanomolar range[62]. As with paclitaxel, cell killing by sagopilone is biphasic dose dependent. Low concentration treatment (2.5 nmol/L) resulted in aberrant cell division, aneuploidy and cell cycle arrest in G1 whereas high concentration treatment (40 nmol/L) led to mitotic arrest[63]. The gene expression profiles of low and high concentration phenotypes were substantially different from each other. Low concentration sagopilone led to p53 (TP53) transactivation in A549 lung cancer cells resulting in cell cycle arrest. Incubation with high concentrations of sagopilone was associated with upregulation of several genes involved in spindle assembly checkpoint and mitosis, indicating mitotic arrest. For both phenotypes, differences in the ability to undergo apoptosis were observed. Regarding resistance, sagopilone was tested in more than 100 different human tumor cell lines and a line resistant to the drug could not be found[64]. Epothilones and taxanes share the same binding site in the interior of the microtubule. Electron microscopy and molecular modeling suggest, however, that they do not share a common pharmacophore[65]. The interaction of epothilones with microtubules is functionally distinct from that of taxanes[66].
Major compounds investigated in clinical trials were natural epothilone B (patupilone), the semisynthetic epothilone B derivative, ixabepilone (BMS-247550), and the fully synthetic epothilone B derivative, sagopilone. In class D, natural epothilone D (KOS-862) and the epothilone D derivative, KOS-1584, were studied clinically. Ixabepilone is the only epothilone that was approved by the FDA in 2007 for the treatment of advanced breast cancer, either alone or in combination with capecitabine. The EMA declined approval in 2008 stating an insufficient benefit/risk ratio. Major side effects were limiting the use of the epothilones, particularly neurotoxicity (ixabepilone, sagopilone) or gastric toxicity (patupilone) and led to the discontinuation of development for patupilone and sagopilone. Ixabepilone is still on the market.
Drug resistance
The use of vinca alkaloids and taxanes remains one of the major pillars in chemotherapy for a variety of tumor types. However, the majority of patients will develop progressive disease after initial response to therapy. Drug resistance therefore represents a major hurdle for the improvement of overall response and survival of cancer patients. In the following chapters resistance aspects will be covered for vinca alkaloids, taxanes and epothilones.
Drug resistance has a multitude of facets. Essentially it means that the tumor is not or not sufficiently or no longer responding to the therapy. The span of cancer treatment outcomes ranges from no response in 1st line drug exposure (primary resistance) via partial response to complete response with minimal residual disease to complete response without minimal residual disease (cure). In nearly all cases, retreatment is necessary, which could either mean repeating the first approach or moving to a different drug or combination of drugs. Even among taxane-naıve patients, primary resistance to taxanes is a critical factor for disease progression. More than one-third of patients with metastatic breast cancer do not respond to first-line anthracyclines or taxanes[67]. Cortes reported taxane resistance rates of up to 55% in anthracycline-pretreated patients and up to one-third in anthracycline-naıve patients[68]. Second-line, the same spectrum of outcomes can be expected. In case of no response with a different drug, cross-resistance is happening. Figure 2 summarizes some of the various possible scenarios.
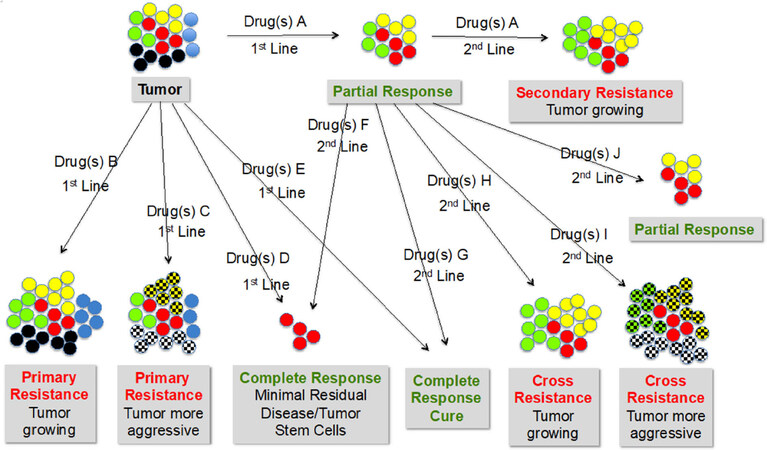
Figure 2. Heterogeneity of tumors and types of drug responses and resistance starting from no response at 1st line (primary resistance) via partial response to complete response with or without residual disease. In 2nd line treatment with a different drug the same type of reactions can be expected. In that case, no response means cross-resistance of the two different drugs is happening. Color codes: Red circles: cancer stem cells; other colors: different tumor cell types; cross-hatched circles: aggressive/metastasizing cell types
Tumor heterogeneity
The major assumption for the scenarios described in Figure 2 is the heterogeneity of tumors[69]. Each patient has his/her own tumor with different characteristics and therefore different therapy outcomes. The variabilities include but are not limited to different genetic, epigenetic, transcriptomic and proteomic properties. The genotypic changes include mutations, gene amplifications, deletions, chromosomal rearrangements, transpositions of the genetic elements, translocations and microRNA alterations[70]. The situation is even worse insofar as the same patient does not exhibit the same tumor over time and, even more important, the tumor at any time point x is a mixture of different cancer cells, even though clonally derived, with different characteristics and different sensitivity/resistance to treatment[71]. Genomic instability generates a great level of intercellular genetic heterogeneity in cancer.
Types of resistance
Primary resistance can originate from a variety of possible mechanisms. It could either mean that the drug does not reach the target either at all or in sufficiently high concentrations in order to be able to elicit a cytotoxic effect. Or it does reach the target but it does not show any activity because the receptor is not recognized or not functioning properly.
According to Cree and Charlton[72], resistance to anti-cancer drugs can be acquired by several mechanisms within neoplastic cells, defined as: (1) alteration of drug targets; (2) expression of drug pumps; (3) expression of detoxification mechanisms; (4) reduced susceptibility to apoptosis; (5) increased ability to repair DNA damage; and (6) altered proliferation. Not all of these mechanisms are important for ATAs. Since tumors are not homogeneous but a mixture of different clones, development of resistance is not a straightforward process but a universe of different activities at different levels. In many cases rapid resistance originates from multiple non-mutational, non-genetic mechanisms[73].
Pharmacokinetics and metabolism
The pharmacokinetics of the different drugs is an important factor in determining whether the substance is able to enter the cancer cell in sufficiently high concentrations and to approach the binding site. Table 2 provides a summary of the pharmacokinetics of vinca alkaloids, taxanes, and epothilones.
Main pharmacokinetic parameters of anti-tubulin agents in humans
| Drug | t1/2 | Cl(tot) | Vd | BBB | P-gp | Metabolism | Ref. |
|---|---|---|---|---|---|---|---|
| Vinblastine | 25 (23-85) h | 0.74 L/h/kg (863 mL/min) | 27.3 L/kg (2,047 L) | - | + | CYP3A41 | [74] |
| Vincristine | 23-85 h | 146 mL/min | 215 L | - | + | CYP3A4/52 | [75] |
| Vindesine | 20-24 h | NA | 8.11 L/kg (608 L) | - | + | CYP3A42 | [76] |
| Vinorelbine | 28-44 h | 0.97-1.26 L/h/kg (1,280-1,680 mL/min) | 25.4-40.1 L/kg (1,905-3,007 L) | + | + | CYP3A3 | [77] |
| Vincristine Liposomal | 7.7 ± 3.2 h | 345 ± 177 (148-783) mL/h (6 ± 3 mL/min) | Vd = 3.6 ± 1.9 L Vdss = 2.9 ± 1.2 L | - | + | CYP3A42 | [41] |
| Vinflunine | 40 h | 40 L/h (667 mL/min) | 2,422 ± 676 L | - | + | Esterases, CYP3A42 | [78] |
| Paclitaxel* | 13-53 h | 12.2-23.8 L/h/m2 (351-685 mL/min) | 227-688 L/m2 (392-1,190 L) | - | + | CYP2C8, CYP3A42 | [79] |
| Docetaxel | 11 h | 21 L/h/m2 (605 mL/min) | 113 L | - | + | CYP3A4/5 | [79] |
| Nab-Paclitaxel | 27 h | 15 L/h/m2 (432 mL/min) | 632 L/m2 (1,093 L) | - | + | CYP2C8, CYP3A4 | [79] |
| Cabazitaxel | 95 h | 48.5 L/h (761 mL/min) | 4,864 L | (+) | (-) | CYP2C8, CYP3A4, CYP3A54 | [79] |
| Patupilone | 137 ± 70 h | 14.0 ± 5.3 L/h (233 ± 88 mL/min) | 224,2 ± 926 L | + | - | Esterases, CYP3A4/3A | [80] |
| Ixabepilone | 35/52 h | 616 mL/min | 21-26 L/kg (1,575-1,950 L) | + | - | CYP2D6, CYP2C19, CYP3A4, CYP3A52 | [81] |
| Sagopilone | 53 h | 1,359 mL/min | 4,607 L | + | - | Esterases, CYP3A4/3A, CYP2C192 | [62] |
| KOS-862 | 9.1 ± 2.2 h | 9.3 ± 3.2 L/h/m2 (155 ± 53 mL/min/m2 = 268 ± 92 mL/min) | 119 ± 41 L/m2 (206 ± 71 L) | + | - | CYP3A4, CYP2C9, CYP2C19, CYP2B62 | [82] |
| KOS-1584 | 21.9 ± 8.8 h | 11 ± 6.2 L/h/m2 (183 ± 103 L/min/m2 = 317 ± 178 mL/min) | 327 ± 161 L/m2 (566 ± 279 L) | + | - | CYP3A42 | [83] |
All ATAs are extensively metabolized, mostly by P450 oxidizing enzyme systems, in particular CYP2C8 and CYP3A4. Any co-administered inducers of these enzyme systems, e.g., phenobarbital or carbamazepine, will therefore reduce the cancer drug concentrations in the blood significantly and thereby decrease or even eliminate the anti-tumor activity. This should, of course, be an avoidable case of “resistance” to the drug. On the other hand, any co-administered enzyme inhibitors, such as ketoconazole or fluoxetine will increase the drug concentrations and thereby could increase the efficacy, but at the same time increase toxicity.
The metabolism of vincristine, docetaxel, cabazitaxel, and ixabepilone by CYP3A5 is clinically relevant since this enzyme is polymorphically expressed. CYP3A5 is expressed in 10%-20% of Caucasians and approximately in 70% of African-Americans[84]. Differences in efficacy and tolerability therefore can be expected and may explain cases of “resistance” or drop-out due to side-effects. Likewise, CYP2C19, which metabolizes ixabepilone, sagopilone and KOS-862 exhibits genetic polymorphism. Approximately 3%-5% of Europeans and 15%-20% of Asians are poor metabolizers with no CYP2C19 function. Another example is CYP2C9, which is involved in the metabolism of KOS-862 and where allele frequencies for CYP2C9*2 range from 0% in Asians to 19% in Caucasians. The combined action of more than one of these polymorphically expressed enzymes as is the case with KOS-862 complicates the situation further.
ATAs in general do not require high peak concentrations of drug but rather long exposure of microtubules at a sufficiently high drug level[41]. Parameters for estimating this exposure are the terminal half-life of drug disposition in the blood, the total clearance and the volume of distribution [Table 2]. With 137 h, patupilone has the longest half-life and vincristine liposomal with 7.7 h has the shortest one. However, the latter number is misleading, since it had been determined after a long-lasting plateau of high drug concentration[85]. Therefore, the shortest half-life falls to KOS-862 with 9.1 h. The total clearance, where low values are preferred, is lowest for liposomal vincristine (6 mL/min) and largest for sagopilone (1,359 mL/min) and vinorelbine (1,280-1,680 mL/min). The largest volumes of distribution are exhibited by cabazitaxel (4,864 L) and sagopilone (4,607 L). Problems in this type of evaluation are the very high variability in PK parameters between patients, the non-linearity of PK in some cases, e.g., paclitaxel, and, the major point, that the decisive criterion is the concentration at the target receptor and not the concentration in blood. However, it might be assumed that a longer half-life will also lead to higher and longer-lasting accumulation of drug within the cancer cells. A further complicating factor is that some of the drugs, e.g., cabazitaxel, form active metabolites, which additionally contribute to activity. Cabazitaxel has three active metabolites, one of them being docetaxel. Paclitaxel has a volume of distribution of 392-1,190 L. It does not pass the blood-brain barrier. Cabazitaxel’s volume of distribution (4,864 L) is the highest of all ATAs. This drug is able to pass the blood-brain barrier as does sagopilone, with a Vd of 4,607 L. Probably, the high neurotoxicity of sagopilone can be explained by this effect. On the other hand, neurotoxicity is not an issue for cabazitaxel. In the case of cabazitaxel, the reason for not leading to neurotoxicity could be that the tubulin isotype distribution in brain is different from that in other tissues, with βIII-tubulin being predominant to which the taxanes do not bind whereas the epothilones do[86-89]. As can be seen from Table 2, passage of the blood-brain barrier goes parallel with being able to overcome P-glycoprotein (P-gp) resistance, which will be discussed in detail later in this review. The explanation is that the brain is protected by the same efflux pumps that are responsible for taxane drug resistance, mainly P-gp. Vinorelbine seems to be the only exception. It has been reported to pass the blood-brain barrier and to be subject to P-gp resistance.
ATAs are normally dosed in mg/m2 in order to compensate for differences in body surface, which certainly has advantages over a fixed dose. However, this dose regimen does not take into consideration the very high variability of pharmacokinetic parameters, which is independent of body surface or body weight and which might be much more important. Since all ATAs are extensively metabolized by the P450 system, the metabolic capacity of a patient is a factor that should be considered in a fully individualized dose regimen. Otherwise in patients with over-active enzymes, the bioavailable portion of the administered dose might be too low to be effective. In patients with less active or malfunctioning enzymes, the drug would be too toxic and would lead to discontinuation of therapy. Evaluation of the P450 metabolic status of a patient could lead to individual dose adjustments tailored to the needs of this patient.
Efflux pumps
Taxanes and vinca alkaloids have been shown to be potent weapons in the fight against malignant tumors and are considered first-line therapy for the chemotherapy of many cancers. However, while patients often respond well to the first course of treatment, over time the response to drug exposure diminishes and the tumor may eventually become drug resistant. In some cases resistance can develop even across several classes of anti-cancer drugs, leading to cross or multidrug resistance[90]. The development of drug resistance limits the effectiveness of many anti-cancer agents and is an important contributor to cancer deaths.
In order to be effective, the cytotoxic drug has to be transported into the cancer cell and has to reach sufficiently high concentrations over a sufficiently long period of time. This can be achieved by using a concentration gradient between the exterior and the interior side of the cell membrane and by active transport. The concentration gradient alone is not sufficient for some types of anticancer drugs, such as nucleoside analogs, which need active transport into the tumor cell in order to achieve high intracellular drug concentrations[69,91]. There are three ATP-binding cassette transporter systems available, P-gp, multi-drug resistance-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP/ABCG2)[92]. If the expression of these transporters is reduced, the uptake of the cytotoxic drugs into the cancer cell is diminished in those cases where they depend on active transport, such as nucleosides. If overexpression of the transporters occurs, drugs that had passively passed into the cancer cell by diffusion will be actively pumped out, if they are a substrate of the pumps. Both possibilities, over- and underexpression of active transporters, can lead to drug resistance, depending on the drug class., Since taxanes, vinca alkaloids and epothilones enter the cancer cells only via diffusion, underexpression of these transporters would constitute a beneficial situation. Overexpression of efflux pumps leads to resistance for taxanes and vinca alkaloids but not for epothilones.
A major obstacle in obtaining cytotoxic concentrations in cancer cells is the triggering of pumping systems that eliminate the drugs out of the cells thereby preventing the achievement of concentrations that are high enough to induce apoptosis. The most common form of clinical resistance is overexpression of the MDR1 gene, which encodes the P-gp drug efflux pump. This membrane-associated ATP-binding cassette (ABC) transporter is overexpressed in many tumor cell lines, including tissues of the liver, kidney, and gastrointestinal tract. Overexpression of P-gp, encoded by the MDR1 gene, decreases intracellular drug levels, consequently limiting drug cytotoxicity. It is the most common mechanism involved in the development of resistance associated with poor response to microtubule-targeted agents including taxanes and vinca alkaloids and subsequent treatment failure[93,94]. Epothilones are not affected by P-gp induced resistance[95,96].
Paclitaxel is a substrate for the P-gp drug efflux pump overexpressed in various multi-drug resistant cancer cells[97,98]. Vincristine and vinblastine are also affected by P-gp[71]. P-gp overexpression has been detected in up to 41% of multiple myeloma patient samples[99]. Inhibitors of P-gp, such as cyclosporin A, valspodar, and verapamil, have been tried to counteract overexpression of P-gp but have shown very limited activity in Phase III trials[100]. In an attempt to reverse multi-drug resistance, vincristine-loaded lipid nanoparticles conjugated to an anti-P-gp monoclonal antibody (MRK-16), showed greater cytotoxicity in resistant human myelogenous leukemia cell lines than control non-targeted particles. The response was attributed to the inhibition of the P-gp-mediated efflux of vincristine by MRK-16[101]. Cabazitaxel is a marginal substrate of P-gp[102]. It could be shown that P-gp, which is the main component of the blood-brain barrier, can be saturated by cabazitaxel so that passage of the barrier becomes possible. In vitro, cabazitaxel is not a substrate for the pumps, MRP1, MRP2, BCRP, OCT1 (organic cation transporter 1), OATP1B1 (organic anion transporting polypeptide 1B1) and OATP1B3.
Multidrug resistance protein 7 (MRP7; ABCC10) is an ATP-binding cassette transporter, which is able to transport amphipathic anions and confer resistance to docetaxel and, to a lesser extent, vincristine and paclitaxel[103,104]. An interesting approach is the use of the natural compound, β-elemene, which can reverse multidrug resistance mediated by the ABCB1 transporter[105]. The incubation of KB-C2 cells overexpressing the ABCB1 transporter with β-elemene (100 μmol/L) significantly augmented the antineoplastic efficacy of colchicine, vinblastine and paclitaxel when compared to KB-C2 cells incubated with these drugs alone. Glutathione-S-transferase (GSH) stimulates the ATP-dependent uptake and efflux of vincristine by MRP[106]. Cellular depletion of GSH decreases MRP1-mediated resistance to anticancer drugs.
In summary, all approved taxanes - with cabazitaxel being a special case due to possible saturation of P-gp - and all vinca alkaloids are substrates for the efflux pump, P-gp, leading to resistance, whereas the epothilones are able to overcome this resistance.
Vault proteins
Once a drug has entered the cancer cell, the next obstacles could be vault proteins such as the lung resistance-related protein (LRP), a 150 kDa protein, which is overexpressed in multidrug resistant cells[107]. A 2-week treatment with sodium butyrate induced LRP and conferred resistance to doxorubicin, vincristine, etoposide, gramicidin D, and paclitaxel in SW-620 cells. Vaults are cytoplasmic organelles that were originally isolated in association with coated vesicles, but approximately 5% of the vaults are localized in nuclear pore complexes. Vaults are involved in the vesicular and nucleocytoplasmic transport of drugs. LRP is the major protein of vaults. Entrapment of the cytotoxic drug in vaults does not reduce the intracellular concentration of the drug but it makes it unavailable to target binding. Expression of LRP, but not P-gp or MRP, was an independent prognostic factor for predicting tumor response to standard chemotherapy and survival in patients with advanced ovarian cancer[108].
Detoxification mechanisms
Detoxification of cytotoxic drugs within the cancer cell can be accomplished by proteins of the glutathione S-transferase family (GST), which can be subdivided into three large groups, cytosolic, mitochondrial and microsomal proteins, known as MAPEG proteins[69]. Increased expression of GST leads to intracellular drug levels that are no longer cytotoxic.
Target modifications
Another important line of defense of cancer cells is their ability to camouflage the target, in this case the microtubules. Altered tubulin isotype composition of microtubules has emerged as a feature of aggressive and treatment refractory cancers[109]. This could mean over- or underexpression of tubulin isotypes that are required as drug targets or the expression of isotypes that are not normally detectable in healthy tissue[110]. This behavior correlates with treatment response and patient outcome. Examples for aberrant overexpression include the βI-, βII-, βIII-, βIVa-, and βV-tubulin isotypes, which lead to aggressive clinical outcome and resistance to chemotherapy in many types of cancer. mRNA levels of βII-, βIII- and βV-tubulin isotypes were found to be significantly upregulated in paclitaxel and docetaxel resistant cells and significantly downregulated in vincristine resistant cells[111]. A modification in βIII-tubulin expression is the most extensively studied tubulin isotype mutation and it could be shown that subsequent resistance is a common feature for taxanes and vinca alkaloids.
The β-tubulin binding site of paclitaxel is located within the lumen of the microtubules. The drug needs to get there by diffusion, which is facilitated by the formation of hydrogen bonds with a β-tubulin serine moiety. The formation of this hydrogen bond is possible with all β-tubulin isotypes except βIII and βVI[112]. It was suggested[2] that the presence of the unique residue, Ala218 in βIII-tubulin, may be a key reason why drug binding to βIII-tubulin is inhibited. Overexpression of this isotype therefore leads to resistance. Overexpression of βIII-tubulin has particularly often been described in taxane-resistant ovarian cancer[86-88,113-118]. But it is also seen in other tumor types such as lung, breast and gastric cancers[119].
βIII-tubulin expression is controlled by estrogens in breast cancer cells but is influenced by exposure to hypoxia and poor-nutrient supply in ovarian cancer. Extensive microtubule mutations occur in hypoxic conditions[109]. Hypoxia-inducible factor 1-α (HIF1α) binds to an E-box motif located within the 30 UTR of TUBB3 and induces its expression in ovarian cancer cells[120]. Similarly, HIF1 and HIF2a interactions with overlapping HIF response elements within the TUBB3 3’UTR suppress and induce TUBB3 expression in glioblastoma in normoxia and hypoxia, respectively[121]. The expression of βIII-tubulin is also regulated by the interaction of the TUBB3 transcript with the RNA binding protein Hu antigen (HuR)[122], which is involved in the recruitment of mRNA transcripts to polysomes and has been demonstrated to be involved in HIF1α stabilization. Oxidative stress depolymerizes microtubules and reduces the microtubule assembly rate[123,124]. βIII-Tubulin and the DNA damage repair enzyme excision repair cross-complementation group-1 co-operatively influence patient response to taxane combination therapy[125].
In some but not all cancers, βIII-tubulin expression is purely a prognostic biomarker, predicting poor outcome of patients regardless of chemotherapy treatment. It enhances the incorporation of pro-survival kinases into the cytoskeleton and protects them from degradation. The associations of βIII-tubulin with survival kinase PIM-1, RNA-binding protein HuR, microRNAs are examples highlighting the functional complexity of this protein[126]. Ovarian clear cell adenocarcinoma and estrogen-receptor negative breast cancer, where βIII-tubulin overexpression indicates beneficial outcome of taxane-based chemotherapy, seem to be exceptions[127,128]. On the other hand, microtubules with overexpression of βIII-tubulin exhibit an increased sensitivity to epothilones, particularly to epothilone B[129]. The configuration of the pocket binding epothilone B (patupilone) in βIII-tubulin differs from that present in βI-tubulin, the most abundantly expressed β-tubulin[130,131]. Therefore, cells with high expression of βIII-tubulin appear more sensitive to patupilone[129] explaining the increased effects of epothilones noticed in a large number of clinical trials conducted in patients relapsing after multiple lines of chemotherapy[132]. In other words, whereas overexpression of class βIII-tubulin plays a major role in taxane resistance, epothilones display their highest efficacy in βIII-tubulin overexpressing malignancies.
The expression of altered β-tubulin isotypes is triggered by certain oncogenes. Downregulation of βIII-tubulin by antisense oligonucleotides in paclitaxel-resistant A549-T24 cells resulted in a 40%-50% decrease in both βIII mRNA and protein levels, and was associated with a 39% increase in sensitivity to paclitaxel[133]. The expression of βIII-tubulin is regulated by TUBB3, a specific tubulin gene, which encodes the otherwise neuron-specific βIII-tubulin isotype. TUBB3 expression was increased in a resistant cell line [retinal pigment epithelial (RPE)-20] derived from untransformed RPE cells, but remained unchanged in four other cell lines after paclitaxel treatment[33]. TUBB3 levels alone therefore do not seem to be a good predictor of resistance.
Whereas increased βII-, βIII- and βIV-tubulin are associated with taxane resistance, decreased βIII-tubulin has been reported in vinca-resistant cell lines, together with increased microtubule stability[134]. βIII-Tubulin overexpression induces resistance to paclitaxel and vinorelbine, but does not affect the resistance to colchicine-site binding agents[52].
Mutations in expression of β-tubulin isotypes are associated with altered sensitivity to ATAs. β-tubulin isotypes differentially affect the efficacy of ATAs in a cell type-dependent manner[135]. Overexpression of βII-tubulin and βIII-tubulin isotypes triggers resistance to taxanes in non-small cell lung cancer cells, whereas underexpression of βII-tubulin or βIVb-tubulin sensitizes NSCLC[136,137] and pancreatic cancer cells to vinca alkaloids[138]. Underexpression of βII-tubulin also sensitizes NSCLC cells to epothilone B[136,137]. In ovarian cancer cells on the other hand, suppression of βII-tubulin does not sensitize the cells to taxanes or vinca alkaloids[136]. Conflicting results have been reported by Narvi[139], who showed that βIII-tubulin but not βI-tubulin or βII-tubulin expression affected epothilone sensitivity in NSCLC and breast cancer cell lines. Likewise, overexpression of βIII- or βV-tubulin in CHO cells increases the resistance of these cells to paclitaxel[140,141]. Other studies indicate that overexpression of βI-, βII-, and βIVb-tubulin does not affect ATA resistance in these cells[142]. Overexpression of βIII-tubulin failed to induce resistance to ATAs in prostate cancer[143]. The epothilones do not seem to act in an identical manner but show individual differences. Whereas for ixabepilone βIII-tubulin overexpression reduces the efficacy[144], for sagopilone resistant cell lines could not be found[62].
βIII-Tubulin probably is a target of oncogenic pathways. Its expression is associated with ERG expression and TMPRSS2: ERG rearrangement in prostate cancer[145]. Jordan[144] hypothesizes that altered β-tubulin isotype expression in cancer may protect cells from oxidative stress to trigger chemotherapy resistance and enable tumor progression by increasing the metastatic potential of cancer cells through migration and invasion. Parker et al.[109] report that activation of the epithelial-to-mesenchymal transition (EMT) program is critical in regulating invasion and metastasis[146] and that EMT re-programming is influenced by the tubulin isotype composition. βIII-Tubulin expression correlates with Snail expression levels and modulates the behavior of Snail overexpression during EMT in colon cancer cells[147]. Knockdown of βIII-tubulin expression with RNA interference agents caused modulation of colon cancer cell movement and a decrease in their ability to migrate and invade. Likewise, knockdown of ZEB1, an EMT transcription factor, in human breast cancer cells reduced β-tubulin isotype classes I, III, and IVB mRNA, whereas upregulation of ZEB1 was associated with increases in these isotype classes.
PTMs
Another line of defense against cytotoxic drugs includes regulatory proteins, in particular alterations to tubulin through PTMs that affect regulatory protein binding, and altered expression or PTMs to tubulin-/microtubule-regulatory proteins[51]. The MAPs, tau, stathmin, MAP2 and MAP4 have been extensively investigated as triggers for cancer resistance[148-150]. They bind to and stabilize microtubules against depolymerization. High MAP expression is associated with resistance to taxanes[148]. Tau overexpression was associated with breast tumors that are resistant to paclitaxel[150].
Stathmin and MAP4 affect the sensitivity of a cell towards paclitaxel. The overexpression or activation of stathmin and/or the downregulation or inactivation of MAP4 increases the microtubule dynamics and decreases their stability thereby resulting in resistance. On the other hand, the potency of microtubule destabilizers such as vinca alkaloids should be enhanced. Paclitaxel-resistant cell lines contain “hypostable” microtubules in which the equilibrium between the dimer and polymer is shifted towards the dimer[151-153]. These cells display increased resistance to polymer-binding drugs like paclitaxel, and increased sensitivity towards tubulin dimer-specific agents, such as vinblastine and colchicine. In paclitaxel-resistant cell lines, the equilibrium between weakly and highly dynamic microtubules is shifted towards the highly dynamic ones[154-157]. Two paclitaxel-resistant human ovarian cancer cell lines, 1A9PTX10 and 1A9PTX22, have been investigated. They were resistant to paclitaxel, but hypersensitive to vinblastine[158]. The paclitaxel-resistant cells also retained their sensitivity towards epothilone B.
Micro-RNAs
Micro-RNAs (miRNAs) are another important factor in the establishment of resistance. miRNAs are short RNAs of 20-25 nucleotides that are involved in regulating gene expression, in particular protein-coding gene expression. They act via cleavage, destabilization or reduction of translation of mRNAs[70]. Exosomes function as vehicles delivering miRNAs from donating cells to accepting cells in the tumor microenvironment. A total of 22 miRNAs is concentrated in exosomes and correlates to resistance[159]. Dysfunction of miRNAs contributes to therapeutic resistance. For paclitaxel resistance, the following miRNAs are relevant, miR-17-5P for the target PTEN in ovarian cancer[160], miR-30c for the targets VIM, IL-11 in breast cancer[161], miR-125b for Sema4C and Bak1 in breast cancer, and miR-100 for mTOR, miR-145 for P-gp in ovarian cancer[162], and miR-181a for PTEN in NSCLC[163]. For docetaxel resistance, miR-34a for the targets BCL-2 and CCND1 and miR-129-3p for CP100 have been identified. The mechanisms of miRNAs in drug resistance have been ascribed to the alteration of drug transporters leading to efflux of anticancer agents, modification of autophagy/apoptosis to enhance survival, promotion of growth factors to disturb associated signal pathways and activation of the EMT process and augmentation of cancer stem cell populations to promote metastasis[161]. A review on post-transcriptional gene silencing (RNAi) has been published by Naghizadeh[164].
Apoptosis inhibition
The last step in ATA cytotoxicity involves the death of the cancer cell, either by apoptosis, necrosis or autophagy. Apoptosis is controlled by ligands and cell death receptors such as FAS, TNF-R, caspase-3, -6, -7 and -8 and by linker proteins[69]. Within the mitochondria, Bcl-2 and AKT contribute as anti-apoptotic proteins and Bax, Bak and caspase-9 act as pro-apoptotic proteins. Overexpression of Bcl-2 and AKT and underexpression of Bax and Bcl-xl in cancer cells leads to resistance[165]. Likewise, p53 gene mutations can lead to apoptosis blockade[166].
However, according to Murray et al.[167], little consensus has been generated for reported associations between taxane sensitivity and mutated p53, or taxane resistance and overexpression of Bcl-2, Bcl-xL or NFκB. In vitro data support an association of spindle assembly checkpoint (SAC) defects with resistance. Clinical data have been limited and inconsistent. The most prominent finding is that pharmaceutical down-regulation of HER-2 appears to reverse the taxane resistance. This statement was opposed by Wang[53] who published that proteins related to SACs such as survivin and MDA[148,168,169], cell cycle-related proteins such as p53[144,170], membrane receptors such as ErbB2[148,171], and proteins related to apoptosis such as Bcl-2[148,172,173] are all involved in taxane resistance.
Oncoproteins
Sorcin is one of the most highly expressed calcium-binding proteins. It is an oncoprotein, which is co-amplified with the efflux pump, ABCB1, in multidrug-resistant cells[174]. Overexpression of Sorcin is found in many tumors resistant to taxanes and vinca alkaloids and inversely correlates with response and outcome of chemotherapy[175]. By transporting calcium into endoplasmic reticulum (ER) and mitochondria, Sorcin prevents ER stress and increases cell escape from apoptosis[176-178]. In MD-resistant tumor cells overexpressing Sorcin, the equilibrium between cell life and death is shifted towards proliferation. Silencing of Sorcin reverses MDR in many tumor cell lines[175,179-181]. Another interesting aspect of Sorcin is its ability to directly bind drugs like vincristine or paclitaxel and thereby decreasing their effective concentrations in the cancer cell.
Another example for this type of proteins is S phase kinase associated protein 2 (SKP2), which is an oncoprotein and a multicomponent RING-type E3 ligase that degrades many tumor suppressor proteins, such as p27, p16, p21, p57, E2F-1, TOB1, RBL2, cyclin D/E, BRCA2, FOXO1 and RASSF1A[182]. SKP2 is closely linked to paclitaxel sensitivity. High expression levels of SKP2 are a biomarker for poor cancer prognosis.
Other protein systems
The ubiquitin-proteasome system is essential for protein degradation in many processes, including cancer progression and chemoresistance[183]. F-box proteins, which are part of this system, have the ability to control the degradation of several crucial protein targets associated with drug resistance, the dysregulation of which may lead to induction of chemoresistance in cancer cells[182]. Out of the three F-box categories, FBXW, FBXL and FBXO, FBXW7 is a well-established FBXW subfamily protein, which plays a key role as tumor suppressor in the occurrence of many forms of cancers. Loss of FBXW7 leads to increased resistance of colon cancer cells to paclitaxel[184]. FBXW7 primarily exerts its antitumor functions by regulating the degradation of an entire network of proteins, including cyclin, c-Myc, c-Jun, Notch, presenilin, and myeloid cell leukemia-1 (Mcl-1), many of which have oncogenic functions[182]. For example, the inhibition of Mcl-1 restores sensitivity towards paclitaxel and vincristine-induced cell death. Therefore, the downregulation of FBXW7 protein levels may contribute to tumor progression and chemoresistance. Other targets of FBXW7 include multidrug resistance-associated protein, cryptochrome 2 and inhibitor of growth 5. The tumor suppressor gene p53 directly targets FBXW7 and promotes the transcription of FBXW7 mRNA[185]. FBXW7 is also affected by dysregulation of the miRNA pathway, in particular by miR-25 and miR-223[186,187]. Overexpression of miR-223 decreases FBXW7 expression and the sensitivity to chemotherapy.
Mozzetti et al.[188] reported that the Gli family of transcription factors, which are effectors of Hedgehog signaling, are overexpressed in patupilone resistant cells and that treatment with a specific Gli1 inhibitor (GANT58) made cells more susceptible to treatment, partially reversing drug resistance. Gli1 overexpression also led to an increase in hepatocyte growth factor, which is a stimulator of mitogenesis and cell motility and plays a role in angiogenesis and cancerogenicity.
β-Transducin repeats-containing protein (β-TrCP, FBXW1) degrades nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα) and β-catenin[182]. IκB is the inhibitor of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a tumor suppressor and β-catenin is a downstream protein of the Wnt pathways. βTrCP acts as an oncoprotein in colorectal cancer and as a suppressor in gastric cancer. As a last example in this series, FBXO15 knockdown has been reported to cause P-gp accumulation leading to enhanced vincristine resistance[189].
Sphingolipids
The sphingolipid-mediated sphingosine-1-phosphate (S1P) pathway, including sphingolipid metabolites, regulates multiple cellular processes including proliferation, neovascularization, migration, invasion, and metastasis by controlling cell signal transduction networks that contribute to both tumorigenesis and tumor progression[190]. Ceramide, sphingosine, and S1P, which are rapidly interconverted in response to various stimuli, are the core players of this pathway, promoting growth, motility and angiogenesis (ceramide) or mediating anti-proliferative and cytotoxic stress responses including apoptosis, cell cycle arrest, lethal autophagy, and growth suppression (sphingosine, S1P). Chemotherapy or other stress factors increase the levels of ceramide and sphingosine. In normal cells under normal conditions, sphingosine and S1P levels are tenfold lower than the level of ceramide. S1P can be moved out of the cell by active transport and may affect metastasis by inhibiting or enhancing migration and invasion in a cell-type- and concentration-dependent manner. S1P signaling has been identified in the top three most transcriptionally altered pathways following chemotherapy of ovarian cancer[191]. Increased expressions of ceramide transport protein (CERT), SPHK1, SPHK2, and glucosylceramide synthase have been associated with resistance to paclitaxel and apoptotic response[192]. The level of ceramide is lower in ovarian tumor cells than in normal ovarian tissue and is further attenuated in paclitaxel-resistant compared to paclitaxel-sensitive ovarian cancer cells, skewing the ratio of ceramide: S1P in favor of the anti-apoptotic lipid S1P in tumor cells[193]. Therapeutic approaches should aim to promote ceramide accumulation (e.g., via ceramide analogs or mimetics or by co-administration of paclitaxel and ceramide in nanocarriers) to induce apoptosis and suppress S1P accumulation (e.g., by neutralizing antibodies), to inhibit tumor growth and overcome drug resistance.
Cancer stem cells
The last point that I will address is cancer stem cells (CSCs). CSCs were first found in 2003 by Al-Hajj et al.[194] and Singh et al.[195] who identified them using the cell surface markers CD34+/CD38- in breast and brain cancer samples, based on work by Lapidot et al.[196] on acute myeloid leukemia. CSCs are the prototypes of therapy resistant cells. They make up as few as 1% of the total tumor cells, making them difficult to detect and study[197], but could as well reach 25%[198]. On the other hand, as few as 100 cancer stem cells were able to form tumors in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice[194]. The expression of CSC surface markers is tissue type-specific, even tumor subtype-specific[199]. CSCs are defined by their ability to generate more SCs (self-renewal) and to produce cells that differentiate[200]. Asymmetric cell division achieves both tasks, as one progeny retains SC identity and the other undergoes rounds of cell division and subsequent post-mitotic differentiation. Cancer cells gain stem-like characteristics through EMT. EMT is driven by transcription factors, including SNAI1/2, ZEB1/2, or TWIST1/2, which increase the invasiveness of epithelial cells. Further involved pathways include Wnt, Hedgehog, Hippo, LATS 1/2, and miRNA. CSCs reside in niches, which are specialized microenvironments located within each tissue. Since the ABCB1 and ABCG2 efflux transporter genes are expressed in both normal stem cells and most tumor stem cells[201], any therapy that spares normal stem cells will also spare CSCs. Likewise, inhibition of drug transporters may also cause toxicity of the patient’s normal stem cells, particularly those of the bone marrow.
The expression of a major glycosaminoglycan in the extracellular matrix, hyaluronan (HA), and its receptor, CD44, a cell surface marker for both normal and cancer stem cells, are tightly linked to MDR and tumor progression[202]. In breast and ovarian cancer cell lines, HA-CD44 interaction may activate the stem cell marker, Nanog, which can further activate the expression of pluripotent stem cell regulators (Rex1 and Sox2) and Stat-3-mediated ABCB1 gene expression. In addition, HA-CD44 binding may form a complex with ankyrin, the downstream effector of CD44. This complex formation results in an efflux of chemotherapeutic drugs.
In Figure 2, CSCs are characterized as red circles. Most likely, “minimal residual disease” is nothing else than CSCs. CSCs exhibit the capabilities of self-renewal, differentiation and tumorigencity. There are several theories for their formation. One states that CSCs arise from normal stem cells following genetic or environmental alterations, another one proposes the reverse process that normal somatic cells become malignant and stem-cell like via genetic or environmental alterations. The situation very much reminds me of HIV treatment where standard therapy is able to keep HIV to a minimum, including non-detectable RNA blood levels, and as soon as treatment is stopped, dormant HIV cells start replicating and there is a burst of disease. This is the reason that until now, cure of HIV infection has not been achieved. Current HIV drugs only inhibit proliferation of the virus but do not kill infected cells. In a recent study, we were able to show that HIV-infected cells can be killed by an antibody directed against CD52, which is expressed on normal and on HIV-infected cells[203]. If we apply these findings to cancer, CSCs would be “dormant” cancer cells that survive any tumor treatment and when they awake lead to progression and metastases. If that should be the case, then there would be two ways of fighting CSCs, either the use of as yet unknown drugs that are able to address dormant cells, potentially via their specific cell surface markers and using a kill mechanism that applies to dormancy, or by forcing the cells out of their hibernation and thereby induce replication, which should make them sensitive to conventional ATAs.
Summary and outlook
In summary, although the taxanes of the first generation, paclitaxel and docetaxel, were revolutionary new drugs and soon became blockbusters, they still had quite a number of drawbacks, such as: (1) extremely low water solubility, which required the use of solubilizing agents (polyoxyethylated castor oil or polysorbate) leading to hypersensitivity reactions with the need for premedication and even modification of drug pharmacokinetics; (2) poor metabolic stability resulting in highly variable pharmacokinetics and poor oral bioavailability with the consequence of long-lasting intravenous infusions and becoming substrates of P450 enzyme inducers or inhibitors; (3) serious dose-limiting toxicities such as myelosuppression, cumulative peripheral sensory neuropathy, and allergic reactions; and (4) most importantly, development of resistance due to a number of different mechanisms, the foremost being induction of efflux pumps and target modifications, mainly βIII-tubulin overexpression.
The development of novel formulations, such as nab-paclitaxel, somewhat relieved the issue of low aqueous solubility and surprisingly opened up new indications such as pancreatic cancer but did not change the resistance problems. The development of other formulations, e.g., nanocarriers, liposomes, cyclodextrin-containing systems, etc. is ongoing[204,205] and might improve tolerability of the drugs but, again, most probably not resistance. Currently, the development of new nano-formulations is a very active area but drug combinations of taxanes with compounds of different mechanisms of action appear to be the most active field of research in the fight against resistance.
Cabazitaxel is a 2nd generation taxane with improved characteristics in terms of pharmacokinetics and metabolic stability and moving in the direction of eliminating the P-gp efflux pump resistance. More compounds have been in clinical trials[206], such as tesetaxel, larotaxel, ortataxel, and milataxel, which are orally bioavailable, and - some of them - overcome P-gp efflux pumps, representing steps in the right direction. However, except for tesetaxel, the development of the other compounds has been discontinued due to toxicities. Preclinically, more taxane analogs are currently being investigated, but will not be covered in this review.
Whereas for other anticancer drug classes tumor-targeting drug conjugates have become the new hype, for the taxanes, there is only one compound under investigation, ANG-1005, which is a paclitaxel-Angiopep-2 conjugate[207] aiming at brain tumors. ANG1005 crosses the blood-brain barrier and enters the cancer cells via receptor-mediated transcytosis of the low-density lipoprotein receptor-related protein 1 (LRP1), which is upregulated in some cancers. Within the cancer cell, esterases will then cleave the conjugate and release paclitaxel. If the intracellular drug concentrations are high enough, P-gp overexpression might no longer be relevant for this drug, which is currently preparing for Phase III.
Regarding vinca alkaloids, the situation is similar to the taxanes. A liposomal formulation of vincristine has been developed and approved in order to improve the shortcomings of the original drug, such as solubility issues and dose limiting toxicity. A new compound, vinflunine, was approved in 2009. Likewise, the synthesis of novel vinca compounds with the aim of maximizing potency and minimizing resistance is ongoing and looks quite successful.
The epothilones described above might have been abandoned too early. The toxicity problems observed in the clinical development that led to discontinuation may be attributable in part to studying worst-case scenarios in most of the trials, i.e., heavily pretreated patients that no longer responded to any other drugs and which were then moved to monotherapy with epothilones. There is also still insignificant knowledge why the dose-limiting toxicities of the epothilones were so strikingly different, for example diarrhea for patupilone and neurotoxicity for sagopilone.
Another aspect is that at the time of development of the epothilones, knowledge about biomarkers was still limited and could not be used for patient selection. Epothilones overcome both efflux pump-related resistance and tubulin isotype-related resistance, in particular overexpression of βIII-tubulin, which are the two major stumbling blocks for taxanes. If this knowledge would have been available and been applied to patient selection, maybe the epothilones would have taken another course and not been abandoned. Likewise, the combination of epothilones with other anticancer drugs with different mechanisms of action was not sufficiently taken into consideration. In particular, platinum-containing drugs might have been appropriate partners for combination.
Alternative ATA drug classes that are currently under preclinical or clinical investigation include natural compounds isolated from marine sponges or plants. Laulimalide (fijianolide) was originally obtained from the sponge Cacospongia mycofijiensis[208]. It binds at a microtubule site different from that of other ATAs located on the exterior of the microtubules[209][Figure 1]. Laulimalide is an inhibitor of cellular proliferation in cancer cell lines with IC50 values in the low nanomolar range. The drug causes cells to arrest in the prometaphase preventing formation of bipolar spindles and increasing kinetochore tension in preformed spindles[210]. Laulimalide is also active in multi-drug resistant cancer cell lines overexpressing P-gp or presenting mutated tubulin isotypes[211] and is both effective in taxoid-reistant cell lines and synergistic with taxoids[212].
Peloruside A is a macrocyclic compound isolated from the New Zealand marine sponge, Mycalehentscheli. It has antimitotic, microtubule-stabilizing and antineoplastic activity[213]. It appears to share the same binding site as laulimalide on the exterior of the microtubules. Peloruside A may be particularly useful for anti-angiogenesis therapy due to its high efficacy in blocking endothelial cell migration[214].
Discodermolide (XAA296A) was isolated from the deep-sea sponge, Discodermia dissoluta[215]. The compound is one of the most potent natural promoters of tubulin assembly and competes with paclitaxel for binding, but with higher affinity. Due to its improved water solubility, the formulation problems encountered with paclitaxel can be avoided. Discodermolide is effective in paclitaxel- and in epothilone-resistant cell lines[216] and was investigated in a Phase I clinical study showing good tolerance and non-linear pharmacokinetics[217].
Zampanolide was obtained from the marine sponge, Fasciospongia rimosa[218]. The compound competes with paclitaxel for binding to microtubules[219]. However, in contrast to paclitaxel, zampanolide’s binding is covalent at residues N228 and H229. Alkylation of N228 and H229 was also detected in αβ-tubulin dimers[220]. Efforts to generate a zampanolide-resistant cell line were unsuccessful. Zampanolide could be shown to be an effective inhibitor of migration of human umbilical vein endothelial cells and fibroblast cells (D551) and inhibits cell growth in paclitaxel- and epothilone-resistant cells[221].
Taccalonolides are natural, highly oxygenated pentacyclic steroids of tacca plants (Polynesian arrowroot). The first compound was isolated from Tacca leontopetaloides[222]. Meanwhile a large number of taccalonolides have been isolated from different tacca genus, designated A-Y. Bioassay-guided fractionation led to the identification of taccalonolides A and E as microtubule stabilizers. They retain efficacy in cells containing mutations in the paclitaxel binding site as well as those expressing P-gp, the βIII-isotype of tubulin or the MRP7 drug efflux transporter. Since the taccalonolides A and E were unable to interact directly with microtubules or to enhance the polymerization of purified tubulin and their in vivo efficacy was much higher than in vitro, it was proposed that they actually are prodrugs. In contrast, the significantly more potent natural taccalonolide AF and the semi-synthetic taccalonolide AJ directly interact with microtubules via covalent binding[223]. Taccalonolides are able to displace paclitaxel from its binding site indicating that they bind at or near the luminal and/or pore taxane binding site. Their activity is synergistic with paclitaxel and with laulimalide[224,225].
PM050489 and its dechlorinated analog, PM060184, are natural polyketides and were isolated from the Madagascan sponge Lithoplocamialithistoides[226]. They are highly potent microtubule inhibitors that impair mitosis by binding with nanomolar affinity to unassembled αβ-tubulin dimers. PM050489, possibly acting like a hinge at the association interface between tubulin heterodimers, reshapes Mg2+-induced 42 S tubulin double rings into smaller 19 S single rings made of 7 ± 1 αβ-tubulin dimers. PM060184-resistant mutants of Aspergillusnidulans map to β-tubulin Asn100, suggesting a new binding site different from that of vinblastine at the associating β-tubulin end[227]. PM060184 is currently undergoing clinical trials. A Phase I study with monotherapy has been completed, another one in combination with gemcitabine is ongoing and a Phase II monotherapy study is active but not recruiting.
SSE15206 is a synthetic chalcone derivative that binds to the colchicine binding site and induces G2/M mitotic arrest in a time- and dose-dependent manner in various cell systems due to failure of microtubules to polymerize, finally leading to apoptosis via induction of p53[228]. SSE15206 is not a substrate of P-gp efflux pumps and therefore is able to overcome multi-drug resistance. The drug is still under preclinical investigation.
Conclusion
In conclusion, one could speculate what the best steps in avoiding or overcoming cancer resistance might be. An appropriate starting point is the great heterogeneity of tumors within the same class. As a consequence, the “one fits all” approach for cancer treatment should then be considered obsolete. The future needs to and will move to individualized therapies that have to be established following a thorough analysis of the type and status of the tumor and after having evaluated a host of biomarkers. In an ideal case, the treatment of an individual patient will be individually tailored according to the “molecular signature” of the biomarkers covering protein, RNA and DNA markers. Combinations of synergistic drugs or compounds with orthogonal mechanisms of action and sequences of complementary treatments - kill, sensitize, kill - will have to be considered. The problem of combinations is that cell-line models are not very indicative of activity/resistance if the cells have been passaged for some time. Combinations therefore still might have to be tried out in clinical studies, which certainly is limited by time and costs, in particular if more than two drugs are combined. Special care should also be taken in any future treatment considerations to cover any dormant (cancer stem) cells in the treatment schedule. Resistance can only be overcome if treatment uses the same tricks as the cancer cells do, switch to another pathway if the applied path is blocked, which, of course is very cumbersome, since the number of possible pathways is very large. However, since the development of resistance mechanisms will in future be predictable from genomic and proteomic profiles, and since sophisticated methods to measure and then tackle these mechanisms in patients will increasingly be available, the chances to finally overcome resistance are real.
Declarations
Authors’ contributionsKrause W solely contributed to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 2015;16:711-26.
2. Yang CH, Horwitz SB. Taxol®: the first microtubule stabilizing agent. Int J Mol Sci 2017;18:E1733.
3. Lewis SA, Lee MG, Cowan NJ. Five mouse tubulin isotypes and their regulated expression during development. J Cell Biol 1985;101:852-61.
4. Denoulet P, Eddé B, Gros F. Differential expression of several neurospecific beta-tubulin mRNAs in the mouse brain during development. Gene 1986;50:289-97.
5. Yu I, Garnham CP, Roll-Mecak A. Writing and Reading the Tubulin Code. J Biol Chem 2015;290:17163-72.
6. Janke C, Montagnac G. Causes and consequences of microtubule acetylation. Curr Biol 2017;27:R1287-92.
7. Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol 2008;20:71-6.
9. Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 2003;4:938-47.
10. Cambray-Deakin MA, Burgoyne RD. Posttranslational modifications of α-tubulin: acetylated and detyrosinated forms in axons of rat cerebellum. J Cell Biol 1987;104:1569-74.
11. Paturle L, Wehland J, Margolis RL, Job D. Complete separation of tyrosinated, detyrosinated, and nontyrosinatable brain tubulin subpopulations using affinity chromatography. Biochemistry 1989;28:2698-704.
12. Eddé B, Rossier J, Le Caer JP, Desbruyères E, Gros F, et al. Posttranslational glutamylation of alpha-tubulin. Science 1990;247:83-5.
13. Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010;143:564-78.
14. Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc Natl Acad Sci USA 2005;102:7853-8.
15. Ikegami K, Heier RL, Taruishi M, Takagi H, Mukai M, et al. Loss of β-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc Natl Acad Sci USA 2007;104:3213-8.
16. Farache D, Emorine L, Haren L, Merdes A. Assembly and regulation of γ-tubulin complexes. Open Biol 2018;8:170266.
17. Caplow M, Fee L. Dissociation of the tubulin dimer is extremely slow, thermodynamically very unfavorable, and reversible in the absence of an energy source. Mol Biol Cell 2002;13:2120-31.
18. Hiller G, Weber K. Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Cell 1978;14:795-804.
20. Luduena RF. A hypothesis on the origin and evolution of tubulin. Int Rev Cell Mol Biol 2013;302:41-185.
21. Fees CP, Moore JK. Regulation of microtubule dynamic instability by the carboxy-terminal tail of β-tubulin. Life Sci Alliance 2018;1:e201800054.
22. Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents 2002;2:1-17.
23. Inoué S, Sato H. Cell motility by labile association of molecules. The nature of mitotic spindle fibers and their role in chromosome movement. J Gen Physiol 1967;50:259-92.
25. Cassimeris L. Accessory protein regulation of microtubule dynamics throughout the cell cycle. Curr Opin Cell Biol 1999;11:134-41.
26. Kreis T, Vale R. Guidebook to the cytoskeleton and motor proteins. London/New York: Oxford Univ. Press; 1999.
27. Ohi R, Zanic M. Ahead of the curve: new insights into microtubule dynamics. F1000Res 2016;5:314-22.
29. Pepperkok R, Bre MH, Davoust J, Kreis TE. Microtubules are stabilized in confluent epithelial cells but not in fibroblasts. J Cell Biol 1990;111:3003-12.
30. Saxton WM, Stemple DL, Leslie RJ, Salmon ED, Zavortink M, et al. Tubulin dynamics in cultured mammalian cells. J Cell Biol 1984;99:2175-86.
31. Rusan NM, Fagerstrom CJ, Yvon AM, Wadsworth P. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein-α tubulin. Mol Biol Cell 2001;12:971-80.
32. Correia JJ, Lobert S. Physiochemical aspects of tubulin-interacting antimitotic drugs. Current Pharmaceutical Design 2001;7:1213-28.
33. Visconti R, Grieco D. Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocr Relat Cancer 2017;24:T107-17.
34. Tame MA, Manjón AG, Belokhvostova D, Raaijmakers JA, Medema RH. TUBB3 overexpression has a negligible effect on the sensitivity to taxol in cultured cell lines. Oncotarget 2017;8:71536-47.
35. Alkadi H, Khubeiz MJ, Jbeily R. Colchicine: a review on chemical structure and clinical usage. Infect Disord Drug Targets 2018;18:105-21.
36. Rao CV, Kurkjian CD, Yamada HY. Mitosis-targeting natural compounds for cancer prevention and therapy. Curr Drug Targets 2012;13:1820-30.
37. Downing KH, Nogales E. New insights into microtubule structure and function from the atomic model of tubulin. Eur Biophys J 1998;27:431-6.
38. Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010;9:790-803.
40. Downing KH. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu Rev Cell Dev Biol 2000;16:89-111.
41. Silverman JA, Deitcher SR. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol 2013;71:555-64.
42. Silvestri R. New prospects for vinblastine analogues as anticancer agents. J Med Chem 2013;56:625-7.
43. Carlson RO. New tubulin targeting agents currently in clinical development. Expert Opin Invest Drugs 2008;17:707-22.
44. Blume E. Investigators Seek to Increase Taxol Supply. J National Cancer Institute 1989;81:1122-3.
46. Diaz JF, Andreu JM. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry 1993;32:2747-55.
47. Manfredi JJ, Parness J, Horwitz SB. Taxol binds to cellular microtubules. J Cell Biol 1982;94:688-96.
48. Abal M, Andreu JM, Barasoain I. Taxanes: microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr Cancer Drug Targets 2003;3:193-203.
49. Field JJ, Pera B, Calvo E, Canales A, Zurwerra D, et al. Zampanolide, a potent new microtubule-stabilizing agent, covalently reacts with the taxane luminal site in tubulin α,β-heterodimers and microtubules. Chem Biol 2012;19:686-98.
50. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature 1979;277:665-7.
51. Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of taxol resistance related to microtubules. Oncogene 2003;22:7280-95.
52. Diaz JF, Valpuesta JM, Chacon P, Diakun G, Andreu JM. Changes in microtubule protofilament number induced by Taxol binding to an easily accessible site. Internal microtubule dynamics. J Biol Chem 1998;273:33803-10.
54. Ferlini C, Cicchillitti L, Raspaglio G, Bartollino S, Cimitan S, et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res 2009;69:6906-14.
55. Ganguly A, Yang H, Cabral F. Paclitaxel dependent cell lines reveal a novel drug activity. Mol Cancer Ther 2010;9:2914-23.
56. Komlodi-Pasztor E, Sackett D, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 2012;18:51-63.
57. Bogdan C, Ding A. Taxol, a microtubule-stabilizing antineoplastic agent, induces expression of tumor necrosis factor alpha and interleukin-1 in macrophages. J Leukoc Biol 1992;52:119-21.
58. Bocci G, Di Paolo A, Danesi R. The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 2013;16:481-92.
61. Höfle G, Bedorf N, Steinmetz H, Schemburg D, Gerth K, et al. Epothilone A and B - novel 16-membered macrolides and cytotoxic activity: isolation, crystal structure, and conformation in solution. Angew Chem Int Ed Engl 1996;35:1567-9.
62. Krause W, Klar U. Differences and similarities of epothilones. Current Cancer Therapy Reviews 2011;7:10-36.
63. Winsel S, Berlin A. .
64. Klar U, Hoffmann J, Giurescu M. Sagopilone (ZK-EPO): from a natural product to a fully synthetic clinical development candidate. Expert Opin Investig Drugs 2008;17:1735-48.
65. Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, et al. The binding mode of epothilone A on α, β-tubulin by electron crystallography. Science 2004;305:866-9.
66. Bode CJ, Gupta ML, Reiff EA, Suprenant KA, Georg GI, et al. Epothilone and paclitaxel: unexpected differences in promoting the assembly and stabilization of yeast microtubules. Biochemistry 2002;41:3870-4.
67. Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer 2013;2013:137414.
68. Cortes J, Vidal M. Beyond taxanes: the next generation of microtubule-targeting Agents. Breast Cancer Res Treat 2012;133:821-30.
69. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull 2017;7:339-48.
70. Mansoori B, Mohammadi A, Shirjang S, Baradaran B. MicroRNAs in the Diagnosis and Treatment of Cancer. Immunol Invest 2017;46:880-97.
72. Cree IA, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017;17:10-8.
73. Di Nicolantonio F, Mercer SJ, Knight LA, Gabriel FG, Whitehouse PA, et al. Cancer cell adaptation to chemotherapy. BMC Cancer 2005;5:78-94.
74. BC Cancer. Available from: http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Vinblastine_monograph_1Feb2015.pdf. [Last accessed on 1 Mar 2019].
75. BC Cancer. Available from: http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Vincristine_monograph_1Mar08.pdf. [Last accessed on 1 Mar 2019].
76. Vindesine. Available from: https://hemonc.org/w/images/3/33/Vindesine.pdf. [Last accessed on 26 Feb 2019].
77. BC Cancer. Available from: http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Vinorelbine%20monograph_1Feb2015.pdf. [Last accessed on 1 Mar 2019].
78. AusPAR: Vinflunine ditartrate. Australian Public Assessment Report, 2011. Available from: https://www.tga.gov.au/auspar/auspar-vinflunine-ditartrate. [Last accessed on 26 Feb 2019].
79. FDA Label. Available from: https://www.accessdata.fda.gov/drugsatfda/label. [Last accessed on 28 Feb 2019].
80. Kelly KR, Zollinger M, Lozac’h F, Tan E, Mita A, et al. Metabolism of patupilone in patients with advanced solid tumor malignancies. Invest New Drugs 2013;31:605-15.
81. European Medicines Agency. Withdrawal assessment report for Ixempra. Available from: https://www.ema.europa.eu/documents/withdrawal-report/withdrawal-assessment-report-ixempra_en.pdf. [Last accessed on 28 Feb 2019].
82. Konner J, Grisham RN, Park J, O’Connor OA, Cropp G, et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of KOS-862 (Epothilone D) in patients with advanced solid tumors and lymphoma. Invest New Drugs 2012;30:2294-302.
83. Lam ET, Goel S, Schaaf LJ, Cropp GF, Hannah AL, et al. Phase I dose escalation study of KOS 1584, a novel epothilone, in patients with advanced solid tumors. Cancer Chemother Pharmacol 2012;69:523-31.
84. Xie HG, Wood AJ, Kim RB, Stein CM, Wilkinson GR. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics 2004;5:243-72.
85. Silverman JA, Reynolds L, Deitcher SR. Pharmacokinetics and pharmacodynamics of vincristine sulfate liposome injection (VSLI) in adults with acute lymphoblastic leukemia. J Clin Pharmacol 2013;53:1139-45.
86. Ohishi Y, Oda Y, Basaki Y, Kobayashi H, Wake N, et al. Expression of β-tubulin isotypes in human primary ovarian carcinoma. Gynecol Oncol 2007;105:586-92.
87. Izutsu N, Maesawa C, Shibazaki M, Oikawa H, Shoji T, et al. Epigenetic modification is involved in aberrant expression of class III β-tubulin, TUBB3, in ovarian cancer cells. Int J Oncol 2008;32:1227-35.
88. Du J, Li B, Fang Y, Liu Y, Wang Y, et al. Overexpression of class III beta-tubulin, sox2, and nuclear survivin is predictive of taxane resistance in patients with stage III ovarian epithelial cancer. BMC Cancer 2015;15:536-47.
89. Roque DM, Bellone S, English DP, Buza N, Cocco E, et al. Tubulin-β-III overexpression by uterine serous carcinomas is a marker for poor overall survival after platinum/taxane chemotherapy and sensitivity to epothilones. Cancer 2013;119:2582-92.
90. Ullah MF, Aatif M. The footprints of cancer development: cancer biomarkers. Cancer Treat Rev 2009;35:193-200.
91. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48-58.
92. Safa AR. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr Med Chem Anticancer Agents 2004;4:1-17.
93. Manzoor S, Bilal A, Khan S, Ullah R, Iftikhar S, et al. Identification and characterization of SSE15206, a microtubule depolymerizing agent that overcomes multidrug resistance. Sci Rep 2018;8:3305.
94. Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol 2007;18:3-8.
95. Su DS, Balog A, Meng D, Bertinato P, Danishefsky SJ, et al. Structure-activity Relationships of the epothilones and the first in vivo comparison with paclitaxel. Angew Chem Int Ed Engl 1997;36:2093-6.
96. Chou TC, O’Connor OA, Tong WP, Guan Y, Zhang ZG, et al. The synthesis, discovery, and development of a highly promising class of microtubule stabilization agents: Curative effects of desoxyepothilones B and F against human tumor xenografts in nude mice. Proc Natl Acad Sci USA 2001;98:8113-8.
97. Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, et al. Taxol: mechanisms of action and resistance. J Natl Cancer Inst Monogr 1993:55-61.
98. Jachez B, Nordmann R, Loor F. Restoration of taxol sensitivity of multidrug-resistant cells by the cyclosporine SDZ PSC 833 and the cyclopeptolide SDZ 280-446. J Natl Cancer Inst 1993;85:478-83.
99. Grogan TM, Spier CM, Salmon SE, Matzner M, Rybski J, et al. P-glycoprotein expression in human plasma cell myeloma: correlation with prior chemotherapy. Blood 1993;81:490-5.
100. Dalton WS, Crowley JJ, Salmon SS, Grogan TM, Laufman LR, et al. A phase III randomized study of oral verapamil as a chemosensitizer to reverse drug resistance in patients with refractory myeloma. A Southwest Oncology Group study. Cancer 1995;75:815-20.
101. Koushik OS, Rao YV, Kumar P, Karthikeyan R. Nano drug delivery systems to overcome cancer drug resistance - a review. J Nanomed Nanotechnol 2016;7:378.
102. Cisternino S, Bourasset F, Archimbaud Y, Semiond D, Sanderink G, et al. Nonlinear accumulation in the brain of the new taxoid TXD258 following saturation of P-glycoprotein at the blood-brain barrier in mice and rats. Brit J Pharmacol 2003;138:1367-75.
103. Hopper-Borge E, Xu X, Shen T, Shi Z, Chen ZS, et al. Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res 2009;69:178-84.
104. Lin JH, Yamazaki M. Clinical relevance of P-Glycoprotein in drug therapy. Drug Metab Rev 2003;35:417-54.
105. Guo HQ, Zhang GN, Wang YJ, Zhang YK, Sodani K, et al. β-Elemene, a compound derived from Rhizoma zedoariae, reverses multidrug resistance mediated by theABCB1 transporter. Oncology Reports 2014;31:858-66.
106. Loe DW, Almquist KC, Deeley RG, Cole SP. Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J Biol Chem 1996;271:9675-82.
107. Kitazono M, Sumizawa T, Takebayashi Y, Chen ZS, Furukawa T, et al. Multidrug resistance and the lung resistance-related protein in human colon carcinoma SW-620 cells. J Nat Cancer Inst 1999;91:1647-53.
108. Izquierdo MA, van der Zee AG, Vermorken J, van der Valk P, Belien JA, et al. Drug resistance-associated marker Lrp for prediction of response to chemotherapy and prognoses in advanced ovarian carcinoma. J Natl Cancer Inst 1995;87:1230-6.
109. Parker AL, Teo WS, McCarroll JA, Kavallaris M. An emerging role for tubulin isotypes in modulating cancer biology and chemotherapy resistance. Int J Mol Sci 2017;18:1434-58.
110. Vilmar A, Garcia-Foncillas J, Huarriz M, Santoni-Rugiu E, Sorensen JB. RT-PCR versus immunohistochemistry for correlation and quantification of ERCC1, BRCA1, TUBB3 and RRM1 in NSCLC. Lung Cancer 2012;75:306-12.
111. Iseri OD, Kars MD, Gunduz U. Drug resistant MCF-7 cells have altered expression levels of beta-tubulin isotypes and mutations in TUBB gene. Int J Hematol Oncol 2010;2:75-83.
112. Freedman H, Huzil JT, Luchko T, Luduena RF, Tuszynski JA. Identification and characterization of an intermediate taxol binding site within microtubule nanopores and a mechanism for tubulin isotype binding selectivity. J Chem Inf Model 2009;49:424-36.
113. Kavallaris M, Kuo DYS, Burkhart CA, Regl DL, Norris MD, et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific β-tubulin isotypes. J Clin Invest 1997;100:1282-93.
114. Mozzetti S, Ferlini C, Concolino P, Filippetti F, Raspaglio G, et al. Class III β-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res 2005;11:298-305.
115. Ferrandina G, Zannoni GF, Martinelli E, Paglia A, Gallotta V, et al. Class III beta-tubulin overexpression is a marker of poor clinical outcome in advanced ovarian cancer patients. Clin Cancer Res 2006;12:2774-9.
116. Su D, Smith SM, Preti M, Schwartz P, Rutherford TJ, et al. Stathmin and tubulin expression and survival of ovarian cancer patients receiving platinum treatment with and without paclitaxel. Cancer 2009;115:2453-63.
117. De Donato M, Mariani M, Petrella L, Martinelli E, Zannoni GF, et al. Class III β-tubulin and the cytoskeletal gateway for drug resistance in ovarian cancer. J Cell Physiol 2012;227:1034-41.
118. Roque DM, Buza N, Glasgow M, Bellone S, Bortolomai I, et al. Class III β-tubulin overexpression within the tumor microenvironment is a prognostic biomarker for poor overall survival in ovarian cancer patients treated with neoadjuvant carboplatin/paclitaxel. Clin Exp Metastasis 2014;31:101-10.
119. Parker AL, Turner N, McCarroll JA, Kavallaris M. βIII-Tubulin alters glucose metabolism and stress response signaling to promote cell survival and proliferation in glucose-starved non-small cell lung cancer cells. Carcinogenesis 2016;37:787-98.
120. Raspaglio G, Filippetti F, Prislei S, Penci R, De Maria I, et al. Hypoxia induces class III β-tubulin gene expression by HIF-1α binding to its 3′ flanking region. Gene 2008;409:100-8.
121. Bordji K, Grandval A, Cuhna-Alves L, Lechapt-Zalcman E, Bernaudin M. Hypoxia-inducible factor-2 (HIF-2), but not HIF-1, is essential for hypoxic induction of class III β-tubulin expression in human glioblastoma cells. FEBS J 2014;281:5220-36.
122. Raspaglio G, De Maria I, Filippetti F, Martinelli E, Zannoni GF, et al. Hur regulates β-tubulin isotype expression in ovarian cancer. Cancer Res 2010;70:5891-900.
123. Patel VP, Chu CT. Decreased SIRT2 activity leads to altered microtubule dynamics in oxidatively-stressed neuronal cells: Implications for Parkinson’s disease. Exp Neurol 2014;257:170-81.
124. Drum BML, Yuan C, Li L, Liu QH, Wordeman L, et al. Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J Mol Cell Cardiol 2016;93:32-43.
125. Azuma K, Sasada T, Kawahara A, Takamori S, Hattori S, et al. Expression of ERCC1 and ulin in non-small cell lung cancer patients treated with carboplatin and paclitaxel. Lung Cancer 2009;64:326-33.
126. Karki R, Ferlini C. Class III beta-tubulin, drug resistance, therapeutic approaches in cancers. Atlas Genet Cytogenet Oncol Haematol 2014;18:865-71.
127. Aoki D, Oda Y, Hattori S, Taguchi K, Ohishi Y, et al. Overexpression of class III β-tubulin predicts good response to taxane-based chemotherapy in ovarian clear cell adenocarcinoma. Clin Cancer Res 2009;15:1473-80.
128. Wang Y, Sparano JA, Fineberg S, Stead L, Sunkara J, et al. High expression of class III beta-tubulin predicts good response to neoadjuvant taxane and doxorubicin/cyclophosphamide-based chemotherapy in estrogen receptor-negative breast cancer. Clin Breast Cancer 2013;13:103-8.
129. Mozzetti S, Iantomasi R, de Maria I, Prislei S, Mariani M, et al. Molecular mechanisms of patupilone resistance. Cancer Res 2008;68:10197-204.
130. Magnani M, Ortuso F, Soro S, Alcaro S, Tramontano A, et al. The betaI/betaIII-tubulin isoforms and their complexes with antimitotic agents. Docking and molecular dynamics studies. FEBS J 2006;273:3301-10.
131. Ferlini C, Raspaglio G, Mozzetti S, Cicchillitti L, Filippetti F, et al. The seco-taxane IDN5390 is able to target class III beta-tubulin and to overcome paclitaxel resistance. Cancer Res 2005;65:2397-405.
132. Ferrandina G, Mariani M, Andreoli M, Shahabi S, Scambia G, et al. Novel Drugs Targeting Microtubules: the Role of Epothilones. Curr Pharm Des 2012;18:2793-803.
133. Kavallaris M, Burkhardt CA, Horwitz SB. Antisense oligonucleotides to class III β-tubulin sensitize drug-resistant cells to Taxol. Br J Cancer 1999;80:1020-5.
134. Miyamoto DT, Perlman ZE, Mitchison TJ, Shirasu-Hiza M. Dynamics of the mitotic spindle - potential therapeutic targets. Prog Cell Cycle Res 2003;5:349-60.
135. Kanakkanthara A, Northcote PT, Miller JH. βII-tubulin and βIII-tubulin mediate sensitivity to peloruside A and laulimalide, but not paclitaxel or vinblastine, in human ovarian carcinoma cells. Mol. Cancer Ther 2012;11:393-404.
136. Gan PP, McCarroll JA, Po’uha ST, Kamath K, Jordan MA, et al. Microtubule dynamics, mitotic arrest, and apoptosis: drug-induced differential effects of betaIII-tubulin. Mol Cancer Ther 2010;9:1339-48.
137. Gan PP, Kavallaris M. Tubulin-targeted drug action: functional significance of class ii and class IVb beta-tubulin in vinca alkaloid sensitivity. Cancer Res 2008;68:9817-24.
138. Sharbeen G, McCarroll J, Liu J, Youkhana J, Limbri LF, et al. Delineating the role of βIV-tubulins in pancreatic cancer: βIVb-tubulin inhibition sensitizes pancreatic cancer cells to vinca alkaloids. Neoplasia 2016;18:753-64.
139. Narvi E, Jaakkola K, Winsel S, Oetken-Lindholm C, Halonen P, et al. Altered TUBB3 expression contributes to the epothilone response of mitotic cells. Br J Cancer 2013;108:82-90.
140. Bhattacharya R, Cabral F. A ubiquitous beta-tubulin disrupts microtubule assembly and inhibits cell proliferation. Mol Biol Cell 2004;15:3123-31.
141. Hari M, Yang H, Zeng C, Canizales M, Cabral F. Expression of class III β-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil Cytoskelet 2003;56:45-56.
142. Blade K, Menick DR, Cabral F. Overexpression of class I, II or IVb β-tubulin isotypes in CHO cells is insufficient to confer resistance to paclitaxel. J Cell Sci 1999;112:2213-21.
143. Ranganathan S, McCauley RA, Dexter DW, Hudes GR. Modulation of endogenous beta-tubulin isotype expression as a result of human beta(III)cDNA transfection into prostate carcinoma cells. Br J Cancer 2001;85:735-40.
144. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004;4:253-65.
145. Tsourlakis MC, Weigand P, Grupp K, Kluth M, Steurer S, et al. βIII-tubulin overexpression is an independent predictor of prostate cancer progression tightly linked to ERG fusion status and PTEN deletion. Am J Pathol 2014;184:609-17.
146. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871-90.
147. Sobierajska K, Wieczorek K, Ciszewski WM, Sacewicz-Hofman I, Wawro ME, et al. β-III tubulin modulates the behavior of snail overexpressed during the epithelial-to-mesenchymal transition in colon cancer cells. Biochim Biophys Acta 2016;1863:2221-33.
148. McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta 2008;1785:96-132.
149. Kavallaris M. Microtubules and resistance to tubulin- tubulin-binding agents. Nat Rev Cancer 2010;10:194-204.
150. Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, et al. Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci USA 2005;102:8315-20.
151. Cabral FR, Brady RC, Schibler MJ. A mechanism of cellular resistance to drugs that interfere with microtubule assembly. Ann NY Acad Sci 1986;466:745-56.
152. Cabral F, Barlow SB. Mechanisms by which mammalian cells acquire resistance to drugs that affect microtubule assembly. FASEB J 1989;3:1593-9.
153. Minotti AM, Barlow SB, Cabral F. Resistance to antimitotic drugs in Chinese hamster ovary cells correlated with changes in the level of polymerized tubulin. J Biol Chem 1991;266:3987-94.
154. Derry WB, Wilson L, Jordan MA. Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry 1995;34:2203-11.
155. Wilson L, Jordan MA. Microtubule dynamics: taking aim at a moving target. Chem Biol 1995;2:569-73.
156. Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol 1998;10:123-30.
157. Goncalves A, Braguer D, Kamath K, Martello L, Briand C, et al. Resistance to taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci USA 2001;98:11737-41.
158. Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, et al. Paclitaxel resistant human ovarian cancer cells have mutant beta- tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 1997;272:17118-25.
159. Zhong S, Chen X, Wang D, Zhang X, Shen H, et al. MicroRNA expression profiles of drug-resistance breast cancer cells and their exosomes. Oncotarget 2016;7:19601-9.
160. Fang Y, Xu C, Fu Y. MicroRNA-17-5p induces drug resistance and invasion of ovarian carcinoma cells by targeting PTEN signaling. J Biol Res (Thessalon) 2015;22:12.
161. Hu W, Tan C, He Y, Zhang G, Xu Y, et al. Functional miRNAs in breast cancer drug resistance. Onco Targets Ther 2018;11:1529-41.
162. Zhu X, Li Y, Xie C, Yin X, Liu Y, et al. miR-145 sensitizes ovarian cancer cells to paclitaxel by targeting Sp1 and Cdk6. Int J Cancer 2014;135:1286-96.
163. Li H, Zhang P, Sun X, Sun Y, Shi C, et al. MicroRNA-181a regulates epithelial-mesenchymal transition by targeting PTEN in drug-resistant lung adenocarcinoma cells. Int J Oncol 2015;47:1379-92.
164. Naghizadeh S, Mansoori B, Mohammadi A, Sakhinia E, Baradaran B. Gene silencing strategies in cancer therapy: an update for drug resistance. Curr Med Chem 2018; doi: 10.2174/0929867325666180403141554.
166. de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA 1998;95:282-7.
167. Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat Rev 2012;38:890-903.
168. Kienitz A, Vogel C, Morales I, Muller R, Bastians H. Partial downregulation of MAD1 causes spindle checkpoint inactivation and aneuploidy, but does not confer resistance towards taxol. Oncogene 2005;24:4301-10.
169. Carvalho A, Carmena M, Sambade C, Earnshaw WC, Wheatley SP. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J Cell Sci 2003;116:2987-98.
170. Chen X, Yeung T, Wang W. Enhanced drug resistance in cells coexpressing ErbB2 with EGF receptor or ErbB3. Biochem Biophys Res Commun 2000;277:757-63.
171. Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, et al. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med 1996;2:72-9.
172. Shitashige M, Toi M, Yano T, Shibata M, Matsuo Y, et al. Dissociation of Bax from a Bcl-2/Bax heterodimer triggered by phosphorylation of serine 70 of Bcl-2. J Biochem 2001;130:741-8.
174. Genovese I, Ilari A, Battista T, Chiarini V, Fazi F, et al. Molecular bases of Sorcin-dependent resistance to chemotherapeutic agents. Cancer Drug Resists 2018;1:164-80.
175. Hu Y, Li S, Yang M, Yan C, Fan D, et al. Sorcin silencing inhibits epithelial-to-mesenchymal transition and suppresses breast cancer metastasis in vivo. Breast Cancer Res Treat 2014;143:287-99.
176. Lalioti VS, Ilari A, O’Connell DJ, Poser E, Sandoval IV, et al. Sorcin links calcium signaling to vesicle trafficking, regulates Pololike kinase 1 and is necessary for mitosis. PLoS One 2014;9:e85438.
177. Maddalena F, Sisinni L, Lettini G, Condelli V, Matassa DS, et al. Resistance to paclitxel in breast carcinoma cells requires a quality control of mitochondrial antiapoptotic proteins by TRAP1. Mol Oncol 2013;7:895-906.
178. Maddalena F, Laudiero G, Piscazzi A, Secondo A, Scorziello A, et al. Sorcin induces a drug-resistant phenotype in human colorectal cancer by modulating Ca(2+) homeostasis. Cancer Res 2011;71:7659-69.
179. Colotti G, Poser E, Fiorillo A, Genovese I, Chiarini V, et al. Sorcin, a calcium binding protein involved in the multidrug resistance mechanisms in cancer cells. Molecules 2014;19:13976-89.
180. Gao Y, Li W, Liu X, Gao F, Zhao X. Reversing effect and mechanism of soluble resistance-related calcium-binding protein on multidrug resistance in human lung cancer A549/DDP cells. Mol Med Rep 2015;11:2118-24.
181. Hu Y, Cheng X, Li S, Zhou Y, Wang J, et al. Inhibition of sorcin reverses multidrug resistance of K562/A02 cells and MCF-7/A02 cells via regulating apoptosis-related proteins. Cancer Chemother Pharmacol 2013;72:789-98.
182. Gong J, Zhou Y, Liu D, Huo J. F-box proteins involved in cancer-associated drug resistance (Review). Oncol Lett 2018;15:8891-900.
183. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008;112:1415-24.
184. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011;471:110-4.
185. Yokobori T, Mimori K, Iwatsuki M, Ishii H, Onoyama I, et al. p53-altered FBXW7 expression determines poor prognosis in gastric cancer cases. Cancer Res 2009;69:3788-94.
186. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Molecular Cancer 2018;17:115.
187. Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. J Biol Chem 2010;285:34439-46.
188. Mozzetti S, Martinelli E, Raspaglio G, Prislei S, De Donato M, et al. Gli family transcription factors are drivers of patupilone resistance inovarian cancer. Biochem Pharmacol 2012;84:1409-18.
189. Katayama K, Noguchi K, Sugimoto Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin-proteasome pathway in cancer cells. Cancer Sci 2013;104:694-702.
190. Kreitzburg KM, van Waardenburg RCAM, Yoon KJ. Sphingolipid metabolism and drug resistance in ovarian cancer. Cancer Drug Resist 2018;1:181-97.
191. Dobbin ZC, Katre AA, Steg AD, Erickson BK, Shah MM, et al. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014;5:8750-64.
192. Swanton C, Marani M, Pardo O, Warne PH, Kelly G, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007;11:498-512.
193. Prinetti A, Millimaggi D, D’Ascenzo S, Clarkson M, Bettiga A, et al. Lack of ceramide generation and altered sphingolipid composition are associated with drug resistance in human ovarian carcinoma cells. Biochem J 2006;395:311-8.
194. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. PNAS 2003;100:3983-8.
195. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821-8.
196. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645-8.
197. Liu FS. Mechanisms of chemotherapeutic drug resistance in cancer therapy - a quick review. Taiwan J Obstet Gynecol 2009;48:239-44.
198. Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science 2007;317:37.
199. Mansoori M, Madjd Z, Janani L, Rasti A. Circulating cancer stem cell markers in breast carcinomas: a systematic review protocol. Syst Rev 2017;6:262.
200. Yu Z, Pestellc TG, Lisantic MP, Pestellb RG. Cancer stem cells. Int J Biochem Cell Biol 2012;44:2144-51.
201. Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene 2007;26:1357-60.
202. Bourguignon LYW, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker, Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem 2008;283:17635-51.
203. Ruxrungtham K, Sirivichayakul S, Buranapraditkun S, Krause W. Alemtuzumab-induced elimination of HIV-1-infected immune cells. J Virus Erad 2016;2:12-8.
204. Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol 2006;17:735-49.
205. Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. Nanomed Nanotechnol 2013;4:1000164.
206. Ojima I, Lichtenthal B, Lee S, Wang C, Wang X. Taxane anticancer agents: a patent perspective. Expert Opin Ther Pat 2016;26:1-20.
207. Régina A, Demeule M, Ché C, Lavallée I, Poirier J, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol 2008;155:185-97.
208. Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res 1999;59:653-60.
209. Churchill CDM, Klobukowski M, Tuscynski JA. Analysis of the binding mode of laulimalide to microtubules: establishing a laulimalide-tubulin pharmacophore. J Biomol Struct Dyn 2015;34:1-35.
210. Bennett MJ, Chan GK, Rattner JB, David C, Schriemer DC. Low-dose laulimalide represents a novel molecular probe for investigating microtubule organization. Cell Cycle 2012;11:3045-54.
211. Pryor DE, O’Brate A, Bilcer G, Díaz JF, Wang Y, et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002;41:9109-15.
212. Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, et al. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol 2006;70:1555-64.
213. Kanakkanthara A, Northcote PT, Miller JH. Peloruside A: a lead non-taxoid-site microtubule-stabilizing agent with potential activity against cancer, neurodegeneration, and autoimmune disease. Nat Prod Rep 2016;33:549-61.
214. Ganguly A, Cabral F, Yang H, Patel KD. Peloruside A is a microtubule-stabilizing agent with exceptional anti-migratory properties in human endothelial cells. Oncoscience 2015;2:585-95.
215. Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. Discodermolide: a new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J Org Chem 1990;55:4912-5.
216. Martello LA, McDaid HM, Regl DL, Yang CPH, Meng D, et al. Taxol and discodermolide represent a synergistic drug combination in human carcinoma cell lines. Clin Cancer Res 2000;6:1978-87.
217. Mita A, Lockhart AC, Chen TL, Bochinski K, Curtright J, et al. A phase I pharmacokinetic (PK) trial of XAA296A (Discodermolide) administered every 3 wks to adult patients with advanced solid malignancies. J Clin Oncol 2004;22:2025.
218. Tanaka J, Higa T. Zampanolide, a new cytotoxic macrolide from a marine sponge. Tetrahedron Lett 1996;37:5535-8.
219. Field JJ, Pera B, Calvo E, Canales A, Zurwerra D, et al. Zampanolide, a potent new microtubule stabilizing agent, covalently reacts with the taxane luminal site in both tubulin α,β-heterodimers and microtubules. Chem Biol 2012;19:686-98.
220. Chen QH, Kingston DG. Zampanolide and dactylolide: cytotoxic tubulin-assembly agents and promising anticancer leads. Nat Prod Rep 2014;31:1202-26.
221. Field JJ, Northcote PT, Paterson I, Altmann KH, Díaz JF, et al. Zampanolide, a microtubule-stabilizing agent, is active in resistant cancer cells and inhibits cell migration. Int J Mol Sci 2017;18:E971.
222. Scheuer PJ, Swanholm CE, Madamba LA, Hudgins WR. The constituents of Tacca leontopetaloides. Lloydia 1963;26:133-40.
223. Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, et al. Taccalonolides E and A: plant-derived steroids with microtubule-stabilizing activity. Cancer Res 2003;63:3211-20.
224. Lia J, Risinger AL, Mooberry SL. Taccalonolide microtubule stabilizers. Bioorg Med Chem 2014;22:5091-6.
225. Wang Y, Yu Y, Li GB, Li SA, Wu C, et al. Mechanism of microtubule stabilization by taccalonolide AJ. Nat Commun 2017;8:15787.
226. Martín MJ, Coello L, Fernández R, Reyes F, Rodríguez A, et al. Isolation and first total synthesis of PM050489 and PM060184, two new marine anticancer compounds. J Am Chem Soc 2013;135:10164-71.
227. Pera B, Barasoain I, Pantazopoulou A, Canales A, Matesanz R, et al. New interfacial microtubule inhibitors of marine origin, PM050489/PM060184, with potent antitumor activity and a distinct mechanism. ACS Chem Biol 2013;8:2084-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Krause W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist 2019;2:82-106. http://dx.doi.org/10.20517/cdr.2019.06
AMA Style
Krause W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resistance. 2019; 2(1): 82-106. http://dx.doi.org/10.20517/cdr.2019.06
Chicago/Turabian Style
Krause, Werner. 2019. "Resistance to anti-tubulin agents: From vinca alkaloids to epothilones" Cancer Drug Resistance. 2, no.1: 82-106. http://dx.doi.org/10.20517/cdr.2019.06
ACS Style
Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 82-106. http://dx.doi.org/10.20517/cdr.2019.06
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 67 clicks
Cite This Article 67 clicks

 Like This Article 7
likes
Like This Article 7
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.